The Role of Alpha-Dystrobrevin in Striated Muscle

{kind=link}
{kind=link}
Abstract
:1. Introduction
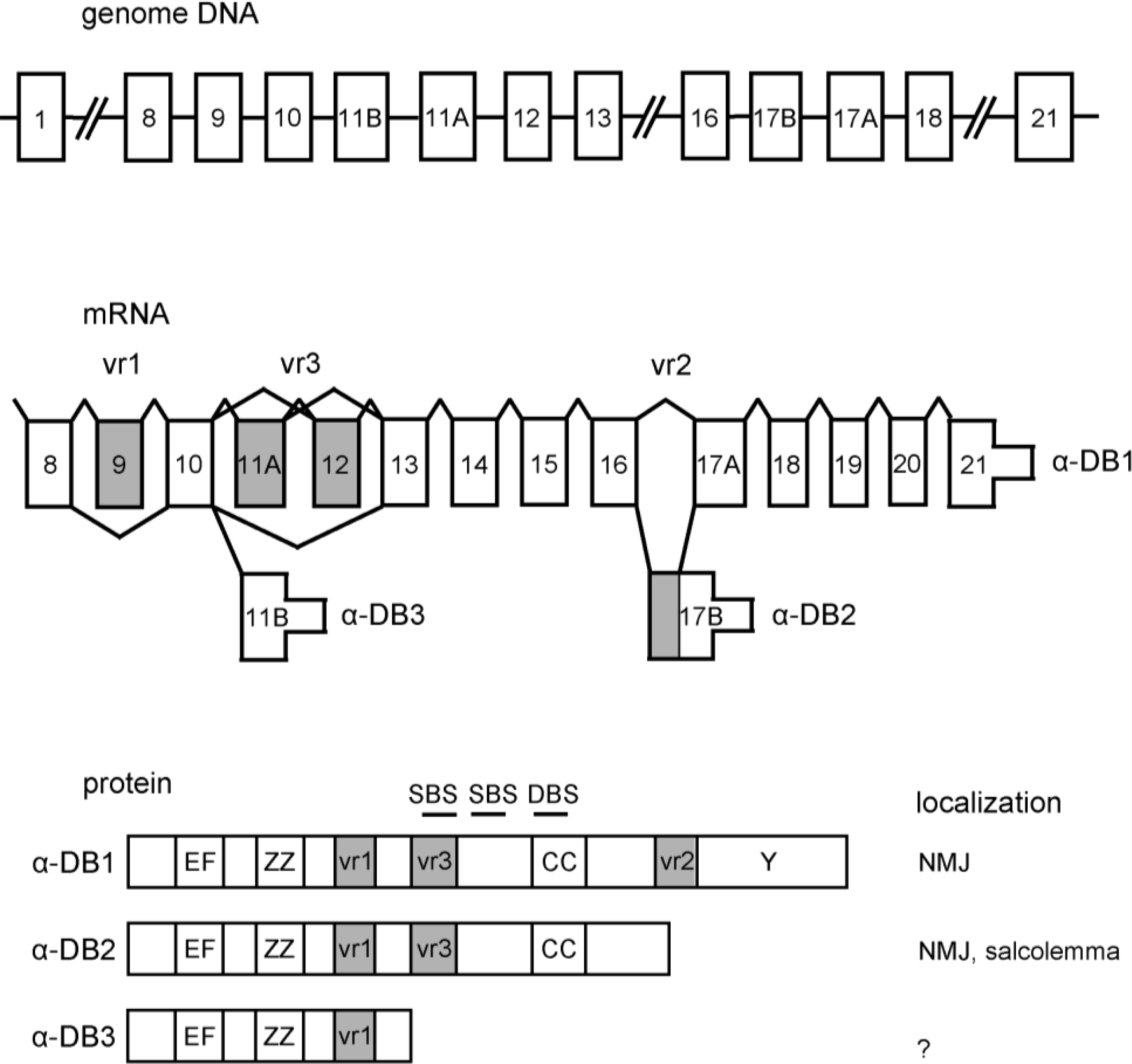
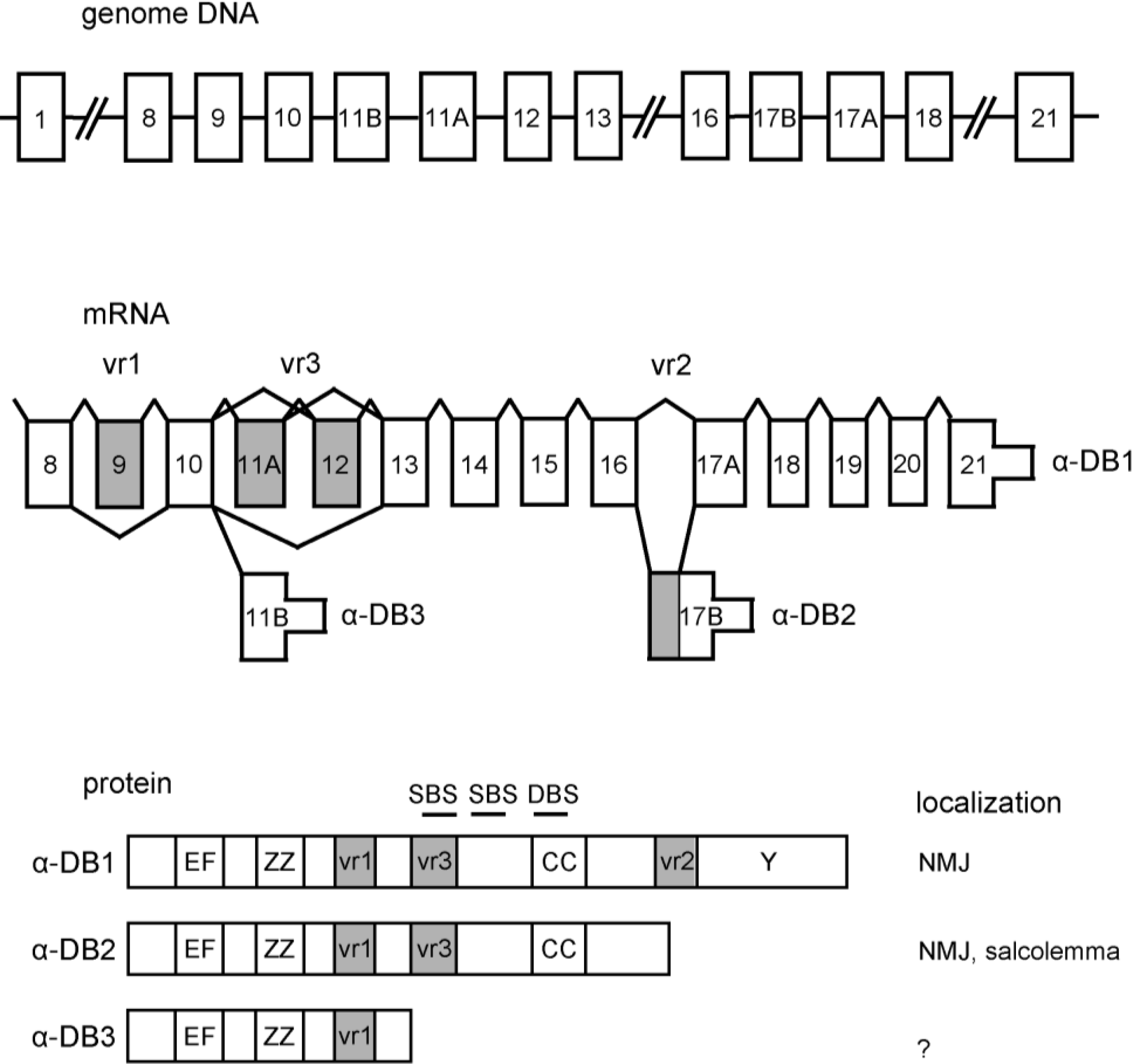
2. α-Dystrobrevin Gene and Transcripts
3. Structure of Dystrobrevin Protein and Localization in Striated Muscle
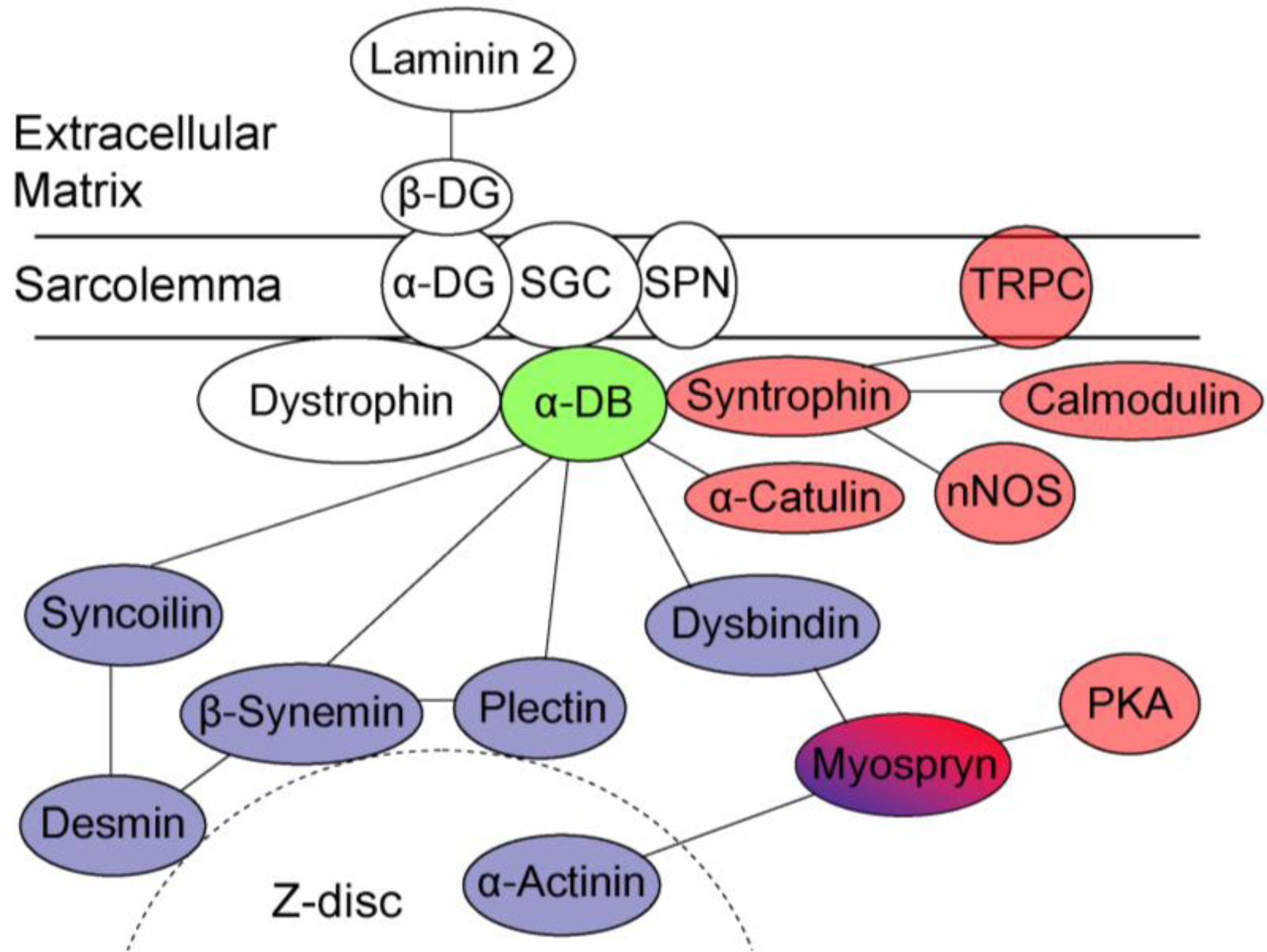
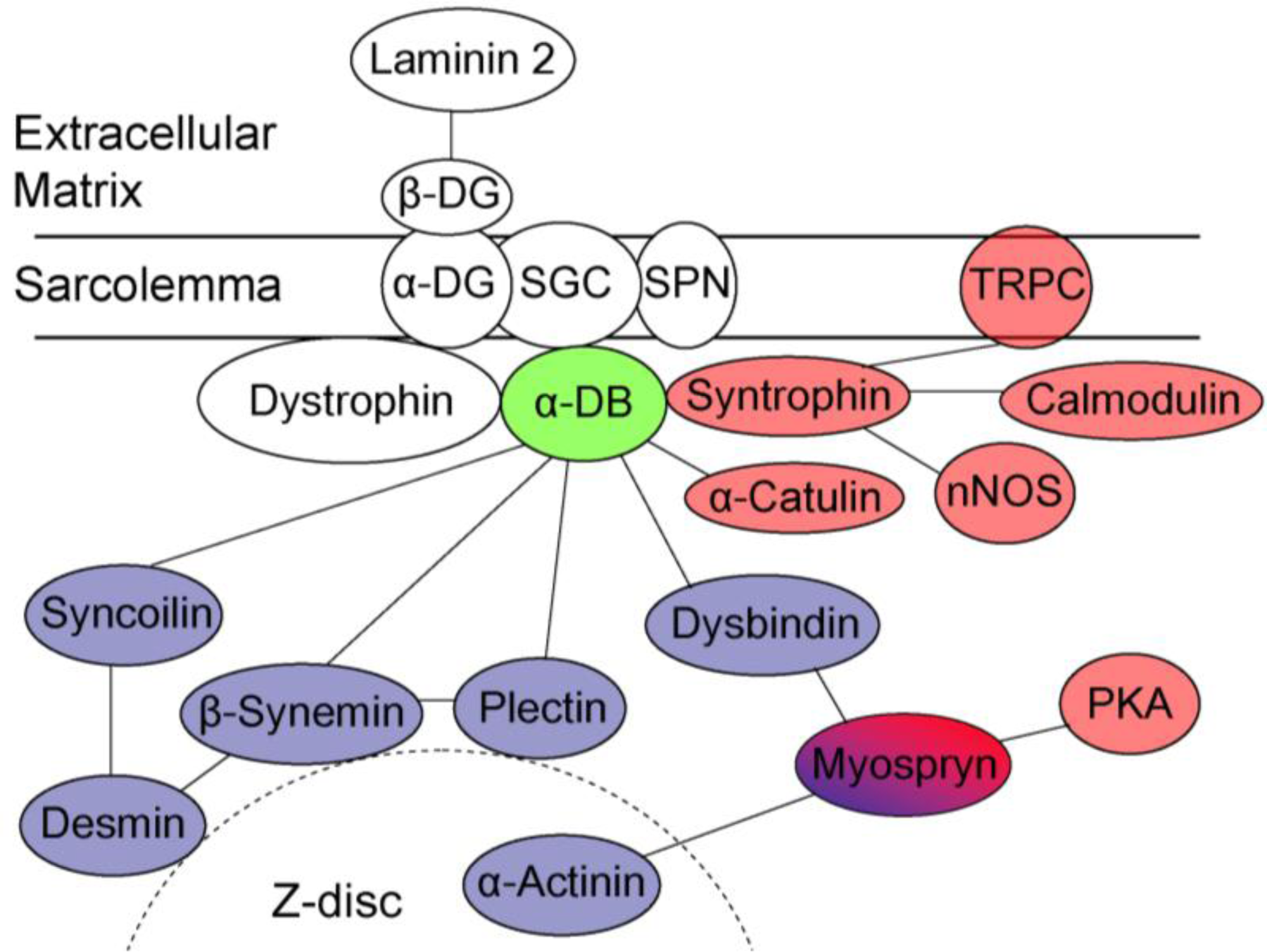
4. The Role of Dystrobrevin for Structural Integrity of Muscle—Interaction with Cytoskeletal Binding Partners
5. The Role of Dystrobrevin in Signaling—Interaction with Syntrophin
6. Dystrobrevin in Muscle Disease
7. Concluding Remarks
Acknowledgments
References
- Wagner, KR; Cohen, JB; Huganir, RL. The 87K postsynaptic membrane protein from Torpedo is a protein-tyrosine kinase substrate homologous to dystrophin. Neuron 1993, 10, 511–522. [Google Scholar]
- Blake, DJ; Tinsley, JM; Davies, KE; Knight, AE; Winder, SJ; Kendrick-Jones, J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteins: Potentials for protein-protein interactions. Trends Biochem. Sci 1995, 20, 133–135. [Google Scholar]
- Sadoulet-Puccio, HM; Khurana, TS; Cohen, JB; Kunkel, LM. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedo electric organ post-synaptic membrane. Hum. Mol. Genet 1996, 5, 489–496. [Google Scholar]
- Blake, DJ; Nawrotzki, R; Peters, MF; Froehner, SC; Davies, KE. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J. Biol. Chem 1996, 271, 7802–7810. [Google Scholar]
- Lapidos, KA; Kakkar, R; McNally, EM. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res 2004, 94, 1023–1031. [Google Scholar]
- Ozawa, E; Noguchi, S; Mizuno, Y; Hagiwara, Y; Yoshida, M. From dystrophinopathy to sarcoglycanopathy: Evolution of a concept of muscular dystrophy. Muscle Nerve 1998, 21, 421–438. [Google Scholar]
- Sadoulet-Puccio, HM; Rajala, M; Kunkel, LM. Dystrobrevin and dystrophin: An interaction through coiled-coil motifs. Proc. Natl. Acad. Sci. USA 1997, 94, 12413–12418. [Google Scholar]
- Yoshida, M; Hama, H; Ishikawa-Sakurai, M; Imamura, M; Mizuno, Y; Araishi, K; Wakabayashi-Takai, E; Noguchi, S; Sasaoka, T; Ozawa, E. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum. Mol. Genet 2000, 9, 1033–1040. [Google Scholar]
- Grady, RM; Grange, RW; Lau, KS; Maimone, MM; Nichol, MC; Stull, JT; Sanes, JR. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol 1999, 1, 215–220. [Google Scholar]
- Sadoulet-Puccio, HM; Feener, CA; Schaid, DJ; Thibodeau, SN; Michels, VV; Kunkel, LM. The genomic organization of human dystrobrevin. Neurogenetics 1997, 1, 37–42. [Google Scholar]
- Peters, MF; Sadoulet-Puccio, HM; Grady, MR; Kramarcy, NR; Kunkel, LM; Sanes, JR; Sealock, R; Froehner, SC. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J. Cell Biol 1998, 142, 1269–1278. [Google Scholar]
- Peters, MF; O’Brien, KF; Sadoulet-Puccio, HM; Kunkel, LM; Adams, ME; Froehner, SC. beta-dystrobrevin, a new member of the dystrophin family. Identification, cloning, and protein associations. J. Biol. Chem 1997, 272, 31561–31569. [Google Scholar]
- Enigk, RE; Maimone, MM. Differential expression and developmental regulation of a novel alpha-dystrobrevin isoform in muscle. Gene 1999, 238, 479–488. [Google Scholar]
- Jones, KJ; Compton, AG; Yang, N; Mills, MA; Peters, MF; Mowat, D; Kunkel, LM; Froehner, SC; North, KN. Deficiency of the syntrophins and alpha-dystrobrevin in patients with inherited myopathy. Neuromuscul. Disord 2003, 13, 456–467. [Google Scholar]
- Nawrotzki, R; Loh, NY; Ruegg, MA; Davies, KE; Blake, DJ. Characterisation of alpha-dystrobrevin in muscle. J. Cell Sci 1998, 111, 2595–2605. [Google Scholar]
- Holzfeind, PJ; Ambrose, HJ; Newey, SE; Nawrotzki, RA; Blake, DJ; Davies, KE. Tissue-selective expression of alpha-dystrobrevin is determined by multiple promoters. J. Biol. Chem 1999, 274, 6250–6258. [Google Scholar]
- Nakamori, M; Kimura, T; Kubota, T; Matsumura, T; Sumi, H; Fujimura, H; Takahashi, MP; Sakoda, S. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology 2008, 70, 677–685. [Google Scholar]
- Bohm, SV; Constantinou, P; Tan, S; Jin, H; Roberts, RG. Profound human/mouse differences in alpha-dystrobrevin isoforms: A novel syntrophin-binding site and promoter missing in mouse and rat. BMC Biol 2009, 7, 85. [Google Scholar]
- Froehner, SC; Adams, ME; Peters, MF; Gee, SH. Syntrophins: Modular adapter proteins at the neuromuscular junction and the sarcolemma. Soc. Gen. Physiol. Ser 1997, 52, 197–207. [Google Scholar]
- Compton, AG; Cooper, ST; Hill, PM; Yang, N; Froehner, SC; North, KN. The syntrophin-dystrobrevin subcomplex in human neuromuscular disorders. J. Neuropathol. Exp. Neurol 2005, 64, 350–361. [Google Scholar]
- Metzinger, L; Blake, DJ; Squier, MV; Anderson, LV; Deconinck, AE; Nawrotzki, R; Hilton-Jones, D; Davies, KE. Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum. Mol. Genet 1997, 6, 1185–1191. [Google Scholar]
- Newey, SE; Howman, EV; Ponting, CP; Benson, MA; Nawrotzki, R; Loh, NY; Davies, KE; Blake, DJ. Syncoilin, a novel member of the intermediate filament superfamily that interacts with alpha-dystrobrevin in skeletal muscle. J. Biol. Chem 2001, 276, 6645–6655. [Google Scholar]
- Mizuno, Y; Thompson, TG; Guyon, JR; Lidov, HG; Brosius, M; Imamura, M; Ozawa, E; Watkins, SC; Kunkel, LM. Desmuslin, an intermediate filament protein that interacts with alpha-dystrobrevin and desmin. Proc. Natl. Acad. Sci. USA 2001, 98, 6156–6161. [Google Scholar]
- Benson, MA; Newey, SE; Martin-Rendon, E; Hawkes, R; Blake, DJ. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J. Biol. Chem 2001, 276, 24232–24241. [Google Scholar]
- Lazarides, E. Intermediate filaments as mechanical integrators of cellular space. Nature 1980, 283, 249–256. [Google Scholar]
- Robson, RM; Huiatt, TW; Bellin, RM. Muscle intermediate filament proteins. Methods Cell Biol 2004, 78, 519–553. [Google Scholar]
- Poon, E; Howman, EV; Newey, SE; Davies, KE. Association of syncoilin and desmin: linking intermediate filament proteins to the dystrophin-associated protein complex. J. Biol. Chem 2002, 277, 3433–3439. [Google Scholar]
- Zhang, J; Bang, ML; Gokhin, DS; Lu, Y; Cui, L; Li, X; Gu, Y; Dalton, ND; Scimia, MC; Peterson, KL; Lieber, RL; Chen, J. Syncoilin is required for generating maximum isometric stress in skeletal muscle but dispensable for muscle cytoarchitecture. Am. J. Physiol. Cell Physiol 2008, 294, C1175–C1182. [Google Scholar]
- Brown, SC; Torelli, S; Ugo, I; de Biasia, F; Howman, EV; Poon, E; Britton, J; Davies, KE; Muntoni, F. Syncoilin upregulation in muscle of patients with neuromuscular disease. Muscle Nerve 2005, 32, 715–725. [Google Scholar]
- Mizuno, Y; Guyon, JR; Watkins, SC; Mizushima, K; Sasaoka, T; Imamura, M; Kunkel, LM; Okamoto, K. Beta-synemin localizes to regions of high stress in human skeletal myofibers. Muscle Nerve 2004, 30, 337–346. [Google Scholar]
- Hijikata, T; Nakamura, A; Isokawa, K; Imamura, M; Yuasa, K; Ishikawa, R; Kohama, K; Takeda, S; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through beta-synemin, alpha-dystrobrevin and actin. J. Cell Sci 2008, 121, 2062–2074. [Google Scholar]
- Benson, MA; Tinsley, CL; Blake, DJ. Myospryn is a novel binding partner for dysbindin in muscle. J. Biol. Chem 2004, 279, 10450–10458. [Google Scholar]
- Durham, JT; Brand, OM; Arnold, M; Reynolds, JG; Muthukumar, L; Weiler, H; Richardson, JA; Naya, FJ. Myospryn is a direct transcriptional target for MEF2A that encodes a striated muscle, alpha-actinin-interacting, costamere-localized protein. J. Biol. Chem 2006, 281, 6841–6849. [Google Scholar]
- Sarparanta, J. Biology of myospryn: What’s known? J. Muscle Res. Cell Motil 2008, 29, 177–180. [Google Scholar]
- Butler, MH; Douville, K; Murnane, AA; Kramarcy, NR; Cohen, JB; Sealock, R; Froehner, SC. Association of the Mr 58,000 postsynaptic protein of electric tissue with Torpedo dystrophin and the Mr 87,000 postsynaptic protein. J. Biol. Chem 1992, 267, 6213–6218. [Google Scholar]
- Ahn, AH; Kunkel, LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J. Cell Biol 1995, 128, 363–371. [Google Scholar]
- Dwyer, TM; Froehner, SC. Direct binding of Torpedo syntrophin to dystrophin and the 87 kDa dystrophin homologue. FEBS Lett 1995, 375, 91–94. [Google Scholar]
- Ahn, AH; Freener, CA; Gussoni, E; Yoshida, M; Ozawa, E; Kunkel, LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J. Biol. Chem 1996, 271, 2724–2730. [Google Scholar]
- Adams, ME; Butler, MH; Dwyer, TM; Peters, MF; Murnane, AA; Froehner, SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 1993, 531–540. [Google Scholar]
- Yang, B; Ibraghimov-Beskrovnaya, O; Moomaw, CR; Slaughter, CA; Campbell, KP. Heterogeneity of the 59-kDa dystrophin-associated protein revealed by cDNA cloning and expression. J. Biol. Chem 1994, 269, 6040–6044. [Google Scholar]
- Peters, MF; Adams, ME; Froehner, SC. Differential association of syntrophin pairs with the dystrophin complex. J. Cell Biol 1997, 138, 81–93. [Google Scholar]
- Piluso, G; Mirabella, M; Ricci, E; Belsito, A; Abbondanza, C; Servidei, S; Puca, AA; Tonali, P; Puca, GA; Nigro, V. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J. Biol. Chem 2000, 275, 15851–15860. [Google Scholar]
- Newey, SE; Benson, MA; Ponting, CP; Davies, KE; Blake, DJ. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr. Biol 2000, 10, 1295–1298. [Google Scholar]
- Adams, ME; Kramarcy, N; Krall, SP; Rossi, SG; Rotundo, RL; Sealock, R; Froehner, SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol 2000, 150, 1385–1398. [Google Scholar]
- Hosaka, Y; Yokota, T; Miyagoe-Suzuki, Y; Yuasa, K; Imamura, M; Matsuda, R; Ikemoto, T; Kameya, S; Takeda, S. Alpha1-syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J. Cell Biol 2002, 158, 1097–107. [Google Scholar]
- Brenman, JE; Chao, DS; Gee, SH; McGee, AW; Craven, SE; Santillano, DR; Wu, Z; Huang, F; Xia, H; Peters, MF; Froehner, SC; Bredt, DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar]
- Hasegawa, M; Cuenda, A; Spillantini, MG; Thomas, GM; Buee-Scherrer, V; Cohen, P; Goedert, M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J. Biol. Chem 1999, 274, 12626–12631. [Google Scholar]
- Oak, SA; Russo, K; Petrucci, TC; Jarrett, HW. Mouse alpha1-syntrophin binding to Grb2: Further evidence of a role for syntrophin in cell signaling. Biochemistry 2001, 40, 11270–11278. [Google Scholar]
- Madhavan, R; Massom, LR; Jarrett, HW. Calmodulin specifically binds three proteins of the dystrophin-glycoprotein complex. Biochem. Biophys. Res. Commun 1992, 185, 753–759. [Google Scholar]
- Iwata, Y; Pan, Y; Yoshida, T; Hanada, H; Shigekawa, M. Alpha1-syntrophin has distinct binding sites for actin and calmodulin. FEBS Lett 1998, 423, 173–177. [Google Scholar]
- Thomas, GD; Sander, M; Lau, KS; Huang, PL; Stull, JT; Victor, RG. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA 1998, 95, 15090–15095. [Google Scholar]
- Dalkilic, I; Kunkel, LM. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev 2003, 13, 231–238. [Google Scholar]
- Kameya, S; Miyagoe, Y; Nonaka, I; Ikemoto, T; Endo, M; Hanaoka, K; Nabeshima, Y; Takeda, S. alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem 1999, 274, 2193–2200. [Google Scholar]
- Wehling, M; Spencer, MJ; Tidball, JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol 2001, 155, 123–131. [Google Scholar]
- Vandebrouck, A; Sabourin, J; Rivet, J; Balghi, H; Sebille, S; Kitzis, A; Raymond, G; Cognard, C; Bourmeyster, N; Constantin, B. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J 2007, 21, 608–617. [Google Scholar]
- Sabourin, J; Lamiche, C; Vandebrouck, A; Magaud, C; Rivet, J; Cognard, C; Bourmeyster, N; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem 2009, 284, 36248–36261. [Google Scholar]
- Treves, S; Anderson, AA; Ducreux, S; Divet, A; Bleunven, C; Grasso, C; Paesante, S; Zorzato, F. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul. Disord 2005, 15, 577–587. [Google Scholar]
- Lyssand, JS; Whiting, JL; Lee, KS; Kastl, R; Wacker, JL; Bruchas, MR; Miyatake, M; Langeberg, LK; Chavkin, C; Scott, JD; Gardner, RG; Adams, ME; Hague, C. α-Dystrobrevin-1 recruits α-catulin to the α1D-adrenergic receptor/dystrophin-associated protein complex signalosome. Proc. Natl. Acad. Sci. USA 2010, 107, 21854–21859. [Google Scholar]
- Janssens, B; Staes, K; van Roy, F. Human alpha-catulin, a novel alpha-catenin-like molecule with conserved genomic structure, but deviating alternative splicing. Biochim. Biophys. Acta 1999, 1447, 341–347. [Google Scholar]
- Ichida, F; Tsubata, S; Bowles, KR; Haneda, N; Uese, K; Miyawaki, T; Dreyer, WJ; Messina, J; Li, H; Bowles, NE; Towbin, JA. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar]
- Stollberger, C; Winkler-Dworak, M; Blazek, G; Finsterer, J. Left ventricular hypertrabeculation/noncompaction with and without neuromuscular disorders. Int. J. Cardiol 2004, 97, 89–92. [Google Scholar]
- Ichida, F. Left ventricular noncompaction. Circ. J 2009, 73, 19–26. [Google Scholar]
- Grady, RM; Zhou, H; Cunningham, JM; Henry, MD; Campbell, KP; Sanes, JR. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin—glycoprotein complex. Neuron 2000, 25, 279–293. [Google Scholar]
- Grady, RM; Akaaboune, M; Cohen, AL; Maimone, MM; Lichtman, JW; Sanes, JR. Tyrosine-phosphorylated and nonphosphorylated isoforms of alpha-dystrobrevin: Roles in skeletal muscle and its neuromuscular and myotendinous junctions. J. Cell Biol 2003, 160, 741–752. [Google Scholar]
- Bunnell, TM; Jaeger, MA; Fitzsimons, DP; Prins, KW; Ervasti, JM. Destabilization of the dystrophin-glycoprotein complex without functional deficits in alpha-dystrobrevin null muscle. PLoS One 2008, 3, e2604. [Google Scholar]
- Mankodi, A; Takahashi, MP; Jiang, H; Beck, CL; Bowers, WJ; Moxley, RT; Cannon, SC; Thornton, CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar]
- Kimura, T; Nakamori, M; Lueck, JD; Pouliquin, P; Aoike, F; Fujimura, H; Dirksen, RT; Takahashi, MP; Dulhunty, AF; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet 2005, 14, 2189–2200. [Google Scholar]
- Osborne, RJ; Thornton, CA. RNA-dominant diseases. Hum. Mol. Genet 2006, 15, R162–R169. [Google Scholar]
- Nakamori, M; Kimura, T; Fujimura, H; Takahashi, MP; Sakoda, S. Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve 2007, 36, 251–257. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakamori, M.; Takahashi, M.P. The Role of Alpha-Dystrobrevin in Striated Muscle. Int. J. Mol. Sci. 2011, 12, 1660-1671. https://doi.org/10.3390/ijms12031660
Nakamori M, Takahashi MP. The Role of Alpha-Dystrobrevin in Striated Muscle. International Journal of Molecular Sciences. 2011; 12(3):1660-1671. https://doi.org/10.3390/ijms12031660
Chicago/Turabian StyleNakamori, Masayuki, and Masanori P. Takahashi. 2011. "The Role of Alpha-Dystrobrevin in Striated Muscle" International Journal of Molecular Sciences 12, no. 3: 1660-1671. https://doi.org/10.3390/ijms12031660