Combining Molecular Docking and Molecular Dynamics to Predict the Binding Modes of Flavonoid Derivatives with the Neuraminidase of the 2009 H1N1 Influenza A Virus

Abstract
:1. Introduction
2. Results and Discussion
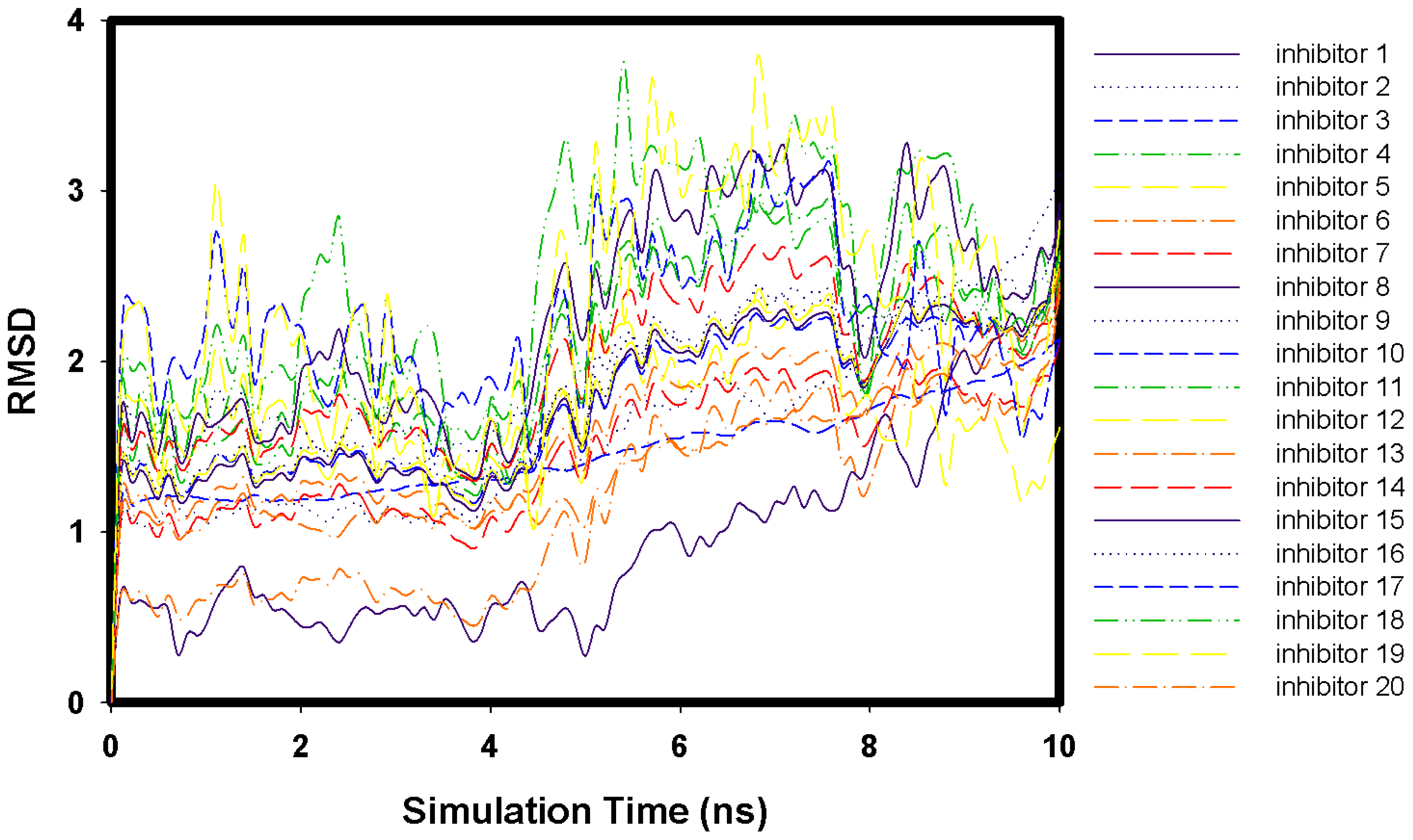
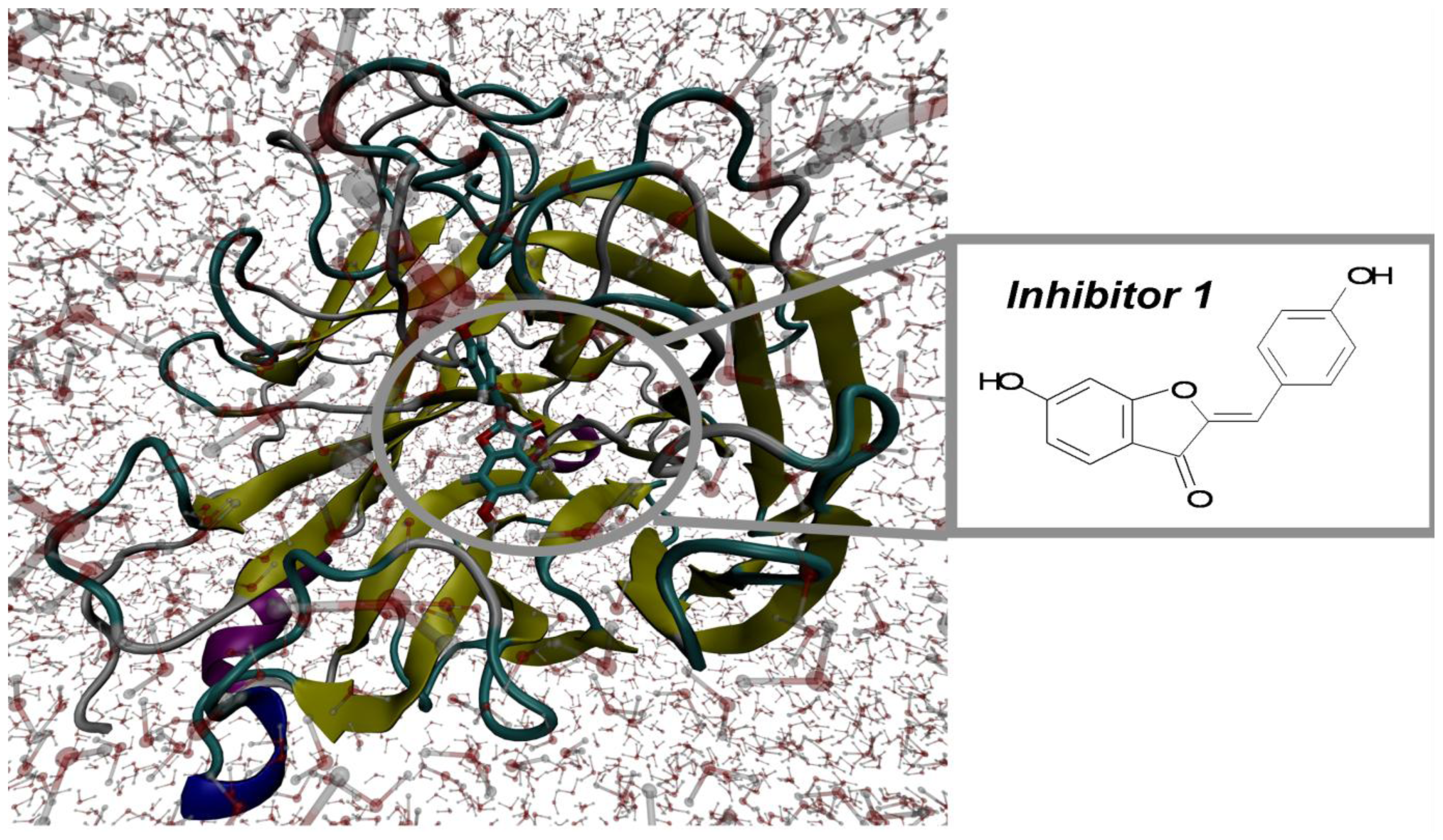
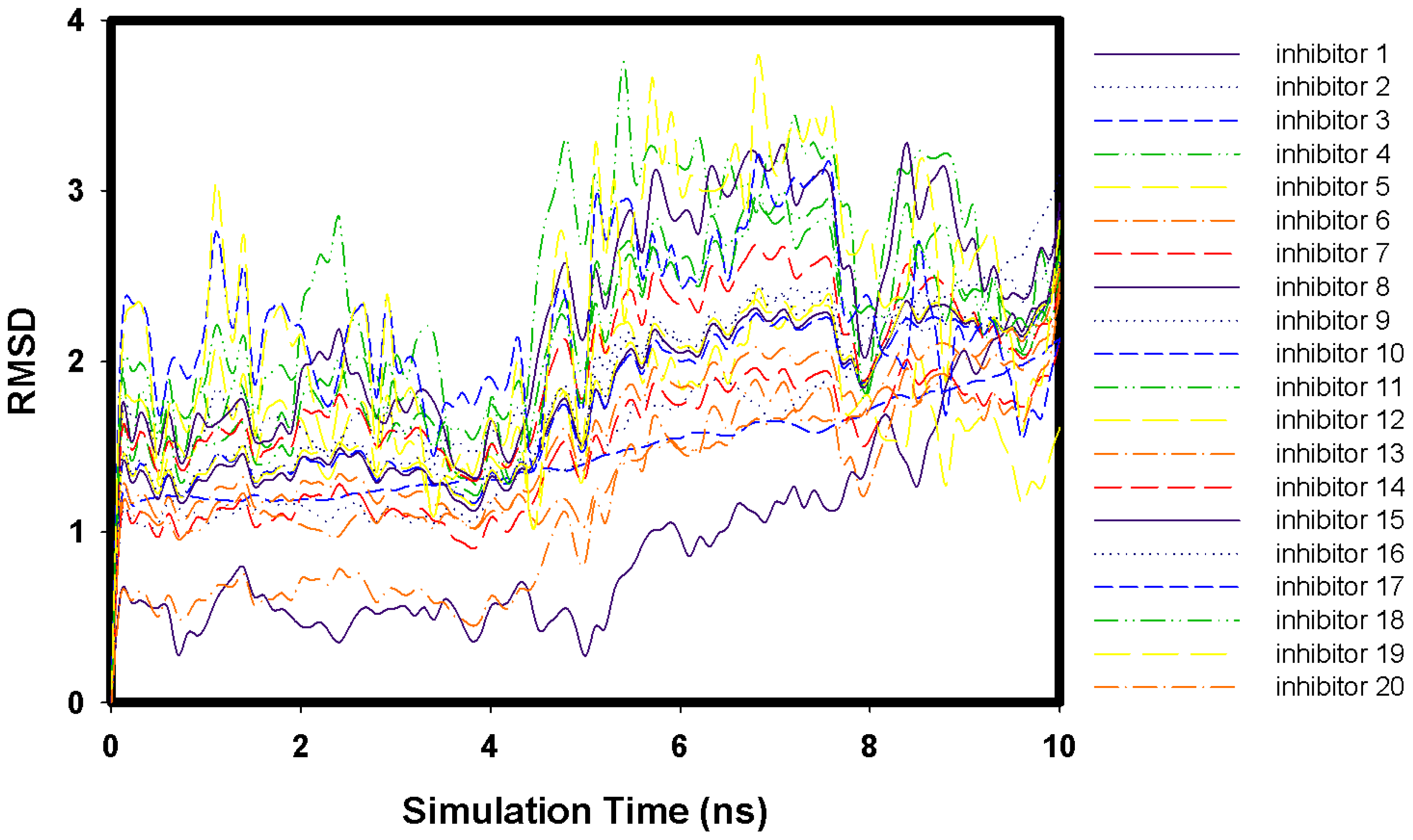
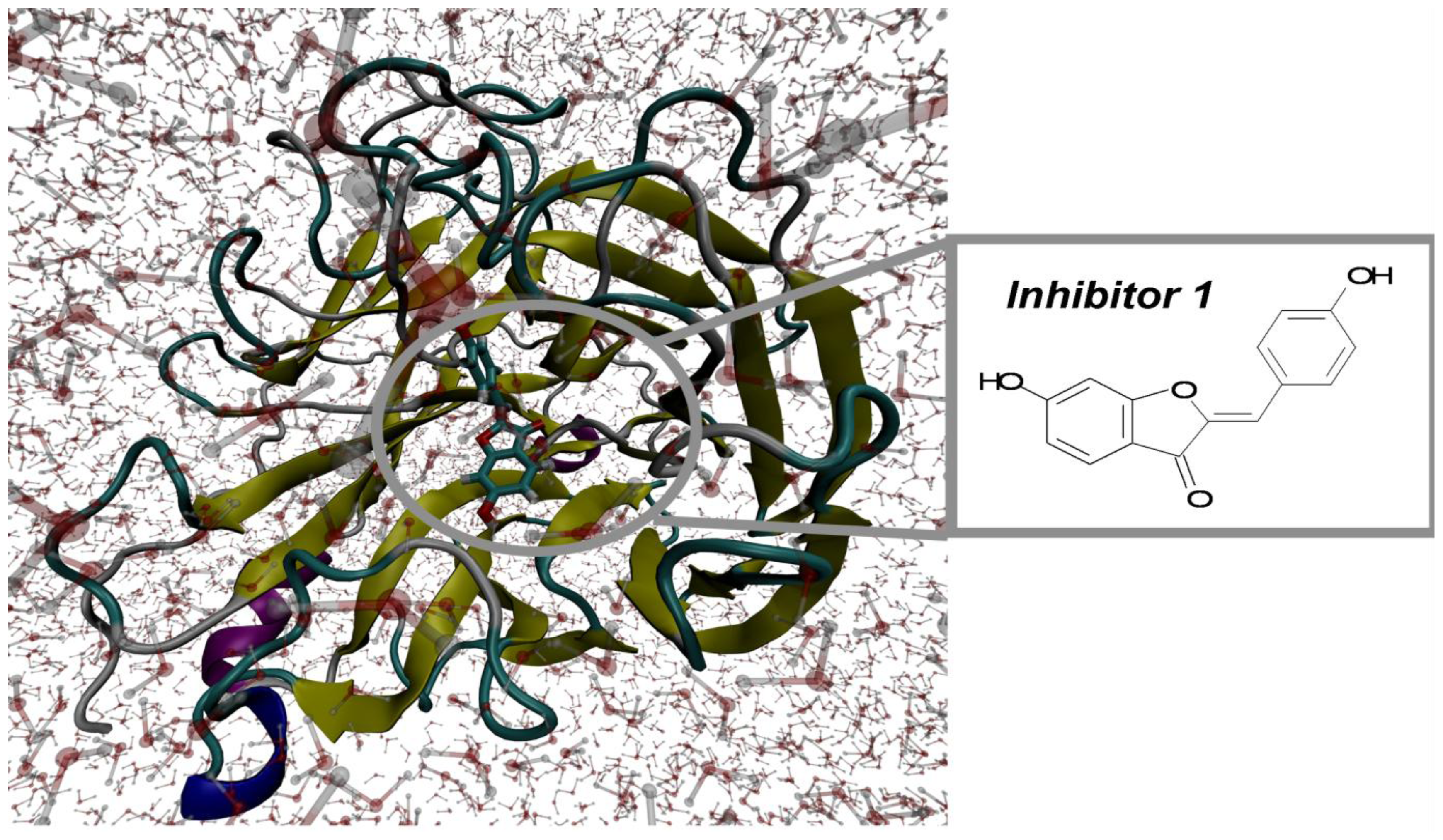
2.1. Molecular Docking and MD Simulation
2.2. Key Residues of 2009 H1N1 Neuraminidase
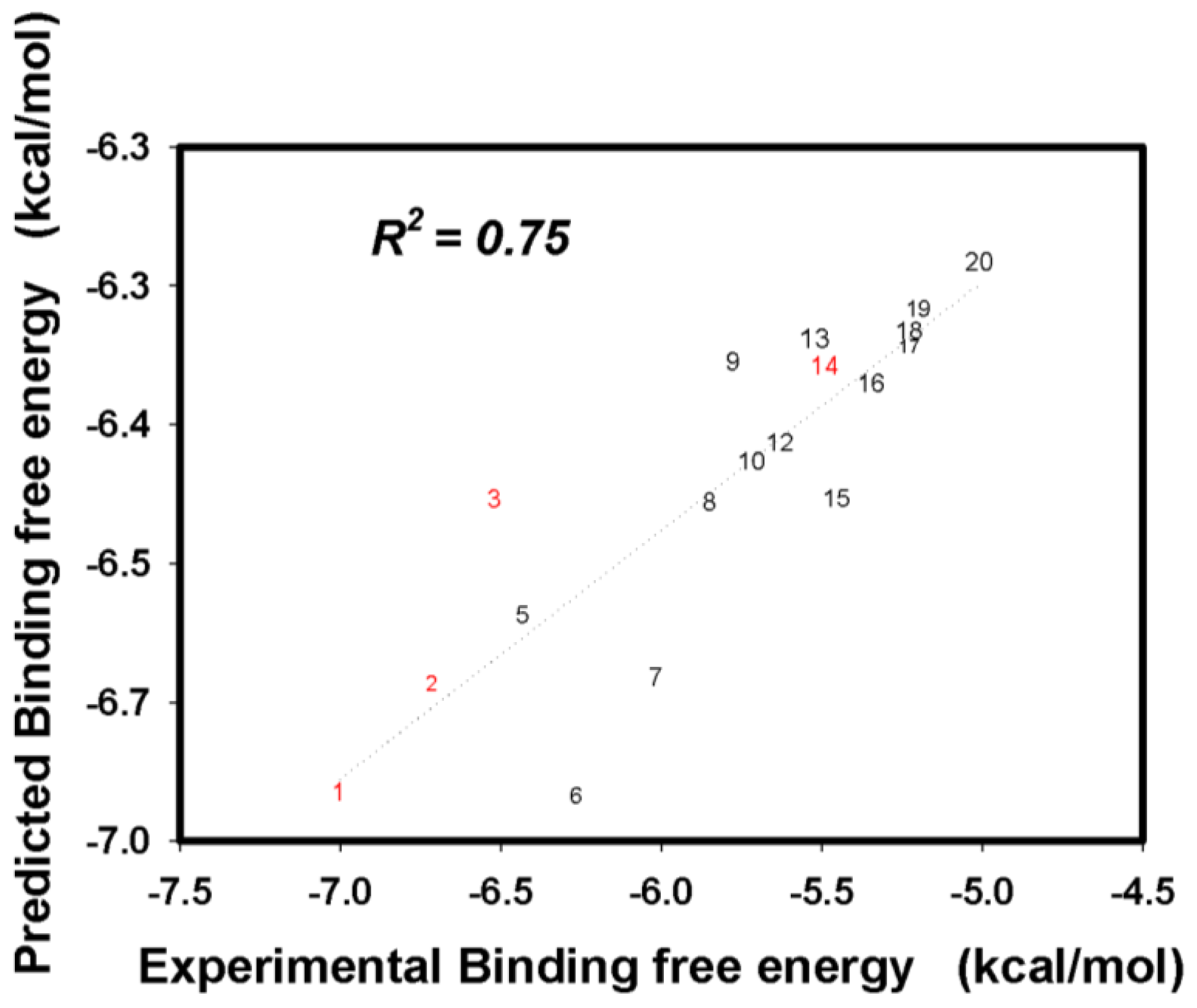
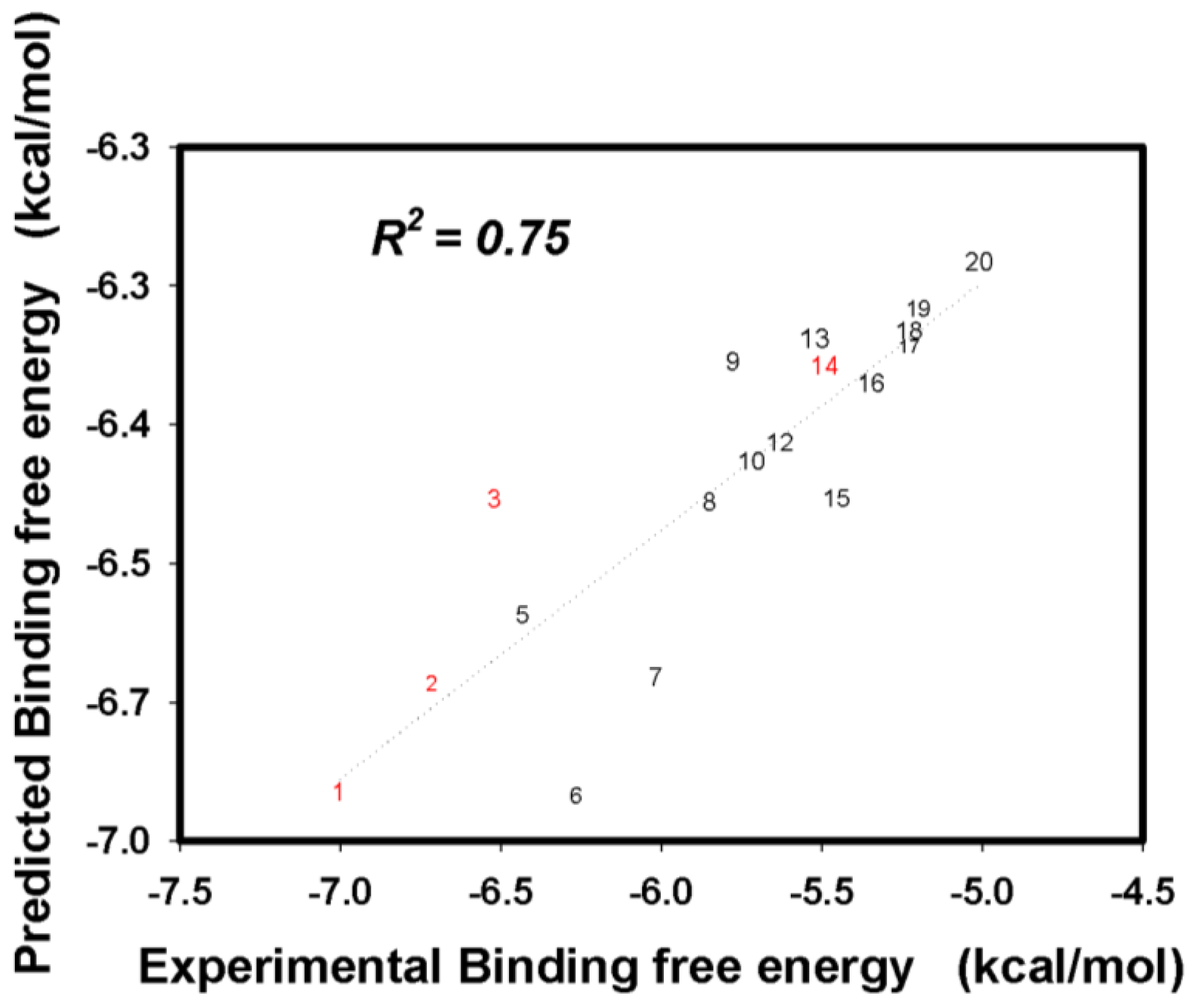
2.3. Flavonoid Derivatives Binding Free Energies
2.4. Free Energies Contribution Analysis of Hydrophobic and Hydrophilic Nature of 20 Flavonoids
3. Materials and Methods
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation
3.3. Functionally Important Residues of H1N1 Influenza Neuraminidase
3.4. Binding Free Energies Calculations (Solvated Interaction Energies Method)
4. Conclusions
Supplementary Information
ijms-13-04496-s001.pdfAcknowledgments
- Supporting InformationThe 2D structures, experimental IC50 and experimental binding free energies of 20 flavonoid derivatives are listed in Table S1. The functional residues analysis of the 20 flavonoid derivatives are listed in Figures S1–S20. Binding free energies analysis of hydrophobic/hydrophilic nature of 20 flavonoids are listed in Tables S2–S3 and Figures S21–S23.
References
- Ginting, T.E.; Shinya, K.; Kyan, Y.; Makino, A.; Matsumoto, N.; Kaneda, S.; Kawaoka, Y. Amino acid changes in hemagglutinin contribute to the replication of oseltamivir-resistant H1N1 influenza viruses. J. Virol 2011. [Google Scholar] [CrossRef]
- Van der Vries, E.; Veldhuis Kroeze, E.J.; Stittelaar, K.J.; Linster, M.; van der Linden, A.; Schrauwen, E.J.A.; Leijten, L.M.; van Amerongen, G.; Schutten, M.; Kuiken, T.; et al. Multidrug resistant 2009 A/H1N1 influenza clinical isolate with a neuraminidase I223R mutation retains its virulence and transmissibility in ferrets. PLoS Pathog 2011, 7. [Google Scholar] [CrossRef]
- Abed, Y.; Boivin, G.; Yoshida, R.; Kodama, M.; Hernandez, J.E. Parenteral peramivir treatment for oseltamivir-resistant 2009 pandemic influenza A H1N1 viruses. J. Infect. Dis 2011, 204, 1641–1642. [Google Scholar]
- Nomura, N.; Sakoda, Y.; Endo, M.; Yoshida, H.; Yamamoto, N.; Okamatsu, M.; Sakurai, K.; Hoang, N.; Nguyen, L.; Chu, H.; et al. Characterization of avian influenza viruses isolated from domestic ducks in Vietnam in 2009 and 2010. Arch. Virol 2012, 157, 247–257. [Google Scholar]
- Vavricka, C.J.; Li, Q.; Wu, Y.; Qi, J.; Wang, M.; Liu, Y.; Gao, F.; Liu, J.; Feng, E.; He, J.; et al. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog 2011, 7. [Google Scholar] [CrossRef]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar]
- Jennifer, L.M.-B. Resistance of influenza viruses to neuraminidase inhibitors—a review. Antivir. Res 2000, 47, 1–17. [Google Scholar]
- Wang, S.-Q.; Du, Q.-S.; Chou, K.-C. Study of drug resistance of chicken influenza A virus (H5N1) from homology-modeled 3D structures of neuraminidases. Biochem. Biophys. Res. Commun 2007, 354, 634–640. [Google Scholar]
- Wang, Y.-T.; Chan, C.-H.; Su, Z.-Y.; Chen, C.-L. Homology modeling, docking, and molecular dynamics reveal HR1039 as a potent inhibitor of 2009 A(H1N1) influenza neuraminidase. Biophys. Chem 2010, 147, 74–80. [Google Scholar]
- Liu, H.; Yao, X.; Wang, C.; Han, J. In silico identification of the potential drug resistance sites over 2009 influenza A (H1N1) virus neuraminidase. Mol. Pharm 2010, 7, 894–904. [Google Scholar]
- Amaro, R.E.; Swift, R.V.; Votapka, L.; Li, W.W.; Walker, R.C.; Bush, R.M. Mechanism of 150-cavity formation in influenza neuraminidase. Nat. Commun 2011, 2. [Google Scholar] [CrossRef]
- Pan, D.; Sun, H.; Bai, C.; Shen, Y.; Jin, N.; Liu, H.; Yao, X. Prediction of zanamivir efficiency over the possible 2009 Influenza A (H1N1) mutants by multiple molecular dynamics simulations and free energy calculations. J. Mol. Model 2011, 17, 2465–2473. [Google Scholar]
- Chavan, S.; Bhayye, S.; Sobhia, M. Molecular dynamics directed CoMFA studies on carbocyclic neuraminidase inhibitors. Mol. Divers 2011, 15, 979–987. [Google Scholar]
- Mai, B.K.; Li, M.S. Neuraminidase inhibitor R-125489–A promising drug for treating influenza virus: Steered molecular dynamics approach. Biochem. Biophys. Res. Commun 2011, 410, 688–691. [Google Scholar]
- Liu, A.-L.; Wang, H.-D.; Lee, S.M.; Wang, Y.-T.; Du, G.-H. Structure–activity relationship of flavonoids as influenza virus neuraminidase inhibitors and their in vitro anti-viral activities. Bioorg. Med. Chem 2008, 16, 7141–7147. [Google Scholar]
- Wei, F.; Ma, S.-C.; Ma, L.-Y.; But, P.P.-H.; Lin, R.-C.; Khan, I.A. Antiviral flavonoids from the seeds of aesculus chinensis. J. Nat. Prod 2004, 67, 650–653. [Google Scholar]
- Li, Y.; Leung, K.-T.; Yao, F.; Ooi, L.S.M.; Ooi, V.E.C. Antiviral flavans from the leaves of pithecellobium clypearia. J. Nat. Prod 2006, 69, 833–835. [Google Scholar]
- Miki, K.; Nagai, T.; Nakamura, T.; Tuji, M.; Koyama, K.; Kinoshita, K.; Furuhata, K.; Yamada, H.; Takahashi, K. Synthesis and evaluation of influenza virus sialidase inhibitory activity of hinokiflavone-sialic acid conjugates. Heterocycles 2008, 75, 879–885. [Google Scholar]
- Miki, K.; Nagai, T.; Suzuki, K.; Tsujimura, R.; Koyama, K.; Kinoshita, K.; Furuhata, K.; Yamada, H.; Takahashi, K. Anti-influenza virus activity of biflavonoids. Bioorg. Med. Chem. Lett 2007, 17, 772–775. [Google Scholar]
- Maron, D. Flavonoids for reduction of atherosclerotic risk. Curr. Atheroscler. Rep 2004, 6, 73–78. [Google Scholar]
- Huxley, R.R.; Neil, H.A.W. The relation between dietary flavonol intake and coronary heart disease mortality: A meta-analysis of prospective cohort studies. Eur. J. Clin. Nutr 2003, 57, 904–908. [Google Scholar]
- Hodgson, J.M.; Croft, K.D. Dietary flavonoids: Effects on endothelial function and blood pressure. J. Sci. Food Agric 2006, 86, 2492–2498. [Google Scholar]
- Ruiz-Ortega, M.; Esteban, V.; Egido, J. The regulation of the inflammatory response through nuclear factor-κB pathway by angiotensin IV extends the role of the renin angiotensin system in cardiovascular diseases. Trends Cardiovasc. Med 2007, 17, 19–25. [Google Scholar]
- Pignatelli, P.; Pulcinelli, F.M.; Celestini, A.; Lenti, L.; Ghiselli, A.; Gazzaniga, P.P.; Violi, F. The flavonoids quercetin and catechin synergistically inhibit platelet function by antagonizing the intracellular production of hydrogen peroxide. Am. J. Clin. Nutr 2000, 72, 1150–1155. [Google Scholar]
- Hirvonen, T.; Pietinen, P.; Virtanen, M.; Ovaskainen, M.-L.; Häkkinen, S.; Albanes, D.; Virtamo, J. Intake of flavonols and flavones and risk of coronary heart disease in male smokers. Epidemiology 2001, 12, 62–67. [Google Scholar]
- Raso, G.M.; Meli, R.; Di Carlo, G.; Pacilio, M.; di Carlo, R. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 expression by flavonoids in macrophage J774A.1. Life Sci 2001, 68, 921–931. [Google Scholar]
- Welton, A.F.; Hurley, J.; Will, P. Flavonoids and arachidonic acid metabolism. Prog. Clin. Biol. Res 1988, 280, 301–312. [Google Scholar]
- Saragusti, A.C.; Ortega, M.G.; Cabrera, J.L.; Estrin, D.A.; Marti, M.A.; Chiabrando, G.A. Inhibitory effect of quercetin on matrix metalloproteinase 9 activity Molecular mechanism and structure—activity relationship of the flavonoid—enzyme interaction. Eur. J. Pharmacol 2010, 644, 138–145. [Google Scholar]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol 2000, 10, 322–328. [Google Scholar]
- Lanzarotti, E.; Biekofsky, R.R.; Estrin, D.A.; Marti, M.A.; Turjanski, A.G. Aromatic—Aromatic interactions in proteins: Beyond the dimer. J. Chem. Inf. Model 2011, 51, 1623–1633. [Google Scholar]
- Espinoza-Fonseca, L.M.; García-Machorro, J. Aromatic–aromatic interactions in the formation of the MDM2-p53 complex. Biochem. Biophys. Res. Commun 2008, 370, 547–551. [Google Scholar]
- Gauto, D.F.; Di Lella, S.; Guardia, C.M.A.; Estrin, D.A.; Martií, M.A. Carbohydrate-binding proteins: Dissecting ligand structures through solvent environment occupancy. J. Phys. Chem. B 2009, 113, 8717–8724. [Google Scholar]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R.A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc 2008, 130, 2817–2831. [Google Scholar]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Energetics of displacing water molecules from protein binding sites: Consequences for ligand optimization. J. Am. Chem. Soc 2009, 131, 15403–15411. [Google Scholar]
- Di Lella, S.; Martí, M.A.; Álvarez, R.M.S.; Estrin, D.A.; Ricci, J.C.D. Characterization of the galectin-1 carbohydrate recognition domain in terms of solvent occupancy. J. Phys. Chem. B 2007, 111, 7360–7366. [Google Scholar]
- Gauto, D.F.; di Lella, S.; Estrin, D.A.; Monaco, H.L.; Martí, M.A. Structural basis for ligand recognition in a mushroom lectin: Solvent structure as specificity predictor. Carbohydr. Res 2011, 346, 939–948. [Google Scholar]
- Mercader, A.G.; Pomilio, A.B. QSAR study of flavonoids and biflavonoids as influenza H1N1 virus neuraminidase inhibitors. Eur. J. Med. Chem 2010, 45, 1724–1730. [Google Scholar]
- Li, Q.; Qi, J.; Zhang, W.; Vavricka, C.J.; Shi, Y.; Wei, J.; Feng, E.; Shen, J.; Chen, J.; Liu, D.; et al. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat. Struct. Mol. Biol 2010, 17, 1266–1268. [Google Scholar]
- Grienke, U.; Schmidtke, M.; von Grafenstein, S.; Kirchmair, J.; Liedl, K.R.; Rollinger, J.M. Influenza neuraminidase: A druggable target for natural products. Nat. Prod. Rep 2012, 29, 11–36. [Google Scholar]
- Nervall, M.; Hanspers, P.; Carlsson, J.; Boukharta, L.; Åqvist, J. Predicting binding modes from free energy calculations. J. Med. Chem 2008, 51, 2657–2667. [Google Scholar]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA–An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des 2004, 18, 167–173. [Google Scholar]
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: Review on chemdraw, chemwindow, ISIS/draw, and chemsketch. J. Chem. Inf. Comput. Sci 2004, 44, 1886–1890. [Google Scholar]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol 2008, 82, 10493–10501. [Google Scholar]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. NNScore 2.0: A neural-network receptor–ligand scoring function. J. Chem. Inf. Model 2011, 51, 2897–2903. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [Google Scholar]
- Bren, U.; Hodošček, M.; Koller, J. Development and validation of empirical force field parameters for netropsin. J. Chem. Inf. Model 2005, 45, 1546–1552. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem 1993, 14, 1347–1363. [Google Scholar]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys 2010, 12, 7821–7839. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T., III; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, J.A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668–1688. [Google Scholar]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng 1995, 8, 127–134. [Google Scholar]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. exploring the parameter space. J. Chem. Inf. Model 2007, 47, 122–133. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Hydrogen bonding–related residues | Non-bonding contact-related residues |
|---|---|---|
| 1(A) | Trp179, Thr226, Asn295 | Tyr402 |
| 2(A) | Glu277, Asn295 | Null |
| 3(A) | Glu119, Asn295 | Null |
| 4(B) | Glu119, Ser180, Glu228, Asp294, Val346 | Null |
| 5(B) | Val346 | Null |
| 6(B) | Arg152, Glu228, Glu277 Asn295 | Null |
| 7(B) | Asn295 | Trp179, Ile223 |
| 8(B) | Glu119, Ser180, Glu228, Asp294, Val346 | Null |
| 9(B) | Arg152, Glu228, Val346 | Null |
| 10(B) | Ser180, Thr226, Glu228, Asp294 | Null |
| 11(B) | Glu228, Glu277 | Null |
| 12(B) | Asp151, Glu277, Asn325, Val346, Ser366 | Null |
| 13(B) | Trp179, Asn295 | Null |
| 14(A) | Asn295 | Null |
| 15(B) | Arg152, Trp179, Asn295 | Null |
| 16(B) | Glu119, Asp151, Ser366, Ser367, Tyr402 | Ile223, Arg368 |
| 17(B) | Trp179 | Null |
| 18(B) | Asp151, Arg152, Asn344 | Null |
| 19(B) | Asp151, Trp179, Glu228, Asn295 | Pro326 |
| 20(B) | Arg152, Trp179, Ser247, Glu277 | Null |
| Inhibitors | Energy (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| EC | Evwd | ΔMSA | ΔGbindR | ΔGbind (SIE) | ΔGbind (Experiment) | |
| 1 | −20.7 | −9.95 | 6.12 | −6.98 | −6.825 | −7.002 |
| 2 | −20.9 | −7.36 | 5.29 | −5.64 | −6.435 | −6.714 |
| 3 | −15.2 | −6.69 | 4.83 | −5.71 | −5.771 | −6.518 |
| 4 | −20.3 | −5.31 | 5.31 | −5.91 | −6.186 | −6.430 |
| 5 | −16.9 | −6.12 | 5.81 | −7.41 | −6.071 | −6.296 |
| 6 | −24.1 | −7.26 | 6.43 | −6.39 | −6.837 | −6.265 |
| 7 | −20.9 | −7.36 | 5.11 | −5.43 | −6.413 | −6.014 |
| 8 | −16.9 | −4.81 | 6.57 | −5.94 | −5.779 | −5.847 |
| 9 | −12.9 | −4.64 | 5.11 | −5.33 | −5.277 | −5.773 |
| 10 | −15.4 | −4.73 | 5.59 | −6.11 | −5.633 | −5.717 |
| 11 | −13.9 | −4.91 | 6.19 | −6.82 | −5.567 | −5.629 |
| 12 | −12.2 | −6.71 | 3.91 | −6.08 | −5.503 | −5.608 |
| 13 | −11.4 | −5.73 | 4.41 | −4.91 | −5.193 | −5.519 |
| 14 | −12.4 | −5.12 | 5.25 | −5.51 | −5.291 | −5.489 |
| 15 | −15.4 | −4.73 | 5.65 | −6.41 | −5.766 | −5.450 |
| 16 | −9.3 | −8.73 | 6.81 | −5.56 | −5.353 | −5.343 |
| 17 | −12.9 | −4.32 | 4.81 | −5.09 | −5.221 | −5.226 |
| 18 | −12.7 | −4.21 | 4.61 | −4.84 | −5.163 | −5.225 |
| 19 | −11.6 | −4.29 | 4.82 | −5.11 | −5.084 | −5.199 |
| 20 | −10.1 | −4.33 | 4.99 | −4.97 | −4.916 | −5.007 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, S.-J.; Chong, F.-C. Combining Molecular Docking and Molecular Dynamics to Predict the Binding Modes of Flavonoid Derivatives with the Neuraminidase of the 2009 H1N1 Influenza A Virus. Int. J. Mol. Sci. 2012, 13, 4496-4507. https://doi.org/10.3390/ijms13044496
Lu S-J, Chong F-C. Combining Molecular Docking and Molecular Dynamics to Predict the Binding Modes of Flavonoid Derivatives with the Neuraminidase of the 2009 H1N1 Influenza A Virus. International Journal of Molecular Sciences. 2012; 13(4):4496-4507. https://doi.org/10.3390/ijms13044496
Chicago/Turabian StyleLu, Shih-Jen, and Fok-Ching Chong. 2012. "Combining Molecular Docking and Molecular Dynamics to Predict the Binding Modes of Flavonoid Derivatives with the Neuraminidase of the 2009 H1N1 Influenza A Virus" International Journal of Molecular Sciences 13, no. 4: 4496-4507. https://doi.org/10.3390/ijms13044496