Alzheimer’s Disease-Associated Neurotoxic Peptide Amyloid-Β Impairs Base Excision Repair in Human Neuroblastoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Cytotoxicity Assay
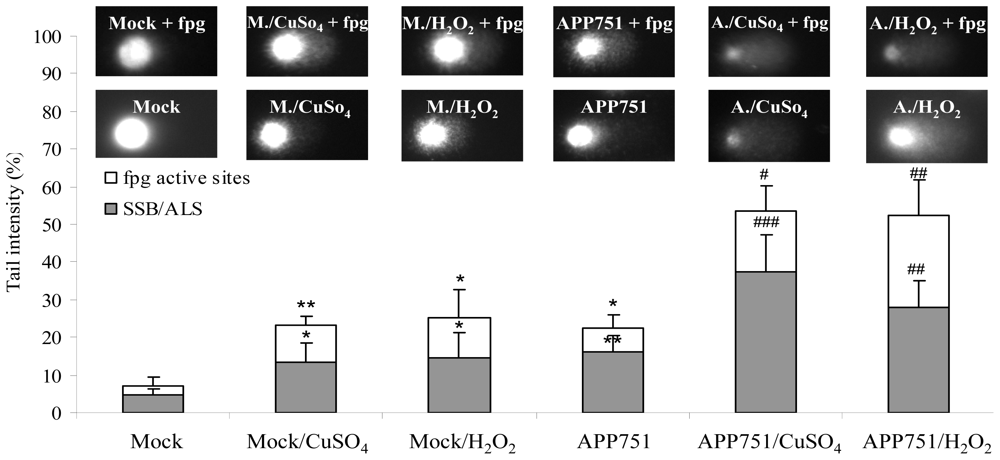
2.1.2. Nuclear DNA Damage Is Increased in APP751-Expressing Cells
2.1.3. Mitochondrial DNA Damage Is Increased in APP751-Expressing Cells
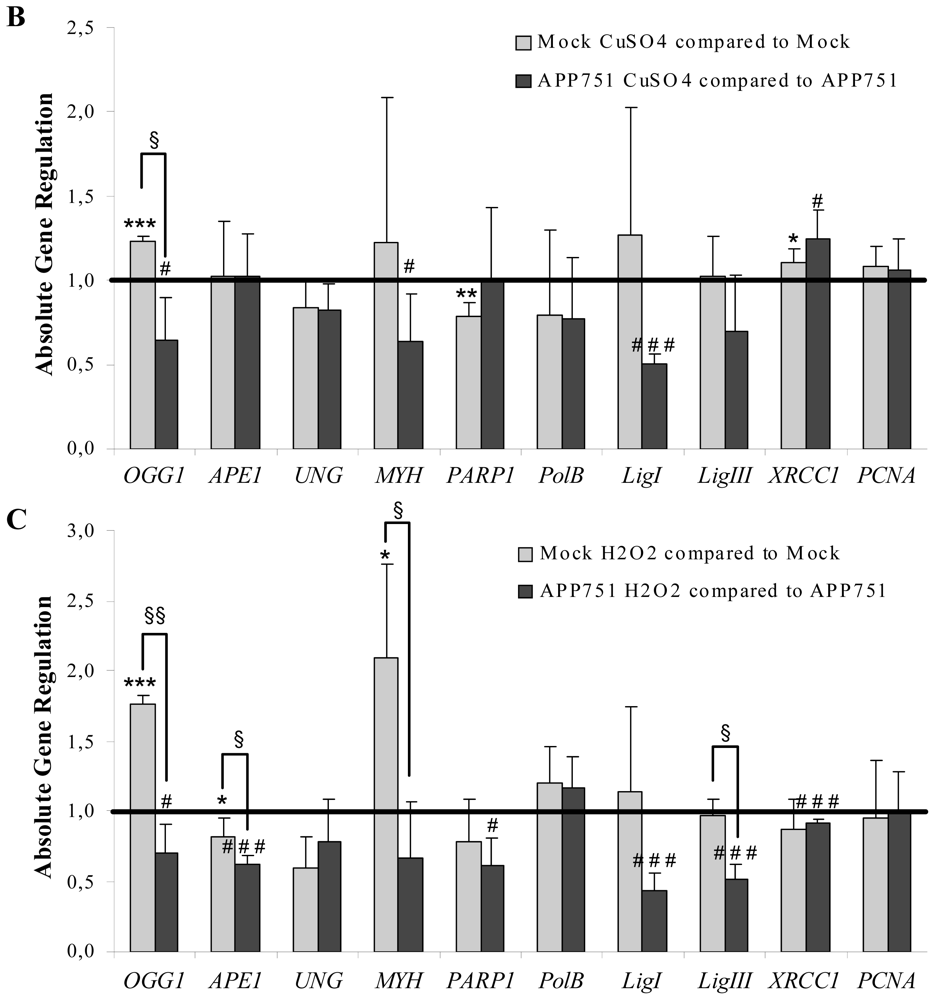
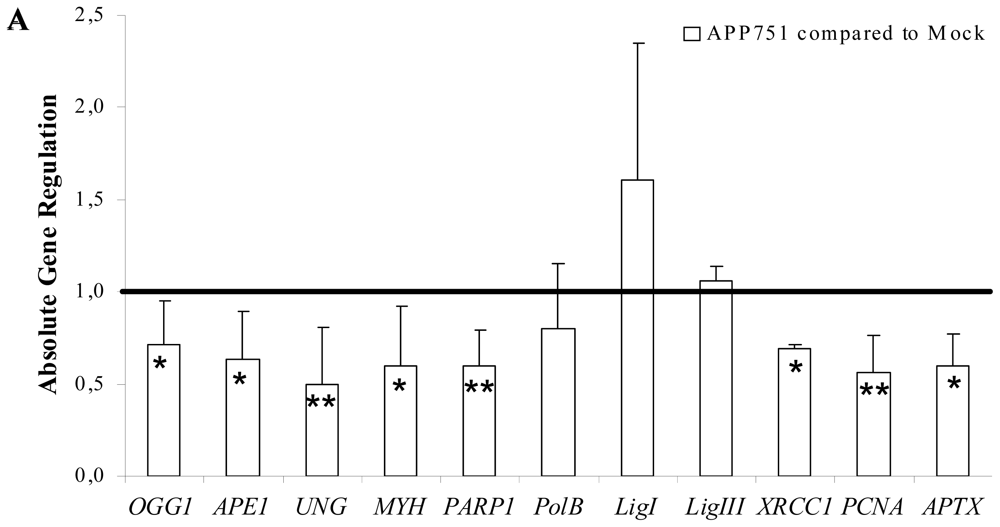
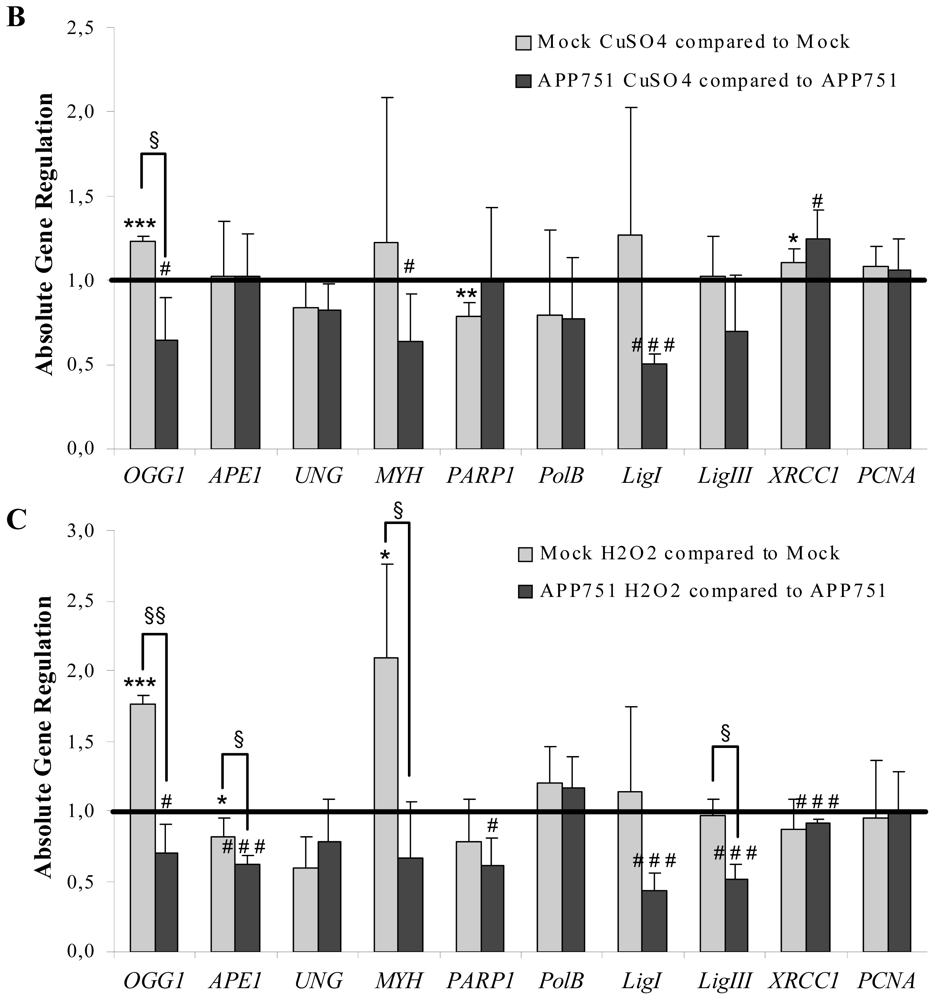
2.1.4. Aβ Secretion Leads to an Overall Downregulation of BER Genes
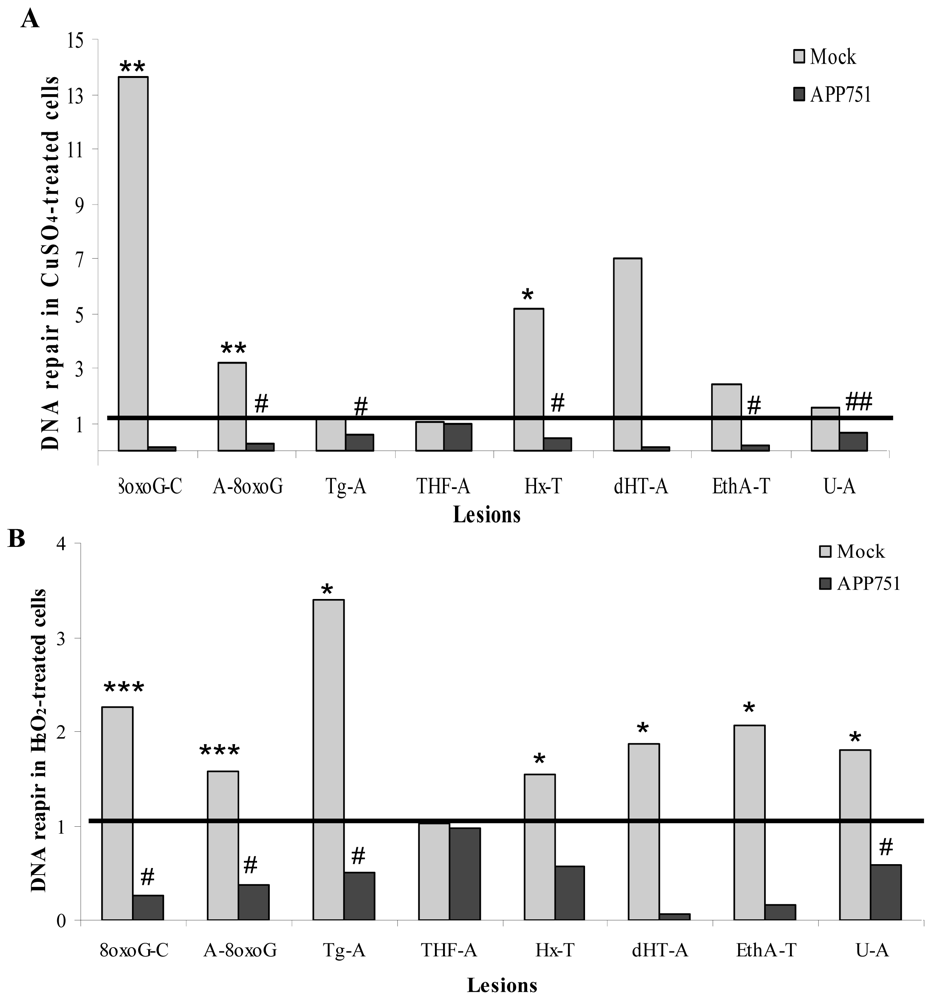
2.1.5. Aβ Secretion Decreases 8oxoG Excision Capacity
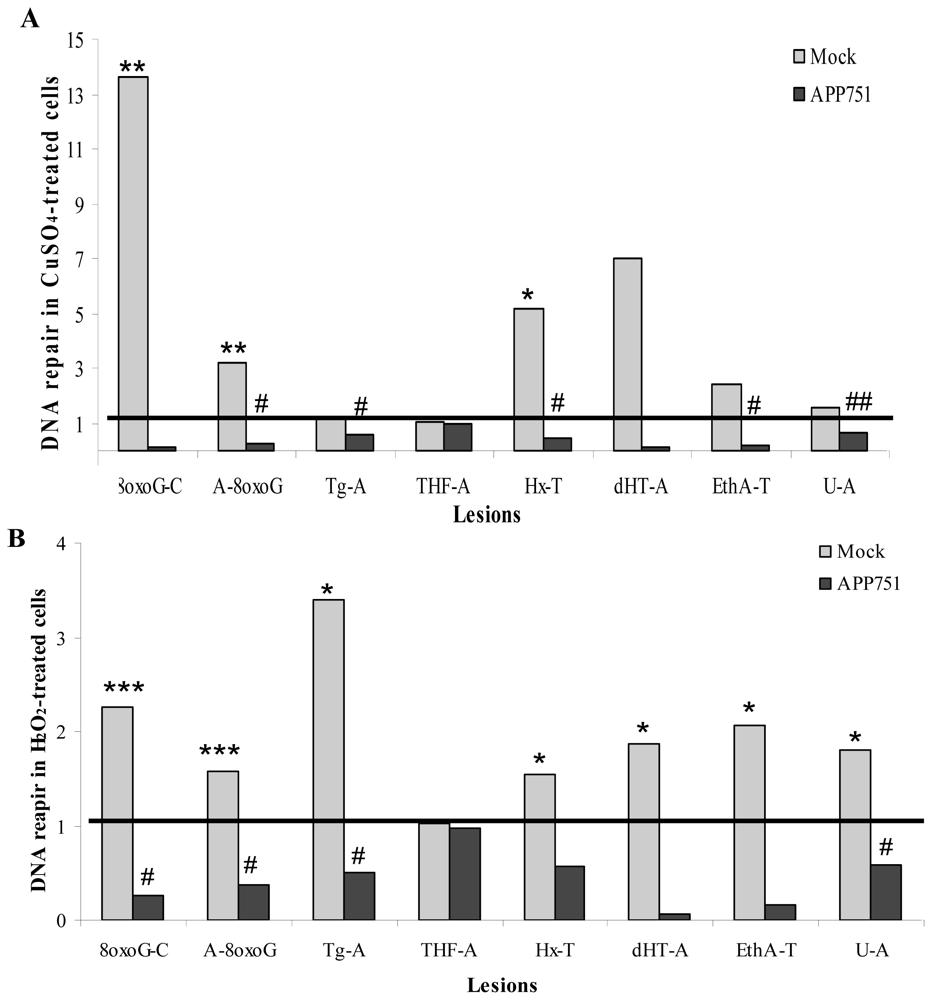
2.1.6. Evaluation of Small Damaged Base Excision Capacity Using a Multiplex Assay: ODN Biochip
2.2. Discussion
3. Experimental Section
3.1. Culture of SKNSH-SY5Y Neuroblastoma Cells (Mock and APP751-Expressing)
3.2. Cytotoxicity Assay
3.3. Preparation of Frozen Pellets
3.4. Quantification of the Common Deletion in Mitochondrial DNA
3.4.1. Lysate Preparation
3.4.2. qPCR Amplification of Nuclear, Total Mitochondrial and Deleted Mitochondrial DNA
3.5. Alkaline Comet Assay
3.6. Reverse Transcription (RT) and Real-Time Quantitative PCR (qPCR) Analysis
3.7. Comet-Based DNA Repair Assays
3.7.1. Substrate Preparation
3.7.2. Preparation of Whole-Cell Extracts
3.7.3. Measurement of DNA Repair Capacities
3.8. Oligonucleotide (ODN) Biochip
3.8.1. Preparation of Nuclear Extracts
3.8.2. Preparation of Lesion ODN Biochip
3.8.3. Excision Reaction
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar]
- Edelberg, H.K.; Wei, J.Y. The biology of Alzheimer’s disease. Mech. Ageing Dev 1996, 91, 95–114. [Google Scholar]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 1997, 20, 154–159. [Google Scholar]
- Forman, M.S.; Cook, D.G.; Leight, S.; Doms, R.W.; Lee, V.M. Differential effects of the swedish mutant amyloid precursor protein on beta-amyloid accumulation and secretion in neurons and nonneuronal cells. J. Biol. Chem 1997, 272, 32247–32253. [Google Scholar]
- Park, S.Y.; Kim, H.S.; Cho, E.K.; Kwon, B.Y.; Phark, S.; Hwang, K.W.; Sul, D. Curcumin protected PC12 cells against beta-amyloid-induced toxicity through the inhibition of oxidative damage and tau hyperphosphorylation. Food Chem. Toxicol 2008, 46, 2881–2887. [Google Scholar]
- Migliore, L.; Coppede, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res 2008, 667, 82–97. [Google Scholar]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol 1999, 56, 1449–1452. [Google Scholar]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy samples of Alzheimer’s cortex show increased peroxidation in vitro. J. Neurochem 1990, 55, 342–345. [Google Scholar]
- Smith, M.A.; Rudnicka-Nawrot, M.; Richey, P.L.; Praprotnik, D.; Mulvihill, P.; Miller, C.A.; Sayre, L.M.; Perry, G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J. Neurochem 1995, 64, 2660–2666. [Google Scholar]
- Kadioglu, E.; Sardas, S.; Aslan, S.; Isik, E.; Esat Karakaya, A. Detection of oxidative DNA damage in lymphocytes of patients with Alzheimer’s disease. Biomarkers 2004, 9, 203–209. [Google Scholar]
- Mecocci, P.; Polidori, M.C.; Ingegni, T.; Cherubini, A.; Chionne, F.; Cecchetti, R.; Senin, U. Oxidative damage to DNA in lymphocytes from AD patients. Neurology 1998, 51, 1014–1017. [Google Scholar]
- Morocz, M.; Kalman, J.; Juhasz, A.; Sinko, I.; McGlynn, A.P.; Downes, C.S.; Janka, Z.; Rasko, I. Elevated levels of oxidative DNA damage in lymphocytes from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 47–53. [Google Scholar]
- Wang, J.; Xiong, S.; Xie, C.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem 2005, 93, 953–962. [Google Scholar]
- Herrup, K.; Neve, R.; Ackerman, S.L.; Copani, A. Divide and die: Cell cycle events as triggers of nerve cell death. J. Neurosci 2004, 24, 9232–9239. [Google Scholar]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 2007, 35, 7497–7504. [Google Scholar]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem 1998, 71, 2034–2040. [Google Scholar]
- Lovell, M.A.; Gabbita, S.P.; Markesbery, W.R. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem 1999, 72, 771–776. [Google Scholar]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppede, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Aging 2005, 26, 567–573. [Google Scholar]
- Moreira, P.I.; Nunomura, A.; Nakamura, M.; Takeda, A.; Shenk, J.C.; Aliev, G.; Smith, M.A.; Perry, G. Nucleic acid oxidation in Alzheimer disease. Free Radic. Biol. Med 2008, 44, 1493–1505. [Google Scholar]
- Krokan, H.E.; Nilsen, H.; Skorpen, F.; Otterlei, M.; Slupphaug, G. Base excision repair of DNA in mammalian cells. FEBS Lett 2000, 476, 73–77. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar]
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res 2007, 35, 5545–5555. [Google Scholar]
- Shao, C.; Xiong, S.; Li, G.M.; Gu, L.; Mao, G.; Markesbery, W.R.; Lovell, M.A. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer’s disease brain. Free Radic. Biol. Med 2008, 45, 813–819. [Google Scholar]
- Weissman, L.; de Souza-Pinto, N.C.; Mattson, M.P.; Bohr, V.A. DNA base excision repair activities in mouse models of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 2080–2081. [Google Scholar]
- Nunomura, A.; Tamaoki, T.; Tanaka, K.; Motohashi, N.; Nakamura, M.; Hayashi, T.; Yamaguchi, H.; Shimohama, S.; Lee, H.G.; Zhu, X.; et al. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol. Dis 2010, 37, 731–737. [Google Scholar]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med 1997, 23, 134–147. [Google Scholar]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar]
- Hung, Y.H.; Bush, A.I.; Cherny, R.A. Copper in the brain and Alzheimer’s disease. J. Biol. Inorg. Chem 2010, 15, 61–76. [Google Scholar]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Begard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis 2005, 20, 625–637. [Google Scholar]
- Zhang, D.L.; Chen, Y.Q.; Jiang, X.; Ji, T.T.; Mei, B. Oxidative damage increased in presenilin1/presenilin2 conditional double knockout mice. Neurosci. Bull 2009, 25, 131–137. [Google Scholar]
- Du, Y.; Wooten, M.C.; Gearing, M.; Wooten, M.W. Age-associated oxidative damage to the p62 promoter: implications for Alzheimer disease. Free Radic. Biol. Med 2009, 46, 492–501. [Google Scholar]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol 2000, 130, 184–208. [Google Scholar]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimers Dis 2006, 10, 145–163. [Google Scholar]
- Peinnequin, A.; Poyot, T.; Dib, A.; Aubourg, A.; Mouret, C.; Demeilliers, C. Direct quantification of mitochondrial DNA and its 4.9-kb common deletion without DNA purification. Anal. Biochem 2011, 409, 298–300. [Google Scholar]
- Mao, G.; Pan, X.; Zhu, B.B.; Zhang, Y.; Yuan, F.; Huang, J.; Lovell, M.A.; Lee, M.P.; Markesbery, W.R.; Li, G.M.; et al. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res 2007, 35, 2759–2766. [Google Scholar]
- Kruman, I.I.; Schwartz, E.; Kruman, Y.; Cutler, R.G.; Zhu, X.; Greig, N.H.; Mattson, M.P. Suppression of uracil-DNA glycosylase induces neuronal apoptosis. J. Biol. Chem 2004, 279, 43952–43960. [Google Scholar]
- Vasko, M.R.; Guo, C.; Kelley, M.R. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair Amst 2005, 4, 367–379. [Google Scholar]
- Tan, Z.; Sun, N.; Schreiber, S.S. Immunohistochemical localization of redox factor-1 Ref-1 in Alzheimer’s hippocampus. Neuroreport 1998, 9, 2749–2752. [Google Scholar]
- Boerrigter, M.E.; van Duijn, C.M.; Mullaart, E.; Eikelenboom, P.; van der Togt, C.M.; Knook, D.L.; Hofman, A.; Vijg, J. Decreased DNA repair capacity in familial, but not in sporadic Alzheimer’s disease. Neurobiol. Aging 1991, 12, 367–370. [Google Scholar]
- Fujimura, M.; Morita-Fujimura, Y.; Sugawara, T.; Chan, P.H. Early decrease of XRCC1, a DNA base excision repair protein, may contribute to DNA fragmentation after transient focal cerebral ischemia in mice. Stroke 1999, 30, 2456–2462, discussion 2463. [Google Scholar]
- Fujimura, M.; Morita-Fujimura, Y.; Noshita, N.; Yoshimoto, T.; Chan, P.H. Reduction of the DNA base excision repair protein, XRCC1, may contribute to DNA fragmentation after cold injury-induced brain trauma in mice. Brain Res 2000, 869, 105–111. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med 2006, 12, 440–450. [Google Scholar]
- Van de Loosdrecht, A.A.; Beelen, R.H.; Ossenkoppele, G.J.; Broekhoven, M.G.; Langenhuijsen, M.M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar]
- Sauvaigo, S.; Petec-Calin, C.; Caillat, S.; Odin, F.; Cadet, J. Comet assay coupled to repair enzymes for the detection of oxidative damage to DNA induced by low doses of gamma-radiation: Use of YOYO-1, low-background slides, and optimized electrophoresis conditions. Anal. Biochem 2002, 303, 107–109. [Google Scholar]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett 2004, 26, 509–515. [Google Scholar]
- Millau, J.F.; Raffin, A.L.; Caillat, S.; Claudet, C.; Arras, G.; Ugolin, N.; Douki, T.; Ravanat, J.L.; Breton, J.; Oddos, T.; et al. A microarray to measure repair of damaged plasmids by cell lysates. Lab Chip 2008, 8, 1713–1722. [Google Scholar]
- Sauvaigo, S.; Guerniou, V.; Rapin, D.; Gasparutto, D.; Caillat, S.; Favier, A. An oligonucleotide microarray for the monitoring of repair enzyme activity toward different DNA base damage. Anal. Biochem 2004, 333, 182–192. [Google Scholar]



© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Forestier, A.; Douki, T.; Sauvaigo, S.; De Rosa, V.; Demeilliers, C.; Rachidi, W. Alzheimer’s Disease-Associated Neurotoxic Peptide Amyloid-Β Impairs Base Excision Repair in Human Neuroblastoma Cells. Int. J. Mol. Sci. 2012, 13, 14766-14787. https://doi.org/10.3390/ijms131114766
Forestier A, Douki T, Sauvaigo S, De Rosa V, Demeilliers C, Rachidi W. Alzheimer’s Disease-Associated Neurotoxic Peptide Amyloid-Β Impairs Base Excision Repair in Human Neuroblastoma Cells. International Journal of Molecular Sciences. 2012; 13(11):14766-14787. https://doi.org/10.3390/ijms131114766
Chicago/Turabian StyleForestier, Anne, Thierry Douki, Sylvie Sauvaigo, Viviana De Rosa, Christine Demeilliers, and Walid Rachidi. 2012. "Alzheimer’s Disease-Associated Neurotoxic Peptide Amyloid-Β Impairs Base Excision Repair in Human Neuroblastoma Cells" International Journal of Molecular Sciences 13, no. 11: 14766-14787. https://doi.org/10.3390/ijms131114766