Molecular Diagnosis of 5α-Reductase Type II Deficiency in Brazilian Siblings with 46,XY Disorder of Sex Development

 and
and
Abstract
:1. Introduction
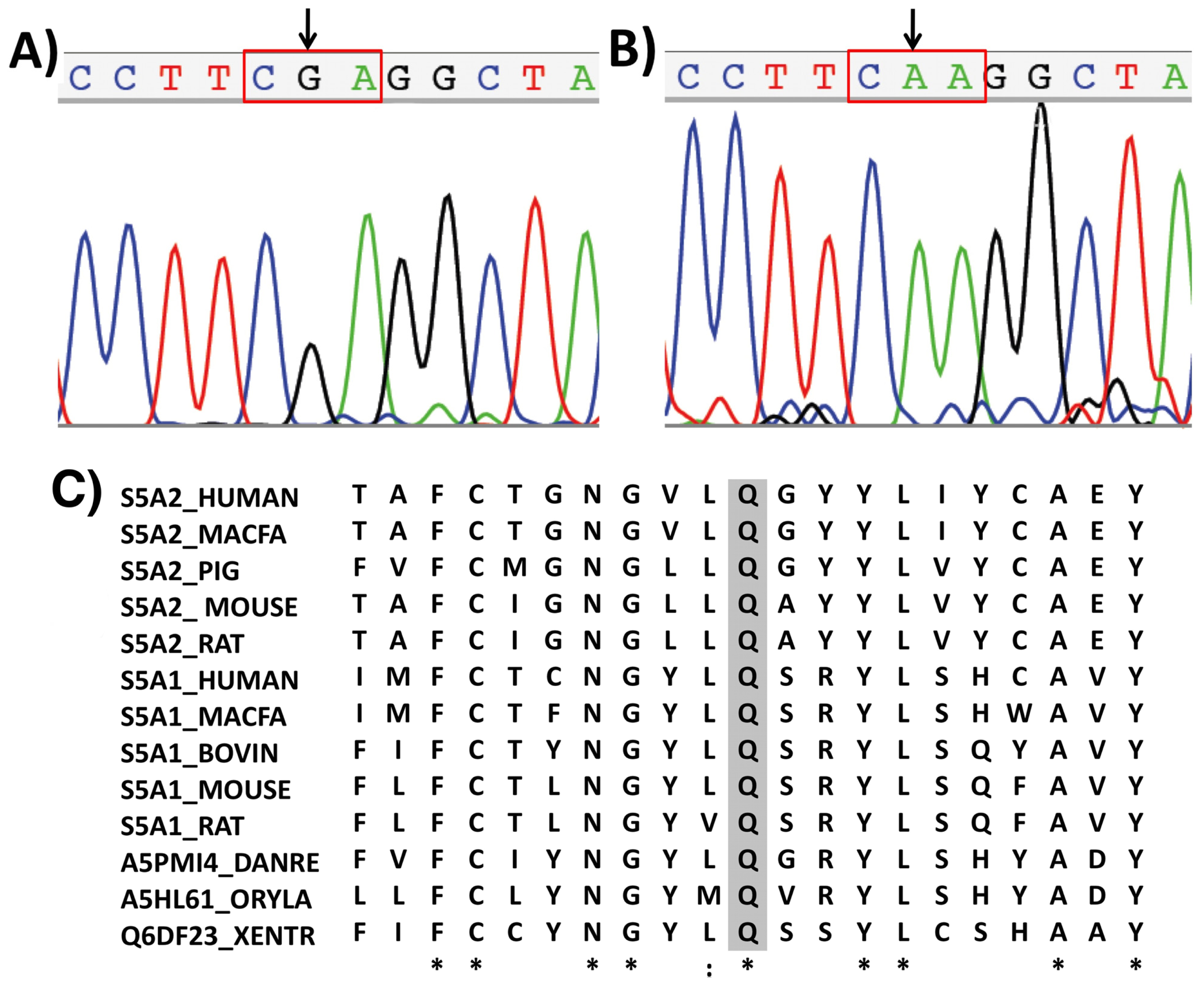
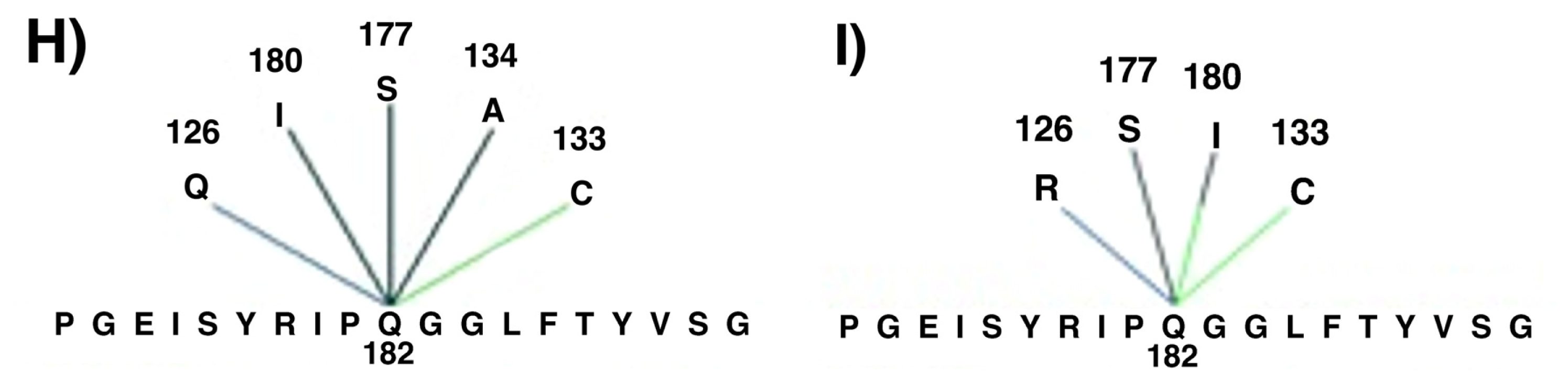
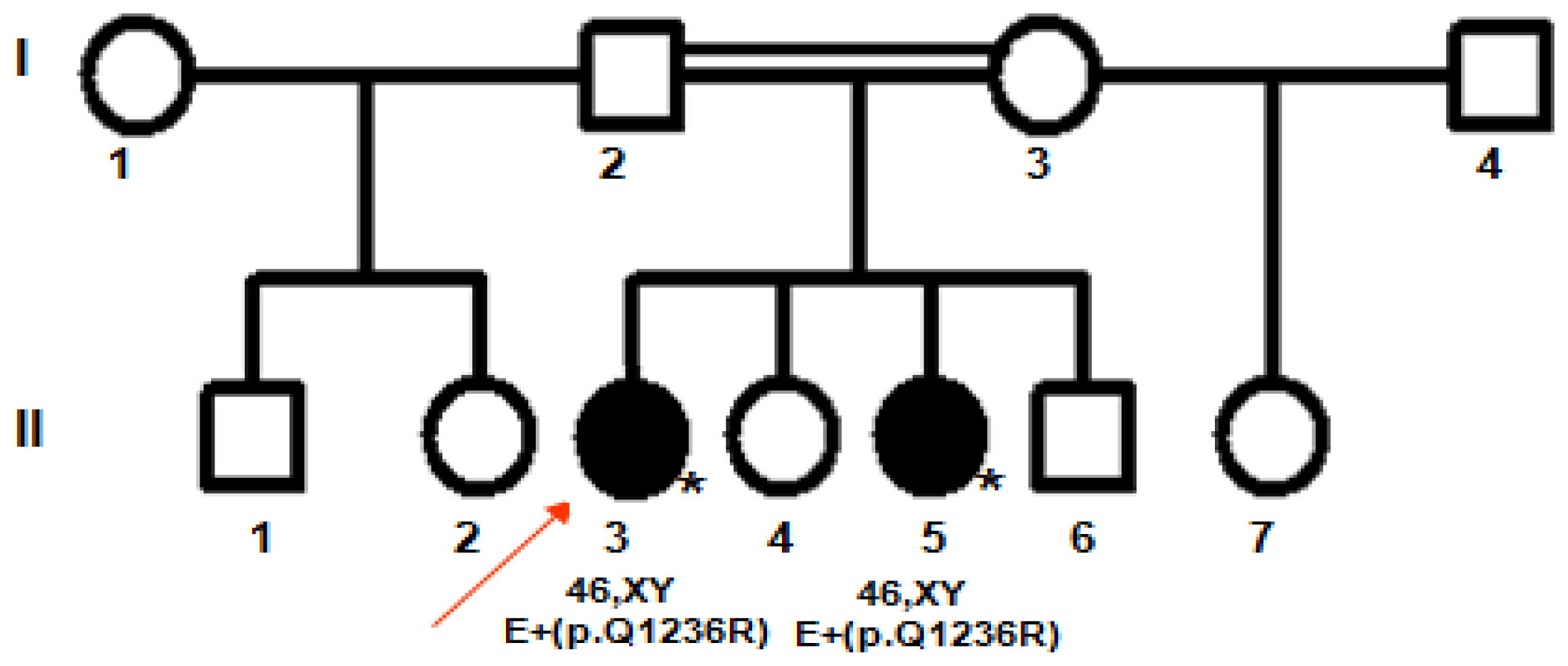
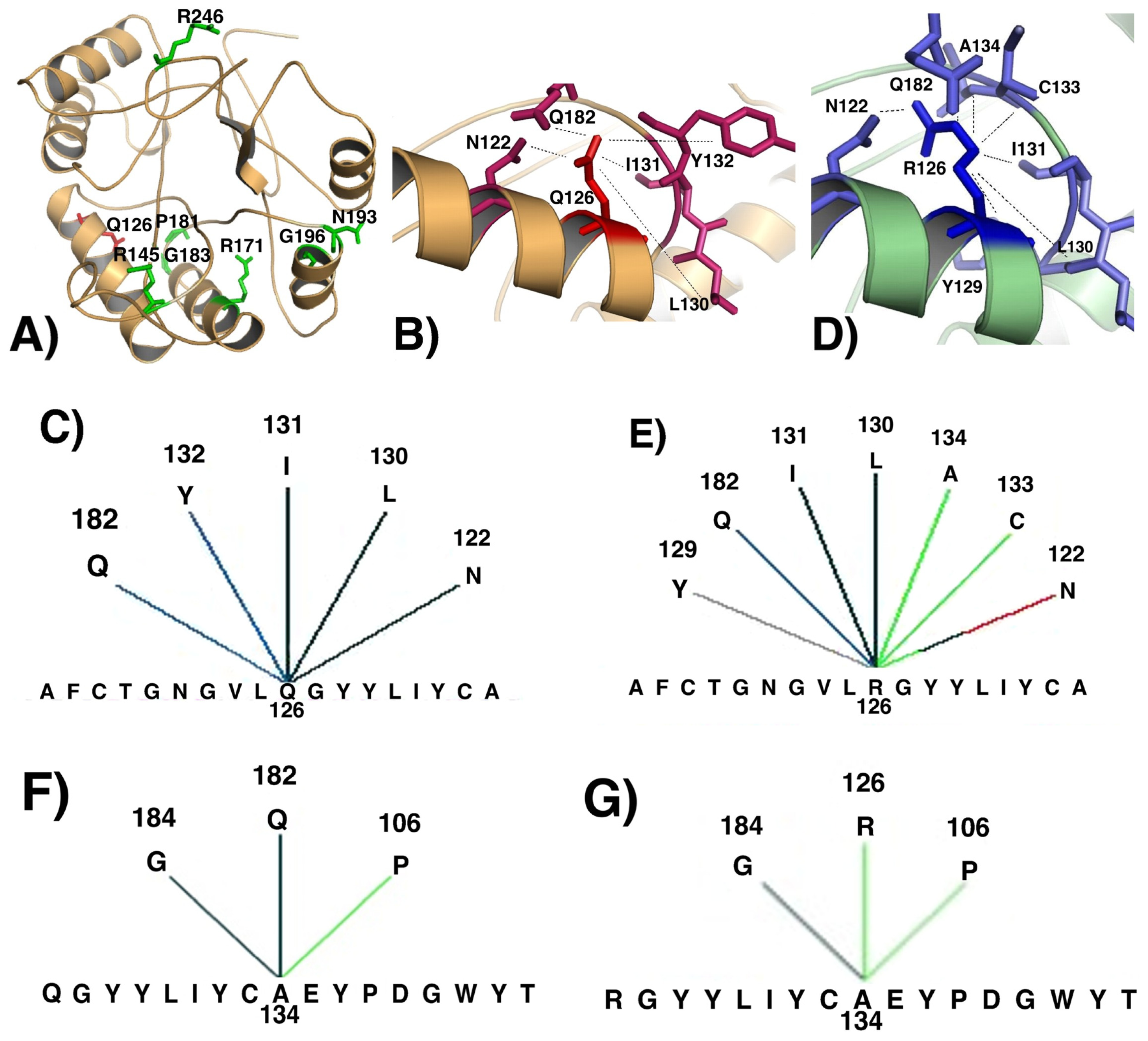
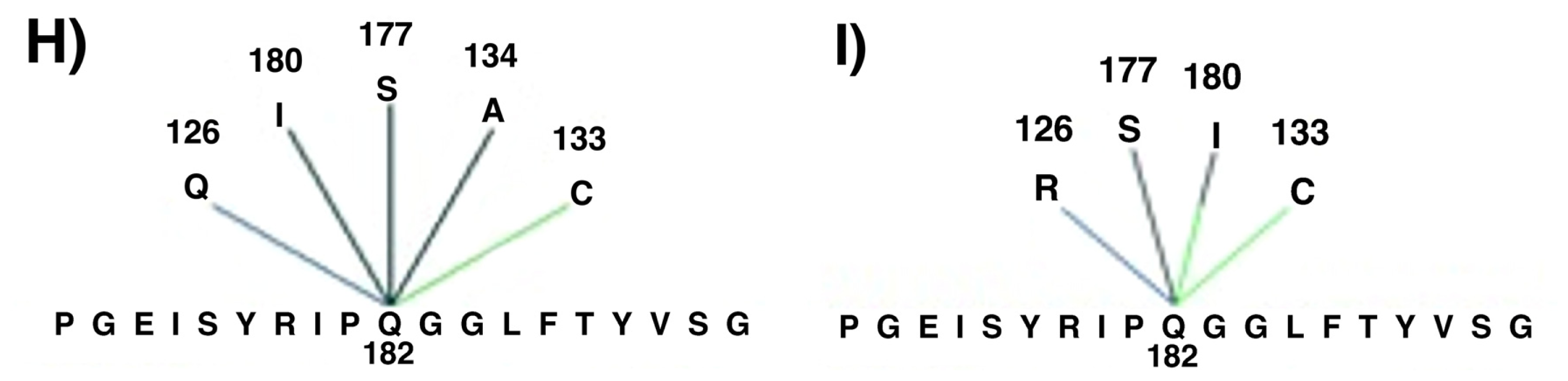
2. Results and Discussion
3. Experimental Section
3.1. Patients
3.2. Methods
4. Conclusions
Acknowledgments
References
- Russell, D.W.; Wilson, J.D. Steroids 5α-reductase: Two genes/two enzymes. Annu. Rev. Biochem 1994, 63, 25–61. [Google Scholar]
- Andersson, S.; Berman, D.M.; Jenkins, E.P.; Russel, D.W. Deletion of steroid 5α-reductase 2 gene in male peseudohermaphroditism. Nature 1991, 354, 159–161. [Google Scholar]
- Pasterski, V.; Prentice, P.; Hughes, I.A. Consequences of the Chicago consensus on disorders of sex development (DSD): Current practices in Europe. Arch. Dis. Child 2010, 95, 618–623. [Google Scholar]
- Thipgen, A.E.; Davis, D.L.; Milatovich, A.; Mendonça, B.B.; Imperator-Mcgingley, J.; Griffin, J.E.; Francke, U.; Wilson, J.D.; Russel, D.W. Molecular genetics of 5α-reductase deficiency. J. Clin. Invest 1992, 90, 799–809. [Google Scholar]
- Wilson, J.D. Characterization of the testicular abnormality in 5a-reductase deficiency. J. Clin. Endocrinol. Metabol 1986, 63, 1091–1099. [Google Scholar]
- Sinnecker, G.H.G.; Hiort, O.; Dibbelt, L.; Albers, N.; Dorr, H.G.; Hauss, H.; Heinrich, U.; Hemminghaus, M.; Hoepffner, W.; Holder, M.; Schnabel, D.; Kruse, K. Phenotypic classification of male pseudo-hermaphroditism due to steroid 5alfa-reductase 2 deficiency. Am. J. Med. Genet. 1996, 63, 223–230. [Google Scholar]
- Hughes, I.A. Minireview: Sex differentiation. Endocrinology 2001, 142, 3281–3287. [Google Scholar]
- Hackel, C.; Oliveira, L.E.C.; Toralles, B.; Silva, D.N.; Tonini, M.M.O.; Ferraz, L.F.C.; Steinmetz, L.; Damiani, D.; Oliveira, L.C.; Maciel-Guerra, A.T.; Stuchi-Perez, E.G.; Guerra-Junior, G. 5alpha-reductase type 2 deficiency: Experiences from Campinas (SP) and Salvador (BA). Arq. Bras. Endocrinol. Metabol 2005, 49, 37–47. [Google Scholar]
- Wilson, J.D.; Griffin, J.E.; Russell, D.W. Steroid 5alfa-reductase 2 deficiency. Endocr. Rev 1993, 14, 577–593. [Google Scholar]
- Wigley, W.C.; Prihoda, J.S.; Mowszowicz, I.; Mendonça, B.B.; New, M.I.; Wilson, J.D.; Russell, D.W. Natural mutagenesis study of the human steroid 5alfa-reductase 2 isozyme. Biochemistry 1994, 33, 1265–1270. [Google Scholar]
- Griffin, J.I.; Mcphaul, M.J.; Russell, D.W.; Wilson, J.D. The Androgen Resistance Syndromes: Steroid 5 Alfa-Reductase Type 2 Deficiency, Testicular Feminization and Related Disorders. In The Metabolic and Molecular Basis of Inherited Diseases; Mcgraw-Hill: New York, NY, USA, 2001; pp. 4117–4146. [Google Scholar]
- Ferraz, L.F.C.; Baptista, M.T.M.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; Hackel, C. New frameshift mutation in the5alpha-reductase type II gene in a Brazilian patient with 5alpha-reductase deficiency. Am. J. Med. Genet 1999, 87, 221–225. [Google Scholar]
- Forti, G.; Falchetti, A.; Santoro, S.; Davis, D.L.; Wilson, J.D.; Russell, D.W. Steroid 5α-reductase 2 deficiency: Virilization in early infancy may be due to partial function of mutant enzyme. Clin. Endocrinol (Oxf) 1996, 44, 477–482. [Google Scholar]
- Boudon, C.; Lumbroso, S.; Lobaccaro, J.M.; Szarras-Czapnik, M.; Romer, T.E.; Garandeau, P.; Montoya, P.; Sultan, C. Molecular study of the 5alpha-reductase type II gene in three European families with 5alpha-reductase deficiency. J. Clin. Endocrinol. Metabol 1995, 80, 2149–2153. [Google Scholar]
- Mendonça, B.B.; Inácio, M.; Costa, E.M.; Arnhold, I.J.; Silva, F.A.; Nicolau, W.; Bloise, W.; Russel, D.W.; Wilson, J.D. Male pseudohermaphroditism due to steroid 5alpha-reductase 2 deficiency. Diagnosis, psychological evaluation, and management. Medicine (Baltimore) 1996, 75, 64–76. [Google Scholar]
- Hackel, C.; Oliveira, L.E.C.; Ferraz, L.F.C.; Tonini, M.M.O.; Silva, D.N.; Toralles, M.B.; Stuchi-Perez, E.G.; Guerra-Junior, G. New mutations, hotspots and founder effects in Brazilian patients with steroid 5α-reductase deficiency type II. J. Mol. Med 2005, 83, 569–576. [Google Scholar]
- Maimoun, L.; Philibert, P.; Cammas, B.; Audran, F.; Bouchard, P.; Fenichel, P.; Cartigny, M.; Pienkowski, C.; Polak, M.; Skordis, N.; et al. Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: An extensive international experience of 55 patients. J. Clin. Endocrinol. Metabol 2011, 96, 296–307. [Google Scholar]
- Fernández-Cancio, M.; Audí, L.; Andaluz, P.; Torán, N.; Piró, C.; Albisu, M.; Gussinyé, M.; Yeste, D.; Clemente, M.; Martínez-Mora, J.; Blanco, A.; Granada, M.L.; Marco, M.; Ferragut, J.; López-Siguero, J.P.; Beneyto, M.; Carles, C.; Carrascosa, A. SRD5A2 gene mutations and polymorphisms in Spanish 46,XY patients with a disorder of sex differentiation. Int. J. Androl 2011, 34, e526–e535. [Google Scholar]
- Basic Local Alignment Search Tool. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi accessed on 27 May 2010.
- Sanchez, R.; Pieper, U.; Melo, F.; Eswar, N.; Marti-Renom, M.A.; Madhusudhan, M.S.; Mirković, N.; Sali, A. Protein structure modeling for structural genomics. Nat. Struct. Biol 2000, 7, 986–990. [Google Scholar]
- Drury, J.E.; Costanzo, L.D.; Penning, T.M.; Christianson, D.W. Inhibition of human steroid 5beta-reductase (AKR1D1) by finasteride and structure of the enzyme-inhibitor complex. J. Biol. Chem 2009, 284, 19786–19790. [Google Scholar]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem 2003, 72, 137–174. [Google Scholar]
- Neshich, G.; Borro, L.C.; Higa, R.H.; Kuser, P.R.; Yamagishi, M.E.; Franco, E.H.; Krauchenco, J.N.; Fileto, R.; Ribeiro, A.A.; Bezerra, G.B.; et al. The Diamond STING server. Nucleic Acids Res 2005, 33, W29–W35. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon | Forward Primer | Reverse Primer | Ta* (°C) | Fragment size (pb) |
|---|---|---|---|---|
| 1 | GCAGCGGCCCACCGGCGAGGAACA | TGGACGCCGGGAGCAGGGCAGT | 66 | 369 |
| 2 | CAGTGAATCCTAACCTTTCCTCCC | TTGTTAGCTGGGAAGTAGGTGGAG | 59.5 | 243 |
| 3 | AAGCACCACAATCTGGACACAT | CTCCAGGGAAGAGTGAGAGTCTG | 59.5 | 203 |
| 4 | CAATGATTGACCTTCCGATTCTTC | GTTTGGAGAAGAAGAAAGCTACGT | 63 | 238 |
| 5 | TCAGCCACTGCTCCATTATATTTA | TTGACAGTTTTCATCCAGCATTGTG | 59.5 | 171 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calais, F.L.d.; Soardi, F.C.; Petroli, R.J.; Lusa, A.L.G.; Silva, R.B.d.P.e.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; Mello, M.P.d. Molecular Diagnosis of 5α-Reductase Type II Deficiency in Brazilian Siblings with 46,XY Disorder of Sex Development. Int. J. Mol. Sci. 2011, 12, 9471-9480. https://doi.org/10.3390/ijms12129471
Calais FLd, Soardi FC, Petroli RJ, Lusa ALG, Silva RBdPe, Maciel-Guerra AT, Guerra-Júnior G, Mello MPd. Molecular Diagnosis of 5α-Reductase Type II Deficiency in Brazilian Siblings with 46,XY Disorder of Sex Development. International Journal of Molecular Sciences. 2011; 12(12):9471-9480. https://doi.org/10.3390/ijms12129471
Chicago/Turabian StyleCalais, Flávia Leme de, Fernanda Caroline Soardi, Reginaldo José Petroli, Ana Letícia Gori Lusa, Roberto Benedito de Paiva e Silva, Andréa Trevas Maciel-Guerra, Gil Guerra-Júnior, and Maricilda Palandi de Mello. 2011. "Molecular Diagnosis of 5α-Reductase Type II Deficiency in Brazilian Siblings with 46,XY Disorder of Sex Development" International Journal of Molecular Sciences 12, no. 12: 9471-9480. https://doi.org/10.3390/ijms12129471