Molecular Basis of Medullary Thyroid Carcinoma: The Role of RET Polymorphisms

Abstract
:1. Molecular Basis of Medullary Thyroid Carcinoma
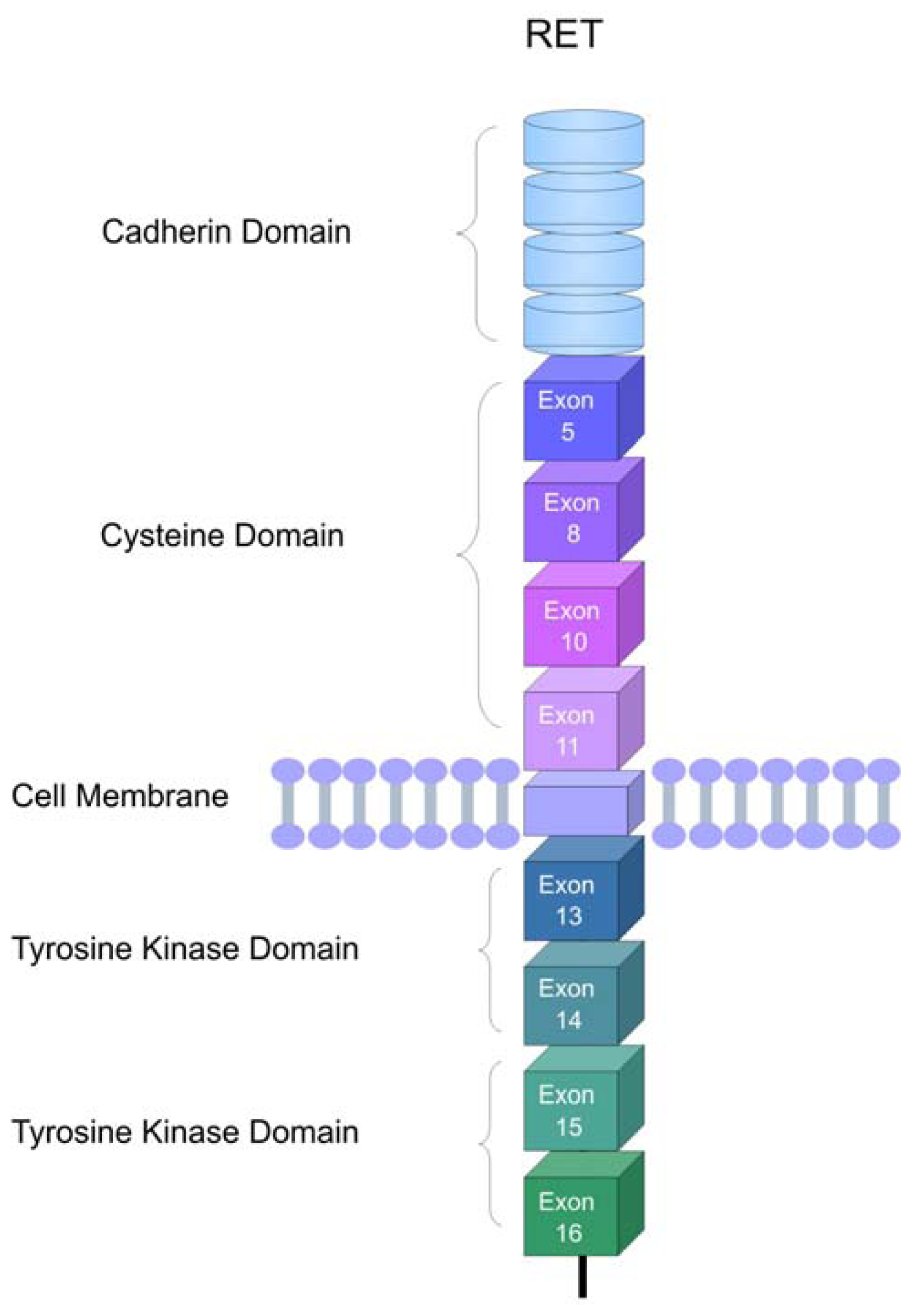
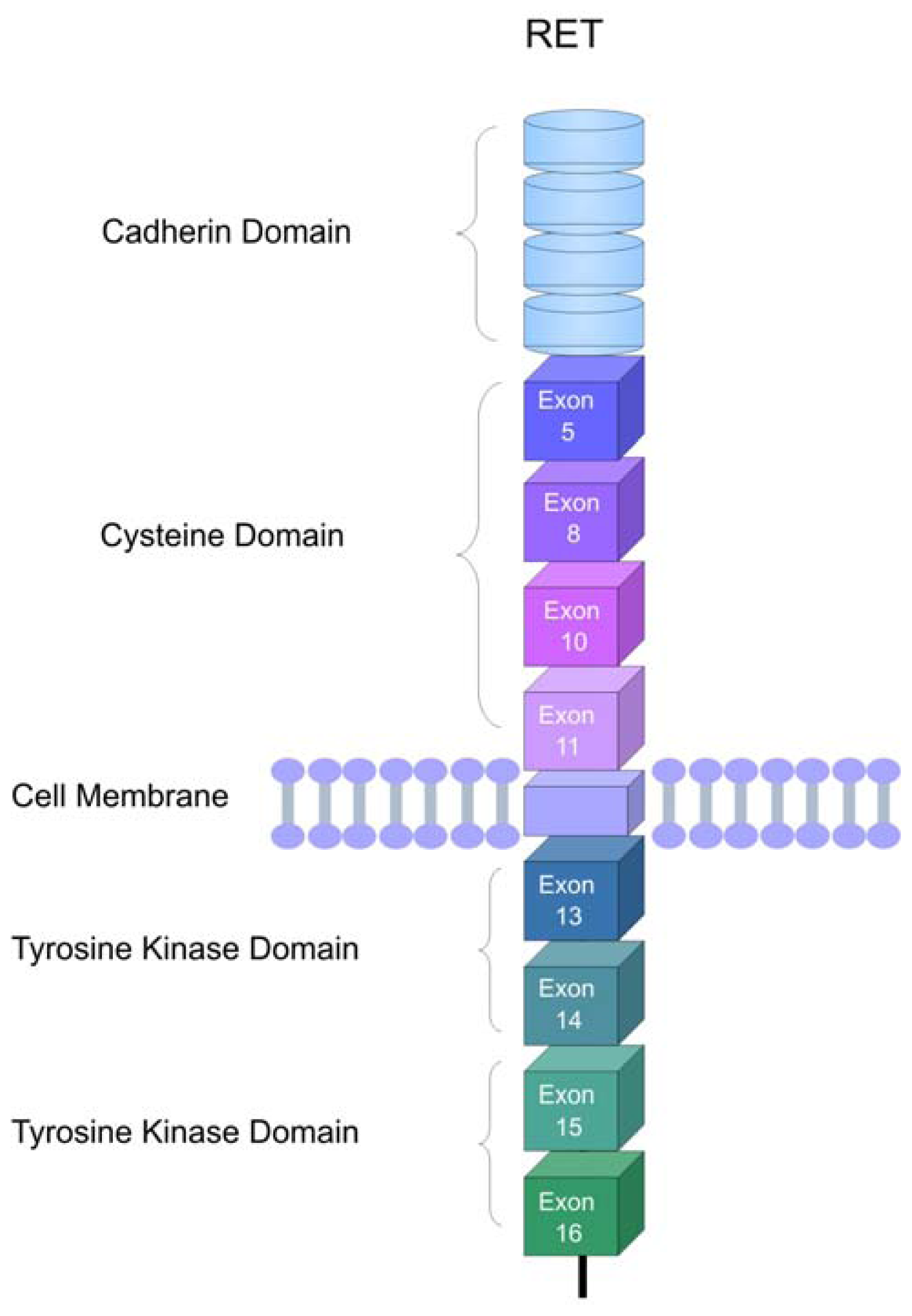
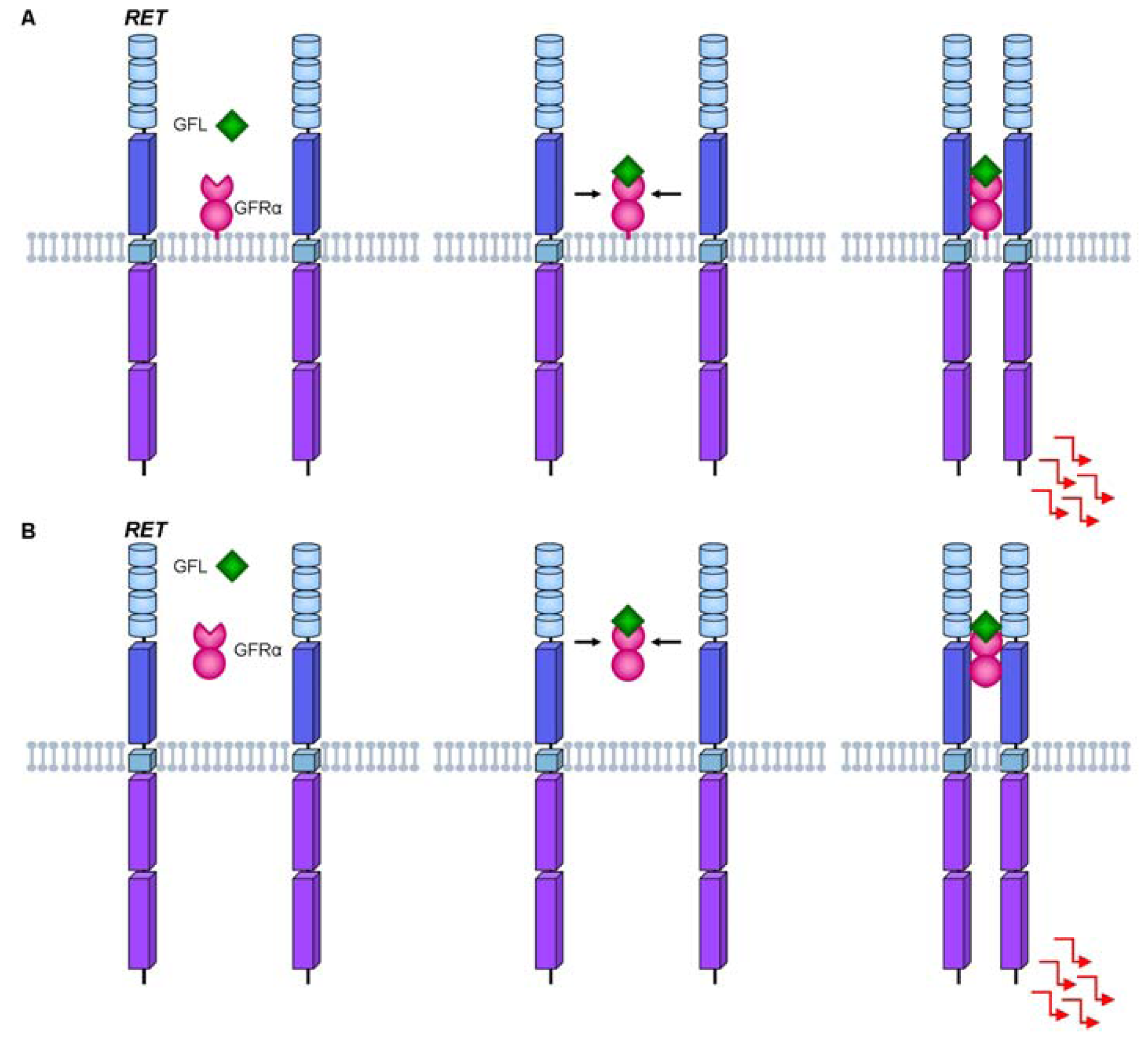
2. The RET Proto-Oncogene
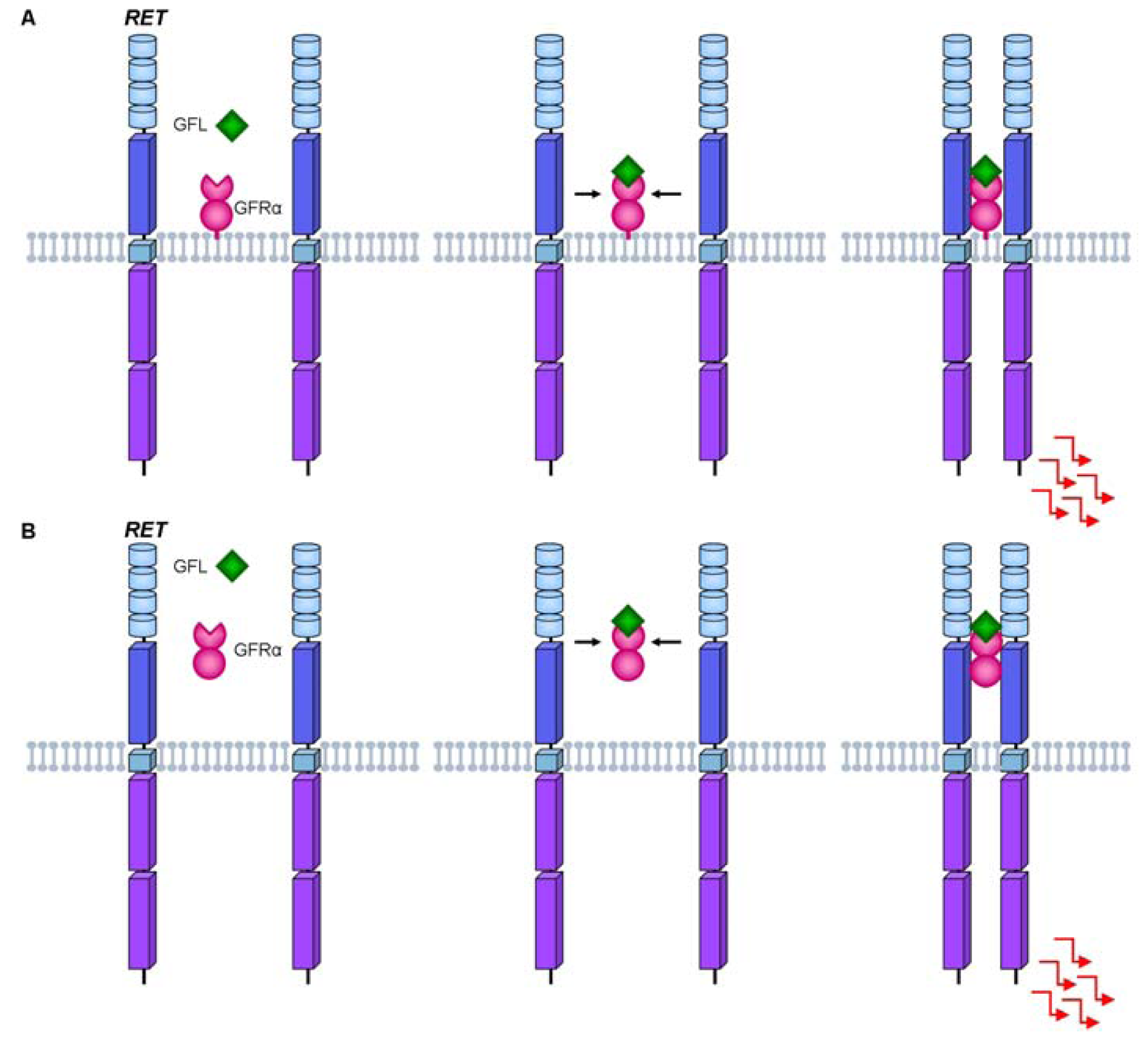
3. RET Protein Activation
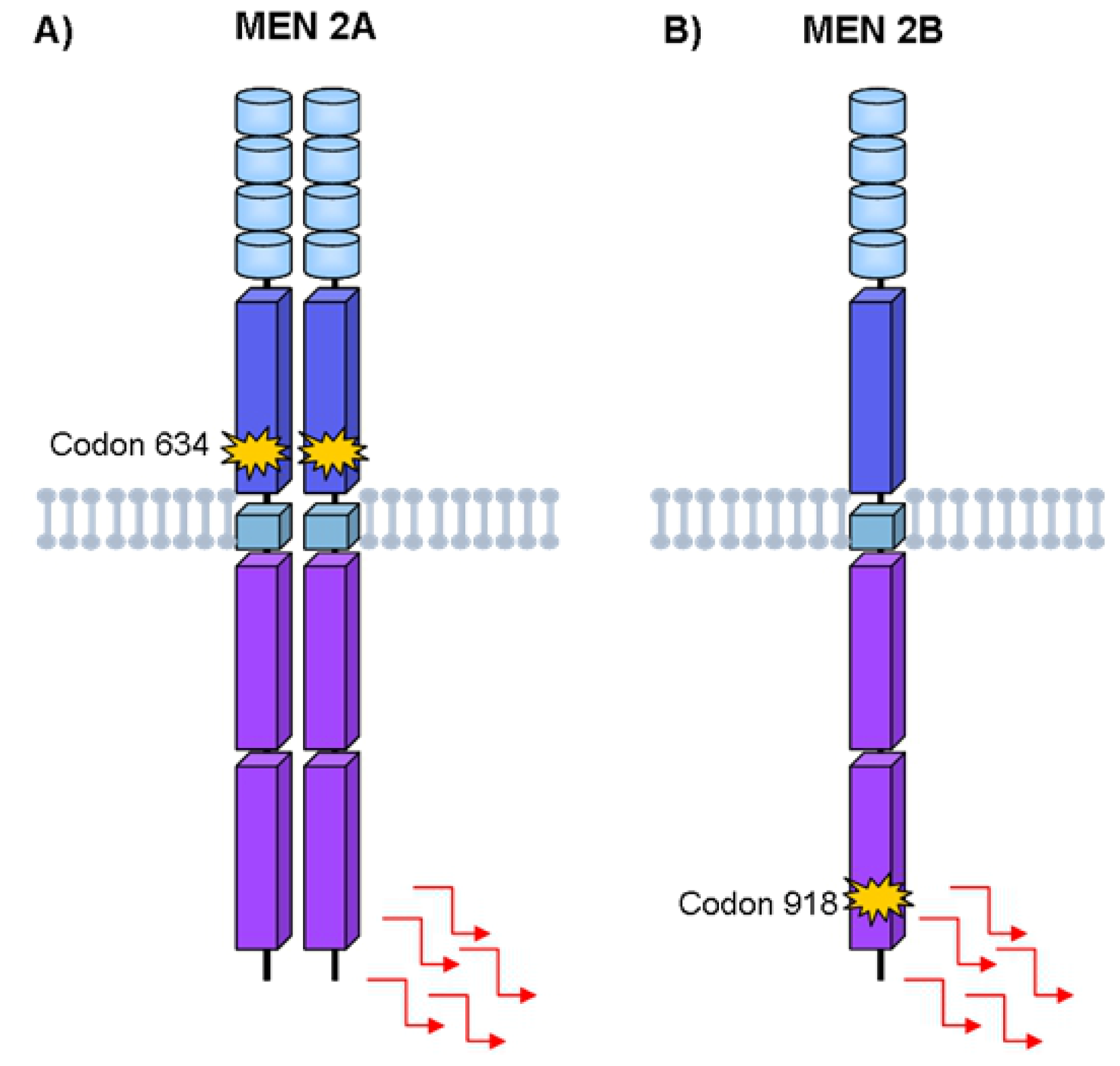
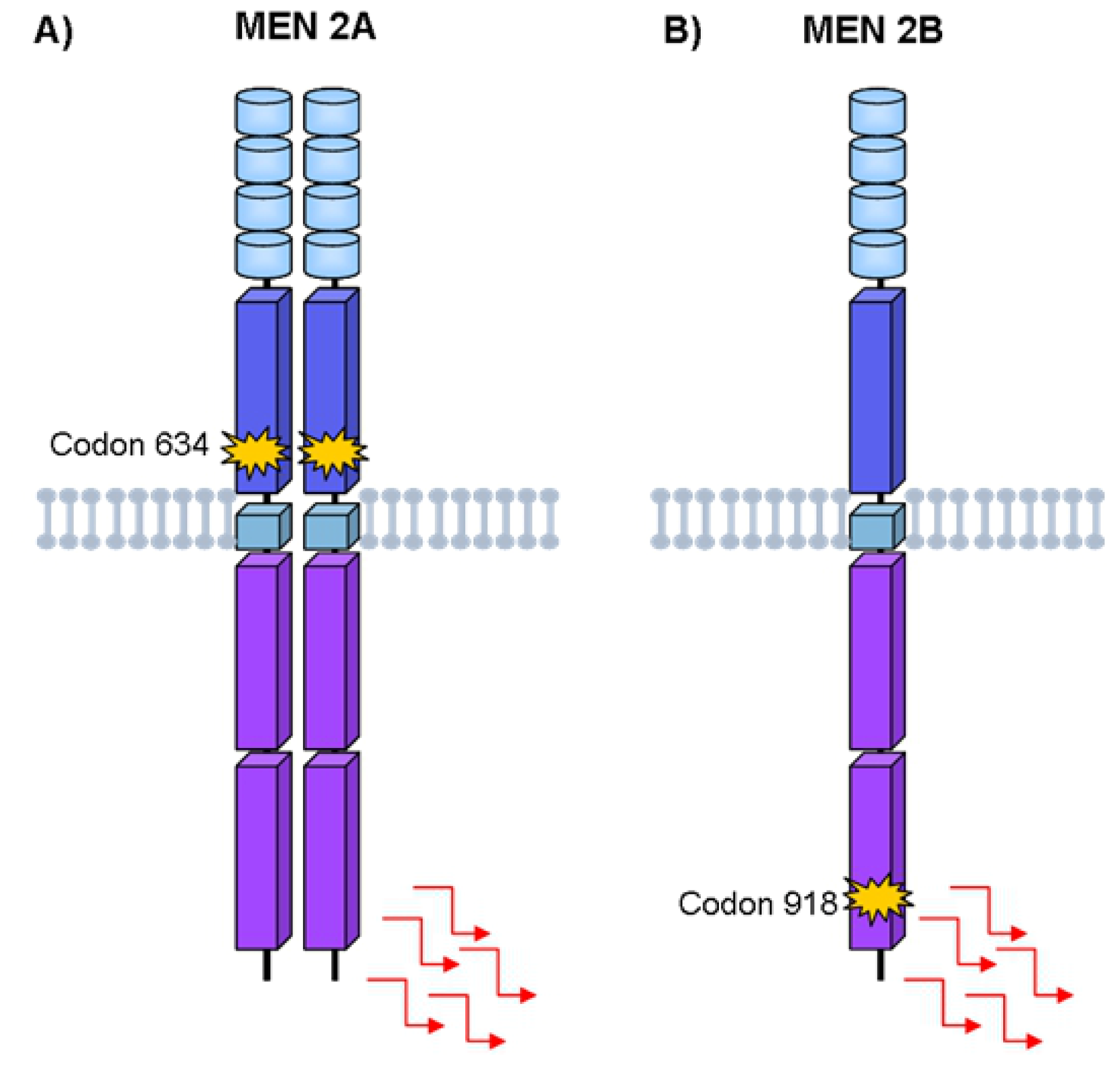
4. Hereditary Medullary Thyroid Carcinoma
Germline RET Mutations and Disease Phenotype
5. Sporadic Medullary Thyroid Carcinoma
Somatic RET Mutations and Disease Phenotype
6. Role of RET Polymorphisms in Medullary Thyroid Carcinoma
6.1. RET G691S and S904S Polymorphisms
6.2. RET L769L Polymorphism
6.3. RET S836S Polymorphism
6.4. Other RET Variants
6.5. Possible Mechanisms of Action for RET Polymorphism in Medullary Thyroid Carcinoma
7. Conclusion
References
- Hazard, J.B.; Hawk, W.A.; Crile, G., Jr. Medullary (solid) carcinoma of the thyroid; a clinicopathologic entity. J. Clin. Endocrinol. Metab 1959, 19, 152–161. [Google Scholar]
- Gilliland, F.D.; Hunt, W.C.; Morris, D.M.; Key, C.R. Prognostic factors for thyroid carcinoma. A population-based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program 1973–1991. Cancer 1997, 79, 564–573. [Google Scholar]
- Raue, F.; Kotzerke, J.; Reinwein, D.; Schroder, S.; Roher, H.D.; Deckart, H.; Hofer, R.; Ritter, M.; Seif, F.; Buhr, H.; et al. Prognostic factors in medullary thyroid carcinoma: evaluation of 741 patients from the German Medullary Thyroid Carcinoma Register. Clin. Investig 1993, 71, 7–12. [Google Scholar]
- Saad, M.F.; Ordonez, N.G.; Rashid, R.K.; Guido, J.J.; Hill, C.S., Jr; Hickey, R.C.; Samaan, N.A. Medullary carcinoma of the thyroid. A study of the clinical features and prognostic factors in 161 patients. Medicine (Baltimore) 1984, 63, 319–342. [Google Scholar]
- Girelli, M.E.; Nacamulli, D.; Pelizzo, M.R.; De Vido, D.; Mian, C.; Piccolo, M.; Busnardo, B. Medullary thyroid carcinoma: clinical features and long-term follow-up of seventy-eight patients treated between 1969 and 1986. Thyroid 1998, 8, 517–523. [Google Scholar]
- Kebebew, E.; Ituarte, P.H.; Siperstein, A.E.; Duh, Q.; Clark, O.H. Medullary thyroid cancer. Curr. Treat. Opt. Oncol 2000, 1, 359–367. [Google Scholar]
- Modigliani, E.; Cohen, R.; Campos, J.M.; Conte-Devolx, B.; Maes, B.; Boneu, A.; Schlumberger, M.; Bigorgne, J.C.; Dumontier, P.; Leclerc, L.; Corcuff, B.; Guilhem, I. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’etude des tumeurs a calcitonine. Clin. Endocrinol. (Oxf) 1998, 48, 265–273. [Google Scholar]
- Da Silva, A.M.; Maciel, R.M.; Da Silva, M.R.; Toledo, S.R.; De Carvalho, M.B.; Cerutti, J.M. A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J. Clin. Endocrinol. Metab 2003, 88, 5438–5443. [Google Scholar]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A., Jr. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet 1993, 2, 851–856. [Google Scholar]
- Dvorakova, S.; Vaclavikova, E.; Duskova, J.; Vlcek, P.; Ryska, A.; Bendlova, B. Exon 5 of the RET proto-oncogene: A newly detected risk exon for familial medullary thyroid carcinoma, a novel germ-line mutation Gly321Arg. J. Endocrinol. Invest 2005, 28, 905–909. [Google Scholar]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; van Amstel, H.K.; Lips, C.J.; Nishisho, I.; Takai, S.I.; et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. J. Am. Med. Assoc 1996, 276, 1575–1579. [Google Scholar]
- Mulligan, L.M.; Eng, C.; Healey, C.S.; Clayton, D.; Kwok, J.B.; Gardner, E.; Ponder, M.A.; Frilling, A.; Jackson, C.E.; Lehnert, H.; et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat. Genet 1994, 6, 70–74. [Google Scholar]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar]
- Eng, C.; Mulligan, L.M.; Healey, C.S.; Houghton, C.; Frilling, A.; Raue, F.; Thomas, G.A.; Ponder, B.A. Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res 1996, 56, 2167–2170. [Google Scholar]
- Marsh, D.J.; Learoyd, D.L.; Andrew, S.D.; Krishnan, L.; Pojer, R.; Richardson, A.L.; Delbridge, L.; Eng, C.; Robinson, B.G. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin. Endocrinol. (Oxf) 1996, 44, 249–257. [Google Scholar]
- Romei, C.; Elisei, R.; Pinchera, A.; Ceccherini, I.; Molinaro, E.; Mancusi, F.; Martino, E.; Romeo, G.; Pacini, F. Somatic mutations of the RET protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J. Clin. Endocrinol. Metab 1996, 81, 1619–1622. [Google Scholar]
- Siqueira, D.R.; Romitti, M.; da Rocha, A.P.; Ceolin, L.; Meotti, C.; Estivalet, A.; Punales, M.K.; Maia, A.L. The RET polymorphic allele S836S is associated with early metastatic disease in patients with hereditary or sporadic medullary thyroid carcinoma. Endocr. Relat. Cancer 2010, 17, 953–963. [Google Scholar]
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, RET, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Sugimura, T.; Tucker, J.; Fertitta, A.; Carrano, A.V.; Nagao, M. Human RET proto-oncogene mapped to chromosome 10q11.2. Oncogene 1989, 4, 1519–1521. [Google Scholar]
- Takahashi, M.; Buma, Y.; Hiai, H. Isolation of RET proto-oncogene cDNA with an amino-terminal signal sequence. Oncogene 1989, 4, 805–806. [Google Scholar]
- Hofstra, R.M.; Landsvater, R.M.; Ceccherini, I.; Stulp, R.P.; Stelwagen, T.; Luo, Y.; Pasini, B.; Hoppener, J.W.; van Amstel, H.K.; Romeo, G.; et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994, 367, 375–376. [Google Scholar]
- Myers, S.M.; Eng, C.; Ponder, B.A.; Mulligan, L.M. Characterization of RET proto-oncogene 3′ splicing variants and polyadenylation sites: A novel C-terminus for RET. Oncogene 1995, 11, 2039–2045. [Google Scholar]
- Takahashi, M.; Asai, N.; Iwashita, T.; Isomura, T.; Miyazaki, K.; Matsuyama, M. Characterization of the RET proto-oncogene products expressed in mouse L cells. Oncogene 1993, 8, 2925–2929. [Google Scholar]
- Jing, S.; Wen, D.; Yu, Y.; Holst, P.L.; Luo, Y.; Fang, M.; Tamir, R.; Antonio, L.; Hu, Z.; Cupples, R.; et al. GDNF-induced activation of the RET protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell 1996, 85, 1113–1124. [Google Scholar]
- Kjaer, S.; Ibanez, C.F. Identification of a surface for binding to the GDNF-GFR alpha 1 complex in the first cadherin-like domain of RET. J. Biol. Chem 2003, 278, 47898–47904. [Google Scholar]
- Yu, T.; Scully, S.; Yu, Y.; Fox, G.M.; Jing, S.; Zhou, R. Expression of GDNF family receptor components during development: implications in the mechanisms of interaction. J. Neurosci 1998, 18, 4684–4696. [Google Scholar]
- Santoro, M.; Carlomagno, F.; Romano, A.; Bottaro, D.P.; Dathan, N.A.; Grieco, M.; Fusco, A.; Vecchio, G.; Matoskova, B.; Kraus, M.H.; et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 1995, 267, 381–383. [Google Scholar]
- Borrello, M.G.; Smith, D.P.; Pasini, B.; Bongarzone, I.; Greco, A.; Lorenzo, M.J.; Arighi, E.; Miranda, C.; Eng, C.; Alberti, L.; et al. RET activation by germline MEN2A and MEN2B mutations. Oncogene 1995, 11, 2419–2427. [Google Scholar]
- Carlson, K.M.; Dou, S.; Chi, D.; Scavarda, N.; Toshima, K.; Jackson, C.E.; Wells, S.A., Jr; Goodfellow, P.J.; Donis-Keller, H. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc. Natl. Acad. Sci. USA 1994, 91, 1579–1583. [Google Scholar]
- Mulligan, L.M.; Gardner, E.; Smith, B.A.; Mathew, C.G.; Ponder, B.A. Genetic events in tumour initiation and progression in multiple endocrine neoplasia type 2. Genes Chromosom. Cancer 1993, 6, 166–177. [Google Scholar]
- Pelizzo, M.R.; Boschin, I.M.; Bernante, P.; Toniato, A.; Piotto, A.; Pagetta, C.; Nibale, O.; Rampin, L.; Muzzio, P.C.; Rubello, D. Natural history, diagnosis, treatment and outcome of medullary thyroid cancer: 37 years experience on 157 patients. Eur. J. Surg. Oncol 2007, 33, 493–497. [Google Scholar]
- Mulligan, L.M.; Marsh, D.J.; Robinson, B.G.; Schuffenecker, I.; Zedenius, J.; Lips, C.J.; Gagel, R.F.; Takai, S.I.; Noll, W.W.; Fink, M.; et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J. Intern. Med 1995, 238, 343–346. [Google Scholar]
- Farndon, J.R.; Leight, G.S.; Dilley, W.G.; Baylin, S.B.; Smallridge, R.C.; Harrison, T.S.; Wells, S.A., Jr. Familial medullary thyroid carcinoma without associated endocrinopathies: a distinct clinical entity. Br. J. Surg 1986, 73, 278–281. [Google Scholar]
- Frank-Raue, K.; Buhr, H.; Dralle, H.; Klar, E.; Senninger, N.; Weber, T.; Rondot, S.; Hoppner, W.; Raue, F. Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: impact of individual RET genotype. Eur. J. Endocrinol 2006, 155, 229–236. [Google Scholar]
- Machens, A.; Gimm, O.; Hinze, R.; Hoppner, W.; Boehm, B.O.; Dralle, H. Genotype-phenotype correlations in hereditary medullary thyroid carcinoma: oncological features and biochemical properties. J. Clin. Endocrinol. Metab 2001, 86, 1104–1109. [Google Scholar]
- Punales, M.K.; Graf, H.; Gross, J.L.; Maia, A.L. RET codon 634 mutations in multiple endocrine neoplasia type 2: variable clinical features and clinical outcome. J. Clin. Endocrinol. Metab 2003, 88, 2644–2649. [Google Scholar]
- Donis-Keller, H. The RET proto-oncogene and cancer. J. Intern. Med 1995, 238, 319–325. [Google Scholar]
- Frank-Raue, K.; Hoppner, W.; Frilling, A.; Kotzerke, J.; Dralle, H.; Haase, R.; Mann, K.; Seif, F.; Kirchner, R.; Rendl, J.; et al. Mutations of the RET protooncogene in German multiple endocrine neoplasia families: relation between genotype and phenotype. German Medullary Thyroid Carcinoma Study Group. J. Clin. Endocrinol. Metab 1996, 81, 1780–1783. [Google Scholar]
- Verga, U.; Fugazzola, L.; Cambiaghi, S.; Pritelli, C.; Alessi, E.; Cortelazzi, D.; Gangi, E.; Beck-Peccoz, P. Frequent association between MEN 2A and cutaneous lichen amyloidosis. Clin. Endocrinol. (Oxf) 2003, 59, 156–161. [Google Scholar]
- Milos, I.N.; Frank-Raue, K.; Wohllk, N.; Maia, A.L.; Pusiol, E.; Patocs, A.; Robledo, M.; Biarnes, J.; Barontini, M.; Links, T.P.; et al. Age-related neoplastic risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germ line RET Cys634Trp (TGC>TGG) mutation. Endocr. Relat. Cancer 2008, 15, 1035–1041. [Google Scholar]
- Frank-Raue, K.; Rybicki, L.A.; Erlic, Z.; Schweizer, H.; Winter, A.; Milos, I.; Toledo, S.P.; Toledo, R.A.; Tavares, M.R.; Alevizaki, M.; et al. Risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germline RET mutations located in exon 10. Hum. Mutat 2011, 32, 51–58. [Google Scholar]
- Gimm, O.; Marsh, D.J.; Andrew, S.D.; Frilling, A.; Dahia, P.L.; Mulligan, L.M.; Zajac, J.D.; Robinson, B.G.; Eng, C. Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J. Clin. Endocrinol. Metab 1997, 82, 3902–3904. [Google Scholar]
- Smith, D.P.; Houghton, C.; Ponder, B.A. Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 1997, 15, 1213–1217. [Google Scholar]
- Menko, F.H.; van der Luijt, R.B.; de Valk, I.A.; Toorians, A.W.; Sepers, J.M.; van Diest, P.J.; Lips, C.J. Atypical MEN type 2B associated with two germline RET mutations on the same allele not involving codon 918. J. Clin. Endocrinol. Metab 2002, 87, 393–397. [Google Scholar]
- Miyauchi, A.; Futami, H.; Hai, N.; Yokozawa, T.; Kuma, K.; Aoki, N.; Kosugi, S.; Sugano, K.; Yamaguchi, K. Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn. J. Cancer Res 1999, 90, 1–5. [Google Scholar]
- Zedenius, J.; Larsson, C.; Bergholm, U.; Bovee, J.; Svensson, A.; Hallengren, B.; Grimelius, L.; Backdahl, M.; Weber, G.; Wallin, G. Mutations of codon 918 in the RET proto-oncogene correlate to poor prognosis in sporadic medullary thyroid carcinomas. J. Clin. Endocrinol. Metab 1995, 80, 3088–3090. [Google Scholar]
- Hofstra, R.M.; Fattoruso, O.; Quadro, L.; Wu, Y.; Libroia, A.; Verga, U.; Colantuoni, V.; Buys, C.H. A novel point mutation in the intracellular domain of the RET protooncogene in a family with medullary thyroid carcinoma. J. Clin. Endocrinol. Metab 1997, 82, 4176–4178. [Google Scholar]
- Bolino, A.; Schuffenecker, I.; Luo, Y.; Seri, M.; Silengo, M.; Tocco, T.; Chabrier, G.; Houdent, C.; Murat, A.; Schlumberger, M.; et al. RET mutations in exons 13 and 14 of FMTC patients. Oncogene 1995, 10, 2415–2419. [Google Scholar]
- Dabir, T.; Hunter, S.J.; Russell, C.F.; McCall, D.; Morrison, P.J. The RET mutation E768D confers a late-onset familial medullary thyroid carcinoma—only phenotype with incomplete penetrance: implications for screening and management of carrier status. Fam. Cancer 2006, 5, 201–204. [Google Scholar]
- Eng, C.; Smith, D.P.; Mulligan, L.M.; Healey, C.S.; Zvelebil, M.J.; Stonehouse, T.J.; Ponder, M.A.; Jackson, C.E.; Waterfield, M.D.; Ponder, B.A. A novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in a family with FMTC. Oncogene 1995, 10, 509–513. [Google Scholar]
- Kloos, R.T.; Eng, C.; Evans, D.B.; Francis, G.L.; Gagel, R.F.; Gharib, H.; Moley, J.F.; Pacini, F.; Ringel, M.D.; Schlumberger, M.; et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19, 565–612. [Google Scholar]
- Heshmati, H.M.; Gharib, H.; van Heerden, J.A.; Sizemore, G.W. Advances and controversies in the diagnosis and management of medullary thyroid carcinoma. Am. J. Med 1997, 103, 60–69. [Google Scholar]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Renzini, G.; Molinaro, E.; Agate, L.; Vivaldi, A.; Faviana, P.; Basolo, F.; et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J. Clin. Endocrinol. Metab 2008, 93, 682–687. [Google Scholar]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Domingues, R.; Santos, J.R.; Cid, M.O.; Bugalho, M.J.; Leite, V. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br. J. Cancer 2009, 100, 1777–1783. [Google Scholar]
- Beldjord, C.; Desclaux-Arramond, F.; Raffin-Sanson, M.; Corvol, J.C.; De Keyzer, Y.; Luton, J.P.; Plouin, P.F.; Bertagna, X. The RET protooncogene in sporadic pheochromocytomas: Frequent MEN 2-like mutations and new molecular defects. J. Clin. Endocrinol. Metab 1995, 80, 2063–2068. [Google Scholar]
- van der Harst, E.; de Krijger, R.R.; Bruining, H.A.; Lamberts, S.W.; Bonjer, H.J.; Dinjes, W.N.; Proye, C.; Koper, J.W.; Bosman, F.T.; Roth, J.; et al. Prognostic value of RET proto-oncogene point mutations in malignant and benign, sporadic phaeochromocytomas. Int. J. Cancer 1998, 79, 537–540. [Google Scholar]
- Robledo, M.; Gil, L.; Pollan, M.; Cebrian, A.; Ruiz, S.; Azanedo, M.; Benitez, J.; Menarguez, J.; Rojas, J.M. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 2003, 63, 1814–1817. [Google Scholar]
- Wiench, M.; Wygoda, Z.; Gubala, E.; Wloch, J.; Lisowska, K.; Krassowski, J.; Scieglinska, D.; Fiszer-Kierzkowska, A.; Lange, D.; Kula, D.; et al. Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. J. Clin. Oncol 2001, 19, 1374–1380. [Google Scholar]
- Eng, C.; Mulligan, L.M.; Smith, D.P.; Healey, C.S.; Frilling, A.; Raue, F.; Neumann, H.P.; Pfragner, R.; Behmel, A.; Lorenzo, M.J.; et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosom. Cancer 1995, 12, 209–212. [Google Scholar]
- Gimm, O.; Neuberg, D.S.; Marsh, D.J.; Dahia, P.L.; Hoang-Vu, C.; Raue, F.; Hinze, R.; Dralle, H.; Eng, C. Over-representation of a germline RET sequence variant in patients with sporadic medullary thyroid carcinoma and somatic RET codon 918 mutation. Oncogene 1999, 18, 1369–1373. [Google Scholar]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Sculli, M.; Lari, R.; Barale, R.; Pacini, F.; Pinchera, A. RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J. Clin. Endocrinol. Metab 2004, 89, 3579–3584. [Google Scholar]
- Rocha, A.P.; Magalhães, P.R.; Maia, A.L.; Maciel, L.Z. Polimorfismos Genéticos: Implicações na Patogênese do Carcinoma Medular de Tireóide. Arq. Bras. Endocrinol. Metab 2007, 5, 723–730. [Google Scholar]
- Cebrian, A.; Lesueur, F.; Martin, S.; Leyland, J.; Ahmed, S.; Luccarini, C.; Smith, P.L.; Luben, R.; Whittaker, J.; et al. Polymorphisms in the initiators of RET (rearranged during transfection) signaling pathway and susceptibility to sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab 2005, 90, 6268–6274. [Google Scholar]
- Fugazzola, L.; Muzza, M.; Mian, C.; Cordella, D.; Barollo, S.; Alberti, L.; Cirello, V.; Dazzi, D.; Girelli, M.E.; Opocher, G.; et al. RET genotypes in sporadic medullary thyroid cancer: Studies in a large Italian series. Clin. Endocrinol. (Oxf) 2008, 69, 418–425. [Google Scholar]
- Lesueur, F.; Cebrian, A.; Robledo, M.; Niccoli-Sire, P.; Svensson, K.A.; Pinson, S.; Leyland, J.; Whittaker, J.; Pharoah, P.D.; Ponder, B.A. Polymorphisms in RET and its coreceptors and ligands as genetic modifiers of multiple endocrine neoplasia type 2A. Cancer Res 2006, 66, 1177–1180. [Google Scholar]
- Baumgartner-Parzer, S.M.; Lang, R.; Wagner, L.; Heinze, G.; Niederle, B.; Kaserer, K.; Waldhausl, W.; Vierhapper, H. Polymorphisms in exon 13 and intron 14 of the RET protooncogene: Genetic modifiers of medullary thyroid carcinoma? J. Clin. Endocrinol. Metab 2005, 90, 6232–6236. [Google Scholar]
- Sharma, B.P.; Saranath, D. RET gene mutations and polymorphisms in medullary thyroid carcinomas in Indian patients. J. Biosci 2011, 36, 603–611. [Google Scholar]
- Sromek, M.; Czetwertynska, M.; Skasko, E.; Zielinska, J.; Czapczak, D.; Steffen, J. The frequency of selected polymorphic variants of the RET gene in patients with medullary thyroid carcinoma and in the general population of central Poland. Endocr. Pathol 2010, 21, 178–185. [Google Scholar]
- Wohllk, N.; Soto, E.; Bravo, M.; Becker, P. Polimorfismos G691S, L769L y S836S del proto-oncogen RET no se asocian a mayor riesgo de cáncer medular tiroideo esporádico en pacientes chilenos. Rev. Méd. Chile 2005, 133, 397–402. [Google Scholar]
- Magalhães, P.K.; de Castro, M.; Elias, L.L.; Soares, E.G.; Maciel, L.M. Polymorphisms in the RET proto-oncogene and the phenotypic presentation of familial medullary thyroid carcinoma. Thyroid 2004, 14, 848–852. [Google Scholar]
- Berard, I.; Kraimps, J.L.; Savagner, F.; Murat, A.; Renaudin, K.; Nicolli-Sire, P.; Bertrand, G.; Moisan, J.P.; Bezieau, S. Germline-sequence variants S836S and L769L in the RE arranged during Transfection (RET) proto-oncogene are not associated with predisposition to sporadic medullary carcinoma in the French population. Clin. Genet 2004, 65, 150–152. [Google Scholar]
- Ruiz, A.; Antinolo, G.; Fernandez, R.M.; Eng, C.; Marcos, I.; Borrego, S. Germline sequence variant S836S in the RET proto-oncogene is associated with low level predisposition to sporadic medullary thyroid carcinoma in the Spanish population. Clin. Endocrinol. (Oxf) 2001, 55, 399–402. [Google Scholar]
- Tamanaha, R.; Camacho, C.P.; Pereira, A.C.; da Silva, A.M.; Maciel, R.M.; Cerutti, J.M. Evaluation of RET polymorphisms in a six-generation family with G533C RET mutation: specific RET variants may modulate age at onset and clinical presentation. Clin. Endocrinol. (Oxf) 2009, 71, 56–64. [Google Scholar]
- Griseri, P.; Sancandi, M.; Patrone, G.; Bocciardi, R.; Hofstra, R.; Ravazzolo, R.; Devoto, M.; Romeo, G.; Ceccherini, I. A single-nucleotide polymorphic variant of the RET proto-oncogene is underrepresented in sporadic Hirschsprung disease. Eur. J. Hum. Genet 2000, 8, 721–724. [Google Scholar]
- Borrego, S.; Wright, F.A.; Fernandez, R.M.; Williams, N.; Lopez-Alonso, M.; Davuluri, R.; Antinolo, G.; Eng, C. A founding locus within the RET proto-oncogene may account for a large proportion of apparently sporadic Hirschsprung disease and a subset of cases of sporadic medullary thyroid carcinoma. Am. J. Hum. Genet 2003, 72, 88–100. [Google Scholar]
- Lu, H.; Huan, C. Transcription factor NFAT, its role in cancer development, and as a potential target for chemoprevention. Curr. Cancer Drug Targets 2007, 7, 343–353. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| RET variant | Author | Cases | Controls | P | Frequency (cases vs. controls) | Genotyping platform | Conclusion | Population |
|---|---|---|---|---|---|---|---|---|
| G691S (rs1799939) | Robledo (2003) | 198 | 653 | 0.037 | – | sequencing | Associated with the presence of MTC in younger individuals. | Spanish |
| Lesueur (2006) | 384 | – | – | – | Taqman | N/A | European | |
| Tamanah a (2009) | 77 a | 100 | 0.048 | 0; 4 | RFLP | Underrepresented in G533C-arriers. | Brazilian | |
| Sharma b (2011) | 51 | 50 | NS | 49; 48 | sequencing | N/A | Indian | |
| L769L (rs1800861) | Sharma b (2011) | 51 | 50 | NS | 45; 58 | sequencing | N/A | Indian |
| S836S (rs1800862) | Tamanah a (2009) | 77 a | 100 | 0.008 | 16.9; 4 | RFLP | Over-represented in G533C-carriers. | Brazilian |
| Siqueira (2010) | 88 | – | – | 7.95; – | RFLP | Associated with early onset and increased risk for metastatic disease. | Brazilian | |
| Sharma b (2011) | 51 | 50 | NS | 25; 22 | sequencing | N/A | Indian | |
| S904S (rs1800863) | Lesueur (2006) | 384 | – | – | – | Taqman | N/A | European |
| Sharma b (2011) | 51 | 50 | NS | 25; 22 | sequencing | N/A | Indian | |
| Tamanah a (2009) | 77 a | 100 | 0.048 | 0; 4 | RFLP | Underrepresented in G533C-carriers. | Brazilian | |
| IVS1–126 G>T (rs2565206) | Tamanah a (2009) | 77 a | 100 | 0.002 | 1.3; 0 | RFLP | Associated with younger age at diagnosis. | Brazilian |
| IVS8+82 A>G; 85–86 insC (rs3026750) | Tamanah a (2009) | 77 a | – | 0·019 | – | RFLP | Associated with lymph node metastases. Could induce abnormal splicing. | Brazilian |
| RET variant | Author | Cases | Controls | P | Frequency (cases vs. controls) | Genotyping platform | Conclusion | Population |
|---|---|---|---|---|---|---|---|---|
| G691S/S904S (rs1799939)/(rs1800863) | Elisei (2004) | 106 | 106 | 0.029 | 27.8; 18.8 | RFLP | Higher frequency in MTC patients. Does not influence RET mRNA expression | European |
| Cebrian a (2005) | 120 | 528 | 0.004 | 27; 18 | TaqMan | Associated with higher risk for development of MTC. Does not affect the splicing of RET | British | |
| Wohllk (2005) | 50 | 50 | NS | 25; 25 | sequencing | N/A | Chilean | |
| L769L (rs1800861) | Wiench (2001) | 116 b | – | 0.04 b | 36; 15 | sequencing | Associated with the presence of MTC in younger individuals | Polish |
| Sromek (2010) | 217 | 420 | 0.039c | 48.3; 39.5 c | Sequencing | Associated with the presence of MTC in younger individuals (in homozygosis). Could influence RET mRNA structure | Polish | |
| Berard (2004) | 184 | 174 | NS | 22.3; 25.9 | sequencing | N/A | French | |
| Wohllk (2005) | 50 | 50 | NS | 24; 23 | sequencing | N/A | Chilean | |
| S836S (rs1800862) | Gimm (1999) | 50 | 70 | 0.03 | 9; 3.7 | RFLP | More frequent in MTC patients | German- American |
| Ruiz (2001) | 32 | 250 | 0.04 | 9.3; 3.6 | RFLP | Associated with higher risk for development of MTC | Spanish | |
| Siqueira (2010) | 81 | 80 | 0.01 | 10.5; 3.2 | RFLP | Associated with early onset and increased risk for metastatic disease | Brazilian | |
| Berard (2004) | 184 | 174 | NS | 6.5; 5.2 | sequencing | N/A | French | |
| Wohllk (2005) | 50 | 50 | NS | 6; 1 | sequencing | N/A | Chilean | |
| S904S (rs1800863) | Wohllk (2005) | 50 | 50 | NS | 27; 28 | sequencing | N/A | Chilean |
| Cebrian (2005) | 125 | 528 | 0.005 | 26.4; 15.5 | TaqMan | Associated with higher risk for development of MTC | British | |
| STOP+388pb G>A (rs3026782 | Cebrian (2005) | 123 | 522 | 0.005 | 26.4; 15.5 | TaqMan | Associated with higher risk for development of MTC | British |
| A45A G>A (rs1800858) | Cebrian (2005) | 126 | 525 | 0.04 | 21; 27.9 | TaqMan | Suggest protective effect | British |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ceolin, L.; Siqueira, D.R.; Romitti, M.; Ferreira, C.V.; Maia, A.L. Molecular Basis of Medullary Thyroid Carcinoma: The Role of RET Polymorphisms. Int. J. Mol. Sci. 2012, 13, 221-239. https://doi.org/10.3390/ijms13010221
Ceolin L, Siqueira DR, Romitti M, Ferreira CV, Maia AL. Molecular Basis of Medullary Thyroid Carcinoma: The Role of RET Polymorphisms. International Journal of Molecular Sciences. 2012; 13(1):221-239. https://doi.org/10.3390/ijms13010221
Chicago/Turabian StyleCeolin, Lucieli, Débora R. Siqueira, Mírian Romitti, Carla V. Ferreira, and Ana Luiza Maia. 2012. "Molecular Basis of Medullary Thyroid Carcinoma: The Role of RET Polymorphisms" International Journal of Molecular Sciences 13, no. 1: 221-239. https://doi.org/10.3390/ijms13010221