Fabrication and Intermolecular Interactions of Silk Fibroin/Hydroxybutyl Chitosan Blended Nanofibers

Abstract
:1. Introduction
2. Results and Discussion
2.1. Morphology of SF/HBC Nanofibers
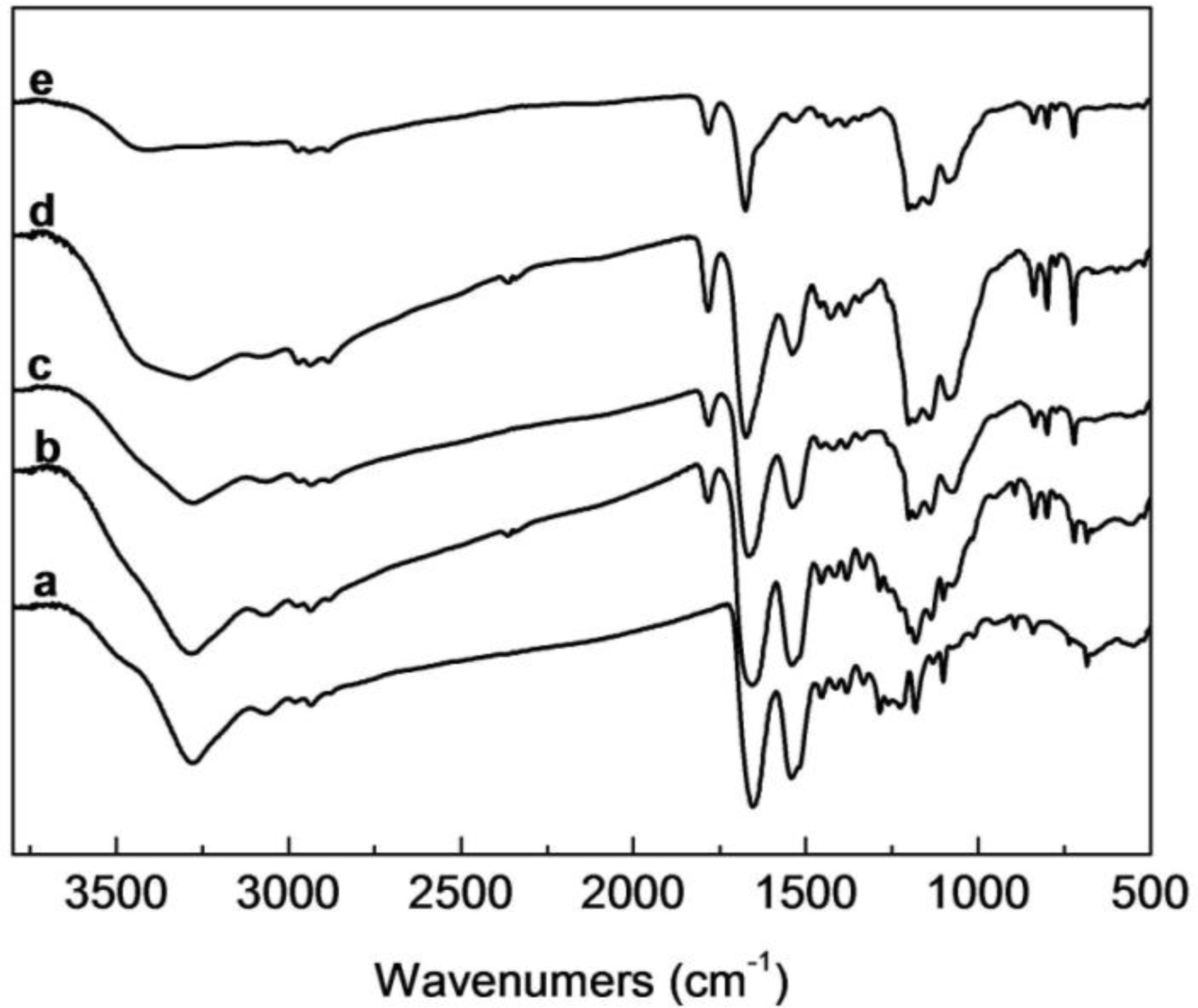
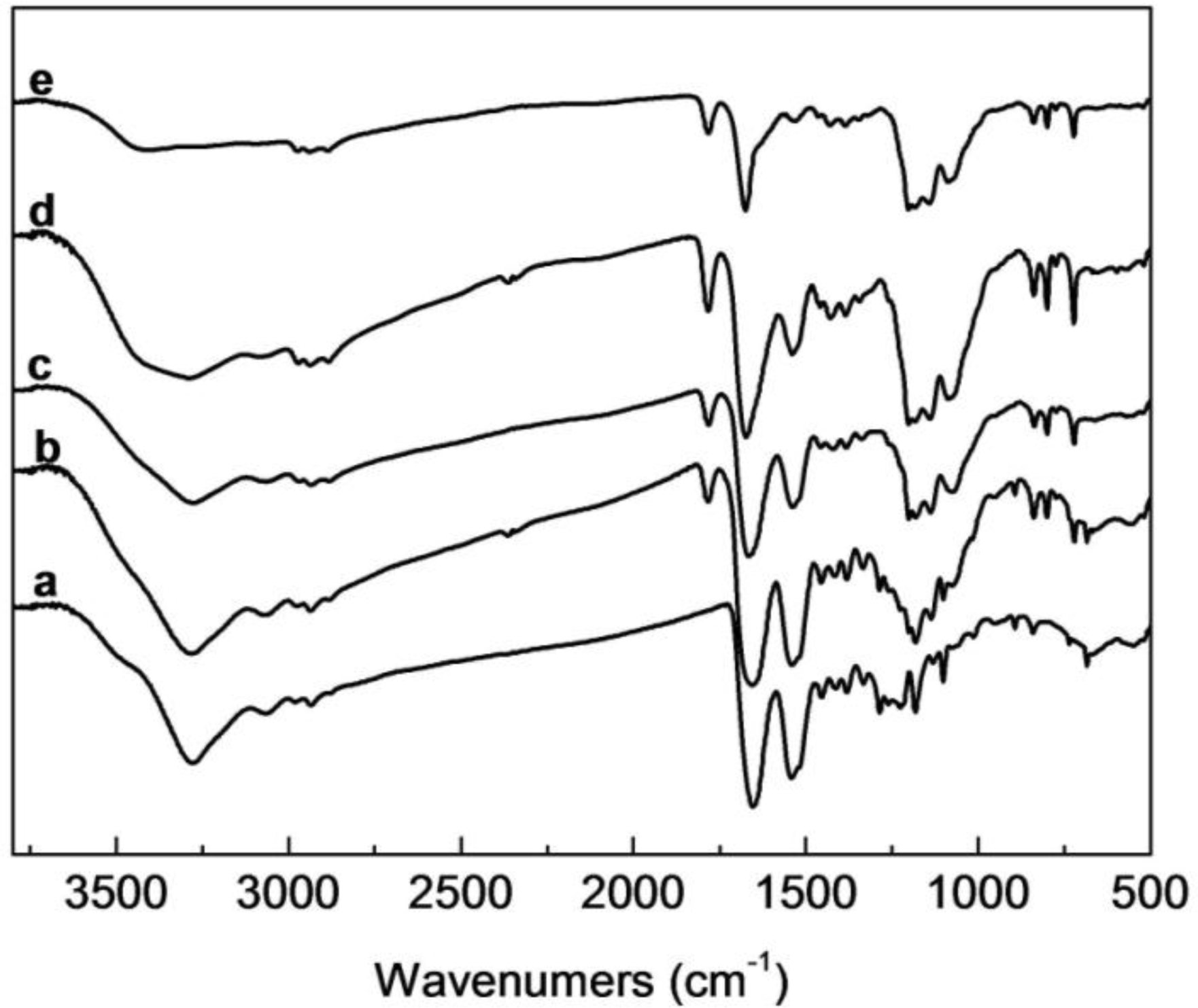
2.2. FTIR Analysis of SF/HBC Nanofibers
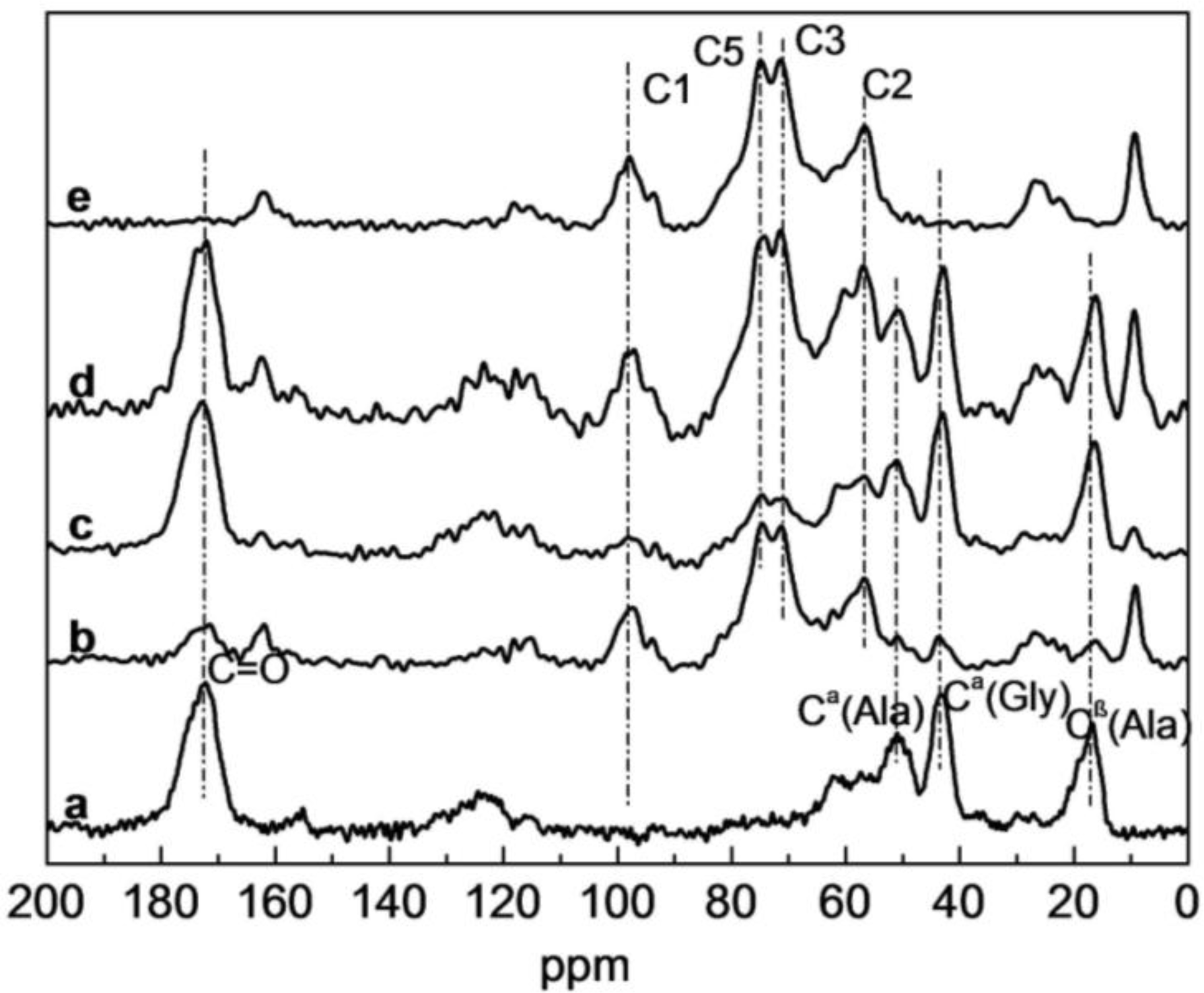
2.3. Solid-state 13C NMR Analysis of SF/HBC Nanofibers
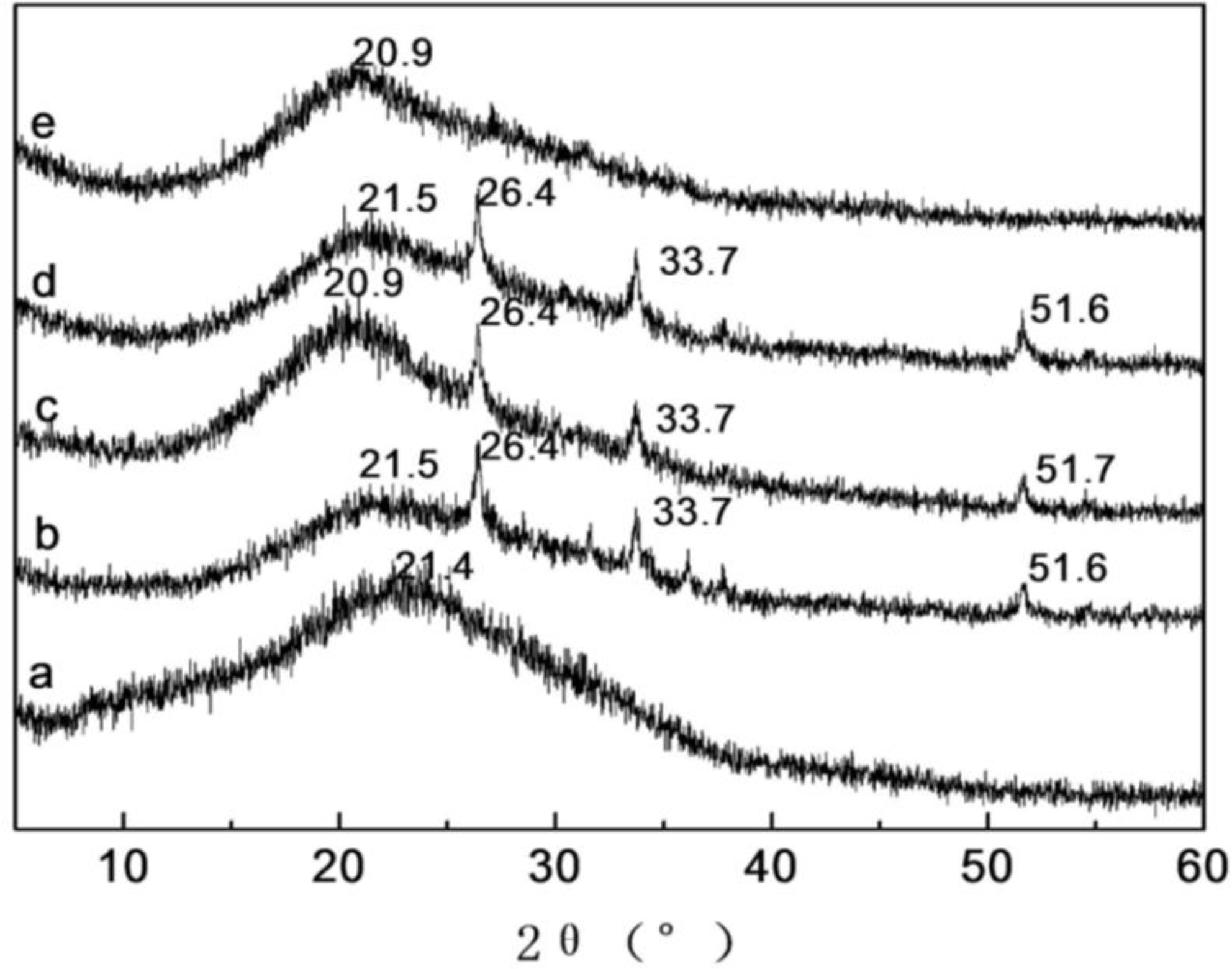
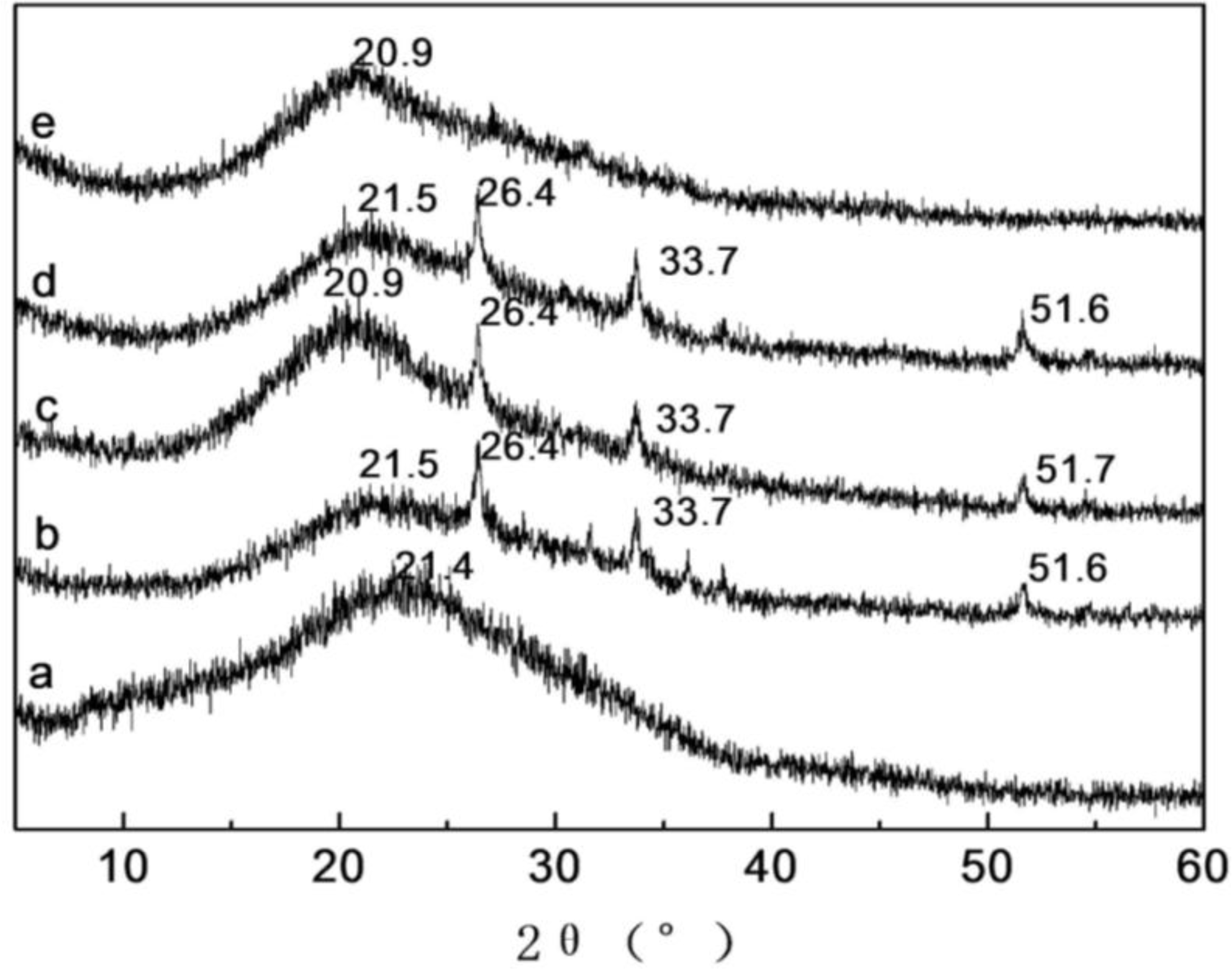
2.4. X-ray Diffraction Analysis of SF/HBC Nanofibers
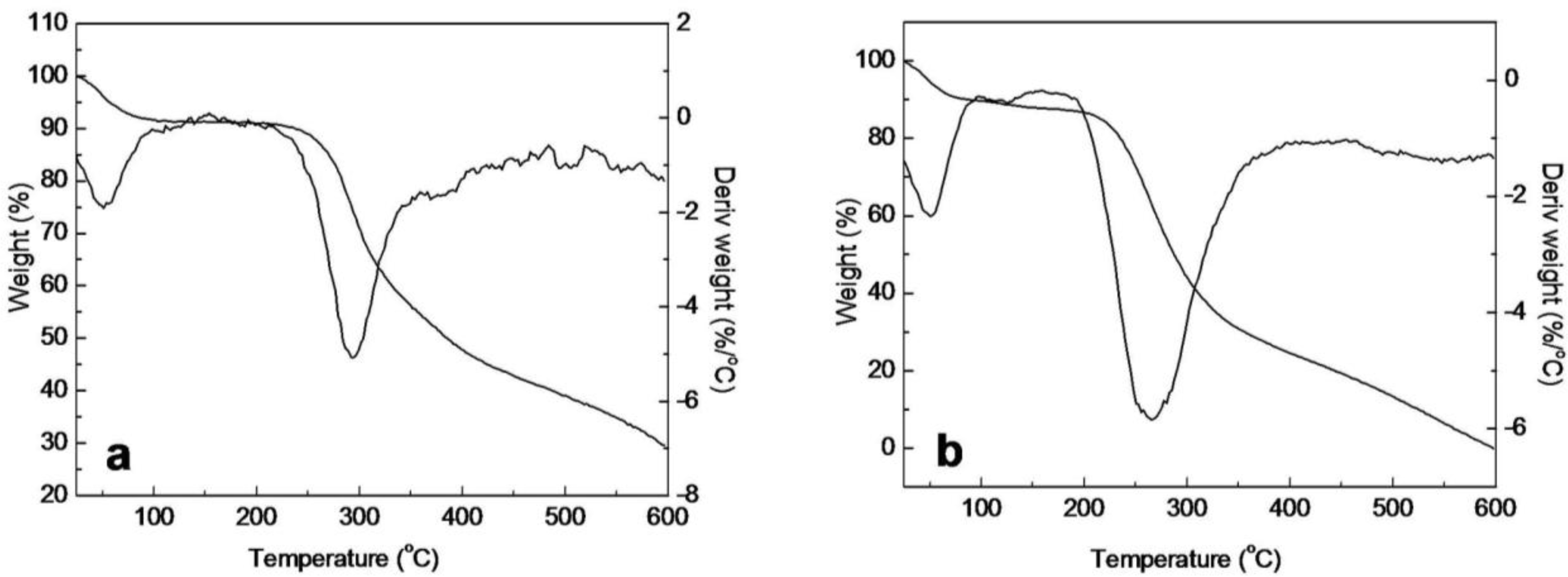
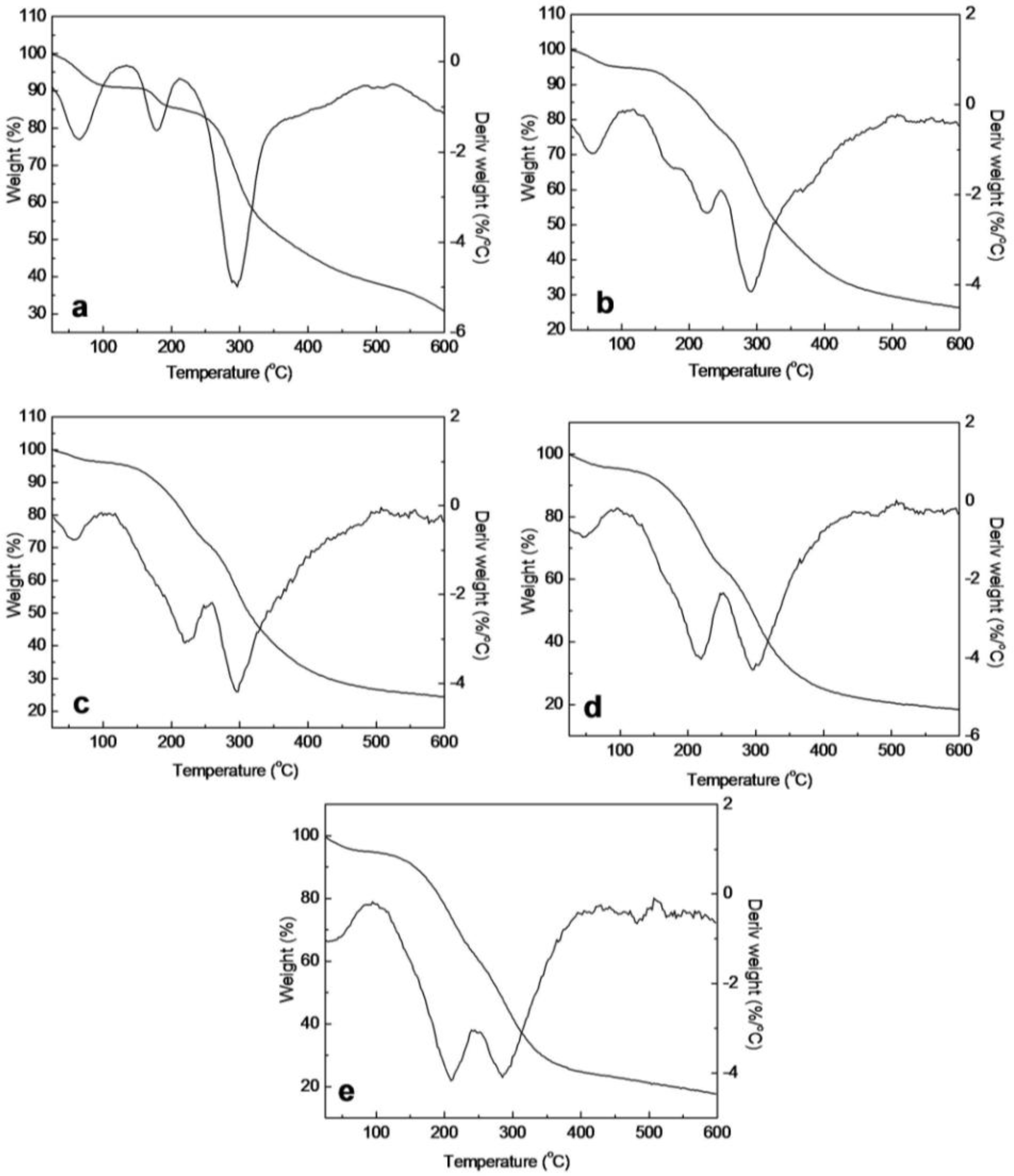
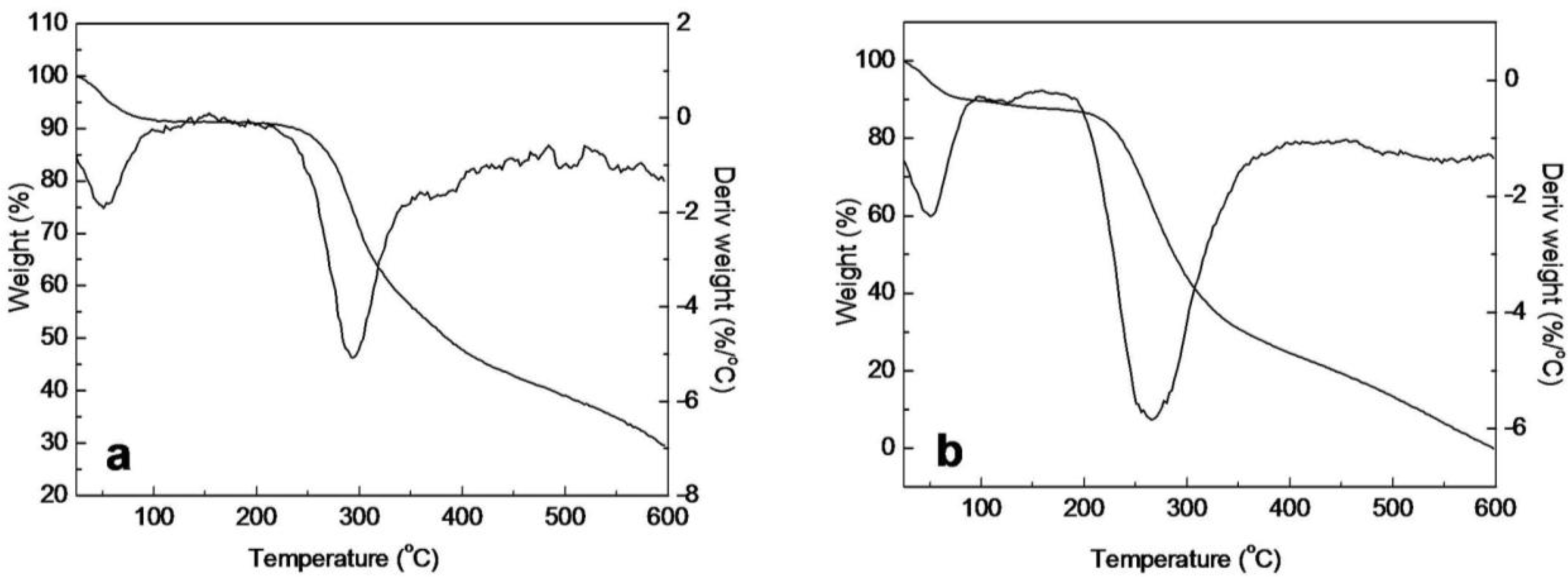
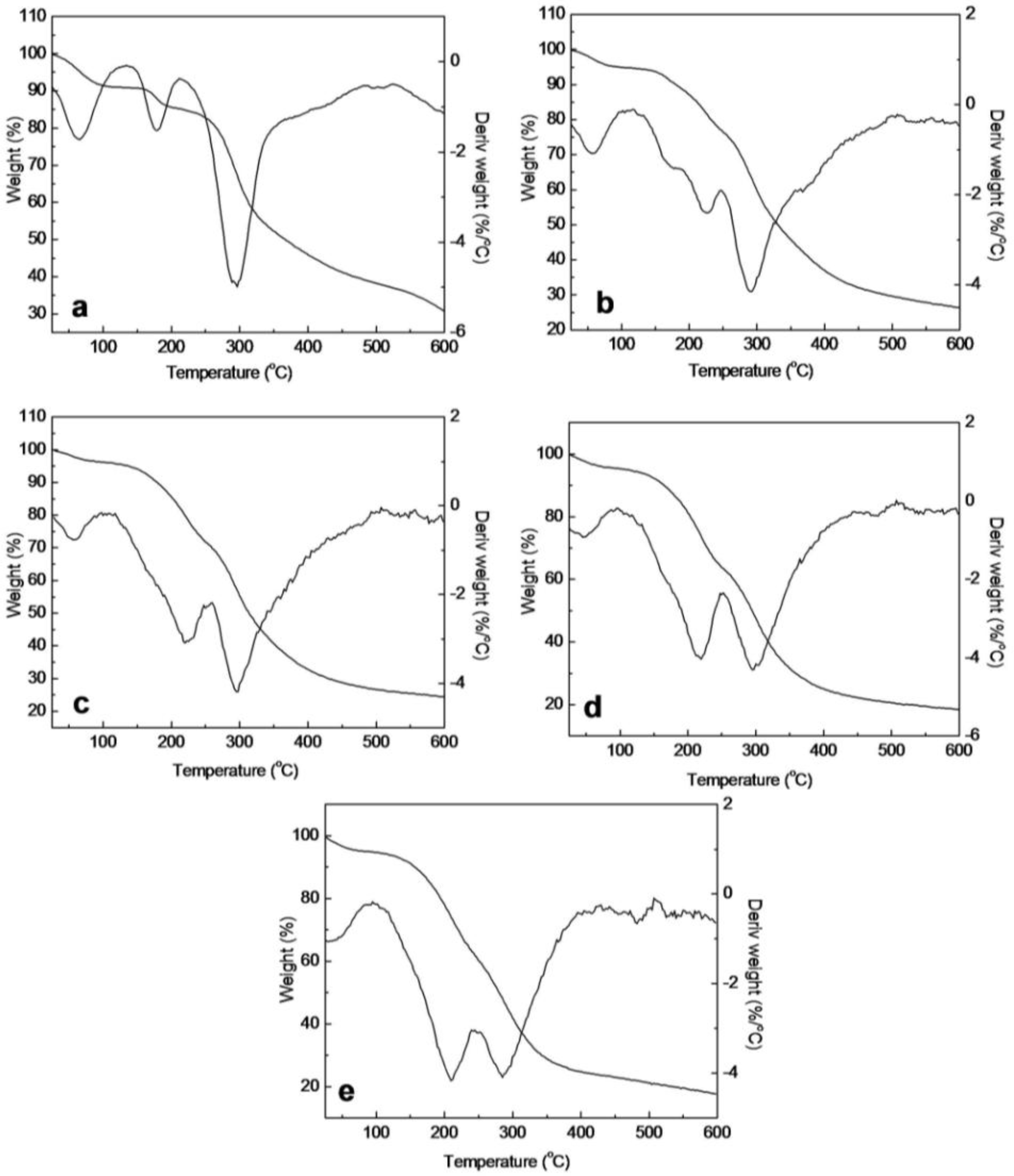
2.5. Thermal Analysis of SF/HBC Nanofibers
3. Experimental Section
3.1. Materials
3.2. Preparation of Regenerated SF
3.3. Electrospinning of SF/HBC Nanofibers
3.4. Characterization
4. Conclusions
Acknowledgments
References
- Langer, R; Vacanti, JP. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar]
- Xu, CY; Inai, R; Kotaki, M; Ramakrishna, S. Aligned biodegradable nanofibrous structure: a potential scaffold for blood vessel engineering. Biomaterials 2004, 25, 877–886. [Google Scholar]
- Min, BM; Lee, G; Kim, SH; Nam, YS; Lee, TS; Park, WH. Electrospinning of silk fibroin nanofibers and its effect on the adhesion and spreading of normal human keratinocytes and fibroblasts in vitro. Biomaterials 2004, 25, 1289–1297. [Google Scholar]
- Wang, M; Jin, HJ; David, L; Rutledge, GC. Mechanical properties of electrospun silk fibers. Macromolecules 2004, 37, 6856–6864. [Google Scholar]
- Santin, M; Motta, A; Freddi, G; Cannas, M. In vitro evaluation of the inflammatory potential of the silk fibroin. J. Biomed. Mater. Res 1999, 46, 382–389. [Google Scholar]
- Horan, RL; Antle, K; Collette, AL; Huang, YZ; Huang, J; Moreau, JE; Volloch, V; Kaplan, DL; Altman, GH. In vitro degradation of silk fibroin. Biomaterials 2005, 26, 3385–3393. [Google Scholar]
- Vepari, C; Kaplan, DL. Silk as a biomaterial. Prog. Polym. Sci 2007, 32, 991–1007. [Google Scholar]
- Numata, K; Subramanian, B; Currie, HA; Kaplan, DL. Bioengineered silk protein-based gene delivery systems. Biomaterials 2009, 30, 5775–5784. [Google Scholar]
- Jiang, X; Zhao, J; Wang, S; Sun, X; Zhang, X; Chen, J; Kaplan, DL; Zhang, Z. Mandibular repair in rats with premineralized silk scaffolds and BMP-2-modified bMSCs. Biomaterials 2009, 30, 4522–4532. [Google Scholar]
- Mandal, BB; Kundu, SC. Cell proliferation and migration in silk fibroin 3D scaffolds. Biomaterials 2009, 30, 2956–2965. [Google Scholar]
- Strand, SP; Lelu, S; Reitan, NK; De Lange Davies, C; Artursson, P; Varum, KM. Molecular design of chitosan gene delivery systems with an optimized balance between polyplex stability and polyplex unpacking. Biomaterials 2010, 31, 975–987. [Google Scholar]
- Chen, MC; Wong, HS; Lin, KJ; Chen, HL; Wey, SP; Sonaje, K; Lin, YH; Chu, CY; Sung, HW. The characteristics, biodistribution and bioavailability of a chitosan-based nanoparticulate system for the oral delivery of heparin. Biomaterials 2009, 30, 6629–6637. [Google Scholar]
- Tchemtchoua, VT; Atanasova, G; Aqil, A; Maquet, V; Jerôme, C; Poumay, Y; Colige, A. Development of a procedure to simultaneously isolate RNA, DNA, and proteins from characterizing cells invading or cultured on chitosan scaffolds. Anal. Biochem 2009, 393, 145–147. [Google Scholar]
- Thanou, BI; Florea, M; Geldof, HE; Junginger, HE; Borchard, G. Quaternized chitosan oligomers as novel gene delivery vectors in epithelial cell lines. Biomaterials 2002, 23, 153–159. [Google Scholar]
- She, ZD; Jin, CR; Huang, Z; Zhang, BF; Feng, QL; Xu, YX. Silk fibroin/chitosan scaffold: Preparation, characterization and culture with HepG2 cell. J. Mater. Sci. Mater. Med 2008, 19, 3545–3553. [Google Scholar]
- Yang, MC; Wang, SS; Chou, NK; Chi, NH; Huang, YY; Chang, YL; Shieh, MJ; Chung, ZW. The cardiomyogenic differentiation of rat mesenchymal stem cells on silk fibroin-polysaccharide cardiac patches in vitro. Biomaterials 2009, 30, 3757–3765. [Google Scholar]
- Li, WJ; Laurencin, CT; Caterson, EJ; Tuan, RS; Ko, FK. Electrospun nanofibrous structure: A novel scaffold for tissue engineering. J. Biomed. Mater. Res 2002, 60, 613–621. [Google Scholar]
- Dang, JM; Leong, KW. Myogenic induction of aligned mesenchymal stem cell sheets by culture on thermally responsive electrospun nanofibers. Adv. Mater 2007, 19, 2775–2779. [Google Scholar]
- Chen, BY; Dang, JY; Tan, TL; Fang, N; Chen, WN; Leong, KW; Chan, V. Dynamics of smooth muscle cell deadhesion from thermosensitivehydroxybutyl chitosan. Biomaterials 2007, 28, 1503–1514. [Google Scholar]
- Dang, JM; Sun, DD; Shin-Ya, Y; Sieber, AN; Kostuik, JP; Leong, KW. Temperature-responsive hydroxybutyl chitosan for the culture of mesenchymal stem cells and intervertebral disk cells. Biomaterials 2006, 27, 406–418. [Google Scholar]
- Sionkowsk, A; Wisniewski, M; Skopinsk, J; Kennedy, CJ; Wess, TJ. Molecular interactions in collagen and chitosan blends. Biomaterials 2004, 25, 795–801. [Google Scholar]
- Chen, X; Li, WJ; Yu, TY. Conformation transition of silk fibroin induced by blending chitosan. J. Appl. Polym. Sci. Part B: Polym. Phys 1997, 35, 2293–2296. [Google Scholar]
- Kweon, A; Ha, HC; Um, IC; Park, YH. Physical properties of silk fibroin/chitosan blend films. J. Appl. Polym. Sci 2001, 80, 928–934. [Google Scholar]
- Huang, CB; Chen, SL; Lai, CL. Electrospun polymer nanofibres with small diameters. Nanotechnology 2006, 17, 1558–1563. [Google Scholar]
- Zong, XH; Kim, K; Fang, DF; Ran, SF; Hsiao, BS; Chu, B. Structure and process relationship of electrospun bioabsorbable nanofiber membranes. Polymer 2002, 43, 4403–4412. [Google Scholar]
- Magoshi, J; Mizuide, M; Magoshi, Y. Physical properties and structure of silk—VI conformational changes in silk fibroin induced by immersion in water at 2 to 130 °C. J. Polym. Sci. Polym. Phys. Edit 1979, 17, 515–520. [Google Scholar]
- Chen, X; Shao, ZZ; Marinkovic, NS; Miller, LM; Zhou, P; Chance, MR. Conformation transition kinetics of regenerated Bombyx mori silk fibroin membrane monitored by time-resolved FTIR spectroscopy. Biophys. Chem 2001, 89, 25–34. [Google Scholar]
- Fernández Cervera, M; Heinämäki, J; Räsänen, M; Maunu, SL; Karjalainen, M; Nieto Acosta, OM; Iraizoz Colartea, A; Yliruusi, J. Solid-state characterization of chitosans derived from lobster chitin. Carbon. Polym 2004, 58, 401–408. [Google Scholar]
- Asakura, T; Iwadate, M; Demura, M; Williamson, MP. Structural analysis of silk with 13C NMR chemical shift contour plots. Int. J. Biol. Macromol 1999, 24, 167–171. [Google Scholar]
- Asakura, AK; Tabeta, R; Saito, H. Conformational characterization of Bombyx mori silk fibroin in the solid state by high-frequency carbon-13 cross polarization-magic angle spinning NMR, X-ray diffraction, and infrared spectroscopy. Macromolecules 1985, 18, 1841–1845. [Google Scholar]
- Du, CH; Zhu, BK; Chen, JY; Xu, YY. Metal ion permeation properties of silk fibroin/chitosan blend membranes. Polym. Int 2006, 55, 377–382. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Content of HBC | Crystallinities (%) | Content of HBC | Crystallinities (%) |
|---|---|---|---|
| 0 | 16.9 | 80 | 24.9 |
| 20 | 25.0 | 100 | 14.6 |
| 50 | 26.1 |
| Raw SF | Electrospun SF | Electrospun SF/HBC blends | Electrospun HBC | Raw HBC | |||
|---|---|---|---|---|---|---|---|
| 80:20 | 50:50 | 20:80 | |||||
| First stage (°C) | 24–215 | 24–147 | 25–122 | 27–122 | 25–122 | 25–122 | 25–185 |
| Tmax1 (°C) | 48.9 | 65.5 | 54.1 | 53.0 | 50.2 | 41.1 | 47.9 |
| Loss mass (%) | 9.05 | 9.15 | 5.36 | 4.36 | 5.53 | 6.05 | 12.74 |
| Second stage (°C) | 215–600 | 147–210 | 122–250 | 122–251 | 122–250 | 122–250 | 185–600 |
| Tmax2 (°C) | 289.5 | 177.2 | 223.5 | 221.3 | 219.8 | 210.7 | 263.3 |
| Loss mass (%) | 61.72 | 5.54 | 18.08 | 23.88 | 30.60 | 44.43 | 87.26 |
| Third stage (°C) | — | 210–600 | 250–600 | 251–600 | 250–600 | 250–600 | — |
| Tmax3 (°C) | — | 293.7 | 291.4 | 293.3 | 300.4 | 284.6 | — |
| Loss mass (%) | — | 54.34 | 50.14 | 47.32 | 45.43 | 42.53 | — |
| Residues (%) | 29.2 | 30.97 | 26.42 | 24.44 | 18.44 | 17.99 | 0 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, K.-H.; Yu, Q.-Z.; Mo, X.-M. Fabrication and Intermolecular Interactions of Silk Fibroin/Hydroxybutyl Chitosan Blended Nanofibers. Int. J. Mol. Sci. 2011, 12, 2187-2199. https://doi.org/10.3390/ijms12042187
Zhang K-H, Yu Q-Z, Mo X-M. Fabrication and Intermolecular Interactions of Silk Fibroin/Hydroxybutyl Chitosan Blended Nanofibers. International Journal of Molecular Sciences. 2011; 12(4):2187-2199. https://doi.org/10.3390/ijms12042187
Chicago/Turabian StyleZhang, Kui-Hua, Qiao-Zhen Yu, and Xiu-Mei Mo. 2011. "Fabrication and Intermolecular Interactions of Silk Fibroin/Hydroxybutyl Chitosan Blended Nanofibers" International Journal of Molecular Sciences 12, no. 4: 2187-2199. https://doi.org/10.3390/ijms12042187