Importance of the Long-Chain Fatty Acid Beta-Hydroxylating Cytochrome P450 Enzyme YbdT for Lipopeptide Biosynthesis in Bacillus subtilis Strain OKB105

Abstract
:1. Introduction
2. Results
2.1. In-Frame Mutation of ybdT in B. subtilis Strain OKB105
2.2. YbdT Activity of OKB105 and NHY1 Cells
2.3. Effect of the ybdT Gene Mutation on Surfactin Synthesis
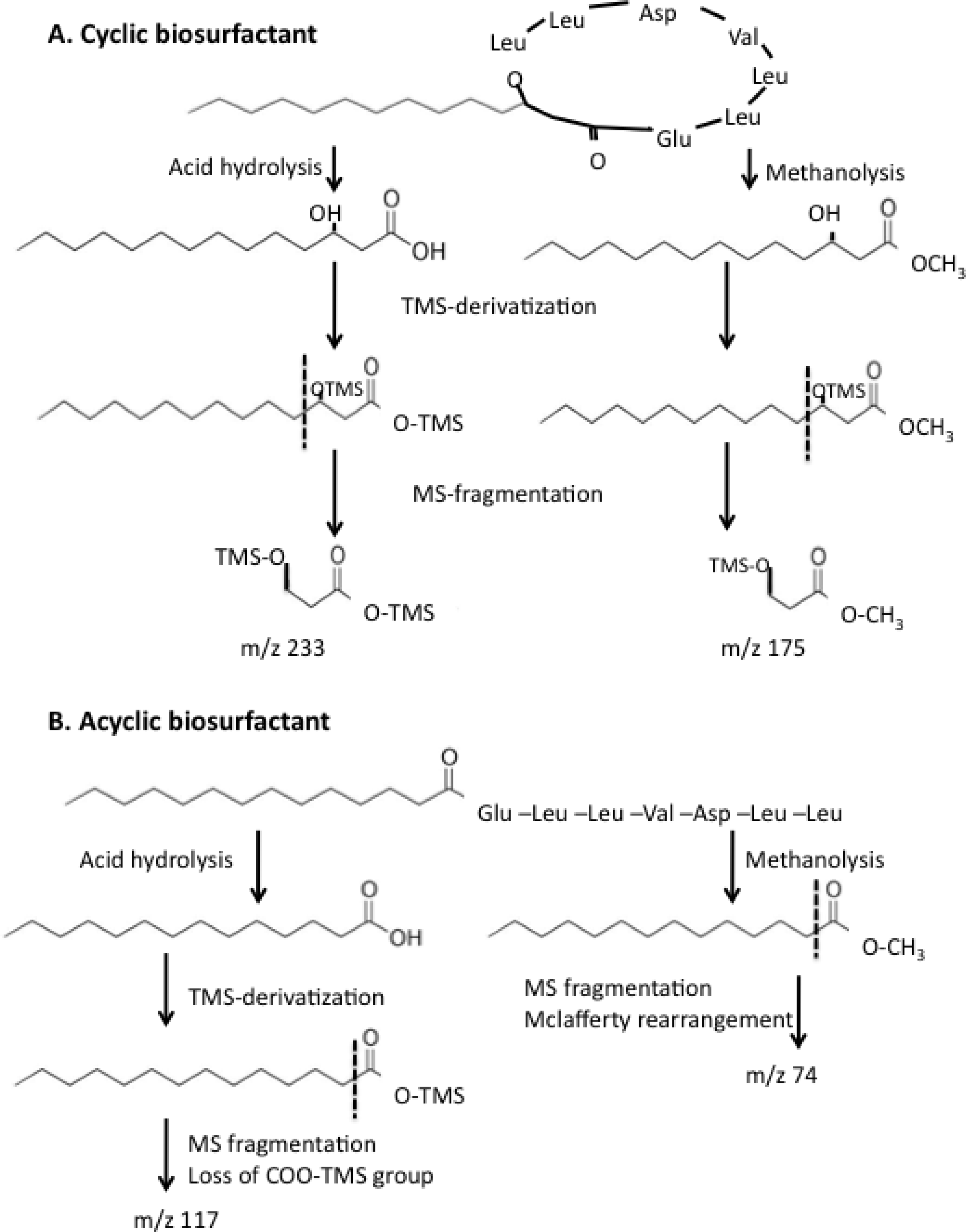
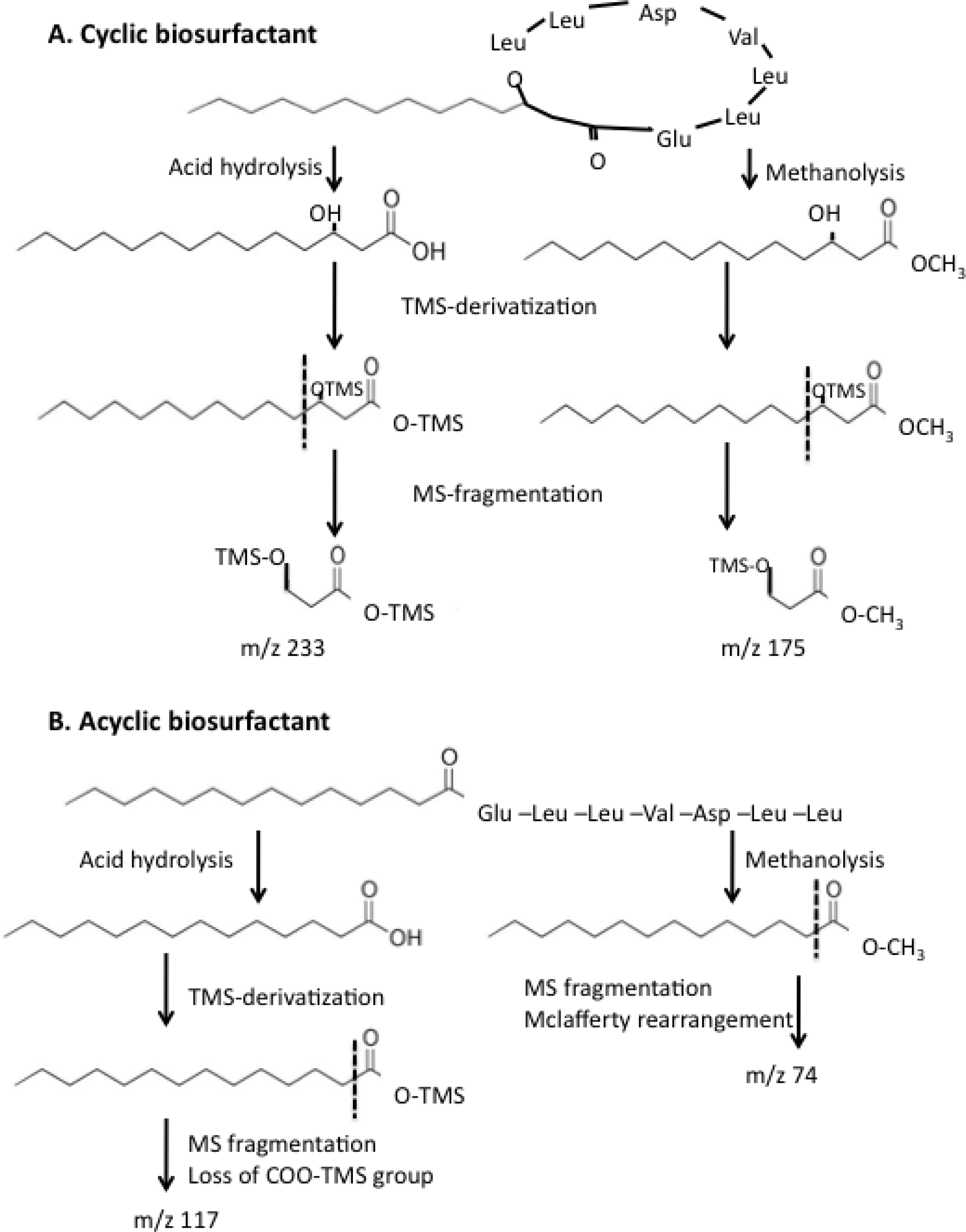
2.4. Effect of ybdT Gene Mutation on Biosurfactant Structure
3. Discussion
4. Experimental Section
4.1. Strains, Plasmids and Growth Conditions
4.2. PCR Construct Preparation
4.3. Transformation
4.4. Expression of srf (Surfactin Synthetase) Gene
4.5. Assay of YbdT Activity
4.6. Biosurfactant Synthesis in Cell-Free Extracts from l-Amino Acids and 3-Hydroxymyristoyl-CoA
4.7. Enzymatic Preparation of 3-Hydroxymyristoyl-CoA
4.8. Purification and Quantification of the 3-Hydroxymyristoyl CoA
4.9. Biosurfactant Synthesis by Cell-Free Extracts from l-amino Acids, Myristic Acid and CoA
4.10. Determination of the Biosurfactant Structure
5. Conclusions
Acknowledgments
References
- Banat, IM. Biosurfactants production and possible uses in microbial enhanced oil recovery and oil pollution remediation: a review. Bioresour. Technol 1995, 51, 1–12. [Google Scholar]
- Banat, IM; Makkar, RS; Cameotra, SS. Potential commercial applications of microbial surfactants. Appl. Microbiol. Biotechnol 2000, 53, 495–508. [Google Scholar]
- Georgiou, G; Lin, SC; Sharma, MM. Surface-active compounds from microorganisms. Biotechnology 1999, 10, 60–65. [Google Scholar]
- Ron, EZ; Rosenberg, E. Natural roles of biosurfactants. Environ. Microbiol 2001, 3, 229–236. [Google Scholar]
- Seydlová, G; Svobodová, J. Review of surfactin chemical properties and the potential biomedical applications. Cent. Eur. J. Med 2008, 3, 123–133. [Google Scholar]
- Arima, K; Kakinuma, A; Tamura, G. Surfactin, a crystaline peptidelipid surfactant produced by Bacillus subtilis: Isolation, characterization, and its inhibition of fibrin clot formation. Biochem. Biophys. Res. Commun 1968, 31, 488–497. [Google Scholar]
- Grangemard, I; Bonmatin, JM; Bernillon, J; Das, BC; Peypoux, F. Lichenysins G, a novel family of lipopeptide biosurfactants from Bacillus licheniformis IM 1307: production, isolation and structural evaluation by NMR and mass spectrometry. J. Antibiot 1999, 52, 363–373. [Google Scholar]
- Jenny, K; Käppeli, O; Fiechter, A. Biosurfactants from Bacillus licheniformis: structural analysis and characterization. Appl. Microbiol. Biotechnol 1991, 36, 5–13. [Google Scholar]
- Peypoux, F; Bonmatin, JM; Wallach, J. Recent trends in the biochemistry of surfactin. Appl. Microbiol. Biotechnol 1999, 51, 553–563. [Google Scholar]
- Marahiel, MA; Stachelhaus, T; Mootz, HD. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev 1997, 97, 2651–2674. [Google Scholar]
- Rausch, C; Hoof, I; Weber, T; Wohlleben, W; Huson, D. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol. Biol 2007, 7, 78. [Google Scholar]
- Straight, PD; Fischbach, MA; Walsh, CT; Rudner, DZ; Kolter, R. A singular enzymatic megacomplex from Bacillus subtilis. Proc. Nat. Acad. Sci. USA 2007, 104, 305–310. [Google Scholar]
- Strieker, M; Tanovic, A; Marahiel, MA. Nonribosomal peptide synthetases: structures and dynamics. Curr. Opin. Struct. Biol 2010, 20, 234–240. [Google Scholar]
- Walsh, CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science 2004, 303, 1805–1810. [Google Scholar]
- Bruner, SD; Weber, T; Kohli, RM; Schwarzer, D; Marahiel, MA; Walsh, CT; Stubbs, MT. Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure 2002, 10, 301–310. [Google Scholar]
- Cosmina, P; Rodriguez, F; de Ferra, F; Grandi, G; Perego, M; Venema, G; van Sinderen, D. Sequence and analysis of the genetic locus responsible for surfactin synthesis in Bacillus subtilis. Mol. Microbiol 1993, 8, 821–831. [Google Scholar]
- D'Souza, C; Nakano, MM; Corbell, N; Zuber, P. Amino-acylation site mutations in amino acid-activating domains of surfactin synthetase: effects on surfactin production and competence development in Bacillus subtilis. J. Bacteriol 1993, 175, 3502–3510. [Google Scholar]
- Galli, G; Rodriguez, F; Cosmina, P; Pratesi, C; Nogarotto, R; de Ferra, F; Grandi, G. Characterization of the surfactin synthetase multi-enzyme complex. Biochim. Biophys. Acta 1994, 1205, 19–28. [Google Scholar]
- Menkhaus, M; Ullrich, C; Kluge, B; Vater, J; Vollenbroich, D; Kamp, RM. Structural and functional organization of the surfactin synthetase multienzyme system. J. Biol. Chem 1993, 268, 7678–7684. [Google Scholar]
- Nakano, MM; Corbell, N; Besson, J; Zuber, P. Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol. Gen. Genet 1992, 232, 313–321. [Google Scholar]
- Vollenbroich, D; Mehta, N; Zuber, P; Vater, J; Kamp, RM. Analysis of surfactin synthetase subunits in srfA mutants of Bacillus subtilis OKB105. J. Bacteriol 1994, 176, 395–400. [Google Scholar]
- Steller, S; Sokoll, A; Wilde, C; Bernhard, F; Franke, P; Vater, J. Initiation of surfactin biosynthesis and the role of the SrfD-thioesterase protein. Biochemistry 2004, 43, 11331–11343. [Google Scholar]
- Kraas, FI; Helmetag, V; Wittmann, M; Strieker, M; Marahiel, MA. Functional Dissection of Surfactin Synthetase Initiation Module Reveals Insights into the Mechanism of Lipoinitiation. Chem. Biol 2010, 17, 872–880. [Google Scholar]
- Bonmatin, JM; Genest, M; Labbé, H; Ptak, M. Solution three-dimensional structure of surfactin: A cyclic lipopeptide studied by 1H-NMR, distance geometry, and molecular dynamics. Biopolymers 1994, 34, 975–986. [Google Scholar]
- Stachelhaus, T; Marahiel, MA. Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science 1995, 269, 5571–5574. [Google Scholar]
- Yakimov, MM; Fredrickson, HL; Timmis, KN. Effect of heterogeneity of hydrophobic moieties on surface activity of lichenysin A, a lipopeptide biosurfactant from Bacillus licheniformis BAS50. Biotechnol. Appl. Biochem 1996, 23, 13–18. [Google Scholar]
- Schneider, A; Stachelhaus, T; Marahiel, MA. Targeted alteration of the substrate specificity of peptide synthetases by rational module swapping. Mol. Gen. Genet 1998, 257, 308–318. [Google Scholar]
- Deleu, M; Bouffioux, O; Razafindralambo, H; Paquot, M; Hbid, C; Thonart, P; Jacques, P; Brasseur, R. Interaction of Surfactin with Membranes: A Computational Approach. Langmuir 2003, 19, 3377–3385. [Google Scholar]
- Youssef, NH; Duncan, KE; McInerney, MJ. Importance of 3-hydroxyfatty acid composition of lipopeptides for biosurfactant activity. Appl. Environ. Microbiol 2005, 71, 7690–7695. [Google Scholar]
- von Wachenfeldt, C; Hedenrstedt, L. Respiratory cytochromes, other heme proteins, and heme biosynthesis. In Bacillus subtilis and Its Closest Relatives: from Genes to Cells; Sonenshein, AL, Hoch, JA, Losick, R, Eds.; ASM Press: Washington, DC, USA, 2002; pp. 163–180. [Google Scholar]
- Hlavica, P; Lehnerer, M. Oxidative biotransformation of fatty acids by cytochromes P450: Predicted key structural elements orchestrating substrate specificity, regioselectivity and catalytic efficiency. Curr. Drug Metabol 2010, 11, 85–104. [Google Scholar]
- Cryle, MJ; Matovic, NJ; de Voss, JJ. Products of Cytochrome P450BioI (CYP107H1)-Catalyzed Oxidation of Fatty Acids. Org. Lett 2003, 5, 3341–3344. [Google Scholar]
- Cryle, MJ; Schlichting, I. Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450BioI ACP complex. Proc. Natl. Acad. Sci. USA 2008, 105, 15696–15701. [Google Scholar]
- Gustafsson, MCU; Roitel, O; Marshall, KR; Noble, MA; Chapman, SK; Pessegueiro, A; Fulco, AJ; Cheesman, MR; von Wachenfeldt, C; Munro, AW. Expression, purification, and characterization of Bacillus subtilis cytochromes P450 CYP102A2 and CYP102A3: Flavocytochrome homologues of P450 BM3 from Bacillus megaterium. Biochemistry 2004, 43, 5474–5487. [Google Scholar]
- Reddick, JJ; Antolak, SA; Raner, GM. PksS from Bacillus subtilis is a cytochrome P450 involved in bacillaene metabolism. Biochem. Biophys. Res. Commun 2007, 358, 363–367. [Google Scholar]
- Rivolta, C; Soldo, B; Lazarevic, V; Joris, B; Mauel, C; Karamat, D. A 35.7 kb DNA fragment from the Bacillus subtilis chromosome containing a putative 12.3 kb operon involved in hexuronate catabolism and a perfectly symmetrical hypothetical catabolite-responsive element. Microbiology 1998, 144, 877–884. [Google Scholar]
- Belitsky, BR; Gustafsson, MC; Sonenshein, AL; von Wachenfeldt, C. An lrp-like gene of Bacillus subtilis involved in branched-chain amino acid transport. J. Bacteriol 1997, 179, 5448–5457. [Google Scholar]
- Cryle, MJ; Bell, SG; Schlichting, I. Structural and biochemical characterization of the cytochrome P450 CypX (CYP134A1) from Bacillus subtilis: A cyclo-l-leucyl-l-leucyl dipeptide oxidase. Biochemistry 2010, 49, 7282–7296. [Google Scholar]
- Matsunaga, I; Ueda, A; Fujiwara, N; Sumimoto, T; Ichihara, K. Characterization of the ybdT gene product of Bacillus subtilis: novel fatty acid beta-hydroxylating cytochrome P450. Lipids 1999, 34, 841–846. [Google Scholar]
- Lee, D-S; Yamada, A; Sugimoto, H; Matsunaga, I; Ogura, H; Ichihara, K; Adachi, S-I; Park, S-Y; Shiro, Y. Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. J. Biol. Chem 2003, 278, 9761–9767. [Google Scholar]
- Majewski, J; Cohan, FM. DNA sequence similarity requirements for interspecific recombination in Bacillus. Genetics 1999, 153, 1525–1533. [Google Scholar]
- Zar, JH. Two-sample hypotheses. In Biostatistical Analysis, 4th ed; Ryu, T, Ed.; Pentice Hall: Upper Saddle River, NJ, USA, 1999; pp. 122–160. [Google Scholar]
- Christie, WW. The lipid library: Mass spectra of some miscellaneous fatty acids and other lipid derivatives—Archive. Avaiable online: http://lipidlibrary.aocs.org/ms/arch_xyz/index.htm#tmsa (accessed on 1 December 2010).
- Christie, WW. The AOCS lipid library: Methyl esters of fatty acids—Archive of mass spectra. Avaiable online: http://lipidlibrary.aocs.org/ms/arch_me/index.htm (accessed on 1 December 2010).
- Christie, WW. The AOCS lipid library: Mass spectra of methyl esters of fatty acids. Part 1. Normal saturated fatty acids. Avaiable online: http://lipidlibrary.aocs.org/ms/ms03/index.htm (accessed on 1 December 2010).
- Chickos, JS; Way, BA; Wilson, J; Shaharuzzaman, M; Laird, J; Landt, M. Analysis of 3-hydroxydodecanedioic acid for studies of fatty acid metabolic disorders: Preparation of stable isotope standards. J. Clin. Lab. Anal 2002, 16, 115–120. [Google Scholar]
- Peypoux, F; Bonmatin, JM; Labbe, H; Grangemard, I; Das, BC; Ptak, M; Wallach, J; Michel, G. [Ala 4] surfactin, a novel isoform from Bacillus subtilis studied by mass and NMR spectroscopies. Eur. J. Biochem 1994, 224, 89–96. [Google Scholar]
- Kowall, M; Vater, J; Kluge, B; Stein, T; Franke, P; Ziessow, D. Separation and characterization of surfactin isoforms produced by Bacillus subtilis OKB 105. J. Colloid Interface Sci 1998, 204, 1–8. [Google Scholar]
- Bonmatin, J-M; Labbe, H; Grangemard, I; Peypoux, F; Maget-Dana, R; Ptak, M; Michel, G. Production, isolation and characterization of [Leu4]- and [Ile4]-surfactins from Bacillus subtilis. Lett. Pept. Sci 1995, 2, 41–47. [Google Scholar]
- Tang, J-S; Zhao, F; Gao, H; Dai, Y; Yao, Z-H; Hong, K; Li, J; Ye, W-C; Yao, X-S. Characterization and Online Detection of Surfactin Isomers Based on HPLC-MSn Analyses and Their Inhibitory Effects on the Overproduction of Nitric Oxide and the Release of TNF-Œ± and IL-6 in LPS-Induced Macrophages. Mar. Drugs 2010, 8, 2605–2618. [Google Scholar]
- Morikawa, M; Hirata, Y; Imanaka, T. A study on the structure-function relationship of lipopeptide biosurfactants. Biochim. Biophys. Acta 2000, 1488, 211–218. [Google Scholar]
- Ingham, CJ; Furneaux, PA. Mutations in the {beta} subunit of the Bacillus subtilis RNA polymerase that confer both rifampicin resistance and hypersensitivity to NusG. Microbiology 2000, 146, 3041–3049. [Google Scholar]
- Maughan, H; Galeano, B; Nicholson, WL. Novel rpoB mutations conferring rifampin resistance on Bacillus subtilis: global effects on growth, competence, sporulation, and germination. J. Bacteriol 2004, 186, 2481–2486. [Google Scholar]
- Ullrich, C; Kluge, B; Palacz, Z; Vater, J. Cell-free biosynthesis of surfactin, a cyclic lipopeptide produced by Bacillus subtilis. Biochemistry 1991, 30, 6503–6508. [Google Scholar]
- Youssef, NH; Duncan, KE; Nagle, DP; Savage, KN; Knapp, RM; McInerney, MJ. Comparison of methods to detect biosurfactant production by diverse microorganisms. J. Microbiol. Meth 2004, 56, 339–347. [Google Scholar]
- Ho, SN; Hunt, HD; Horton, RM; Pullen, JK; Pease, LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar]
- Horton, RM; Hunt, HD; Ho, SN; Pullen, JK; Pease, LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 1989, 77, 61–68. [Google Scholar]
- Yon, J; Fried, M. Precise gene fusion by PCR. Nucl. Acids Res 1989, 17, 4895. [Google Scholar]
- Cutting, SM; Vander Horn, PB. Genetic analysis. In Molecular Biological Methods for Bacillus; Harwood, CR, Cutting, SM, Eds.; John Wiley & Sons Ltd: Chichester, UK, 1990; Chapter 2, pp. 27–74. [Google Scholar]
- Bradford, MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Milne, KG; Mehlert, A; Ferguson, MAJ. The use of Pseudomonas acyl-CoA synthetase to form acyl-CoAs from dicarboxylic fatty acids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2001, 1531, 1–3. [Google Scholar]
- Alber, BE; Fuchs, G. Propionyl-coenzyme A synthase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Biol. Chem 2002, 277, 12137–12143. [Google Scholar]
- Beuerle, T; Pichersky, E. Enzymatic synthesis and purification of aromatic coenzyme A esters. Anal. Biochem 2002, 302, 305–312. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Cell-Free Extract | Coenzyme A Release (nmol·min−1·ng·protein−1) | 3-Hydroxymyristoyl-CoA Consumption (nmol·min−1·ng·protein−1) | Biosurfactant Production (ng·min−1·ng·protein−1) |
|---|---|---|---|
| OKB105 | 0.018 ± 0.015 b | 0.002 ± 0.002 b | 17.4 ± 5.5 b |
| NHY1 | 0.018 ± 0.017 | 0.001 ± 0.001 | 17.0 ± 1.9 |
| Student’s t-test p-value | 0.98 c | 0.23 c | 0.51 c |
| Fatty Acid (FA) Chain Length | OKB105 Biosurfactant Fatty Acid Composition | NHY1 Biosurfactant Fatty Acid Composition | ||||||
|---|---|---|---|---|---|---|---|---|
| Non-Hydroxylated FA | 3-Hydroxylated FA | Non-Hydroxylated FA | 3-Hydroxylated FA | |||||
| Methanolysis a | Acid Hydrolysis a | Methanolysis | Acid Hydrolysis | Methanolysis | Acid Hydrolysis | Methanolysis | Acid Hydrolysis | |
| 12 | ND c | ND | ND | ND | 8.61 (1) b | 21.2 (1) | ND | ND |
| 13 | 11.6 (1) | ND | 10.26 (3) | 1.87 (2) | 13.06 (1) | ND | 1.99 (1) | ND |
| 14 | 18.23 (2) | 6.18 (2) | 5.15 (2) | 0.63 (1) | 1.92 (1) | 12.6 (2) | 3.57 (1) | 2.18 (2) |
| 15 | 61 (1) | 19.9 (2) | 31.3 (3) | 8.9 (2) | ND | 5.46 (2) | 7.28 (2) | ND |
| 16 | 3.42 (1) | 32.6 (2) | ND | ND | 25.3 (3) | 36.2 (2) | ND | ND |
| 17 | ND | 10.1 (1) | ND | ND | ND | 4.19 (1) | ND | ND |
| 18 | 26.2 (2) | 25.3 (2) | ND | ND | 60.1 (3) | 31 (2) | ND | ND |
| Total | 66.6 (3) | 55.4 (2) | 33.4 (3) | 44.6 (2) | 93.3 (3) | 97.8 (2) | 6.72 (3) | 2.18 (2) |
| Avg. | 60.97 | 39 | 95.6 | 4.4 | ||||
| Deduced FA Chain Length | OKB105 Biosurfactant a | NHY1 Biosurfactant a | ||||
|---|---|---|---|---|---|---|
| M + H+ b | M + Na+ b | M + 2Na+ − H+ b | M + H+ b | M + Na+ b | M + 2Na+ − H+ b | |
| 11 | 1024 | 1004 | ||||
| 12 | 1018 | 1040 | 1016, 1018 | |||
| 13 | 1008 | 1032 | 1030, 1032 | |||
| 14 | 1022 | 1044 | 1044, 1046 | |||
| 15 | 1036 | 1058 | ||||
| 16 | 1050 | 1072 | 1052 | |||
| Primer | Sequence (5′-3′) |
|---|---|
| P1F a | ATGAATGAGCAGATTCCACATGACAAAAGT |
| P2R a | TGCCTCCTAACCTGCCCAATAGCACGCTACCCG |
| P3F a | TGGGCAGGTTAGGAGGCAATGAACTTTAAT |
| P4R a | AATGCCTTCTAAAAGCCAGTCATTAGG |
| P5F a | TGGCTTTTAGAAGGCATTACAATTGAAGTCAT |
| P6R a | ACTTTTTCGTCTGATTCCGCTCATTACGAA |
| Srf1F b | GCGGTAGAAAAACTGCTTGC |
| Srf1R b | ACAGGTTCGTCTGCTTTGCT |
| rpoBF c | TCAACTAGTTCAGTATGGACG |
| rpoBR c | ACCTGGTTCAGGAACATTGTC |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Youssef, N.H.; Wofford, N.; McInerney, M.J. Importance of the Long-Chain Fatty Acid Beta-Hydroxylating Cytochrome P450 Enzyme YbdT for Lipopeptide Biosynthesis in Bacillus subtilis Strain OKB105. Int. J. Mol. Sci. 2011, 12, 1767-1786. https://doi.org/10.3390/ijms12031767
Youssef NH, Wofford N, McInerney MJ. Importance of the Long-Chain Fatty Acid Beta-Hydroxylating Cytochrome P450 Enzyme YbdT for Lipopeptide Biosynthesis in Bacillus subtilis Strain OKB105. International Journal of Molecular Sciences. 2011; 12(3):1767-1786. https://doi.org/10.3390/ijms12031767
Chicago/Turabian StyleYoussef, Noha H., Neil Wofford, and Michael J. McInerney. 2011. "Importance of the Long-Chain Fatty Acid Beta-Hydroxylating Cytochrome P450 Enzyme YbdT for Lipopeptide Biosynthesis in Bacillus subtilis Strain OKB105" International Journal of Molecular Sciences 12, no. 3: 1767-1786. https://doi.org/10.3390/ijms12031767