Structural Analysis of Hypothetical Proteins from Helicobacter pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins

Abstract
:1. H. pylori as a Pathogen
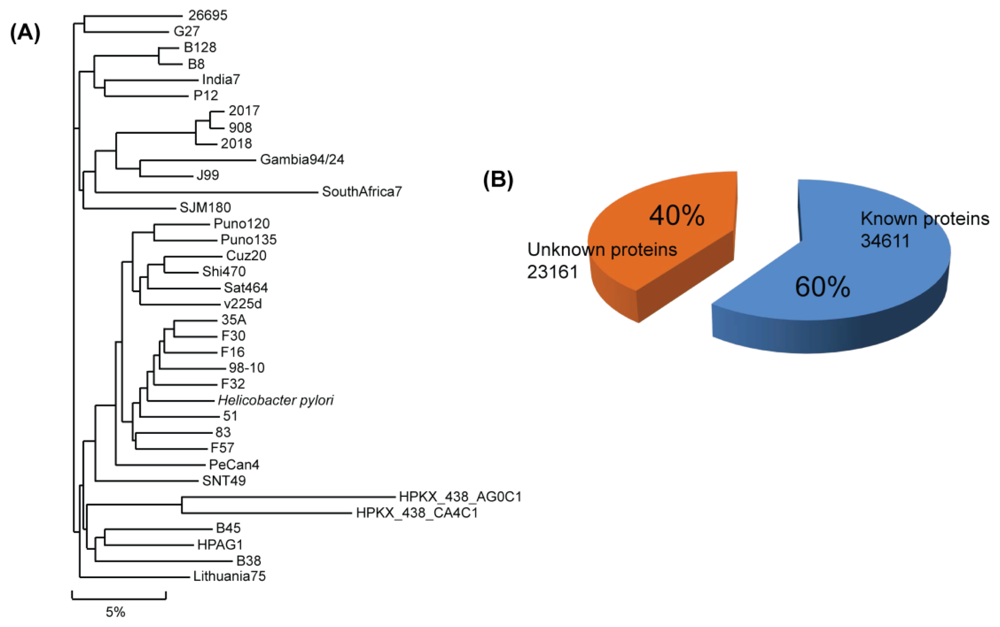
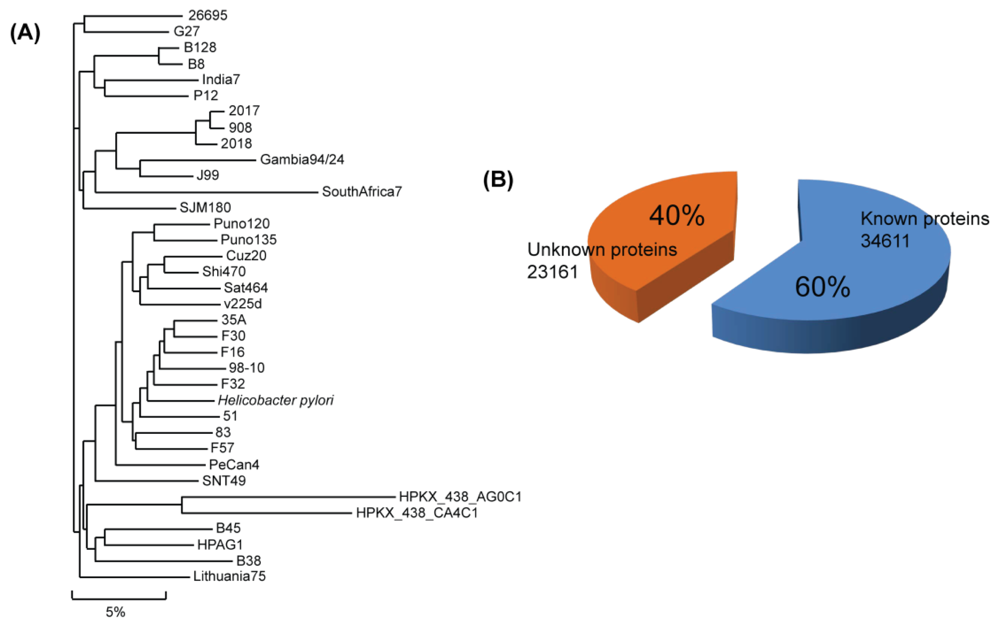
2. H. pylori Genomic Sequence
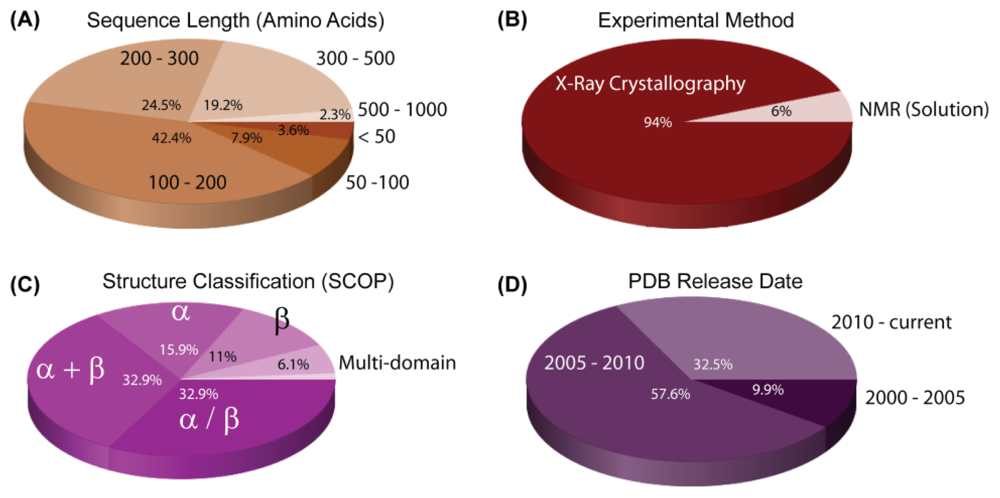
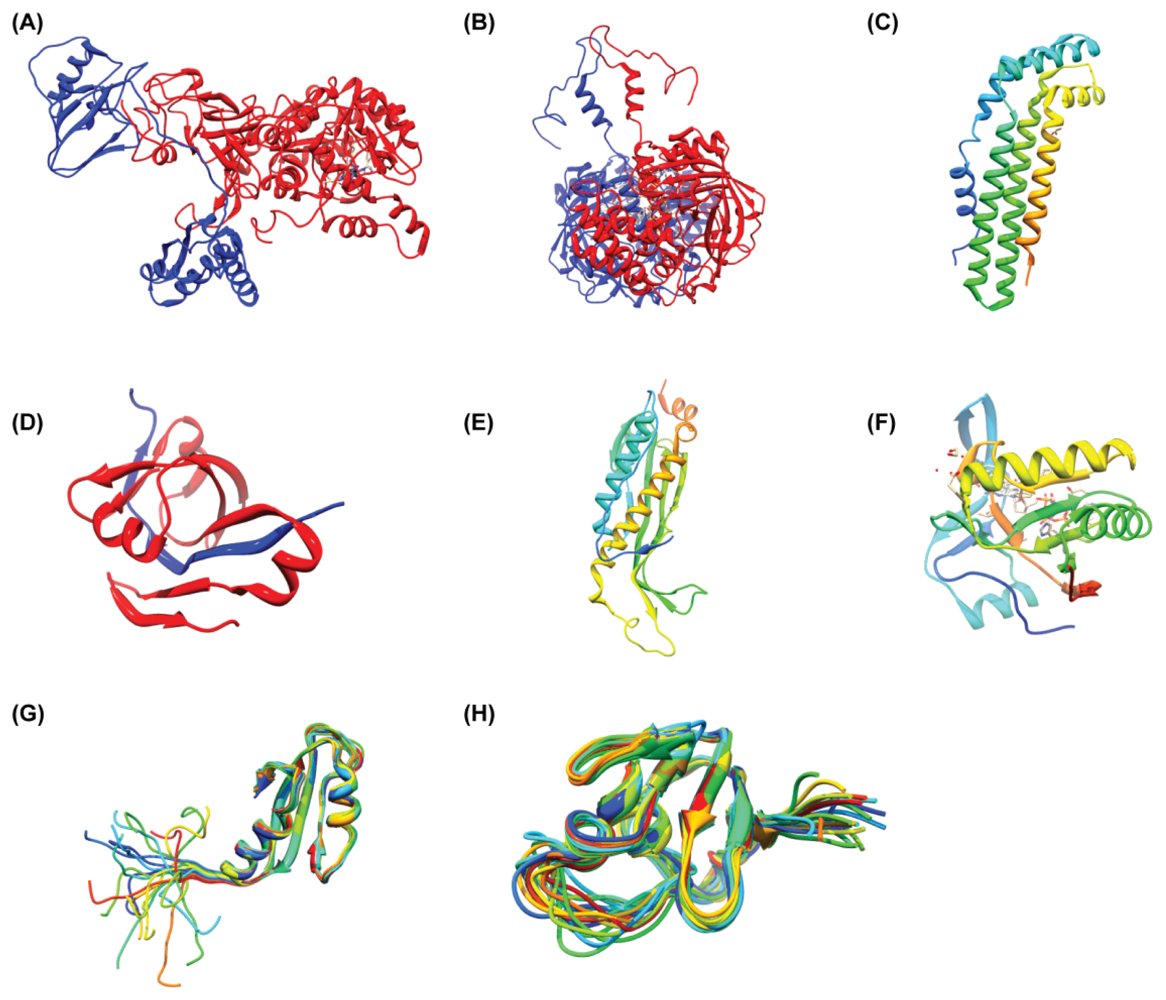
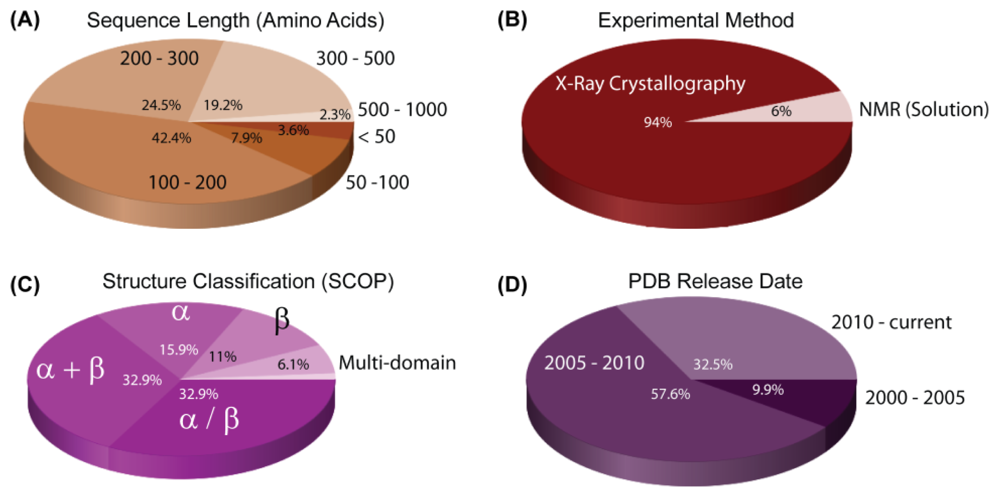
3. Structural Reports on H. pylori Proteins
4. Unknown Proteins in H. pylori and Estimation of Their Function
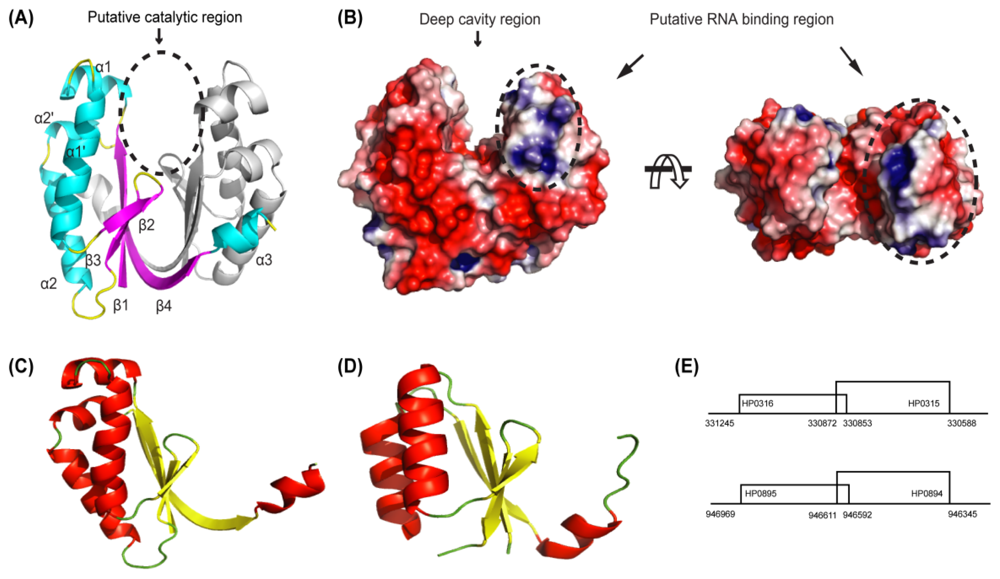
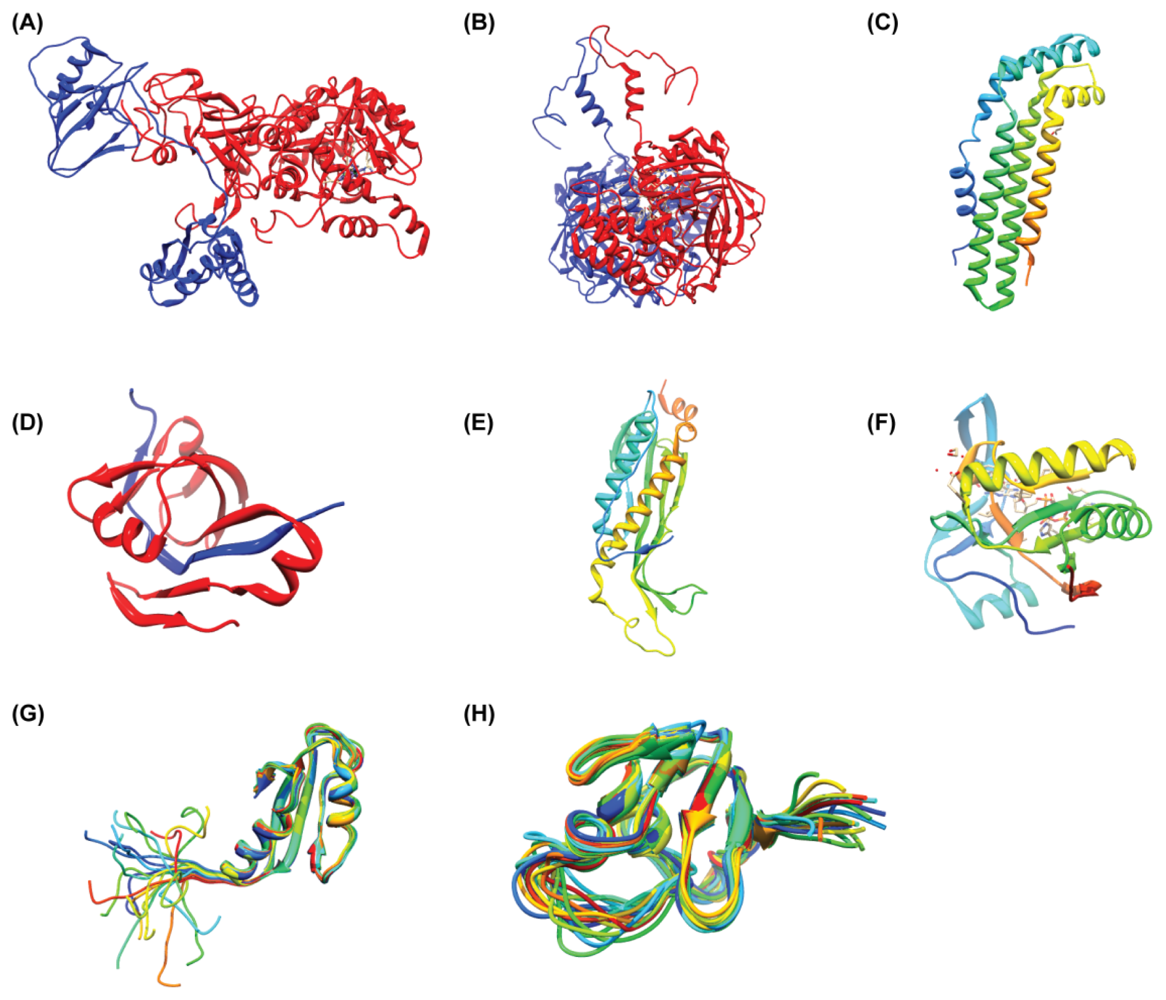
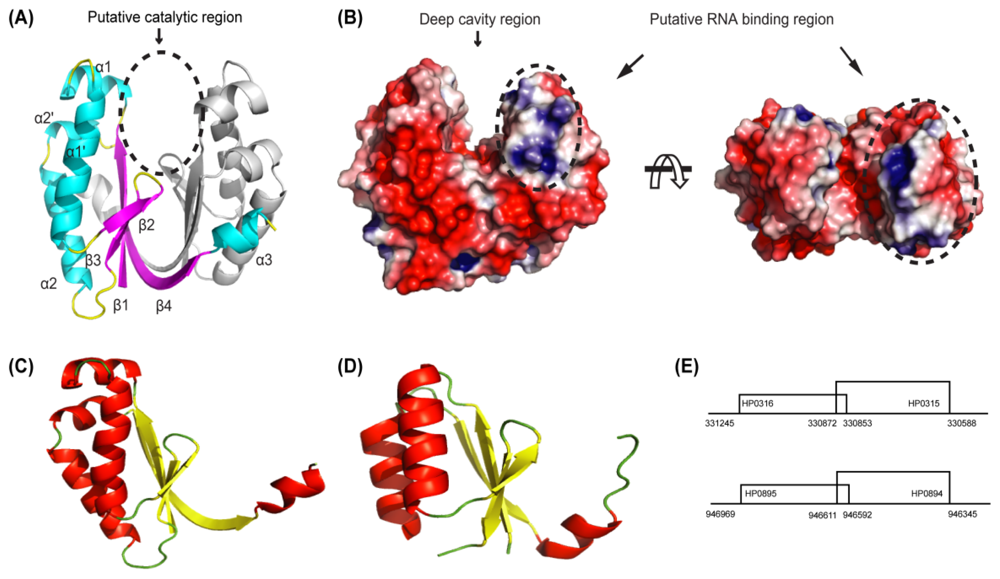
4.1. HP0894–HP0895: Toxin-Antitoxin System in H. pylori
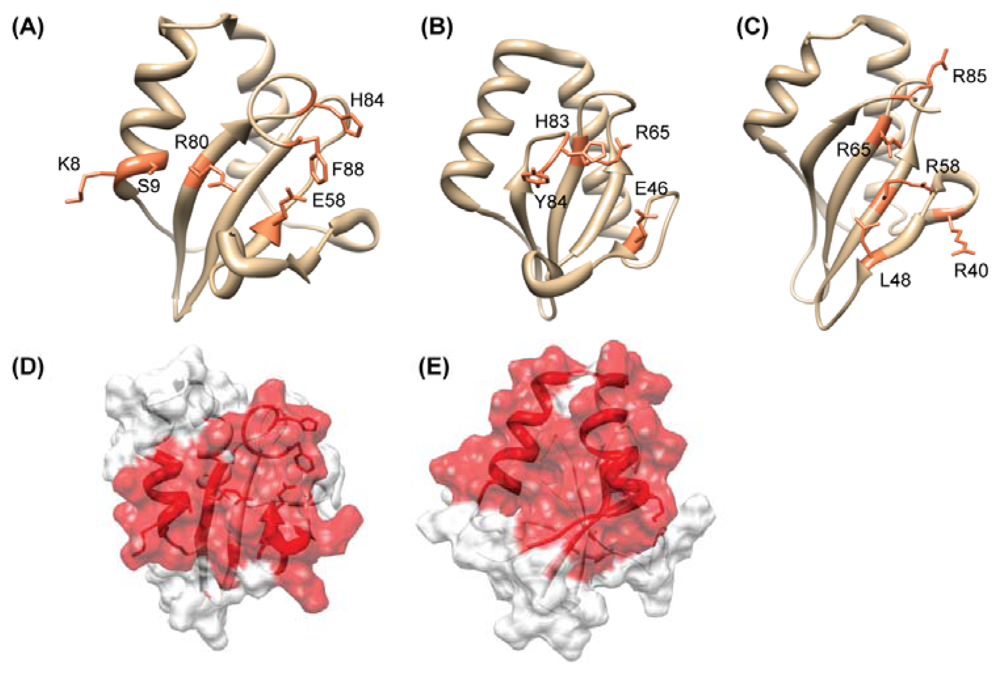
4.2. HP0315: Virulence-Associated Factor, Endoribonuclease
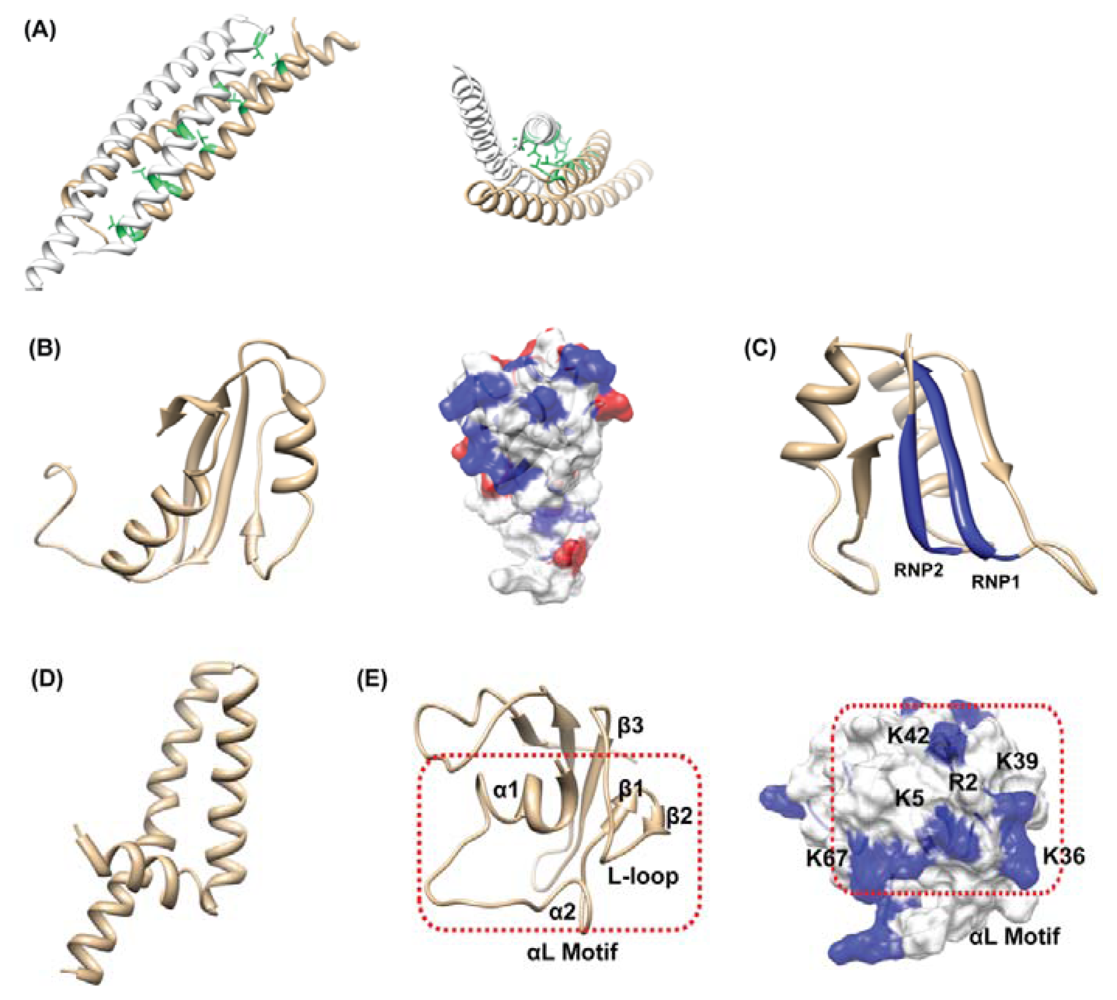
4.3. Others: HP0062, HP0495, HP0827, HP1242, HP1423
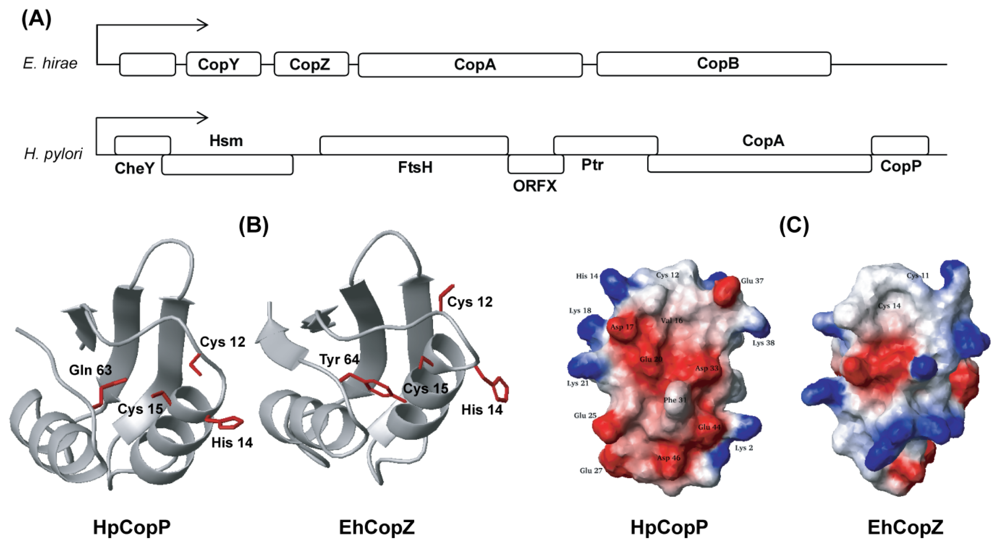
5. Different Characteristic with Known Function
6. Concluding Remarks
Supplementary Information
ijms-13-07109-s001.pdfAcknowledgements
References
- Rothenbacher, D.; Brenner, H. Burden of Helicobacter pylori and H. pylori-related diseases in developed countries: Recent developments and future implications. Microbes Infect 2003, 5, 693–703. [Google Scholar]
- Wotherspoon, A.C.; Doglioni, C.; Diss, T.C.; Pan, L.; Moschini, A.; de Boni, M.; Isaacson, P.G. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993, 342, 575–577. [Google Scholar]
- Peek, R.M., Jr; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med 1991, 325, 1127–1131. [Google Scholar]
- Ferreira, A.C.; Isomoto, H.; Moriyama, M.; Fujioka, T.; Machado, J.C.; Yamaoka, Y. Helicobacter and gastric malignancies. Helicobacter 2008, 13, 28–34. [Google Scholar]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol 2010, 7, 629–641. [Google Scholar]
- El-Omar, E.M. Role of host genes in sporadic gastric cancer. Best Pract. Res. Clin. Gastroenterol 2006, 20, 675–686. [Google Scholar]
- Graham, D.Y. Helicobacter pylori infection in the pathogenesis of duodenal ulcer and gastric cancer: A model. Gastroenterology 1997, 113, 1983–1991. [Google Scholar]
- Graham, D.Y.; Lu, H.; Yamaoka, Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: Facts or medical myths. J. Dig. Dis 2009, 10, 77–84. [Google Scholar]
- Wen, S.; Moss, S.F. Helicobacter pylori virulence factors in gastric carcinogenesis. Cancer Lett 2009, 282, 1–8. [Google Scholar]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori persistence: Biology and disease. J. Clin. Invest 2004, 113, 321–333. [Google Scholar]
- Mahdavi, J.; Sonden, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar]
- Lu, H.; Hsu, P.I.; Graham, D.Y.; Yamaoka, Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology 2005, 128, 833–848. [Google Scholar]
- Backert, S.; Clyne, M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2011, 1, 19–25. [Google Scholar]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar]
- Alm, R.A.; Ling, L.S.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith, D.R.; Noonan, B.; Guild, B.C.; deJonge, B.L.; et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1997, 397, 176–180. [Google Scholar]
- NCBI genome database. Available online: http://www.ncbi.nlm.nih.gov/genome accessed on 31 May 2012.
- Bieker, K.L.; Silhavy, T.J. The genetics of protein secretion in E. coli. Trends Genet 1990, 6, 329–334. [Google Scholar]
- Medigue, C.; Wong, B.C.; Lin, M.C.; Bocs, S.; Danchin, A. The secE gene of Helicobacter pylori. J. Bacteriol 2002, 184, 2837–2840. [Google Scholar]
- Wassarman, K.M.; Repoila, F.; Rosenow, C.; Storz, G.; Gottesman, S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 2001, 15, 1637–1651. [Google Scholar]
- Dong, Q.; Zhang, L.; Goh, K.L.; Forman, D.; O’Rourke, J.; Harris, A.; Mitchell, H. Identification and characterisation of ssrA in members of the Helicobacter genus. Antonie Van Leeuwenhoek 2007, 92, 301–307. [Google Scholar]
- Kazantsev, A.V.; Pace, N.R. Bacterial RNase P: A new view of an ancient enzyme. Nat. Rev. Microbiol 2006, 4, 729–740. [Google Scholar]
- Vogel, J.; Bartels, V.; Tang, T.H.; Churakov, G.; Slagter-Jager, J.G.; Huttenhofer, A.; Wagner, E.G. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 2003, 31, 6435–6443. [Google Scholar]
- Giannakis, M.; Chen, S.L.; Karam, S.M.; Engstrand, L.; Gordon, J.I. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4358–4363. [Google Scholar]
- Raymond, J.; Thiberge, J.M.; Kalach, N.; Bergeret, M.; Dupont, C.; Labigne, A.; Dauga, C. Using macro-arrays to study routes of infection of Helicobacter pylori in three families. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Amieva, M.R.; Covacci, A.; Lowe, T.M.; Merrell, D.S.; Ottemann, K.M.; Stein, M.; Salama, N.R.; Guillemin, K. The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol 2009, 191, 447–448. [Google Scholar]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N.; et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar]
- Oh, J.D.; Kling-Backhed, H.; Giannakis, M.; Xu, J.; Fulton, R.S.; Fulton, L.A.; Cordum, H.S.; Wang, C.; Elliott, G.; Edwards, J.; et al. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: Evolution during disease progression. Proc. Natl. Acad. Sci. USA 2006, 103, 9999–10004. [Google Scholar]
- McClain, M.S.; Shaffer, C.L.; Israel, D.A.; Peek, R.M., Jr.; Cover, T.L. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Devi, S.H.; Taylor, T.D.; Avasthi, T.S.; Kondo, S.; Suzuki, Y.; Megraud, F.; Ahmed, N. Genome of Helicobacter pylori strain 908. J. Bacteriol 2010, 192, 6488–6489. [Google Scholar]
- Farnbacher, M.; Jahns, T.; Willrodt, D.; Daniel, R.; Haas, R.; Goesmann, A.; Kurtz, S.; Rieder, G. Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Fischer, W.; Windhager, L.; Rohrer, S.; Zeiller, M.; Karnholz, A.; Hoffmann, R.; Zimmer, R.; Haas, R. Strain-specific genes of Helicobacter pylori: Genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res 2010, 38, 6089–6101. [Google Scholar]
- Kersulyte, D.; Kalia, A.; Gilman, R.H.; Mendez, M.; Herrera, P.; Cabrera, L.; Velapatino, B.; Balqui, J.; Paredes Puente de la Vega, F.; Rodriguez Ulloa, C.A.; et al. Helicobacter pylori from Peruvian amerindians: Traces of human migrations in strains from remote Amazon, and genome sequence of an Amerind strain. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Mane, S.P.; Dominguez-Bello, M.G.; Blaser, M.J.; Sobral, B.W.; Hontecillas, R.; Skoneczka, J.; Mohapatra, S.K.; Crasta, O.R.; Evans, C.; Modise, T.; et al. Host-interactive genes in Amerindian Helicobacter pylori diverge from their Old World homologs and mediate inflammatory responses. J. Bacteriol 2010, 192, 3078–3092. [Google Scholar]
- Thiberge, J.M.; Boursaux-Eude, C.; Lehours, P.; Dillies, M.A.; Creno, S.; Coppee, J.Y.; Rouy, Z.; Lajus, A.; Ma, L.; Burucoa, C.; et al. From array-based hybridization of Helicobacter pylori isolates to the complete genome sequence of an isolate associated with MALT lymphoma. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Avasthi, T.S.; Devi, S.H.; Taylor, T.D.; Kumar, N.; Baddam, R.; Kondo, S.; Suzuki, Y.; Lamouliatte, H.; Megraud, F.; Ahmed, N. Genomes of two chronological isolates (Helicobacter pylori 2017 and 2018) of the West African Helicobacter pylori strain 908 obtained from a single patient. J. Bacteriol 2011, 193, 3385–3386. [Google Scholar]
- Furuta, Y.; Kawai, M.; Yahara, K.; Takahashi, N.; Handa, N.; Tsuru, T.; Oshima, K.; Yoshida, M.; Azuma, T.; Hattori, M.; et al. Birth and death of genes linked to chromosomal inversion. Proc. Natl. Acad. Sci. USA 2011, 108, 1501–1506. [Google Scholar]
- Lehours, P.; Vale, F.F.; Bjursell, M.K.; Melefors, O.; Advani, R.; Glavas, S.; Guegueniat, J.; Gontier, E.; Lacomme, S.; Alves Matos, A.; et al. Genome sequencing reveals a phage in Helicobacter pylori. MBio 2011, 2. [Google Scholar] [CrossRef]
- Alvi, A.; Devi, S.M.; Ahmed, I.; Hussain, M.A.; Rizwan, M.; Lamouliatte, H.; Megraud, F.; Ahmed, N. Microevolution of Helicobacter pylori type IV secretion systems in an ulcer disease patient over a ten-year period. J. Clin. Microbiol 2007, 45, 4039–4043. [Google Scholar]
- Prouzet-Mauleon, V.; Hussain, M.A.; Lamouliatte, H.; Kauser, F.; Megraud, F.; Ahmed, N. Pathogen evolution in vivo: Genome dynamics of two isolates obtained 9 years apart from a duodenal ulcer patient infected with a single Helicobacter pylori strain. J. Clin. Microbiol 2005, 43, 4237–4241. [Google Scholar]
- Linz, B.; Balloux, F.; Moodley, Y.; Manica, A.; Liu, H.; Roumagnac, P.; Falush, D.; Stamer, C.; Prugnolle, F.; van der Merwe, S.W.; et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007, 445, 915–918. [Google Scholar]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Science 2003, 299, 1582–1585.
- Wirth, T.; Wang, X.; Linz, B.; Novick, R.P.; Lum, J.K.; Blaser, M.; Morelli, G.; Falush, D.; Achtman, M. Distinguishing human ethnic groups by means of sequences from Helicobacter pylori: Lessons from Ladakh. Proc. Natl. Acad. Sci. USA 2004, 101, 4746–4751. [Google Scholar]
- Peterson, J.D.; Umayam, L.A.; Dickinson, T.M.; Hickey, E.K.; White, O. The comprehensive microbial resource. Nucleic Acids Res. 2001, 29, 123–125. [Google Scholar]
- Marais, A.; Mendz, G.L.; Hazell, S.L.; Megraud, F. Metabolism and genetics of Helicobacter pylori: The genome era. Microbiol. Mol. Biol. Rev 1999, 63, 642–674. [Google Scholar]
- Merrell, D.S.; Thompson, L.J.; Kim, C.C.; Mitchell, H.; Tompkins, L.S.; Lee, A.; Falkow, S. Growth phase-dependent response of Helicobacter pylori to iron starvation. Infect. Immun 2003, 71, 6510–6525. [Google Scholar]
- Wen, Y.; Marcus, E.A.; Matrubutham, U.; Gleeson, M.A.; Scott, D.R.; Sachs, G. Acid-adaptive genes of Helicobacter pylori. Infect. Immun 2003, 71, 5921–5939. [Google Scholar]
- Cremades, N.; Velazquez-Campoy, A.; Martinez-Julvez, M.; Neira, J.L.; Perez-Dorado, I.; Hermoso, J.; Jimenez, P.; Lanas, A.; Hoffman, P.S.; Sancho, J. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem. Biol 2009, 4, 928–938. [Google Scholar]
- Han, K.D.; Matsuura, A.; Ahn, H.C.; Kwon, A.R.; Min, Y.H.; Park, H.J.; Won, H.S.; Park, S.J.; Kim, D.Y.; Lee, B.J. Functional identification of toxin-antitoxin molecules from Helicobacter pylori 26695 and structural elucidation of the molecular interactions. J. Biol. Chem 2011, 286, 4842–4853. [Google Scholar]
- Han, K.D.; Park, S.J.; Jang, S.B.; Son, W.S.; Lee, B.J. Solution structure of conserved hypothetical protein HP0894 from Helicobacter pylori. Proteins 2005, 61, 1114–1116. [Google Scholar]
- Yeo, H.J.; Savvides, S.N.; Herr, A.B.; Lanka, E.; Waksman, G. Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol. Cell 2000, 6, 1461–1472. [Google Scholar]
- Protein Data Bank. Available online: http://www.rcsb.org accessed on 1 March 2012.
- Goulding, C.W.; Perry, L.J. Protein production in Escherichia coli for structural studies by X-ray crystallography. J. Struct. Biol 2003, 142, 133–143. [Google Scholar]
- Cussac, V.; Ferrero, R.L.; Labigne, A. Expression of Helicobacter pylori urease genes in Escherichia coli grown under nitrogen-limiting conditions. J. Bacteriol 1992, 174, 2466–2473. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Kannan, S.; Hauth, A.M.; Burger, G. Function prediction of hypothetical proteins without sequence similarity to proteins of known function. Protein Pept. Lett 2008, 15, 1107–1116. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc 2008, 3, 153–162. [Google Scholar]
- Shen, H.B.; Chou, K.C. EzyPred: A top-down approach for predicting enzyme functional classes and subclasses. Biochem. Biophys. Res. Commun 2007, 364, 53–59. [Google Scholar]
- Dobson, P.D.; Cai, Y.D.; Stapley, B.J. Doig, A.J. Prediction of protein function in the absence of significant sequence similarity. Curr. Med. Chem 2004, 11, 2135–2142. [Google Scholar]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 2006, 34, W116–W118. [Google Scholar]
- Holm, L.; Kääriäinen, S.; Rosenström, P.; Schenkel, A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. [Google Scholar]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res 2010, 38, W545–W549. [Google Scholar]
- Kawabata, T.; Nishikawa, K. Protein structure comparison using the markov transition model of evolution. Proteins 2000, 41, 108–122. [Google Scholar]
- Nimrod, G.; Schushan, M.; Steinberg, D.M.; Ben-Tal, N. Detection of functionally important regions in “hypothetical proteins” of known structure. Structure 2008, 16, 1755–1763. [Google Scholar]
- Aloy, P.; Querol, E.; Aviles, F.X.; Sternberg, M.J. Automated structure-based prediction of functional sites in proteins: Applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J. Mol. Biol 2001, 311, 395–408. [Google Scholar]
- Ondrechen, M.J.; Clifton, J.G.; Ringe, D. THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. USA 2001, 98, 12473–12478. [Google Scholar]
- Pazos, F.; Sternberg, M.J. Automated prediction of protein function and detection of functional sites from structure. Proc. Natl. Acad. Sci. USA 2004, 101, 14754–14759. [Google Scholar]
- Pettit, F.K.; Bare, E.; Tsai, A.; Bowie, J.U. HotPatch: A statistical approach to finding biologically relevant features on protein surfaces. J. Mol. Biol 2007, 369, 863–879. [Google Scholar]
- Sierk, M.L.; Pearson, W.R. Sensitivity and selectivity in protein structure comparison. Protein Sci 2004, 13, 773–785. [Google Scholar]
- Altschul, S.F.; Gish, W. Local alignment statistics. Methods Enzymol 1996, 266, 460–480. [Google Scholar]
- Han, K.D.; Park, S.J.; Jang, S.B.; Son, W.S.; Lee, B.J. Solution structure of conserved hypothetical protein HP0894 from Helicobacter pylori. Proteins 2005, 61, 1111–1113. [Google Scholar]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res 2004, 32, W327–W331. [Google Scholar]
- Bateman, A.; Birney, E.; Cerruti, L. The Pfam protein families data base. Nucleic Acids Res 2002, 30, 276–280. [Google Scholar]
- Takagi, H.; Kakuta, Y.; Okada, T.; Yao, M.; Tanaka, I.; Kimura, M. Crystal structure of archaeal toxin-antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat. Struct. Mol. Biol 2005, 12, 327–331. [Google Scholar]
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol 2005, 3, 371–382. [Google Scholar]
- Wilson, D.N.; Nierhaus, K.H. RelBE or not to be. Nat. Struct. Mol. Biol 2005, 12, 282–284. [Google Scholar]
- Han, K.D.; Matsuura, A.; Ahn, H.C.; Kwon, A.R.; Min, Y.H.; Park, H.J.; Won, H.S.; Park, S.J.; Kim, D.Y.; Lee, B.J. Functional identification of toxin-antitoxin molecules from Helicobacter pylori 26695 and structural elucidation of the molecular interactions. J. Biol. Chem 2011, 286, 4842–4853. [Google Scholar]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the MazE/MazF complex: Molecular bases of antidote-toxin recognition. Mol. Cell 2003, 11, 875–884. [Google Scholar]
- Han, K.D.; Park, S.J.; Jang, S.B.; Lee, B.J. Solution structure of conserved hypothetical protein HP0892 from Helicobacter pylori. Proteins 2008, 70, 599–602. [Google Scholar]
- Terry, C.E.; McGinnis, L.M.; Madigan, K.C.; Cao, P.; Cover, T.L.; Liechti, G.W.; Peek, R.M., Jr; Forsyth, M.H. Genomic comparison of cag pathogenicity island (PAI)-positive and -negative Helicobacter pylori strains: Identification of novel markers for cag PAI-positive strains. Infect. Immun. 2005, 73, 3794–3798. [Google Scholar]
- Cheetham, B.F.; Tattersall, D.B.; Bloomfield, G.A.; Rood, J.I.; Katz, M.E. Identification of a gene encoding a bacteriophage-related integrase in a vap region of the Dichelobacter nodosus genome. Gene 1995, 162, 53–58. [Google Scholar]
- Katz, M.E.; Strugnell, R.A.; Rood, J.I. Molecular characterization of a genomic region associated with virulence in Dichelobacter nodosus. Infect. Immun 1992, 60, 4586–4592. [Google Scholar]
- Takai, S.; Hines, S.A.; Sekizaki, T.; Nicholson, V.M.; Alperin, D.A.; Osaki, M.; Takamatsu, D.; Nakamura, M.; Suzuki, K.; Ogino, N.; et al. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect. Immun 2000, 68, 6840–6847. [Google Scholar]
- Tomb, J.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar]
- Katz, M.E.; Strugnell, R.A.; Rood, J.I. Molecular characterization of a genomic region associated with virulence in Dichelobacter nodosus. Infect. Immun 1992, 60, 4586–4592. [Google Scholar]
- Benoit, S.; Benachour, A.; Taouji, S.; Auffray, Y.; Hartke, A. Induction of vap genes encoded by the virulence plasmid of Rhodococcus equi during acid tolerance response. Res. Microbiol 2001, 152, 439–449. [Google Scholar]
- Galli, D.M.; LeBlanc, D.J. Characterization of pVT736-1, a rolling-circle plasmid from the gram-negative bacterium Actinobacillus actinomycetemcomitans. Plasmid 1994, 31, 148–157. [Google Scholar]
- Kwon, A.R.; Kim, J.H.; Park, S.J.; Lee, K.Y.; Min, Y.H.; Im, H.; Lee, I.; Lee, K.Y.; Lee, B.J. Structural and biochemical characterization of HP0315 from Helicobacter pylori as a VapD protein with an endoribonuclease activity. Nucleic Acids Res 2012, 40, 4216–4228. [Google Scholar]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1. [Google Scholar] [CrossRef]
- Jang, S.B.; Kwon, A.R.; Son, W.S.; Park, S.J.; Lee, B.J. Crystal structure of hypothetical protein HP0062 (O24902_HELPY) from Helicobacter pylori at 1.65 A resolution. J. Biochem 2009, 146, 535–540. [Google Scholar]
- Pallen, M.J. The ESAT-6/WXG100 superfamily—And a new Gram-positive secretion system? Trends Microbiol 2002, 10, 209–212. [Google Scholar]
- Plano, G.V.; Day, J.B.; Ferracci, F. Type III export: New uses for an old pathway. Mol. Microbiol 2001, 40, 284–293. [Google Scholar]
- Seo, M.D.; Park, S.J.; Kim, H.J.; Lee, B.J. Solution structure of hypothetical protein, HP0495 (Y495_HELPY) from Helicobacter pylori. Proteins 2007, 67, 1189–1192. [Google Scholar]
- Seo, M.D.; Park, S.J.; Kim, H.J.; Seok, S.H.; Lee, B.J. Backbone 1H, 15N, and 13C resonance assignment and secondary structure prediction of HP0495 from Helicobacter pylori. J. Biochem. Mol. Biol 2007, 40, 839–843. [Google Scholar]
- Jang, S.B.; Ma, C.; Lee, J.Y.; Kim, J.H.; Park, S.J.; Kwon, A.R.; Lee, B.J. NMR solution structure of HP0827 (O25501_HELPY) from Helicobacter pylori: Model of the possible RNA-binding site. J. Biochem 2009, 146, 667–674. [Google Scholar]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.R.; Griffiths-Jones, S.; Howe, K.L.; Marshall, M.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res 2002, 30, 276–280. [Google Scholar]
- Kang, S.J.; Park, S.J.; Jung, S.J.; Lee, B.J. Backbone 1H, 15N, and 13C resonance assignment of HP1242 from Helicobacter pylori. J. Biochem. Mol. Biol 2005, 38, 591–594. [Google Scholar]
- Kang, S.J.; Park, S.J.; Jung, S.J.; Lee, B.J. Solution structure of HP1242 from Helicobacter pylori. Proteins 2005, 61, 1111–1113. [Google Scholar]
- Aravind, L.; Koonin, E.V. Novel predicted RNA-binding domains associated with the translation machinery. J. Mol. Evol 1999, 48, 291–302. [Google Scholar]
- Kim, J.H.; Park, S.J.; Lee, K.Y.; Son, W.S.; Sohn, N.Y.; Kwon, A.R.; Lee, B.J. Solution structure of hypothetical protein HP1423 (Y1423_HELPY) reveals the presence of alphaL motif related to RNA binding. Proteins 2009, 75, 252–257. [Google Scholar]
- Copley, S.D. Enzymes with extra talents: Moonlighting functions and catalytic promiscuity. Curr. Opin. Chem. Biol 2003, 7, 265–272. [Google Scholar]
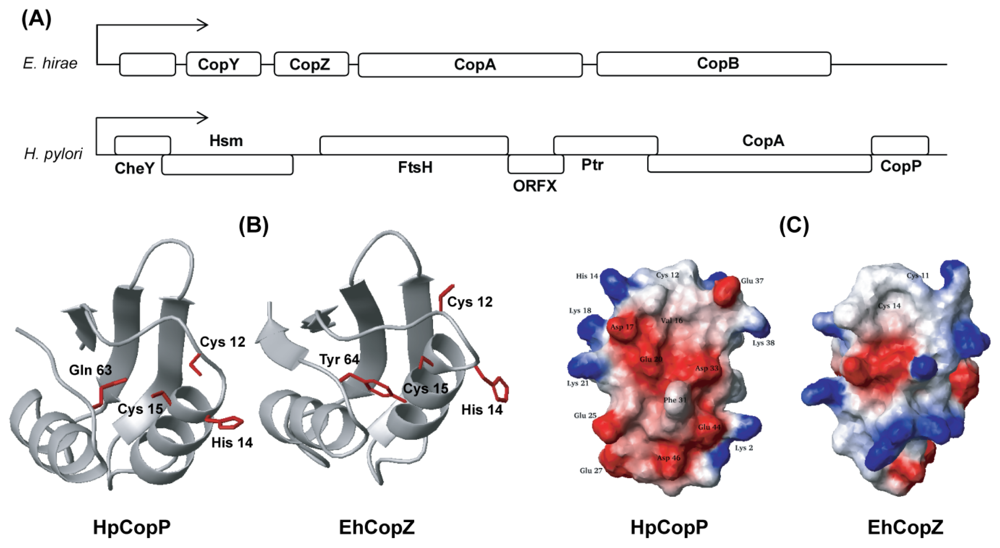
- Odermatt, A.; Suter, H.; Krapf, R.; Solioz, M. Primary structure of two P-type ATPases involved in copper homeostasis in Enterococcus hirae. J. Biol. Chem 1993, 268, 12775–12779. [Google Scholar]
- Odermatt, A.; Solioz, M. Two trans-acting metalloregulatory proteins controlling expression of the copper-ATPases of Enterococcus hirae. J. Biol. Chem 1995, 270, 4349–4354. [Google Scholar]
- Wunderli-Ye, H.; Solioz, M. Effects of promoter mutations on the in vivo regulation of the cop operon of Enterococcus hirae by copper(I) and copper(II). Biochem. Biophys. Res. Commun 1999, 259, 443–449. [Google Scholar]
- Pufahl, R.A.; Singer, C.P.; Peariso, K.L.; Lin, S.; Schmidt, P.J.; Fahrni, C.J.; Culotta, V.C.; Penner-Hahn, J.E.; O’Halloran, T.V. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science 1997, 278, 853–856. [Google Scholar]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Del Conte, R.; Gonnelli, L. Understanding copper trafficking in bacteria: Interaction between the copper transport protein CopZ and the N-terminal domain of the copper ATPase CopA from Bacillus subtilis. Biochemistry 2003, 42, 1939–1949. [Google Scholar]
- Beier, D.; Spohn, G.; Rappuoli, R.; Scarlato, V. Identification and characterization of an operon of Helicobacter pylori that is involved in motility and stress adaptation. J. Bacteriol 1997, 179, 4676–4683. [Google Scholar]
- Bayle, D.; Wangler, S.; Weitzenegger, T.; Steinhilber, W.; Volz, J.; Przybylski, M.; Schafer, K.P.; Sachs, G.; Melchers, K. Properties of the P-type ATPases encoded by the copAP operons of Helicobacter pylori and Helicobacter felis. J. Bacteriol 1998, 180, 317–329. [Google Scholar]
- Solioz, M.; Stoyanov, J.V. Copper homeostasis in Enterococcus hirae. FEMS Microbiol. Rev 2003, 27, 183–195. [Google Scholar]
- Park, S.J.; Jung, Y.S.; Kim, J.S.; Seo, M.D.; Lee, B.J. Structural insight into the distinct properties of copper transport by the Helicobacter pylori CopP protein. Proteins 2008, 71, 1007–1019. [Google Scholar]
- Vandem, B.T.; Cronan, J.E., Jr. Genetics and regulation of bacterial lipid metabolism. Annu. Rev. Microbiol. 1989, 43, 317–343. [Google Scholar]
- Jones, P.J.; Holak, T.A.; Prestegard, J.H. Structural comparison of acyl carrier protein in acylated and sulfhydryl forms by two-dimensional 1H NMR spectroscopy. Biochemistry 1987, 26, 3493–3500. [Google Scholar]
- Cronan, J.E., Jr. Molecular properties of short chain acyl thioesters of acyl carrier protein. J. Biol. Chem. 1982, 257, 5013–5017. [Google Scholar]
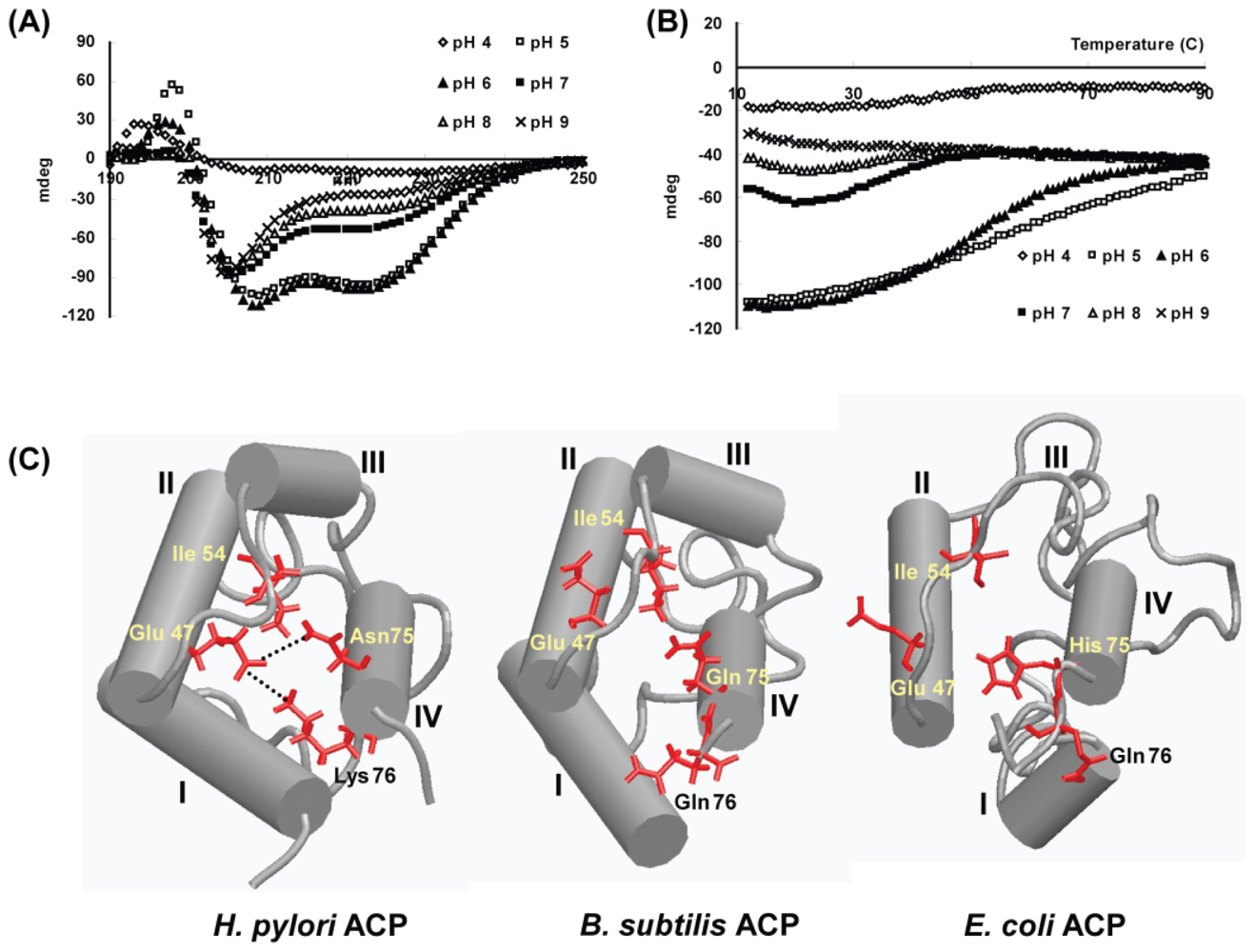
- Park, S.J.; Kim, J.S.; Son, W.S.; Lee, B.J. pH-induced conformational transition of H. pylori acyl carrier protein: Insight into the unfolding of local structure. J. Biochem 2004, 135, 337–346. [Google Scholar]
- Schulz, H. On the structure-function relationship of acyl carrier protein of Escherichia coli. J. Biol. Chem 1975, 250, 2299–2304. [Google Scholar]
- Flaman, A.S.; Chen, J.M.; Van Iderstine, S.C.; Byers, D.M. Site-directed mutagenesis of acyl carrier protein (ACP) reveals amino acid residues involved in ACP structure and acyl-ACP synthetase activity. J. Biol. Chem. 2001, 276, 35934–35939. [Google Scholar]
- Keating, D.H.; Cronan, J.E., Jr. An isoleucine to valine substitution in Escherichia coli acyl carrier protein results in a functional protein of decreased molecular radius at elevated pH. J. Biol. Chem. 1996, 271, 15905–15910. [Google Scholar]
- Keating, M.-M.; Gong, H.; Byers, D.M. Identification of a key residue in the conformational stability of acyl carrier protein. Biochem. Biophys. Acta 2002, 1601, 208–214. [Google Scholar]
- Hanson, A.D.; Pribat, A.; Waller, J.C.; de Crécy-Lagard, V. “Unknown” proteins and “orphan” enzymes: The missing half of the engineering parts list—and how to find it. Biochem. J 2010, 425, 1–11. [Google Scholar]
- Galperin, M.Y.; Koonin, E.V. “Conserved hypothetical” proteins: Prioritization of targets for experimental study. Nucleic Acids Res 2004, 32, 5452–5463. [Google Scholar]
- Galperin, M.Y.; Koonin, E.V. From complete genome sequence to “complete” understanding? Trends Biotechnol 2010, 28, 398–406. [Google Scholar]
- Frishman, D. Protein annotation at genomic scale: The current status. Chem. Rev 2007, 107, 3448–3466. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Gene | Size (Mb) | GC% | Protein (unknown) | Type | Project |
|---|---|---|---|---|---|---|
| Helicobacter pylori | 1480 | 1.57 | 38.9 | 1405 (476) | chr a | Gyeongsang National University College of Medicine and 21c Frontier Human Genome Functional Research Project Helicobacter pylori 52 genome sequencing project |
| Helicobacter pylori 2017 | 1647 | 1.55 | 39.3 | 1593 (525) | chr | Pathogen Biology Laboratory, University of Hyderabad Helicobacter pylori 2017 genome sequencing project |
| Helicobacter pylori 2018 | 1655 | 1.56 | 39.3 | 1603 (459) | chr | Pathogen Biology Laboratory, University of Hyderabad Helicobacter pylori 2018 genome sequencing project |
| Helicobacter pylori 26695 | 1627 | 1.67 | 38.9 | 1573 (1301) | chr | TIGR (The Institute for Genome Research) Causes gastric inflammation and peptic ulcer disease |
| Helicobacter pylori 35A | 1560 | 1.57 | 38.9 | 1470 (362) | chr | Baylor College of Medicine Reference genome for the Human Microbiome Project |
| Helicobacter pylori 51 | 1495 | 1.59 | 38.8 | 1415 (386) | chr | Gyeongsang National University College of Medicine and 21c Frontier Human Functional Genome Research Project Bacterium isolated from duodenal ulcer patient |
| Helicobacter pylori 83 | 1656 | 1.62 | 38.7 | 1609 (445) | chr | Baylor College of Medicine Reference genome for the Human Microbiome Project |
| Helicobacter pylori 908 | 1646 | 1.55 | 39.3 | 1595 (444) | chr | University of Hyderabad, India Helicobacter pylori 908 genome sequencing project |
| Helicobacter pylori B38 | 1571 | 1.58 | 39.2 | 1382 (643) | chr | Institut Pasteur Causes peptic ulcers |
| Helicobacter pylori B8 | 1744 | 1.67 | 38.8 | 1702 (736) | chr | CeBitec, Bielefeld University Helicobacter pylori B8 genome sequencing project |
| 5 | 0.01 | 35.9 | 5 (3) | plsm b | ||
| Helicobacter pylori Cuz20 | 1606 | 1.64 | 38.9 | 1564 (538) | chr | Dept. of Molec. Microbiology, Washington University Medical School, Saint Louis Helicobacter pylori Cuz20 genome sequencing project |
| Helicobacter pylori F16 | 1543 | 1.58 | 38.9 | 1500 (494) | chr | The University of Tokyo Helicobacter pylori F16 genome sequencing project |
| Helicobacter pylori F30 | 1522 | 1.57 | 38.8 | 1479 (470) | chr | The University of Tokyo Helicobacter pylori F30 genome sequencing project. |
| 5 | 0.01 | 34.1 | 5 (1) | plsm | ||
| Helicobacter pylori F32 | 1533 | 1.58 | 38.9 | 1490 (485) | chr | The University of Tokyo Helicobacter pylori F32 genome sequencing project. |
| 1 | 0 | 36.7 | 1 (0) | plsm | ||
| Helicobacter pylori F57 | 1563 | 1.61 | 38.7 | 1520 (498) | chr | The University of Tokyo Helicobacter pylori F57 genome sequencing project. |
| Helicobacter pylori G27 | 1570 | 1.65 | 38.9 | 1493 (470) | chr | University of Oregon Strain used extensively in H. pylori research |
| 11 | 0.01 | 34.9 | 11 (5) | plsm | ||
| Helicobacter pylori Gambia94/24 | 1646 | 1.71 | 39.1 | 1604 (611) | chr | Berg lab, Washington University Medical School Helicobacter pylori Gambia94/24 genome sequencing project |
| 1 | 0 | 37.4 | 1 (1) | plsm | ||
| Helicobacter pylori HPAG1 | 1573 | 1.60 | 39.1 | 1531 (515) | chr | Washington University (WashU) Isolated from a Swedish patient with chronic atrophic gastritis |
| 8 | 0.01 | 36.4 | 8 (5) | plsm | ||
| Helicobacter pylori India7 | 1638 | 1.68 | 38.9 | 1600 (561) | chr | Berg lab, Washington University Medical School Helicobacter pylori Ind7 genome sequencing project |
| Helicobacter pylori J99 | 1534 | 1.64 | 39.2 | 1488 (560) | chr | Astrazeneca-Boston Causes gastric inflammation and peptic ulcer disease |
| Helicobacter pylori Lithuania75 | 1588 | 1.62 | 38.8 | 1546 (522) | chr | Berg lab, Washington University Medical School Helicobacter pylori Lit75 genome sequencing project |
| 19 | 0.02 | 33.7 | 19 (12) | plsm | ||
| Helicobacter pylori P12 | 1624 | 1.67 | 38.8 | 1568 (450) | chr | Max von Pettenkofer-Institut für Hygiene und Medizinische Mikrobiologie, Ludwig-Maximilians-Universität München Clinical isolate |
| 10 | 0.01 | 35.1 | 10 (2) | plsm | ||
| Helicobacter pylori PeCan4 | 1597 | 1.63 | 38.9 | 1555 (529) | chr | Dept. of Molec. Microbiology, Washington University Medical School, Saint Louis Helicobacter pylori PeCan4 genome sequencing project |
| 8 | 0.01 | 32.9 | 8 (0) | plsm | ||
| Helicobacter pylori Puno120 | 1567 | 1.62 | 38.9 | 1525 (518) | chr | Washington University Medical School Helicobacter pylori Puno120 genome sequencing |
| 15 | 0.01 | 35.8 | 15 (13) | plsm | ||
| Helicobacter pylori Puno135 | 1615 | 1.65 | 38.8 | 1573 (532) | chr | Washington University Medical School Genome sequence of Helicobacter pylori strain Puno135 |
| Helicobacter pylori SJM180 | 1623 | 1.66 | 38.9 | 1581 (558) | chr | Dept. of Molec. Microbiology, Washington University Medical School, Saint Louis Helicobacter pylori SJM180 genome sequencing project |
| Helicobacter pylori SNT49 | 1557 | 1.61 | 39 | 1515 (495) | phage | Washington University Medical School Genome sequence of Helicobacter pylori SNT49 |
| 4 | 0 | 37.4 | 4 (3) | plsm | ||
| Helicobacter pylori Sat464 | 1544 | 1.56 | 39.1 | 1502 (504) | chr | Dept. Molec. Microbiology, Washintgton University Medical School in Saint Louis Helicobacter pylori Sat464 genome sequencing project. |
| 6 | 0.01 | 33.5 | 6 (4) | plsm | ||
| Helicobacter pylori Shi470 | 1647 | 1.61 | 38.9 | 1568 (593) | chr | Washington University Medical School Clinical isolate from the Amazon River region |
| Helicobacter pylori SouthAfrica7 | 1585 | 1.65 | 38.4 | 1543 (555) | chr | Berg lab, Washington University Medical Shool Helicobacter pylori SouthAfrica7 genome sequencing project |
| 29 | 0.03 | 33.7 | 29 (19) | plsm | ||
| Helicobacter pylori v225d | 1625 | 1.59 | 39 | 1541 (555) | chr | The Pathosystems Resource Integration Center (PATRIC) Helicobacter pylori v225 genome sequencing |
| 9 | 0.01 | 32.9 | 9 (7) | plsm | ||
| Helicobacter pylori B45 | 27 | 0.02 | 37.3 | 27 (26) | chr S/C c | Karolinska Institute Helicobacter pylori B45 genome sequencing project |
| Helicobacter pylori 98-10 | 1566 | 1.57 | 38.8 | 1527 (1527) | S/C | Vanderbilt University School of Medicine Gastric cancer strain |
| Helicobacter pylori B128 | 1770 | 1.65 | 38.8 | 1731 (1731) | S/C | Vanderbilt University School of Medicine Gastric ulcer strain |
| Helicobacter pylori HPKX_438_AG0C 1 | 2939 | 1.82 | 39.5 | 2898 (1564) | S/C | Washington University Medical School Clinical isolate |
| Helicobacter pylori HPKX_438_CA4C1 | 3962 | 1.57 | 39.2 | 3925 (1548) | S/C | Washington University Medical School Isolate from a patient with gastric carcinoma |
| Total | 59,776 | - | - | 57,872 (23,261) | - | - |
| PDB ID | Chain | Structure | Macromolecule Name | Classification | Scop Fold | Exp. Method | |
|---|---|---|---|---|---|---|---|
| ID | AA | MW | |||||
| 1MW7 | A | 240 | 27161.20 | Hypothetical protein HP0162 | SG a, unknown function | YebC-like | X-ray |
| 1S2X | A | 206 | 23998.70 | Cag-Z | Unknown function | STAT-like | X-ray |
| 1Z8M | A | 88 | 10394.30 | Conserved hypothetical protein HP0894 | SG, unknown function | RelE-like | NMR |
| 1ZHC | A | 76 | 9130.38 | hypothetical protein HP1242 | Unknown function | NMR | |
| 1ZKE | A, B, C, D, E, F | 83 | 56798.00 | Hypothetical protein HP1531 | SG, unknown function | ROP-like | X-ray |
| 2ATZ | A | 180 | 22049.45 | Predicted coding region HP0184 | SG, unknown function | Prim-pol domain | X-ray |
| 2BO3 | A | 94 | 11101.70 | Hypothetical protein HP0242 | SG, unknown function | HP0242-like | X-ray |
| 2EVV | A, B, C, D | 207 | 95692.83 | hypothetical protein HP0218 | SG, unknown function | X-ray | |
| 2F6S | A, B | 201 | 47249.90 | cell filamentation protein, putative | SG, unknown function | Fic-like | X-ray |
| 2G3V | A, B, C, D | 208 | 104975.36 | CAG pathogenicity island protein 13 | Unknown function | X-ray | |
| 2GTS | A | 86 | 10626.50 | hypothetical protein HP0062 | SG, unknown function | Ferritin-like | X-ray |
| 2H9Z | A | 86 | 10205.80 | Hypothetical protein HP0495 | SG, unknown function | Ferredoxin-like | NMR |
| 2I9I | A | 254 | 29526.70 | Hypothetical protein | SG, unknown function | Anticodon-binding domain-like | X-ray |
| 2JOQ | A | 91 | 10673.20 | Hypothetical protein HP0495 | SG, unknown function | Ferredoxin-like | NMR |
| 2K0Z | A | 110 | 12948.60 | Uncharacterized protein HP1203 | SG, unknown function | NMR | |
| 2K6P | A | 92 | 10472.30 | Uncharacterized protein HP1423 | Unknown function | NMR | |
| 2OTR | A | 98 | 11502.60 | Hypothetical protein HP0892 | SG, unknown function | NMR | |
| 2OUF | A | 94 | 11148.60 | Hypothetical protein | SG, unknown function | X-ray | |
| 2UVP | A, B, C, D | 186 | 87079.82 | HOBA, HP1230 | Unknown function | X-ray | |
| 2XRH | A | 100 | 11635.31 | HP0721 | Unknown function | X-ray | |
| 3BGH | A, B | 236 | 55233.49 | Putative neuraminyllactose-binding hemagglutinin homolog | SG, unknown function | X-ray | |
| 3CWX | A, B, C | 176 | 62332.80 | protein CagD | Unknown function | X-ray | |
| 3CWY | A | 176 | 20841.15 | protein CagD | Unknown function | X-ray | |
| 3F42 | A, B | 99 | 22671.87 | protein HP0035 | SG, unknown function | X-ray | |
| 3FX7 | A, B | 94 | 23207.80 | Uncharacterized protein, HP0062 | Unknown function | X-ray | |
| 3KWL | A | 514 | 60116.00 | Uncharacterized protein | Unknown function | X-ray | |
| 3MLG | A, B | 189 | 43924.40 | Uncharacterized protein | Unknown function | X-ray | |
| 3MLI | A, B, C, D | 100 | 47758.96 | Uncharacterized protein HP0242 | Unknown function | X-ray | |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, S.J.; Son, W.S.; Lee, B.-J. Structural Analysis of Hypothetical Proteins from Helicobacter pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins. Int. J. Mol. Sci. 2012, 13, 7109-7137. https://doi.org/10.3390/ijms13067109
Park SJ, Son WS, Lee B-J. Structural Analysis of Hypothetical Proteins from Helicobacter pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins. International Journal of Molecular Sciences. 2012; 13(6):7109-7137. https://doi.org/10.3390/ijms13067109
Chicago/Turabian StylePark, Sung Jean, Woo Sung Son, and Bong-Jin Lee. 2012. "Structural Analysis of Hypothetical Proteins from Helicobacter pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins" International Journal of Molecular Sciences 13, no. 6: 7109-7137. https://doi.org/10.3390/ijms13067109