Novel High-Viscosity Polyacrylamidated Chitosan for Neural Tissue Engineering: Fabrication of Anisotropic Neurodurable Scaffold via Molecular Disposition of Persulfate-Mediated Polymer Slicing and Complexation

 , ,
, ,
Abstract
:1. Introduction
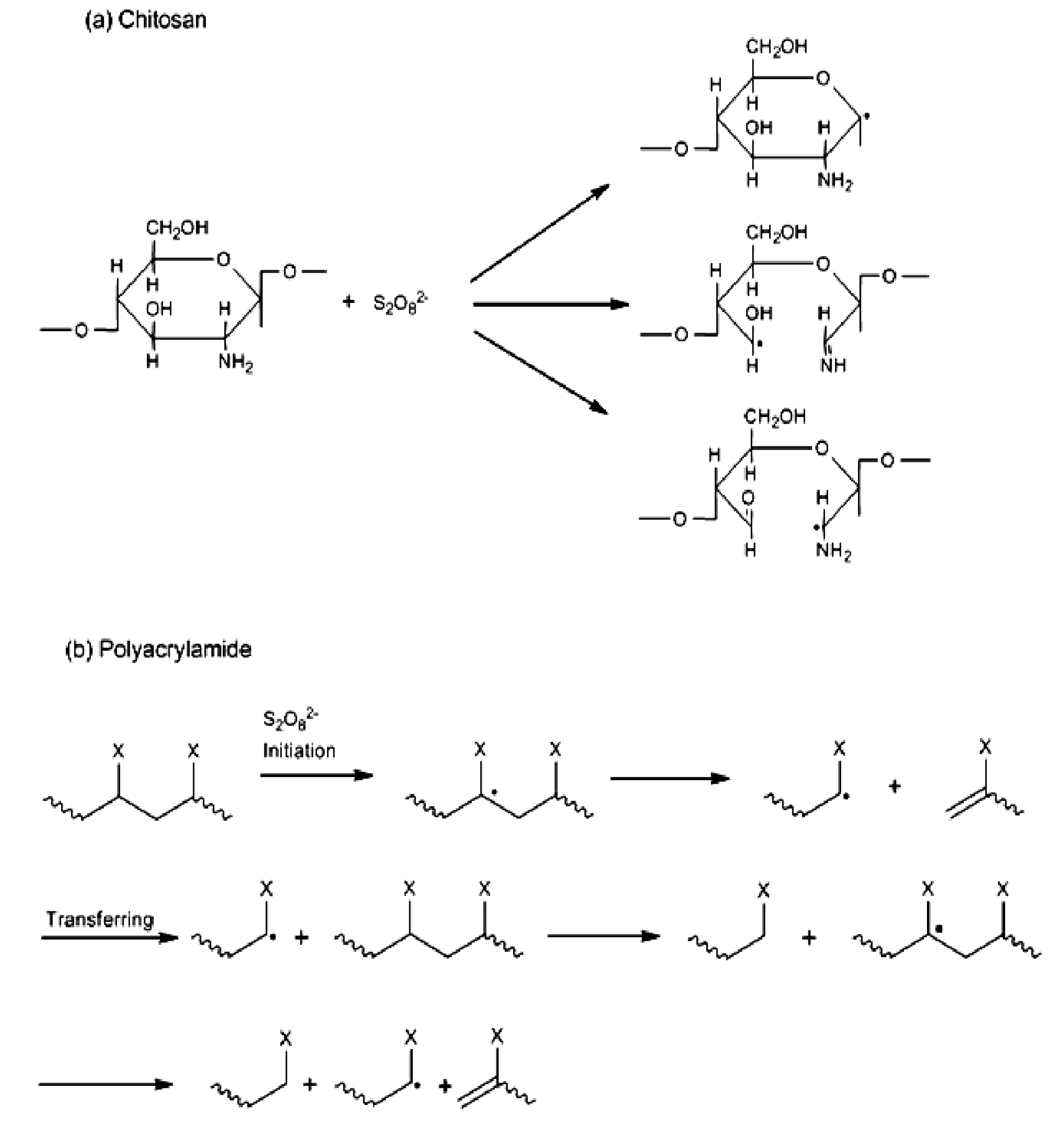
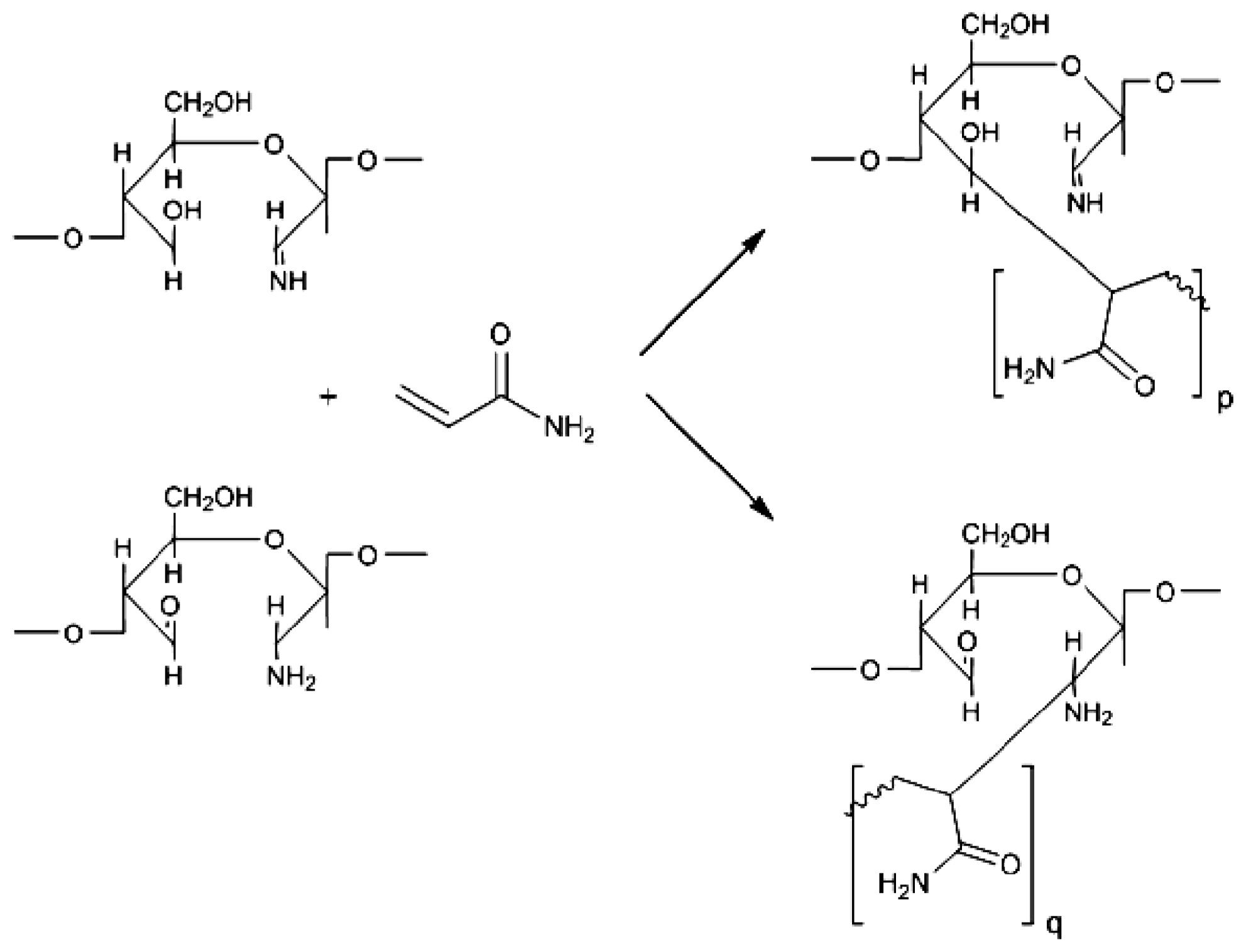
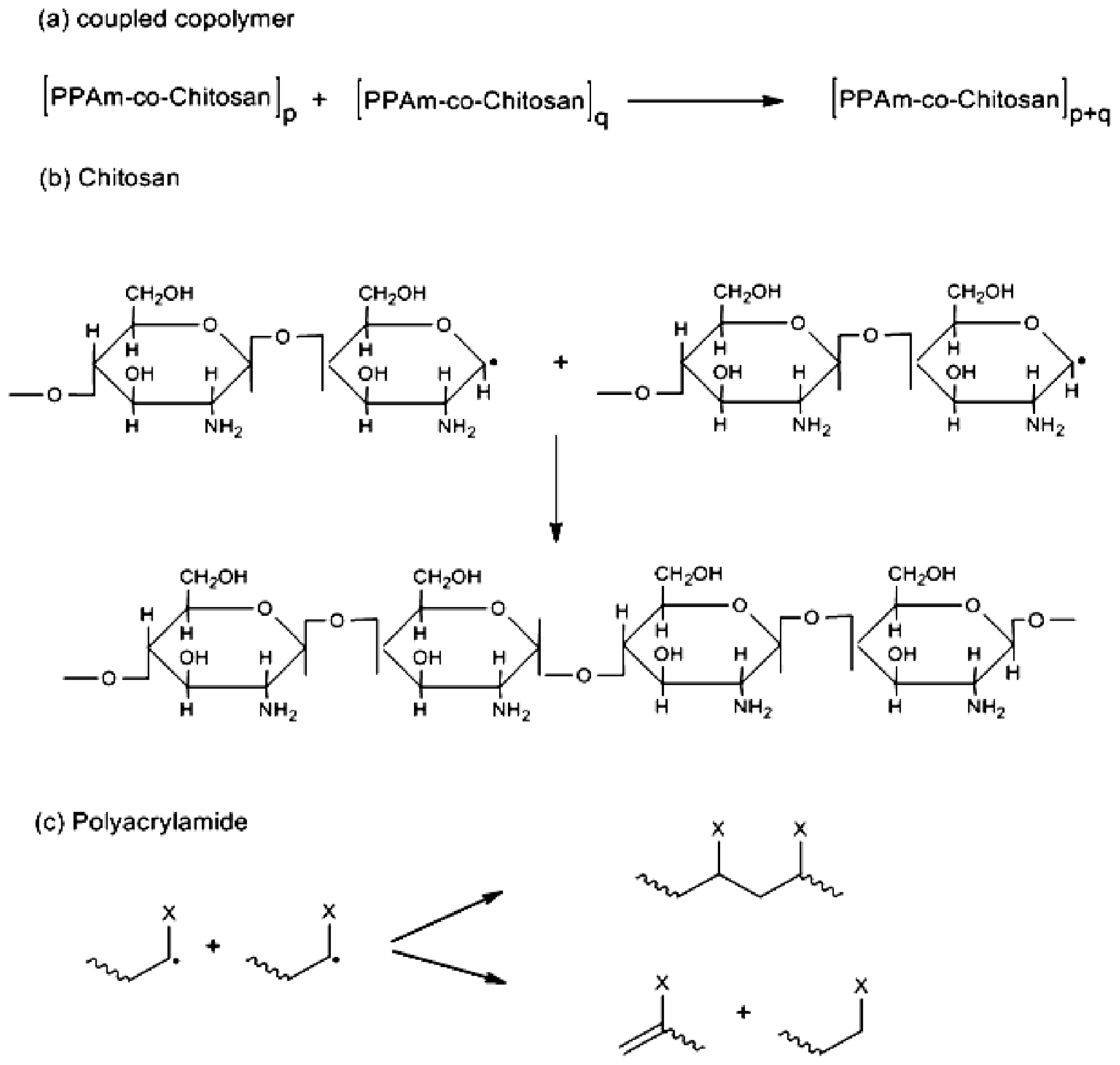
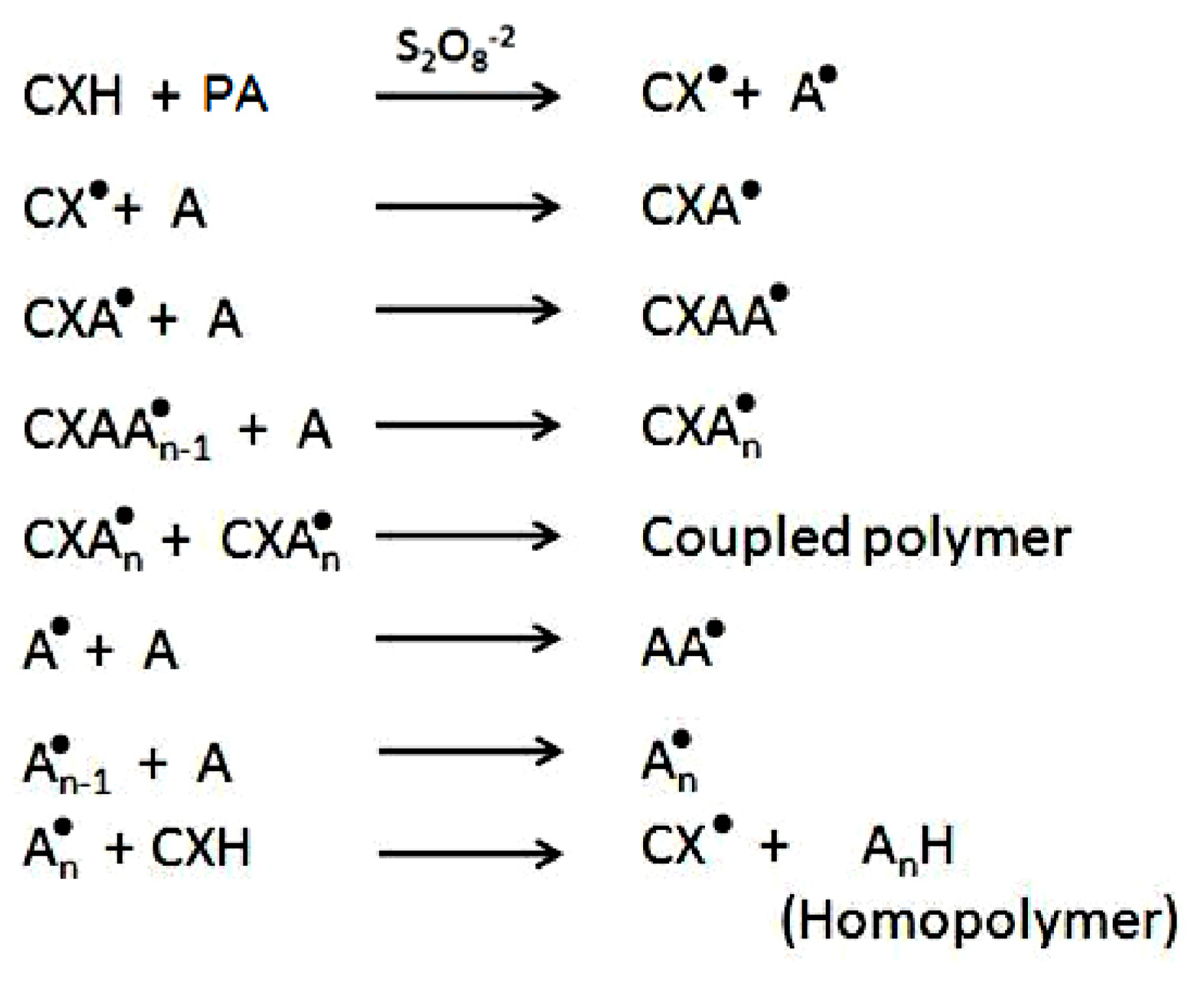
2. Results and Discussion
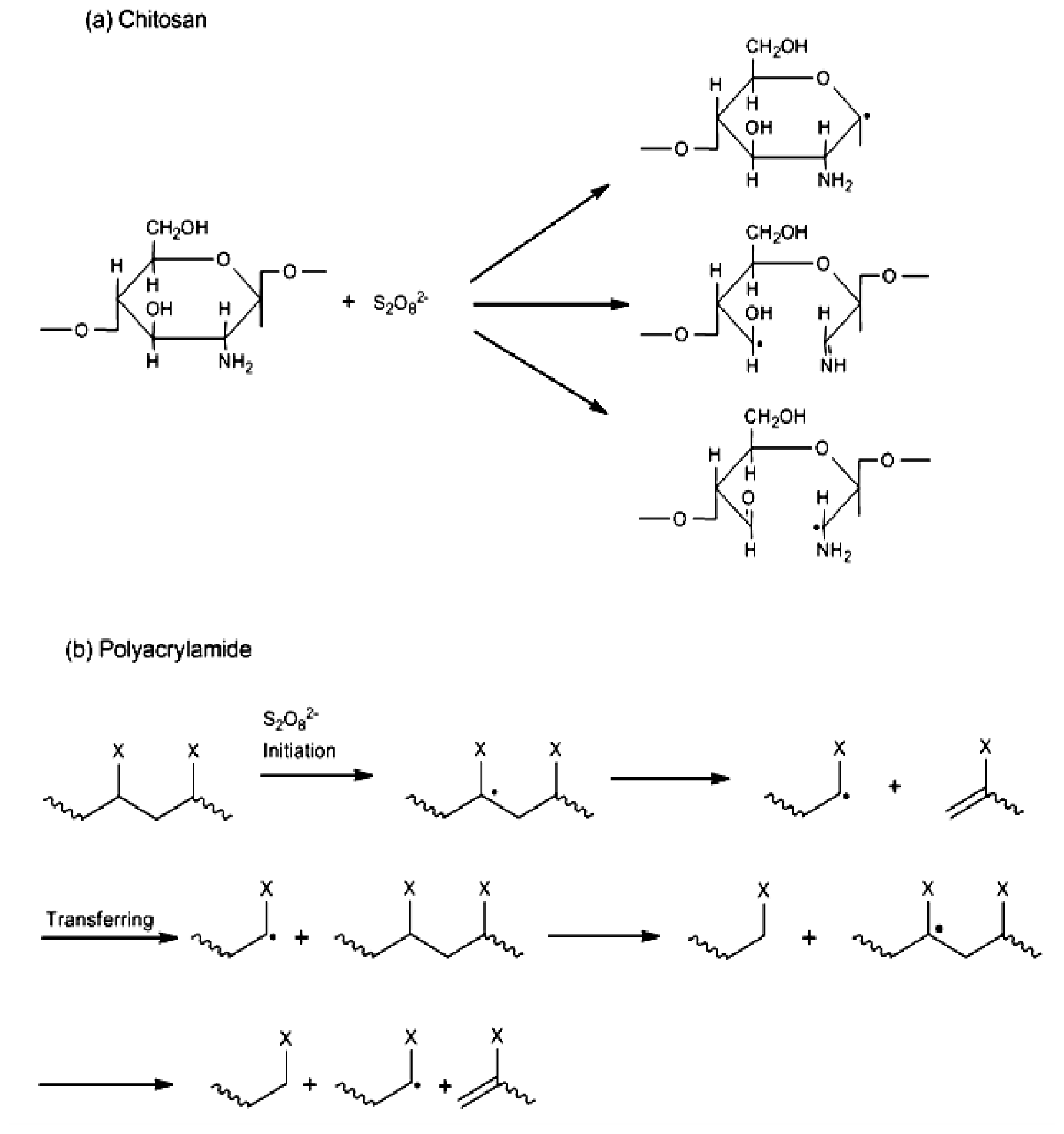
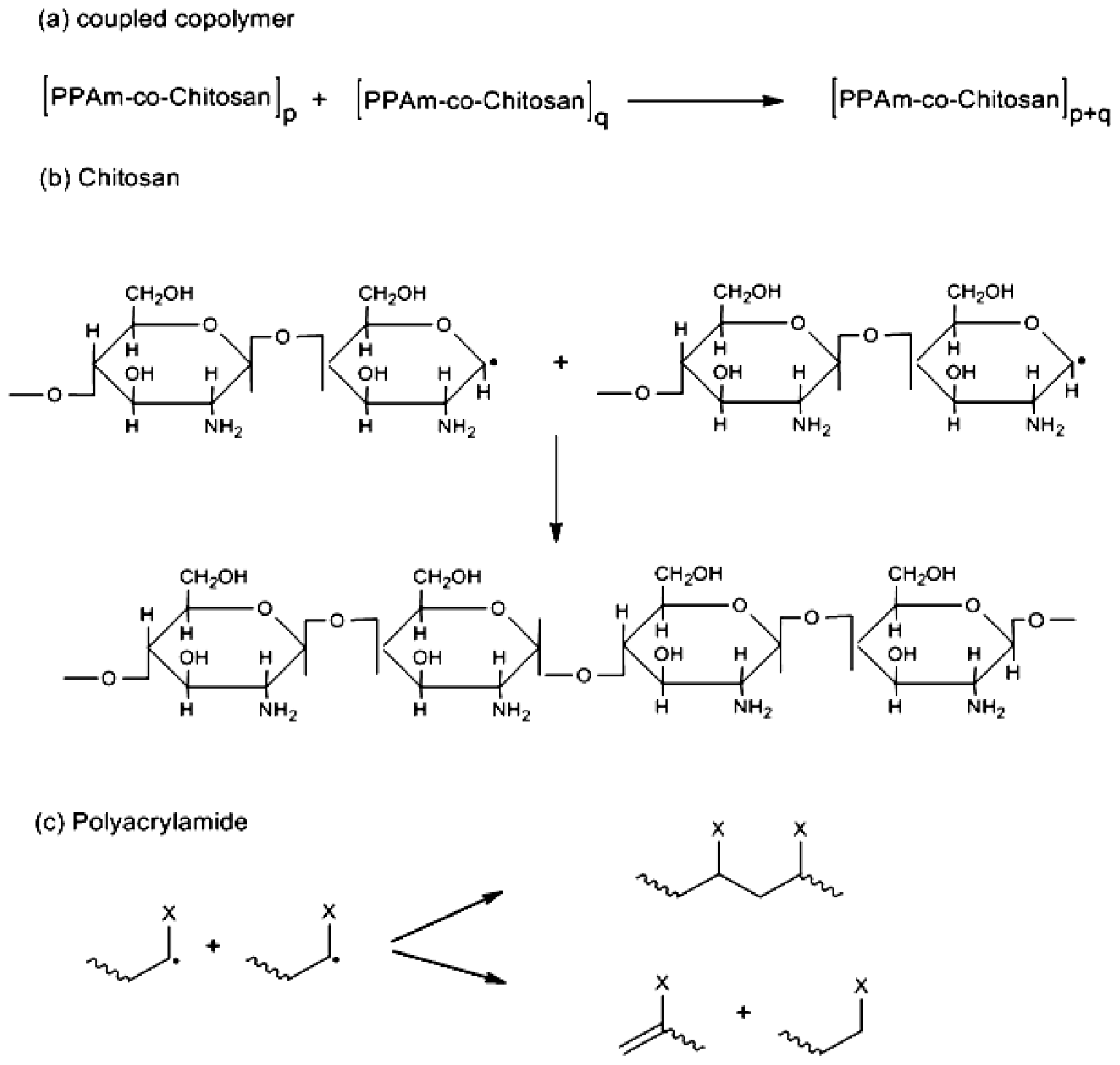
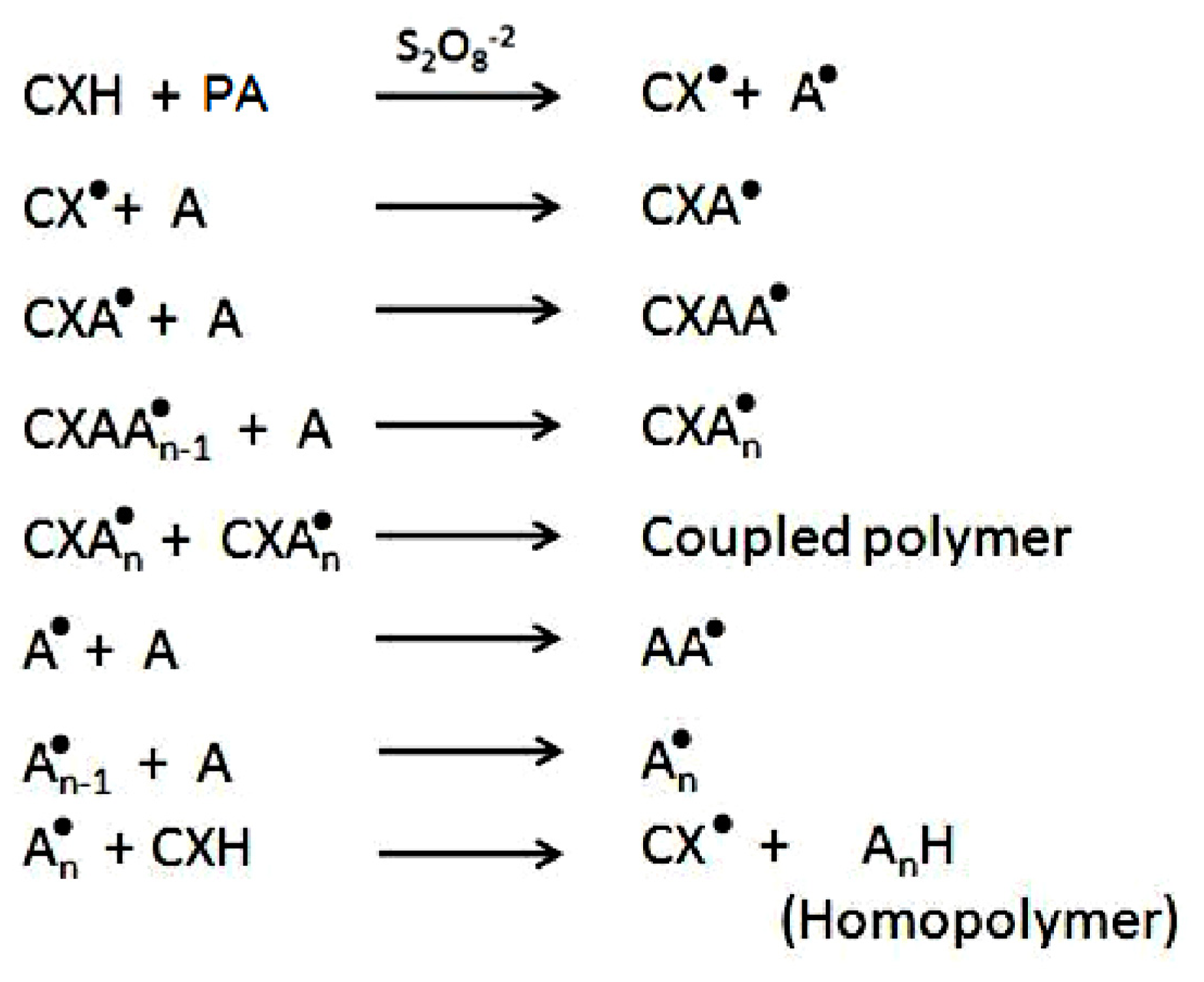
2.1. Copolymer Synthesis, Grafting Parameters and Molecular Mass Analysis
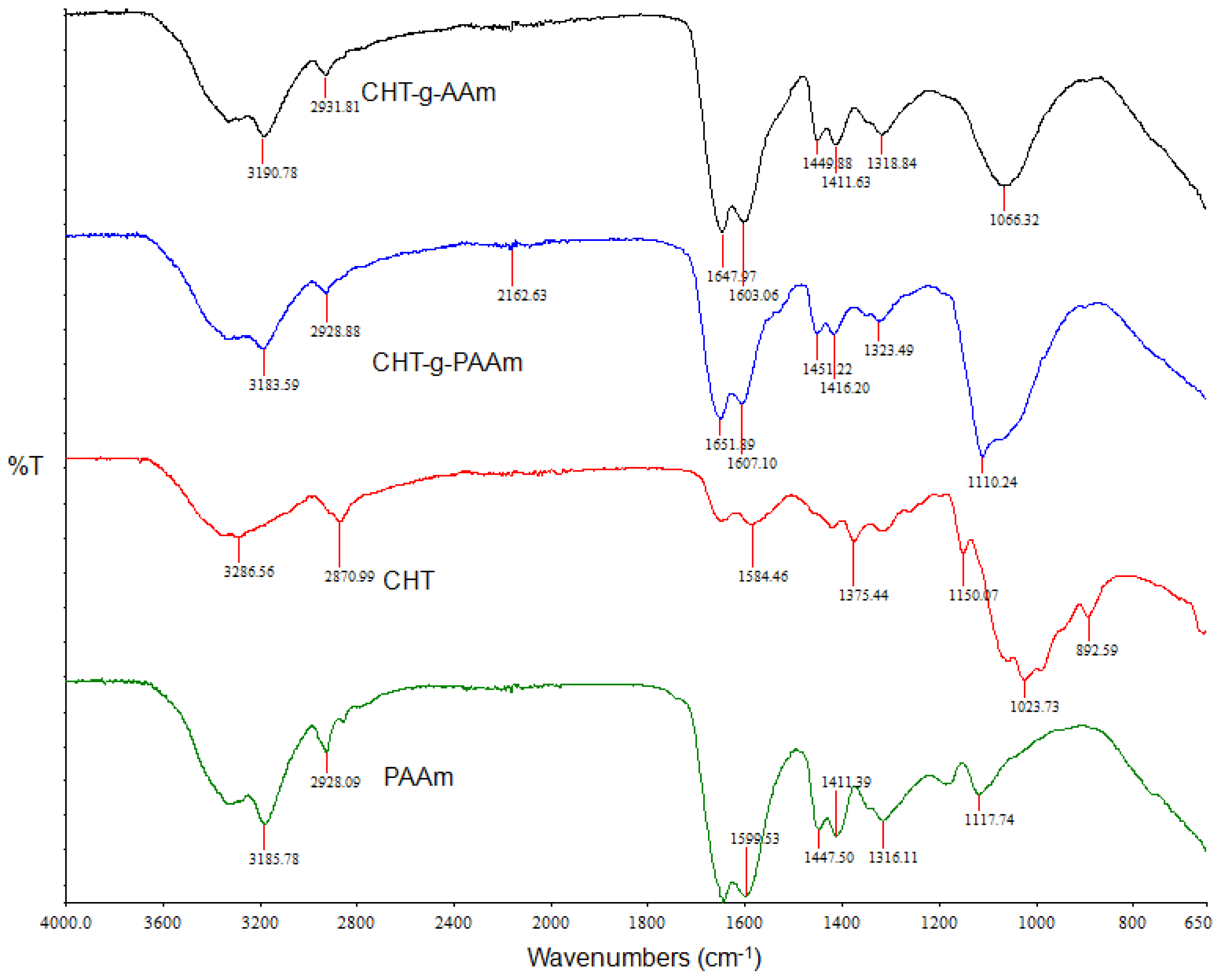
2.2. Investigation of Polymeric Structural Transitions
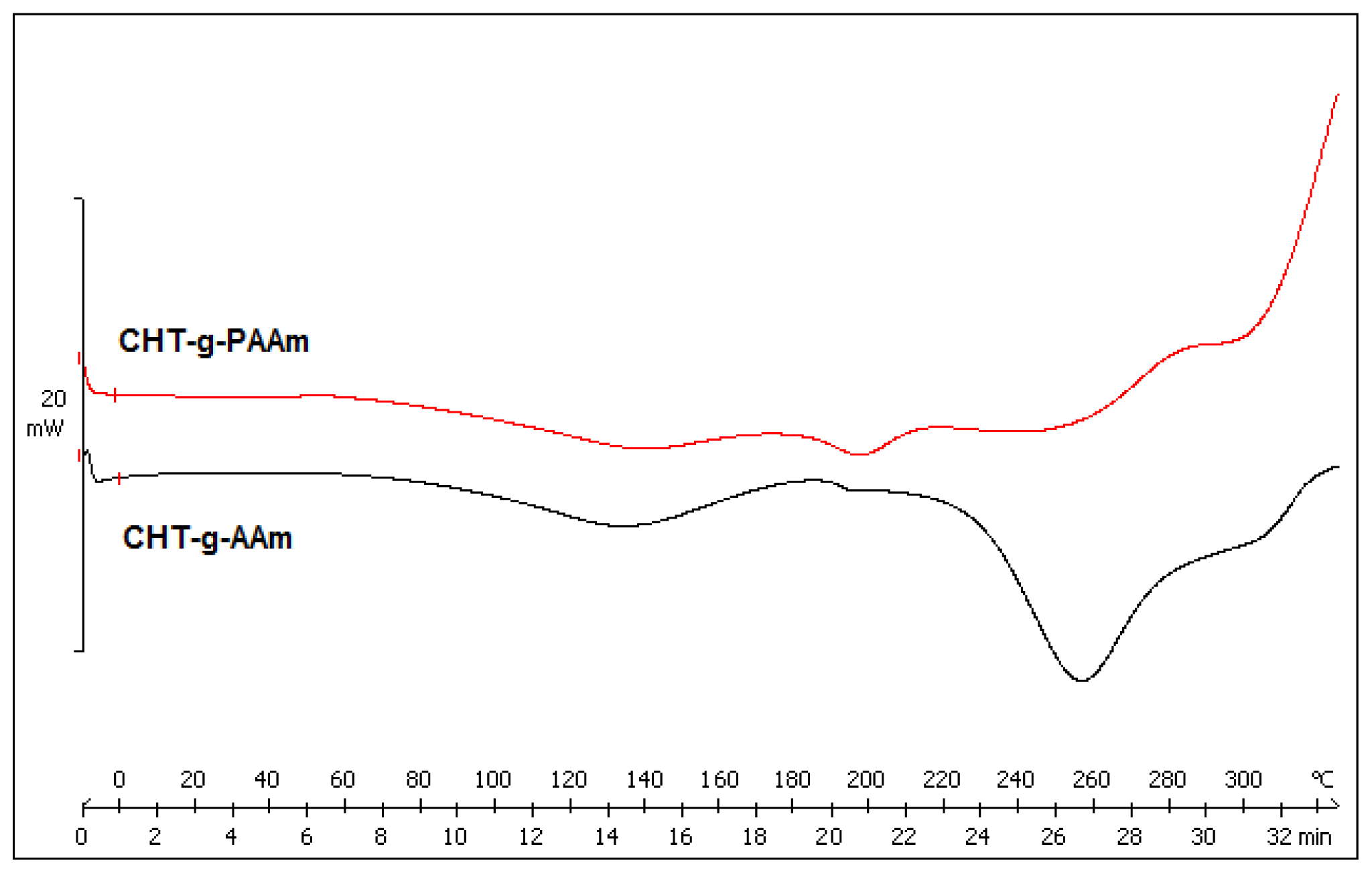
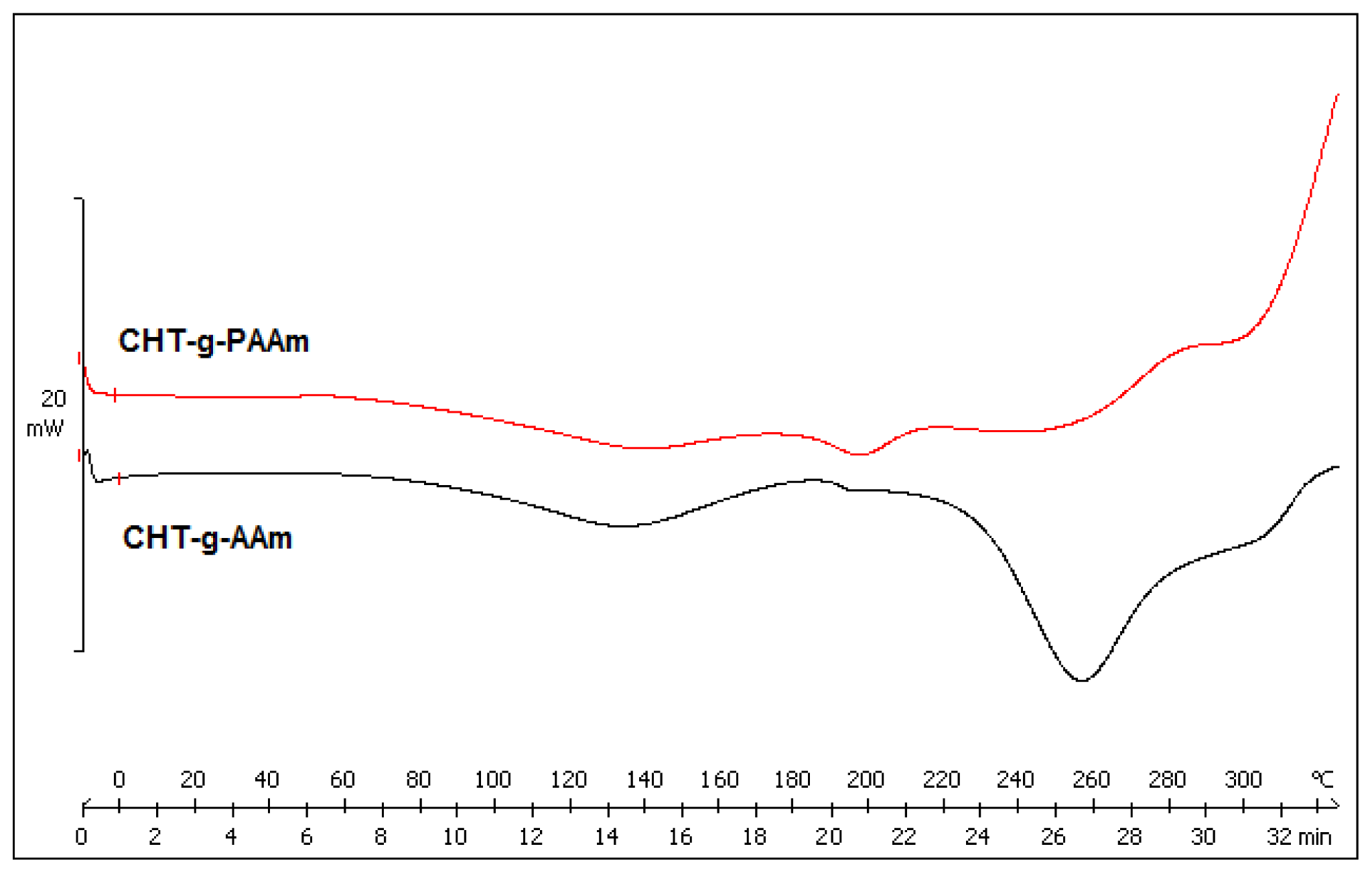
2.3. Comparison of Thermally-Induced Transitions of the Grafted Copolymers
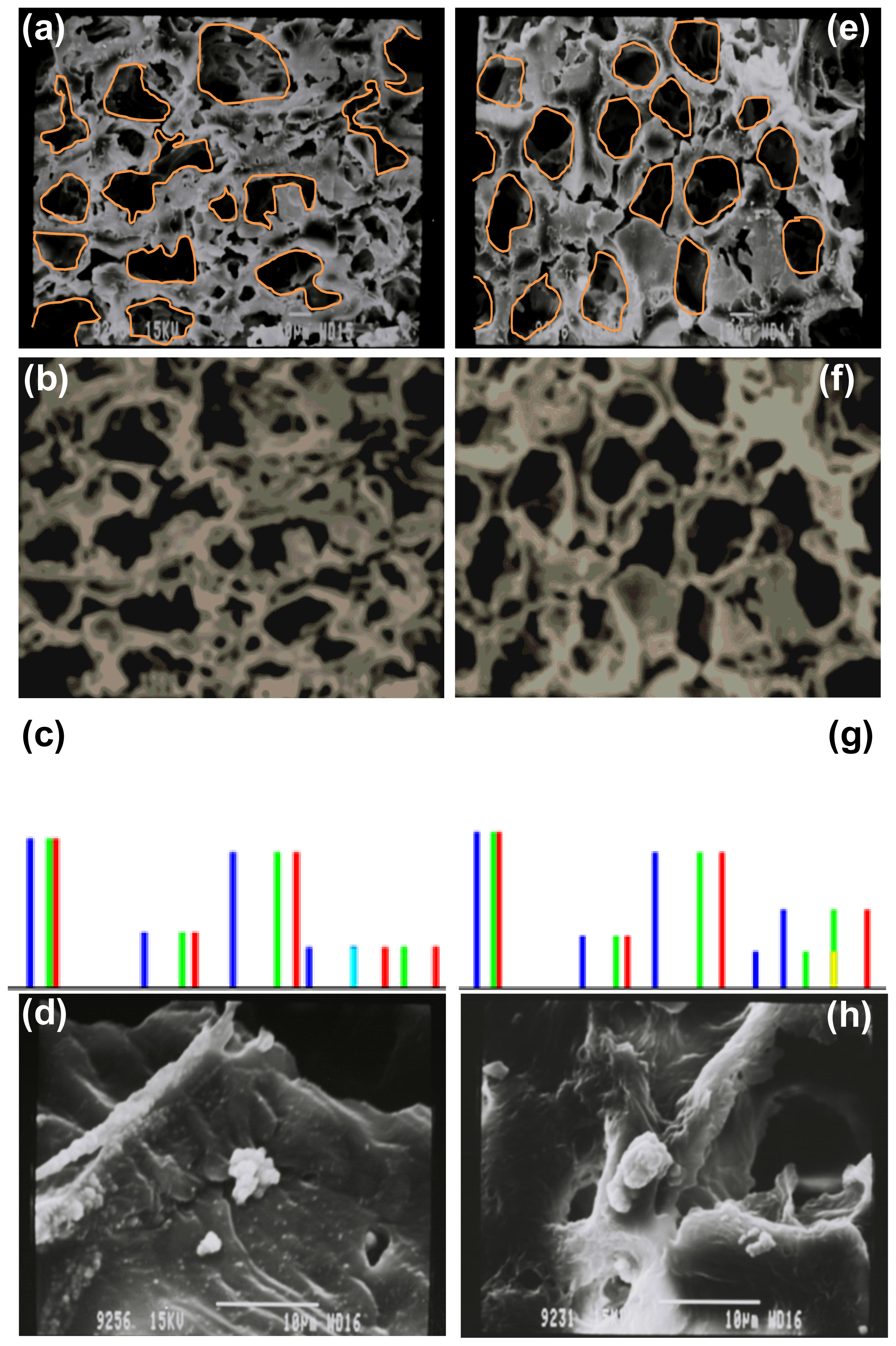
2.4. Morphological Characterization and Quantitative Image Processing Analysis
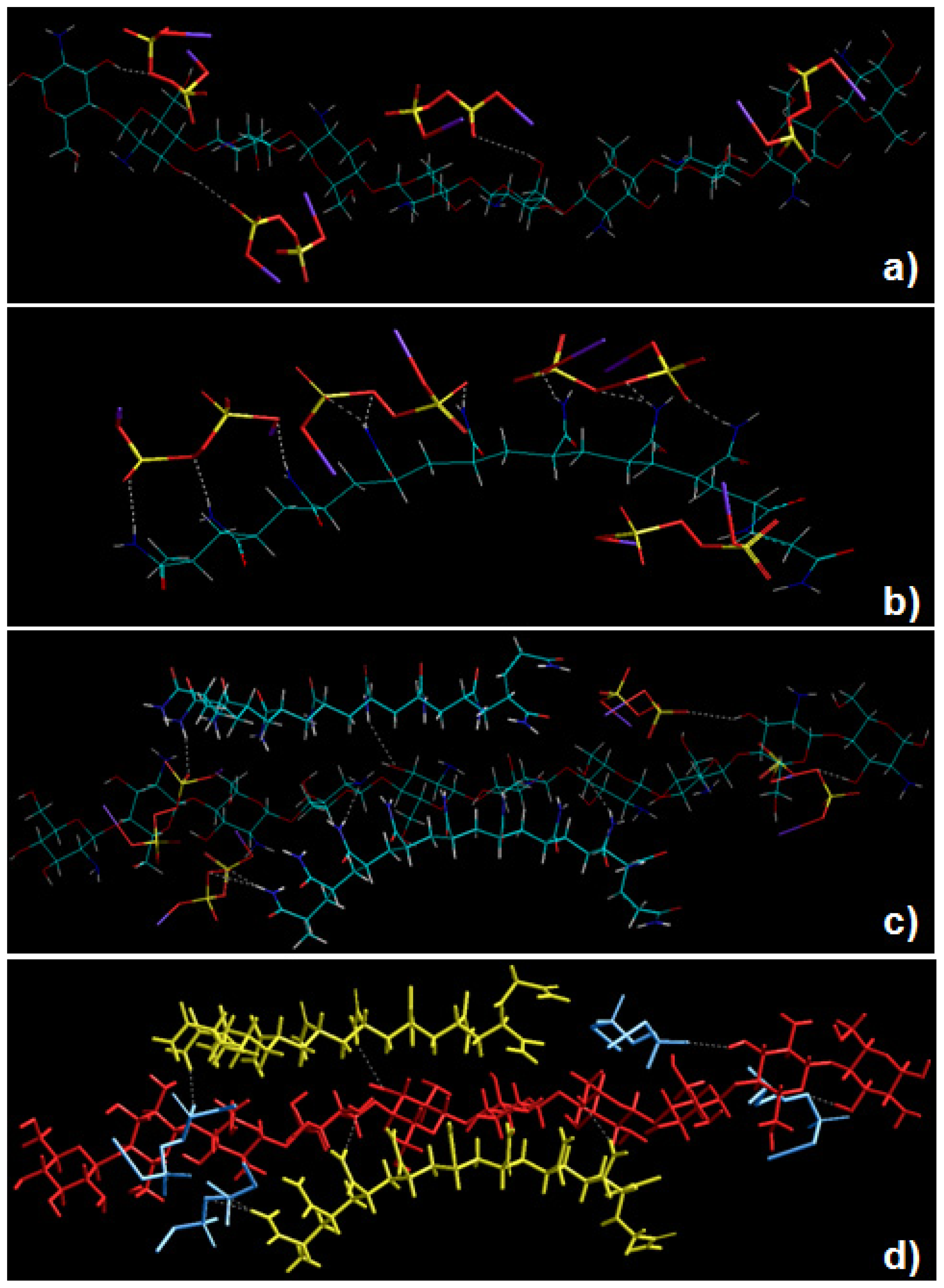
2.5. Static Lattice Atomistic Simulations
3. Experimental Section
3.1. Materials
3.2. Synthesis of Polyacrylamidated Chitosan Using Monomer (AAm-g-CHT)
3.3. Synthesis of Polyacrylamidated Chitosan Using Polymer (PAAm-g-CHT)
3.4. Cell-Culture and Cell-Seeding for Ex Vivo Tissue Engineering Evaluation
3.5. Determination of Grafting Parameters
3.6. Determination of the Approximate Molecular Mass of the Grafted Polymer
3.7. Polymeric Structural Variation Analysis
3.8. Exothermic and Endothermic Mapping of the Grafted Polymers
3.9. Establishment of the Reactional Profile and Mechanisms via SLAS
4. Conclusions
Acknowledgments
References
- Kim, I.Y.; Seo, S.J.; Moon, H.S.; Yoo, M.K.; Park, I.Y.; Kim, B.C.; Cho, C.S. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv 2008, 26, 1–21. [Google Scholar]
- Van Vlierberghe, S.; Dubruel, P.; Schacht, E. Biopolymer-based hydrogels as scaffold for tissue engineering applications: A review. Biomacromolecules 2011, 12, 1387–1408. [Google Scholar]
- Jayakumar, R.; Prabaharan, M.; Reis, R.L.; Mano, J.F. Graft copolymerized chitosan—Present status and applications. Carbohyd. Polym 2005, 62, 142–158. [Google Scholar]
- Alves, N.M.; Mano, J.F. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int. J. Biol. Macromol 2008, 43, 401–414. [Google Scholar]
- Prabaharan, M. Chitosan derivatives as promising materials for controlled drug delivery. J. Biomater. Appl 2008, 23, 5–36. [Google Scholar]
- Andrade, F.; Goycoolea, F.; Chiappetta, D.A.; das Neves, J.; Sosnik, A.; Sarmento, B. Chitosan-grafted copolymers and chitosan-ligand conjugates as matrices for pulmonary drug delivery. Int. J. Carbohyd. Chem. 2011. [Google Scholar] [CrossRef]
- Zohuriaan-Mehr, M.J. Advances in chitin and chitosan modification through graft copolymerization: A comprehensive review. Iran. Polym. J 2005, 14, 235–265. [Google Scholar]
- Cheng, M.; Gong, K.; Li, J.; Gong, Y.; Zhao, N.; Zhang, X. Surface modification and characterization of chitosan film blended with poly-l-lysine. J. Biomater. Appl 2004, 19, 59–75. [Google Scholar]
- Yang, T.H. Recent applications of polyacrylamide as biomaterials. Recent Patents Mater. Sci 2008, 1, 29–40. [Google Scholar]
- Zhu, Y.; Gao, C.; Guan, J.; Shen, J. Promoting the cytocompatibility of polyurethane scaffold via surface photo-grafting polymerization of acrylamide. J. Mater. Sci. Mater. Med 2004, 15, 283–289. [Google Scholar]
- Kumbar, S.G.; Soppimath, K.S.; Aminabhavi, T.M. Synthesis and characterization of polyacrylamide-grafted chitosan hydrogel microspheres for the controlled release of indomethacin. J. Appl. Polym. Sci 2003, 87, 1525–1536. [Google Scholar]
- Joshi, J.M.; Sinha, V.K. Ceric ammonium nitrate induced grafting of polyacrylamide onto carboxymethyl chitosan. Carbohyd. Polym 2007, 67, 427–435. [Google Scholar]
- Singh, V.; Tiwari, A.; Tripathi, D.N.; Sanghi, R. Microwave enhanced synthesis of chitosan-graft-polyacrylamide. Polymer 2006, 47, 254–260. [Google Scholar]
- Singh, V.; Sharma, A.K.; Sanghi, R. Poly(acrylamide) functionalized chitosan: An efficient adsorbent for azo dyes from aqueous solutions. J. Hazard. Mater 2009, 166, 327–335. [Google Scholar]
- Guo, T.Y.; Xia, Y.Q.; Hao, G.J.; Zhang, B.H.; Fu, G.Q.; Yuan, Z.; He, B.L.; Kennedy, J.F. Chemically modified chitosan beads as matrices for adsorptive separation of proteins by molecularly imprinted polymer. Carbohyd. Polym 2005, 62, 214–221. [Google Scholar]
- Li, N.; Bai, R.; Liu, C. Enhanced and selective adsorption of mercury ions on chitosan beads grafted with polyacrylamide via surface-initiated atom transfer radical polymerization. Langmuir 2005, 21, 11780–11787. [Google Scholar]
- Fu, G.; Zhao, J.; Yu, H.; Liu, L.; He, B. Bovine serum albumin-imprinted polymer gels prepared by graft copolymerization of acrylamide on chitosan. React. Funct. Polym 2007, 67, 442–450. [Google Scholar]
- Lin, W.J.; Chen, T.D.; Liu, C.W. Synthesis and characterization of lactobionic acid grafted pegylated chitosan and nanoparticle complex application. Polymer 2009, 50, 4166–4174. [Google Scholar]
- Lu, B.; Xu, X.D.; Zhang, X.Z.; Cheng, S.X.; Zhuo, R.X. Low molecular mass polyethylenimine grafted n-maleated chitosan for gene delivery: Properties and in vitro transfection studies. Biomacromolecules 2008, 9, 2594–2600. [Google Scholar]
- Bonina, P.; Petrova, T.; Manolova, N. pH-Sensitive hydrogels composed of chitosan and polyacrylamide—preparation and properties. J. Bioactive Compat. Polym 2004, 19, 101–116. [Google Scholar]
- Gao, J.; Yu, J.; Wang, W.; Lin, T. The accelerated degradation of aqueous polyacrylamide at low temperature. J. Appl. Polym. Sci 1998, 69, 791–797. [Google Scholar]
- Bawa, P.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Ndesendo, V.M.K.; Kumar, P. A composite polyelectrolyte matrix for controlled oral drug delivery. AAPS PharmSciTech 2011, 12, 227–238. [Google Scholar]
- Yoksan, R.; Akashi, M.; Miyata, M.; Chirachanchai, S. Optimal γ-ray dose and irradiation conditions for producing low-molecular-weight chitosan that retains its chemical structure. Radiat. Res 2004, 161, 471–480. [Google Scholar]
- Fernandes, L.P.; Morais, W.A.; Santos, A.I.B.; de Arau’jo, A.M.L.; dos Santos, D.E.S.; dos Santos, D.S.; Pavinatto, F.J.; Oliveira, O.N.; Dantas, T.N.C.; Pereira, M.R.; et al. The influence of oxidative degradation on the preparation of chitosan nanoparticles. Colloid Polym. Sci 2005, 284, 1–9. [Google Scholar]
- Lin, K.F.; Hsu, C.Y.; Huang, T.S.; Chiu, W.Y.; Lee, Y.H.; Young, T.H. A novel method to prepare chitosan/montmorillonite nanocomposites. J. Appl. Polym. Sci 2005, 98, 2042–2047. [Google Scholar]
- Kang, H.M.; Cai, Y.L.; Liu, P.S. Synthesis, characterization and thermal sensitivity of chitosan-based graft copolymers. Carbohyd. Polym 2006, 341, 2851–2857. [Google Scholar]
- Yuan, B.; Shang, Y.; Lu, Y.B.; Qin, Z.; Jiang, Y.; Chen, A.; Qian, X.; Wang, G.; Yang, H.; Cheng, R. The flocculating properties of chitosan-graft-polyacrylamide flocculants (i)—effect of the grafting ratio. J. Appl. Polym. Sci 2010, 117, 1876–1882. [Google Scholar]
- Sokker, H.H.; El-Sawy, N.M.; Hassan, M.A.; El-Anadouli, B.E. Adsorption of crude oil from aqueous solution by hydrogel of chitosan-based polyacrylamide prepared by radiation induced graft polymerization. J. Hazard. Mater 2011, 190, 359–365. [Google Scholar]
- Alves, N.M.; Ribelles, J.L.G.; Mano, J.F. Study of the molecular mobility in polymers with the thermally stimulated recovery technique—A review. J. Macromol. Sci. C 2005, 45, 99–124. [Google Scholar]
- Sakurai, K.; Maegawa, T.; Takahashi, T. Glass transition temperature of chitosan and miscibility of chitosan/poly(n-vinyl pyrrolidone) blends. Polymer 2004, 41, 7051–7056. [Google Scholar]
- Duarte, A.R.C.; Mano, J.F.; Reis, R.L. Novel 3D scaffolds of chitosan–PLLA blends for tissue engineering applications: Preparation and characterization. J. Supercrit. Fluid 2010, 54, 282–289. [Google Scholar] [Green Version]
- Al-Karawi, A.J.M.; Al-Qaisi, Z.H.J.; Abdullah, H.I.; Al-Mokarama, A.M.A.; Al-Heetimi, D.T.A. Synthesis, characterization of acrylamide grafted chitosan and its use in removal of copper(ii) ions from water. Carbohyd. Polym 2011, 83, 495–500. [Google Scholar]
- Merlin, D.L.; Sivasankar, B. Synthesis and characterization of semi-interpenetrating polymer networks using biocompatible polyurethane and acrylamide monomer. Eur. Polym. J 2009, 45, 165–170. [Google Scholar]
- Nori, A.; Yim, E.K.F.; Hen, S.; Leong, K.W. Cell Substrate Interactions. In Principles of Regenerative Medicine; Atala, A., Lanza, R., Thomson, J.A., Nerem, R.M., Eds.; Academic Press: New York, NY, USA, 2010; pp. 666–685. [Google Scholar]
- Chung, H.J.; Park, T.G. Surface engineered and drug releasing pre-fabricated scaffold for tissue engineering. Adv. Drug Deliv. Rev 2007, 59, 249–262. [Google Scholar]
- Cao, J.; Tan, Y.; Che, Y.; Xin, H. Novel complex gel beads composed of hydrolyzed polyacrylamide and chitosan: An effective adsorbent for the removal of heavy metal from aqueous solution. Bioresour. Technol 2010, 101, 2558–2561. [Google Scholar]
- Krishnamoorthi, S.; Adhikary, P.; Mal, D.; Singh, R.P. Novel polymeric flocculants based on polyacrylamide grafted dextran in kaolin suspension. J. Appl. Polym. Sci 2010, 118, 3539–3544. [Google Scholar]
- Kumar, P.; Pillay, V.; Choonara, Y.E.; Modi, G.; Naidoo, D.; du Toit, L.C. In silico theoretical molecular modeling for alzheimer’s disease: The nicotine-curcumin paradigm in neuroprotection and neurotherapy. Int. J. Mol. Sci 2011, 12, 694–724. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Graft Copolymer | GE a (%) | GR b (%) | IV c [η] [dL/g] | Mvd (×106) |
|---|---|---|---|---|
| CHT-g-AAm | 83 | 178 | 3.901 | 1.22 |
| CHT-g-PAAm | 92 | 263 | 5.231 | 1.63 |
| Compound | Energy (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| Steric energy a | ΔEbindingb | LDF c | Δldf d | H bond e | Ionic f | |
| CHT g | 35.556 | - | 13.323 | - | 0 | −24.697 |
| PAAm h | 10.357 | - | −5.072 | - | −0.035 | 0 |
| KPS4i | 238.832 | - | −5.776 | - | 0 | 0 |
| CHT-KPS4j | 239.153 | −35.235 | −27.503 | −35.05 | −0.0879 | −32.394 |
| PAAm-KPS4k | 207.784 | −51.762 | −39.943 | −29.095 | −3.039 | 0 |
| CHT-PAAm2-KPS4l | 202.903 | −92.199 | −88.755 | −86.158 | −5.927 | −33.481 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kumar, P.; Choonara, Y.E.; Toit, L.C.d.; Modi, G.; Naidoo, D.; Pillay, V. Novel High-Viscosity Polyacrylamidated Chitosan for Neural Tissue Engineering: Fabrication of Anisotropic Neurodurable Scaffold via Molecular Disposition of Persulfate-Mediated Polymer Slicing and Complexation. Int. J. Mol. Sci. 2012, 13, 13966-13984. https://doi.org/10.3390/ijms131113966
Kumar P, Choonara YE, Toit LCd, Modi G, Naidoo D, Pillay V. Novel High-Viscosity Polyacrylamidated Chitosan for Neural Tissue Engineering: Fabrication of Anisotropic Neurodurable Scaffold via Molecular Disposition of Persulfate-Mediated Polymer Slicing and Complexation. International Journal of Molecular Sciences. 2012; 13(11):13966-13984. https://doi.org/10.3390/ijms131113966
Chicago/Turabian StyleKumar, Pradeep, Yahya E. Choonara, Lisa C. du Toit, Girish Modi, Dinesh Naidoo, and Viness Pillay. 2012. "Novel High-Viscosity Polyacrylamidated Chitosan for Neural Tissue Engineering: Fabrication of Anisotropic Neurodurable Scaffold via Molecular Disposition of Persulfate-Mediated Polymer Slicing and Complexation" International Journal of Molecular Sciences 13, no. 11: 13966-13984. https://doi.org/10.3390/ijms131113966