Monitoring Insulin Aggregation via Capillary Electrophoresis

Abstract
:1. Introduction
2. Results and Discussion
2.1. Detection of Insulin Oligomers Using CE with UV Detection
2.2. Effect of Salt Concentration on the Time Course for Insulin Oligomer Formation
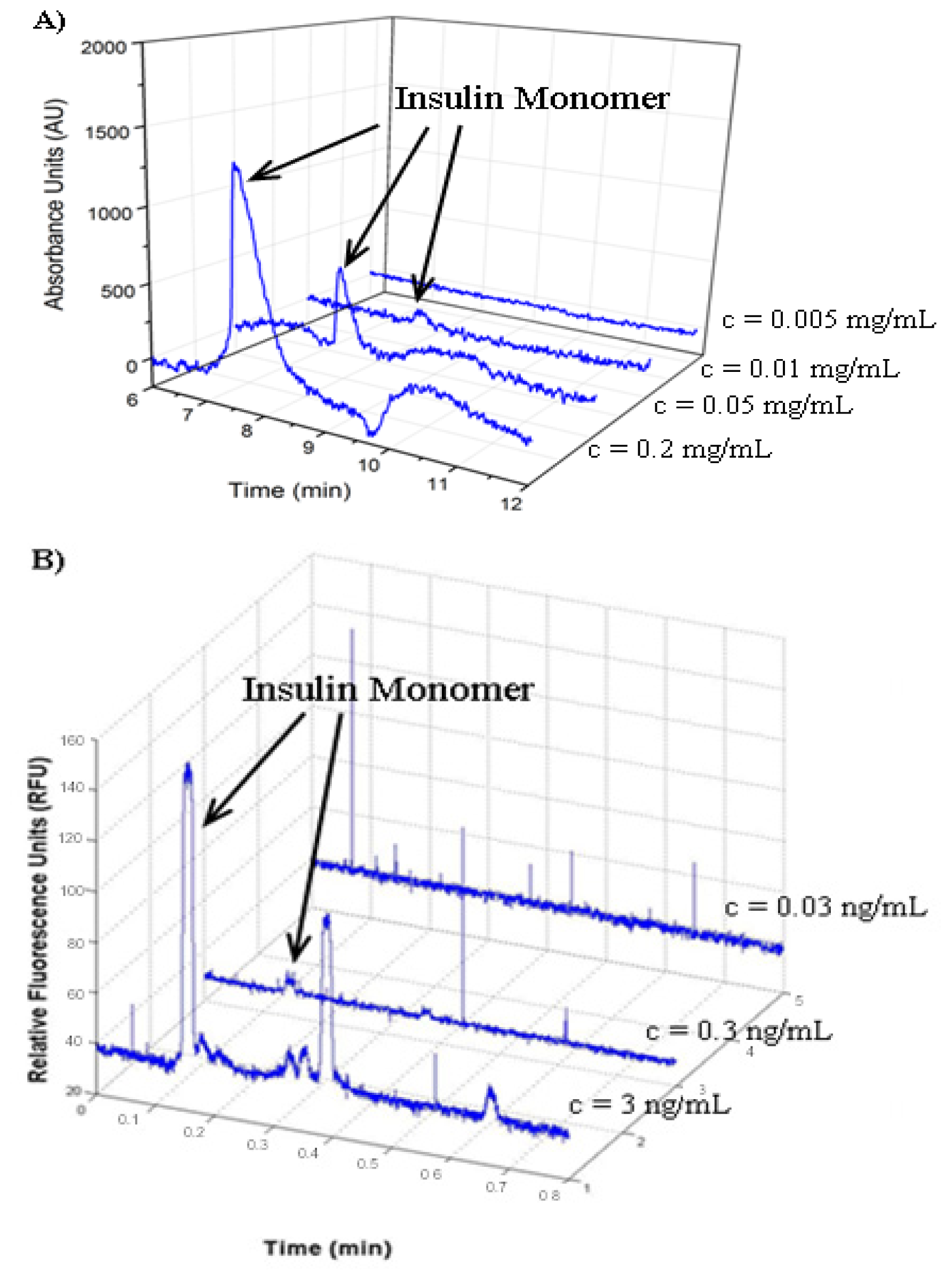
2.3. Determination of Insulin Limit of Detection
2.4. Analysis of FITC Tracer Incorporation into Unlabeled Insulin
3. Materials and Methods
3.1. Materials
3.2. Insulin Preparation
3.3. Electrophoresis Conditions for UV and LIF Studies
3.4. Limit of Detection Studies
3.5. Oligomer Formation Assay
3.6. Statistical Analysis
4. Conclusion
Supplementary Material
ijms-12-09369-s001.pdfAcknowledgements
References
- Hales, C.N. The role of insulin in the regulation of glucose metabolism. Symp. Proc 1971, 30, 282–288. [Google Scholar]
- Blundell, T.; Dodson, G.; Hodgkin, D.; Mercola, D. Insulin: Structure in the crystal and its reflection in chemistry and biology. Adv. Protein Chem 1972, 26, 279–402. [Google Scholar]
- Grudzielanek, S.; Smirnovas, V. Solvation-assisted pressure tuning of insulin fibrillation: From novel aggregation pathways to biotechnological applications. J. Mol. Biol 2006, 356, 497–509. [Google Scholar]
- Waugh, D.F.; Wilhelmson, D.F. Studies of the nucleation and growth reactions of selected types of insulin fibrils. J. Am. Chem. Soc 1953, 75, 2592–2600. [Google Scholar]
- Nielsen, L.; Khurana, R.; Fink, A. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci 1999, 24, 329–332. [Google Scholar]
- Sluzky, V.; Tamada, J.; Klibanov, A.; Langer, R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. USA 1991, 88, 9377–9381. [Google Scholar]
- Jeffrey, P.; Milthorpe, B.; Nichol, L. Polymerization pattern of insulin at pH 7.0. Biochemistry 1976, 15, 4660–4665. [Google Scholar]
- Dische, F.E.; Wernstedt, C. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia 1988, 31, 158–161. [Google Scholar]
- Brange, J.; Andersen, L. Toward understanding insulin fibrillation. J. Pharm. Sci 1997, 86, 517–525. [Google Scholar]
- Shikama, Y.; Kitazawa, J.; Yagihashi, N.; Uehara, O.; Murata, Y.; Yajima, N.; Wada, R.; Yagihashi, S. Localized amyloidosis at the site of repeated insulin injection in a diabetic patient. Intern Med 2010, 49, 397–401. [Google Scholar]
- Mauro, M.; Craparo, E. Kinetics of different processes in human insulin amyloid formation. J. Mol. Biol 2007, 366, 258–274. [Google Scholar]
- Herman, W.; Ilag, L. A clinical trial of continuous subcutaneous insulin infusion versus multiple daily injections in older adults with type 2 diabetes. Diabetes Care 2005, 28, 1568–1573. [Google Scholar]
- Grudzielanek, S.; Velkova, A. Cytotoxicity of insulin within its self-assembly and amyloidogenic pathways. J. Mol. Biol 2007, 370, 372–384. [Google Scholar]
- Carulla, N.; Zhou, M.; Giralt, E.; Robinson, C.; Dobson, C. Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc. Chem. Res 2010, 43, 1072–1079. [Google Scholar]
- Ahmad, A.; Uversky, V.; Hong, D.; Fink, A. Early events in the fibrillation of monomeric insulin. J. Biol. Chem 2005, 280, 42669–42675. [Google Scholar]
- Iurascu, M.; Cozma, C.; Tomczyk, N.; Rontree, J.; Desor, M.; Drescher, M.; Przybylski, M. Structural characterization of β-amyloid oligomer-aggregates by ion mobility mass spectrometry and electron spin resonance spectroscopy. Anal. Bioanal. Chem 2009, 395, 2509–2519. [Google Scholar]
- Maji, S.; Ogorzalek Loo, R.; Inayathullah, M.; Spring, S.; Vollers, S.; Condron, M.; Bitan, G.; Loo, J.; Teplow, D. Amino acid position-specific contributions to amyloid β-protein oligomerization. J. Biol. Chem 2009, 284, 23580–23591. [Google Scholar]
- Palmblad, M.; Westlind-Danielsson, A.; Bergquist, J. Oxidation of methionine 35 attenuates formation of amyloid β-peptide 1–40 oligomers. J. Biol. Chem 2002, 277, 19506–19510. [Google Scholar]
- Bernstein, S.; Wyttenbach, T.; Baumketner, A.; Shea, J.; Bitan, G.; Teplow, D.; Bowers, M. Amyloid β-protein: Monomer structure and early aggregation states of Aβ42 and its pro19 alloform. J. Am. Chem. Soc 2005, 127, 2075–2084. [Google Scholar]
- Bernstein, S.; Dupuis, N.; Lazo, N.; Wyttenbach, T.; Condron, M.; Bitan, G.; Teplow, D.; Shea, J.; Ruotolo, B.; Robinson, C.; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem 2009, 1, 326–331. [Google Scholar]
- Bitan, G.; Kirkitadze, M.; Lomakin, A.; Vollers, S.; Benedek, G.; Teplow, D. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar]
- Murray, M.; Bernstein, S.; Nyugen, V.; Condron, M.; Teplow, D.; Bowers, M. Amyloid β protein: Aβ40 inhibits Aβ42 oligomerization. J. Am. Chem. Soc 2009, 131, 6316–6317. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener 2007, 2. [Google Scholar] [CrossRef]
- Ladiwala, A.; Lin, J.; Bale, S.; Marcelino-Cruz, A.; Bhattacharya, M.; Dordick, J.; Tessier, P. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem 2010, 285, 24228–24237. [Google Scholar]
- Podlisny, M.; Walsh, D.; Selkoe, D. Oligomerization of endogeneous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by congo red. Biochemistry 1998, 37, 3602–3611. [Google Scholar]
- Ward, R.; Jennings, K.; Howlett, D. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β-amyloid peptide. Biochem. J 2000, 348, 137–144. [Google Scholar]
- Zhang, Y.; Wang, X.; He, J.-S.; Bao, F.-X.; Sun, W.-M.; Dai, X.-X.; Wang, X.-B.; Li, Y.-Q.; Zhen, X.-X.; Hu, H.-G.; et al. Preparation and characterization of a monoclonal antibody with high affinity for soluble Aβ oligomers. Hybridoma 2009, 28, 349–354. [Google Scholar]
- Satoh, Y.; Hirakura, Y.; Kirino, Y. β-amyloid peptides inhibit acetylcholine release from cholinergic presynaptic nerve endings isolated from an electric ray. Neurosci. Lett 2001, 302, 97–100. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Stine, W.B., Jr; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar]
- Walsh, D.; Tseng, B.; Rydel, R.; Podlisny, M.; Selkoe, D. The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar]
- Walsh, D.; Hartley, D.; Condron, M.; Selkoe, D.; Teplow, D. In vitro studies of amyloid β-protein fibril assembly and toxicity provide clues to the aetiology of flemish variant (Ala692 → Gly) Alzheimer’s disease. Biochem. J 2001, 355, 869–877. [Google Scholar]
- Walsh, D.; Klyubin, I.; Fadeeva, J.; Cullen, W.; Anwyl, R.; Wolfe, M.; Rowan, M.; Selkoe, D. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar]
- Ryan, D.; Narrow, W.; Federoff, H.; Bowers, W. An improved method for generating consistent soluble amyloid-β oligomer preparations for in vitro neurotoxicity studies. J. Neurosci. Methods 2010, 190, 171–179. [Google Scholar]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B., Jr; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar]
- Kato, M.; Kinoshita, H.; Toyo’oka, T. Analytical method for β-amyloid fibrils using CE-laser induced fluorescence and its application to screening for inhibitors of β-amyloid protein aggregation. Anal. Chem 2007, 79, 4887–4891. [Google Scholar]
- Lee, J.; Ryu, J.; Park, C.B. High-throughput analysis of Alzheimer’s β-amyloid aggregation using a microfluidic self-assembly of monomers. Anal. Chem 2009, 81, 2751–2759. [Google Scholar]
- Picou, R.; Kheterpal, I.; Wellman, A.; Minnamreddy, M.; Ku, G.; Gilman, S.D. Analysis of Aβ(1–40) and Aβ(1–42) monomer and fibrils by capillary electrophoresis. J. Chromatogr. B 2011, 879, 627–632. [Google Scholar]
- Sabella, S.; Quaglia, M.; Lanni, C.; Racchi, M.; Govoni, S.; Caccialanza, G.; Calligaro, A.; Bellotti, V.; Lorenzi, E. Capillary electrophoresis studies on the aggregation process of β-amyloid 1–42 and 1–40 peptides. Electrophoresis 2004, 25, 3186–3194. [Google Scholar]
- Ortner, K.; Buchberger, W. Capillary electrokinetic chromatography of insulin and related synthetic analogues. J. Chromatogr. A 2009, 1216, 2953–2957. [Google Scholar]
- Brambilla, D.; Verpillot, R.; Taverna, M.; De Kimpe, L.; Le Droumagust, B.; Nicolas, J.; Canovi, M.; Gobbi, M.; Mantegazza, F.; Salmona, M.; et al. New method based on capillary electrophoresis with laser-induced fluorescence detection (CE-LIF) to monitor interaction between nanoparticles and the amyloid-β peptide. Anal. Chem 2010, 82, 10083–10089. [Google Scholar]
- Sureshbabu, N.; Kirubagaran, R.; Jayakumar, R. Surfactant-induced conformational transition of amyloid β-peptide. Eur. Biophys. J 2009, 38, 355–367. [Google Scholar]
- Yamamoto, S.; Hasegawa, K.; Yamaguchi, I.; Tsutsumi, S.; Kardos, J.; Goto, Y.; Gejyo, F.; Naiki, H. Low concentrations of sodium dodecyl sulfate induce the extension of β2-microglobulin-related amyloid fibrils at a neutral pH. Biochemistry 2004, 43, 11075–11082. [Google Scholar]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers—What you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar]
- Fukuyama, R.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Aβ40 and Aβ42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aβ42 to Aβ40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol 2000, 43, 155–160. [Google Scholar]
- Lewczuk, P.; Esselmann, H.; Otto, M.; Maler, J.M.; Henkel, A.W.; Henkel, M.K.; Eikenberg, O.; Antz, C.; Krause, W.R.; Reulbach, U.; et al. Neurochemical diagnosis of Alzheimer’s dementia by CSF Aβ42, Aβ42/Aβ40 ratio and total tau. Neurobiol. Aging 2004, 25, 273–281. [Google Scholar]
- Zuberovic, A.; Hanrieder, J.; Wetterhall, M. Proteome profiling of human cerebrospinal fluid: Exploring the potential of capillary electrophoresis with surface modified capillaries for analysis of complex biological samples. Eur. J. Mass Spectrom 2008, 14, 249–260. [Google Scholar]
- Pinto, D.; Arriaga, E.; Dovichi, N. Solid-phase fluorescent labeling reaction of picomole amounts of insulin in very dilute solutions and their analysis by capillary electrophoresis. Electrophoresis 1995, 16, 534–540. [Google Scholar]
- Shihabi, Z.; Friedberg, M. Insulin stacking for capillary electrophoresis. J. Chromatogr. A 1998, 807, 129–133. [Google Scholar]
- Kunkel, A.; Gunter, S.; Watzig, H. Quantitation of insulin by capillary electrophoresis and high-performance liquid chromatography Method comparison and validation. J. Chromatogr. A 1997, 781, 445–455. [Google Scholar]
- Yu, C.; Chin, C. In situ probing of insulin aggregation in chromatography effluents with spectroturbidimetry. J. Colloid Interface Sci 2006, 299, 733–739. [Google Scholar]
- Nayak, A.; Sorci, M.; Krueger, S.; Belfort, G. A universal pathway for amyloid nucleus and precursor formation for insulin. Proteins 2008, 74, 556–565. [Google Scholar]
- Nettleton, E.; Tito, P. Characterization of the oligomeric states of insulin in self-assembly and amyloid fibril formation by mass spectrometry. Biophys. J 2000, 79, 1053–1065. [Google Scholar]
- Vestergaard, B.; Groenning, M.; Roessle, M.; Kastrup, J.S.; van de Weert, M.; Flink, J.M.; Frokjaer, S.; Gajhede, M.; Svergun, D.I. A helical structural nucleus is the primary elongating unit of insulin amyloid fibrils. PLoS Biol 2007, 5, 1089–1097. [Google Scholar]
- Fodera, V.; Librizzi, F.; Leone, M. Secondary nucleation and accessible surface in insulin amyloid fibril formation. J. Phys. Chem 2007, 112, 3853–3858. [Google Scholar]
- Seyrek, E.; Dubin, P.; Tribet, C.; Gamble, E. Ionic strength dependence of protein-polyelectrolyte interactions. Biomacromolecules 2003, 4, 273–282. [Google Scholar]
- Giger, K.; Vanam, R.P.; Seyrek, E.; Dubin, P.L. Suppression of insulin aggregation by heparin. Biomacromolecules 2008, 9, 2338–2344. [Google Scholar]
- LeVine, H. Thioflavine T Interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci 1993, 2, 404–410. [Google Scholar]
- Nielsen, L.; Frokjaer, S. Probing the mechanism of insulin fibril formation with insulin mutants. Biochemistry 2001, 40, 8397–8409. [Google Scholar]
- Ahmad, A.; Millett, I. Partially folded intermediates in insulin fibrillation. Biochemistry 2003, 42, 11404–11416. [Google Scholar]
- Sasahara, K.; Yagi, H.; Sakai, M.; Naiki, H.; Goto, Y. Amyloid nucleation triggered by agitation of β2-microglobulin under acidic and neutral pH conditions. Biochemistry 2008, 47, 2650–2660. [Google Scholar]
- Picotti, P.; Franceschi, G.; Frare, E.; Spolaore, B.; Zambonin, M.; Chiti, F.; Polverino de Laureto, P.; Fontana, A. Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: Insights into the effect of pH on protein fibrillogenesis. J. Mol. Biol 2007, 367, 1237–1245. [Google Scholar]
- Jain, S.; Udgaonkar, J. Evidence for stepwise formation of amyloid fibrils by the mouse prion protein. J. Mol. Biol 2008, 382, 1228–1241. [Google Scholar]
- Lin, M.; Chen, L.; Tsai, H.; Wang, S.; Chang, Y.; Higuchi, A.; Chen, W. Investigation of the mechanism of β-amyloid fibril formation by kinetic and thermodynamic analyses. Langmuir 2008, 24, 5802–5808. [Google Scholar]
- Dick, G.; Sturek, M. Effects of a physiological insulin concetration on the endothelin-sensitive Ca2+ store in porcine coronary artery smooth muscle. Diabetes 1996, 45, 876–880. [Google Scholar]
- Lee, Y.-H.; Maus, R.G.; Smith, B.W.; Winefordner, J.D. Laser-induced fluorescence detection of a single molecule in a capillary. Anal. Chem 1994, 66, 4142–4149. [Google Scholar]
- Skeidsvoll, J.; Ueland, P. Analysis of double-stranded DNA by capillary electrophoresis with laser-induced fluorescence detection using the monomeric dye SYBR green I. Anal. Biochem 1995, 231, 359–365. [Google Scholar]
- Lim, H.B.; Lee, J.J.; Lee, K. Simple and sensitive laser-induced fluorescence detection for capillary electrophoresis and its application to protein separation. Electrophoresis 1995, 16, 674–678. [Google Scholar]
- Ren, J.; Li, B.; Deng, Y.; Cheng, J. Indirect thermo-optical detection for capillary electrophoresis. Talanta 1995, 42, 1891–1895. [Google Scholar]
- Brange, J.; Skelbaek-Pedersen, B. The Physicochemical and Pharmaceutical Aspects of Insulin and Insulin Preparations. In Galenics of Insulin; Springer-Verlag: Berlin, Germany, 1987; pp. 369–319. [Google Scholar]
- Grudzielanek, S.; Jansen, R. Solvational tuning of the unfolding, aggregation and amyloidogenesis of insulin. J. Mol. Biol 2005, 351, 879–894. [Google Scholar]
- Schultz, N.; Kennedy, R. Rapid immunoassays using capillary electrophoresis with fluorescence detection. Anal. Chem 1993, 65, 3161–3165. [Google Scholar]
- Tao, L.; Kennedy, R. On-line competitive immunoassay for insulin based on capillary electrophoresis with laser-induced fluorescence detection. Anal. Chem 1996, 68, 3899–3906. [Google Scholar]
- Nichols, M.; Moss, M.; Rosenberry, T. Growth of β-amyloid(1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 2002, 41, 6115–6127. [Google Scholar]
- Gu, X.; Greiner, E.R.; Mishra, R.; Kodali, R.; Osmand, A.; Finkbeiner, S.; Steffan, J.S.; Thompson, L.M.; Wetzel, R.; Yang, X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009, 64, 828–840. [Google Scholar]
- Morshedi, D.; Ebrahim-Habibi, A.; Moosavi-Movahedi, A.A.; Nemat-Gorgani, M. Chemical modification of lysine residues in lysozyme may dramatically influence its amyloid fibrillation. Biochim. Biophys. Acta (BBA) 2010, 1804, 714–722. [Google Scholar]
- Choi, S.; Connelly, S.; Reixach, N.; Wilson, I.A.; Kelly, J.W. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat. Chem. Biol 2010, 6, 133–139. [Google Scholar]
- DeFelippis, M.R.; Bakaysa, D.L.; Bell, M.A.; Heady, M.A.; Li, S.; Pye, S.; Youngman, K.M.; Radziuk, J.; Frank, B.H. Preparation and characterization of a cocryst. Suspension of [LysB28, ProB29]-human insulin analog. J. Pharm. Sci 1998, 87, 170–176. [Google Scholar]
- Kotzke, G.; Schuett, M.; Missler, U.; Moller, D.E.; Fehm, H.L.; Klein, H.H. Binding of human, porcine and bovine insulin to insulin receptors from human brain, muscle and adipocytes and to expressed recombinant alternatively spliced insulin receptor isoforms. Diabetologia 1995, 38, 757–763. [Google Scholar]
- Moses, S. Bovine Porcine and Human Insulin. A Present Day Comparative Appraisal and Policy Discussions, 1st ed; IDDT (Insulin Dependent Diabetes Trust): Chennai, India, 2002. [Google Scholar]
- Albarghouthi, M.; Stein, T.; Barron, A. Poly-N-hydroxyethylacrylamide as a novel, adsorbed coating for protein separation by capillary electrophoresis. Electrophoresis 2003, 24, 1166–1175. [Google Scholar]
- Davis, T.J.; Soto-Ortega, D.D.; Kotarek, J.A.; Gonzalez-Velasquez, F.J.; Sivakumar, K.; Wu, L.; Wang, Q.; Moss, M.A. Comparative study of inhibition at multiple stages of amyloid-β self assembly provides mechanistic insight. Mol. Pharmacol 2009, 76, 405–413. [Google Scholar]
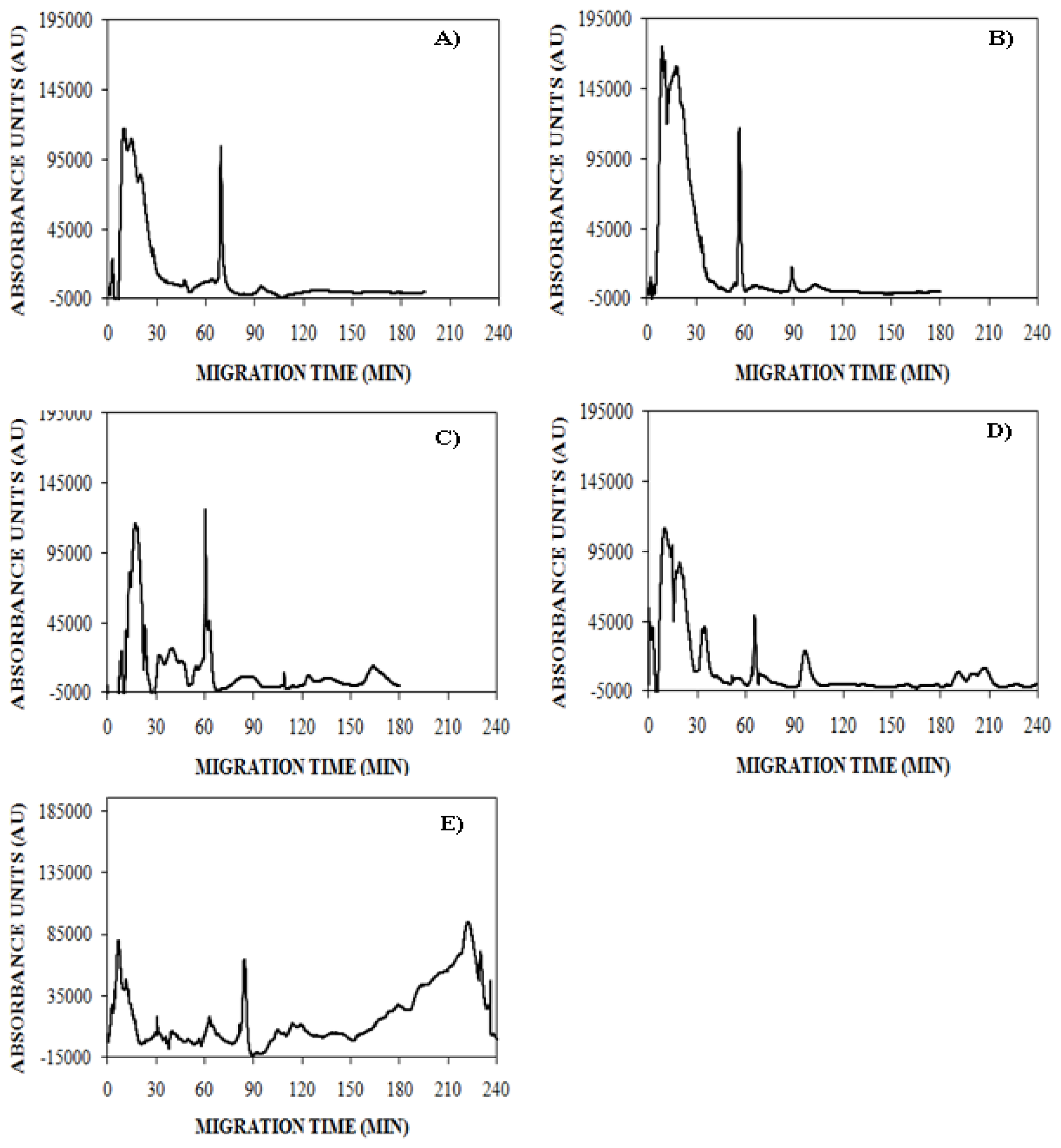
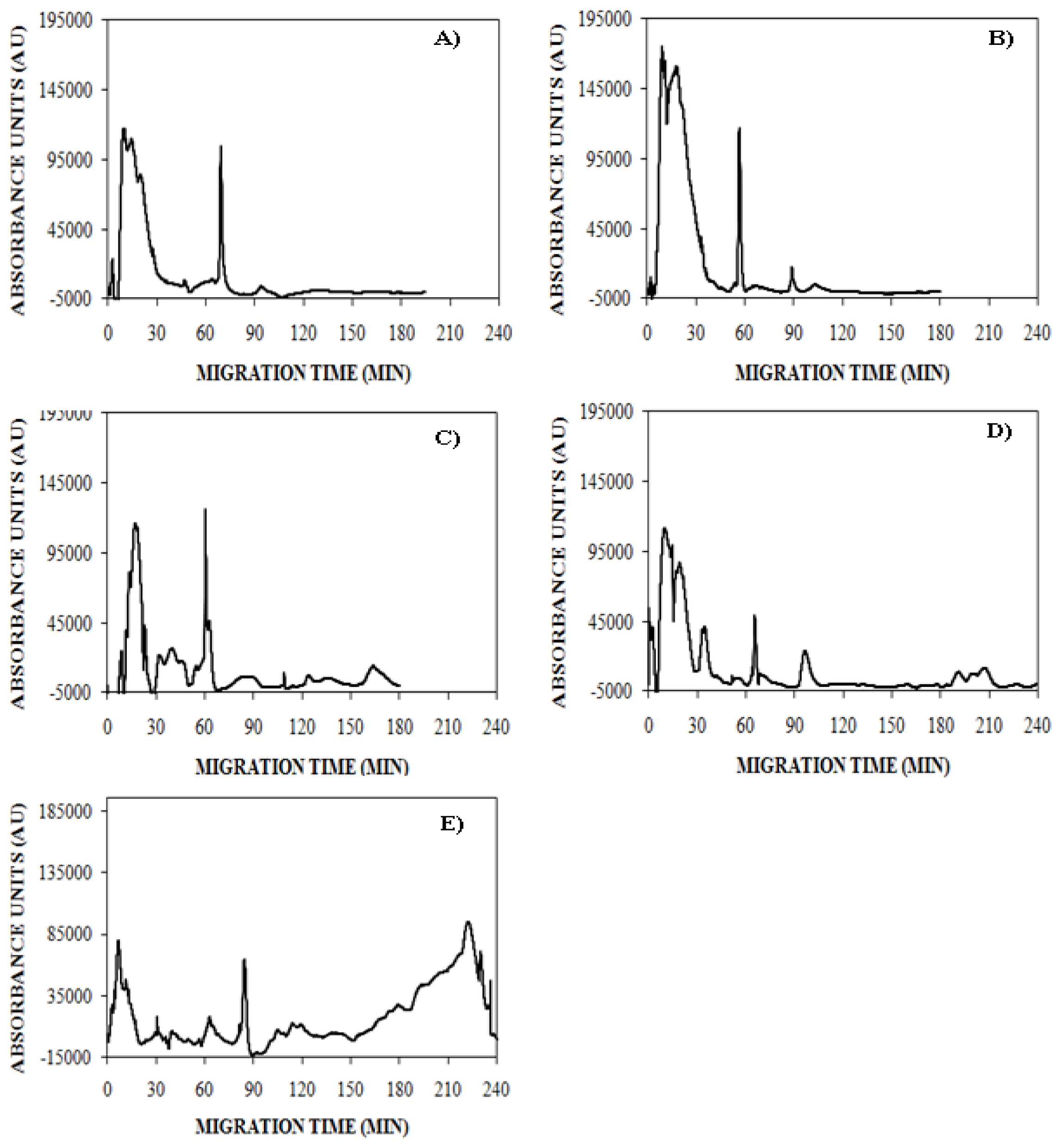
 ), 150 mM (
), 150 mM (
 ), or 250 mM (
), or 250 mM (
 ) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored using UV-CE. CE was performed with sample injection at 0.5 psi for 8 s with 15 kV separation using 1% PHEA separation matrix in a PHEA coated capillary. Migration times were normalized to those observed prior to the onset of aggregation using the peak corresponding to monomer to facilitate comparison between individual runs. Error bars represent SE, n = 3. For the 10 h time point, the migration times of the 150 mM and 250 mM NaCl were both determined to be statistically different from the 100 mM NaCl migration time with a p < 0.1.
), 150 mM (
), or 250 mM (
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored using UV-CE. CE was performed with sample injection at 0.5 psi for 8 s with 15 kV separation using 1% PHEA separation matrix in a PHEA coated capillary. Migration times were normalized to those observed prior to the onset of aggregation using the peak corresponding to monomer to facilitate comparison between individual runs. Error bars represent SE, n = 3. For the 10 h time point, the migration times of the 150 mM and 250 mM NaCl were both determined to be statistically different from the 100 mM NaCl migration time with a p < 0.1.
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored using UV-CE. CE was performed with sample injection at 0.5 psi for 8 s with 15 kV separation using 1% PHEA separation matrix in a PHEA coated capillary. Migration times were normalized to those observed prior to the onset of aggregation using the peak corresponding to monomer to facilitate comparison between individual runs. Error bars represent SE, n = 3. For the 10 h time point, the migration times of the 150 mM and 250 mM NaCl were both determined to be statistically different from the 100 mM NaCl migration time with a p < 0.1.
), 150 mM (
), or 250 mM (
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored using UV-CE. CE was performed with sample injection at 0.5 psi for 8 s with 15 kV separation using 1% PHEA separation matrix in a PHEA coated capillary. Migration times were normalized to those observed prior to the onset of aggregation using the peak corresponding to monomer to facilitate comparison between individual runs. Error bars represent SE, n = 3. For the 10 h time point, the migration times of the 150 mM and 250 mM NaCl were both determined to be statistically different from the 100 mM NaCl migration time with a p < 0.1.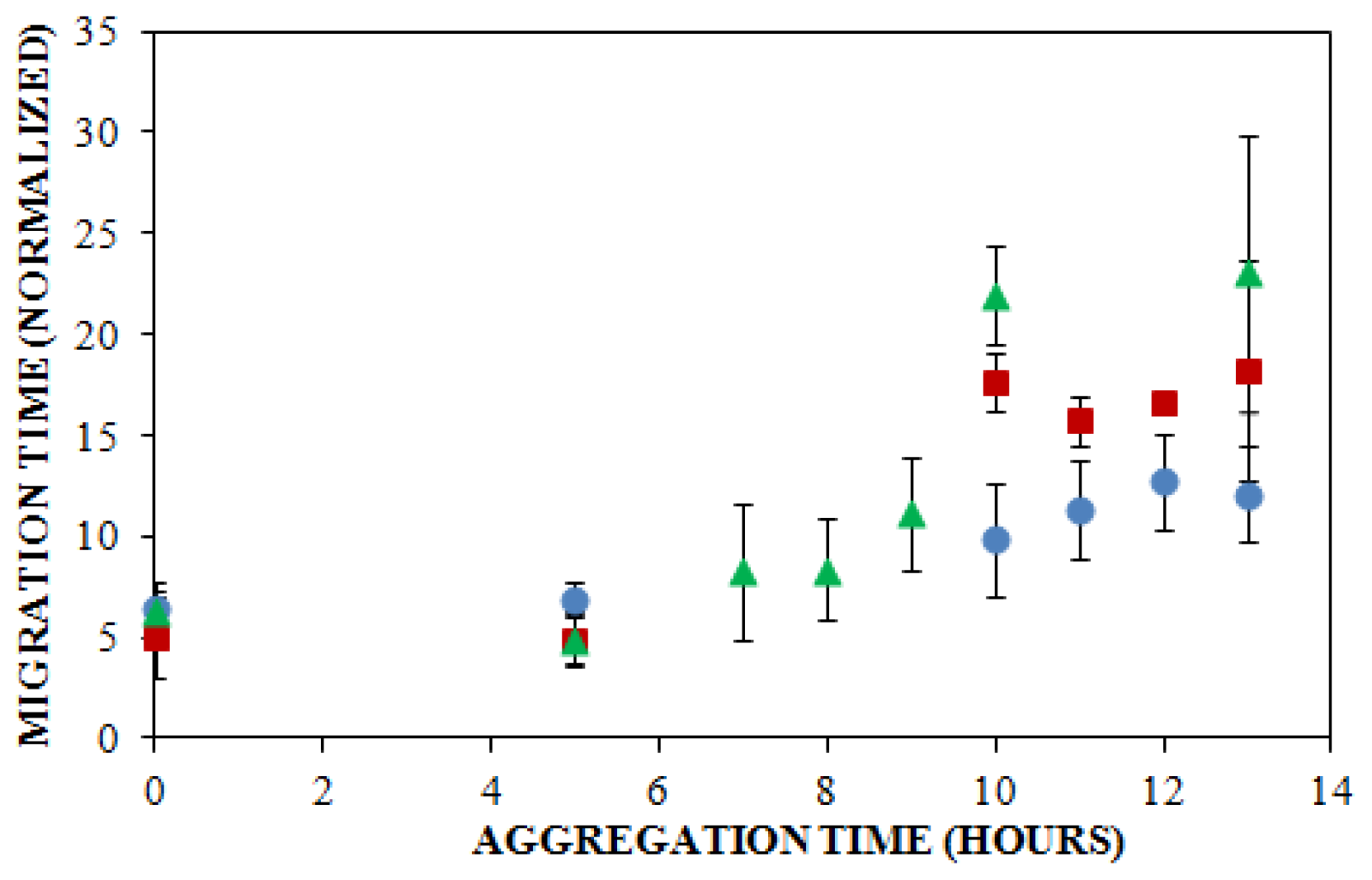 ), 150 mM (
), or 250 mM (
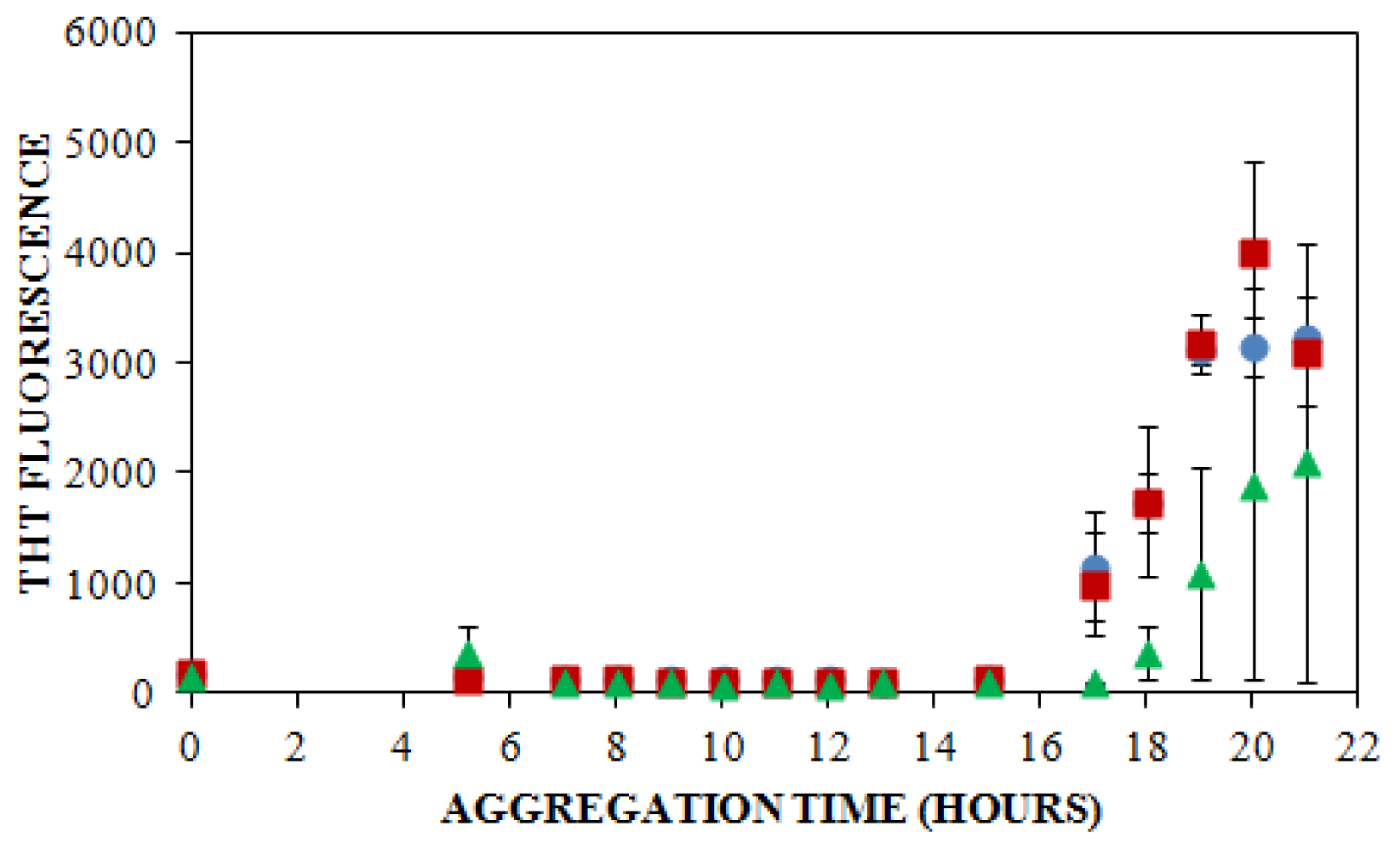
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via ThT fluorescence by periodic dilution into 10 μM ThT. Error bars represent SE. Results are representative of two independent experiments.
), 150 mM (
), or 250 mM (
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via ThT fluorescence by periodic dilution into 10 μM ThT. Error bars represent SE. Results are representative of two independent experiments.
), 150 mM (
), or 250 mM (
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via ThT fluorescence by periodic dilution into 10 μM ThT. Error bars represent SE. Results are representative of two independent experiments.
), 150 mM (
), or 250 mM (
) NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via ThT fluorescence by periodic dilution into 10 μM ThT. Error bars represent SE. Results are representative of two independent experiments. ) and ThT binding (
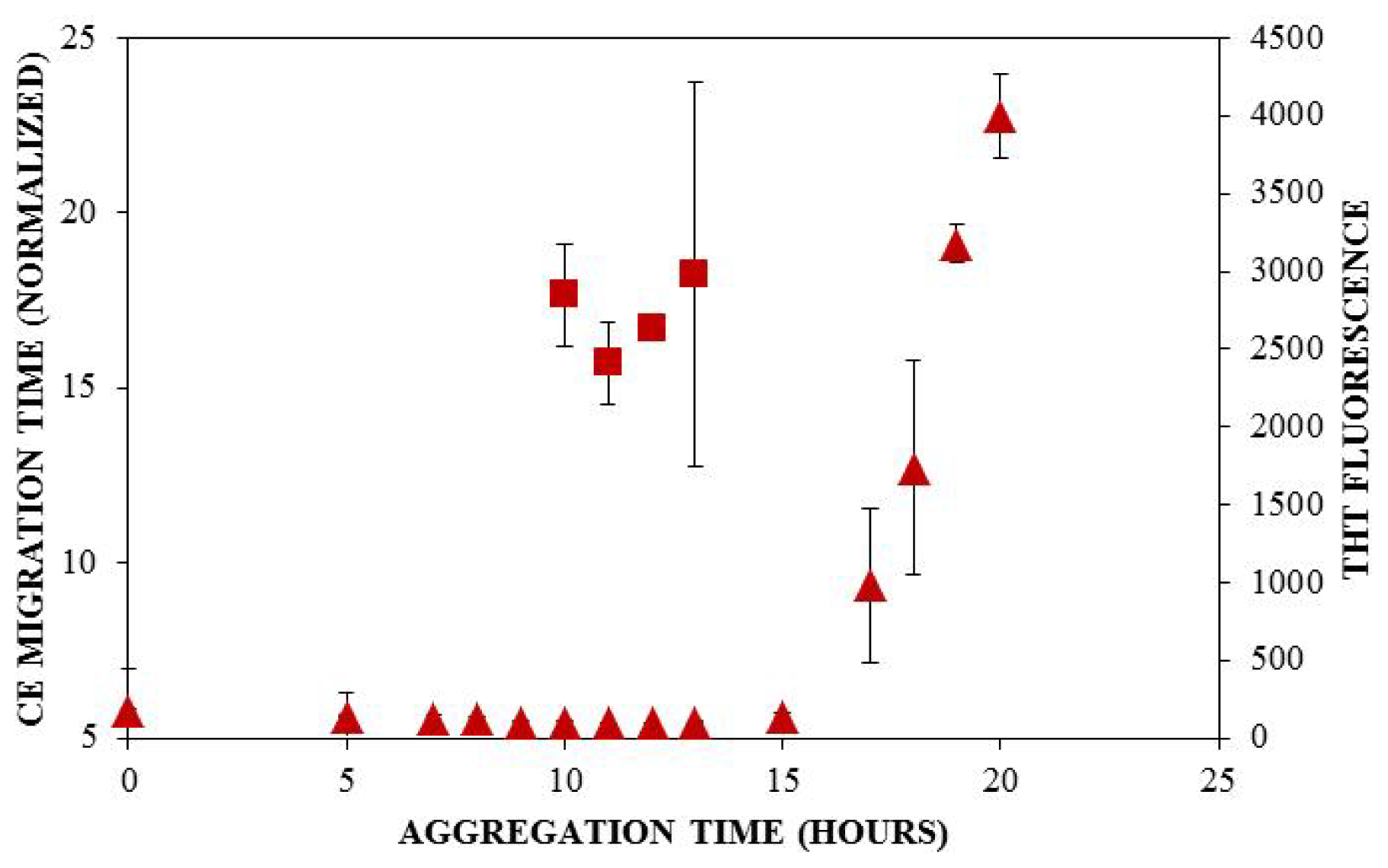
) and ThT binding (
 ). Insulin was diluted to 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via UV-CE or ThT fluorescence as described in Figure 2 and Figure 3, respectively.
) and ThT binding (
). Insulin was diluted to 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via UV-CE or ThT fluorescence as described in Figure 2 and Figure 3, respectively.
). Insulin was diluted to 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via UV-CE or ThT fluorescence as described in Figure 2 and Figure 3, respectively.
) and ThT binding (
). Insulin was diluted to 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Aggregation was induced at 25 °C by continuous agitation (185 rpm) and monitored via UV-CE or ThT fluorescence as described in Figure 2 and Figure 3, respectively.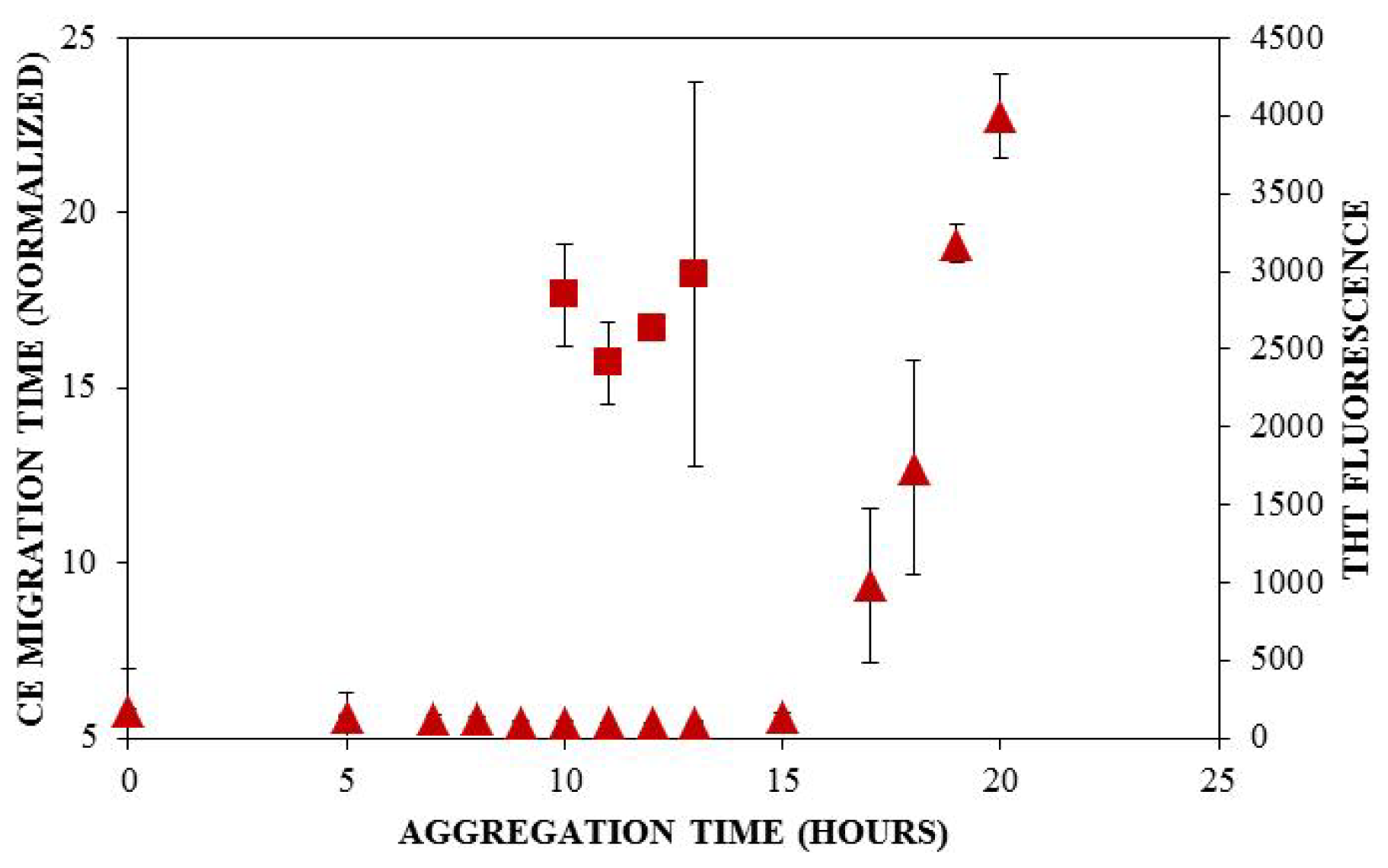
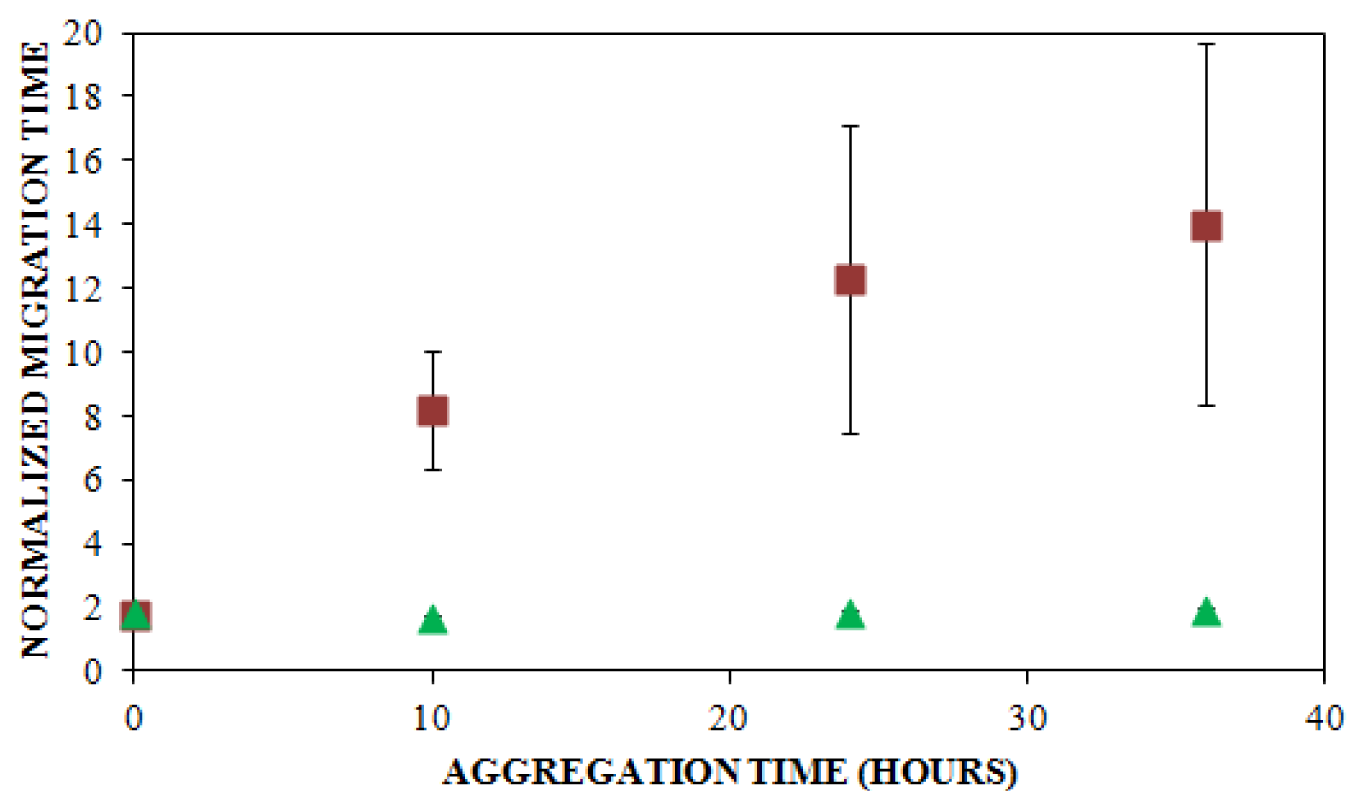
 , n = 6) and UV detection (
, n = 3) were prepared at a concentration of 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Solutions were subjected to agitation (185 rpm), and the formation of aggregates was monitored. LIF-CE was performed with a sample injection at 12 kV for 12 s with 15 kV separation and UV-CE was performed with a sample injection at 10 kV for 12 s with 15 kV separation. Both separations were performed using 1% PHEA separation matrix in PHEA coated capillary. Migration times were normalized as described in Figure 2. Error bars represent SE, n = 3–6. Some error bars lie within symbols. For all time points >0 h, UV data was statistically different from the LIF data with a p < 0.015.
, n = 6) and UV detection (
, n = 3) were prepared at a concentration of 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Solutions were subjected to agitation (185 rpm), and the formation of aggregates was monitored. LIF-CE was performed with a sample injection at 12 kV for 12 s with 15 kV separation and UV-CE was performed with a sample injection at 10 kV for 12 s with 15 kV separation. Both separations were performed using 1% PHEA separation matrix in PHEA coated capillary. Migration times were normalized as described in Figure 2. Error bars represent SE, n = 3–6. Some error bars lie within symbols. For all time points >0 h, UV data was statistically different from the LIF data with a p < 0.015.
, n = 6) and UV detection (
, n = 3) were prepared at a concentration of 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Solutions were subjected to agitation (185 rpm), and the formation of aggregates was monitored. LIF-CE was performed with a sample injection at 12 kV for 12 s with 15 kV separation and UV-CE was performed with a sample injection at 10 kV for 12 s with 15 kV separation. Both separations were performed using 1% PHEA separation matrix in PHEA coated capillary. Migration times were normalized as described in Figure 2. Error bars represent SE, n = 3–6. Some error bars lie within symbols. For all time points >0 h, UV data was statistically different from the LIF data with a p < 0.015.
, n = 6) and UV detection (
, n = 3) were prepared at a concentration of 0.2 mg/mL in 40 mM Tris (pH 8.0) containing 150 mM NaCl. Solutions were subjected to agitation (185 rpm), and the formation of aggregates was monitored. LIF-CE was performed with a sample injection at 12 kV for 12 s with 15 kV separation and UV-CE was performed with a sample injection at 10 kV for 12 s with 15 kV separation. Both separations were performed using 1% PHEA separation matrix in PHEA coated capillary. Migration times were normalized as described in Figure 2. Error bars represent SE, n = 3–6. Some error bars lie within symbols. For all time points >0 h, UV data was statistically different from the LIF data with a p < 0.015.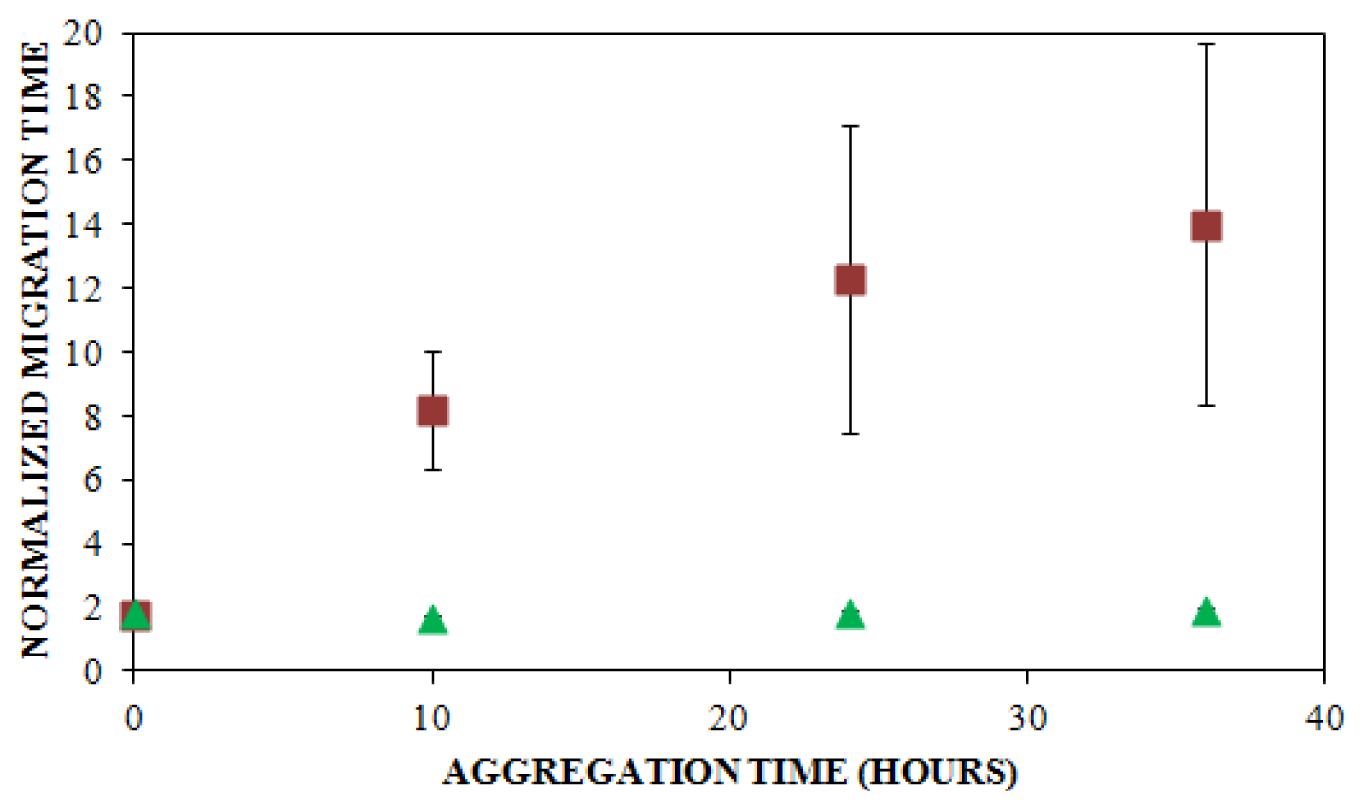

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaCl concentration (mM) | Lag time for CE (h) 1 | Lag time for ThT Binding (h) 2 |
|---|---|---|
| 100 | 6.7 ± 1.7 | 16 ± 1.0 *** |
| 150 | 5.0 ± 0.0 | 15 ± 1.4 *** |
| 250 | 7.6 ± 1.3 | 19 ± 2.4 *** |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pryor, E.; Kotarek, J.A.; Moss, M.A.; Hestekin, C.N. Monitoring Insulin Aggregation via Capillary Electrophoresis. Int. J. Mol. Sci. 2011, 12, 9369-9388. https://doi.org/10.3390/ijms12129369
Pryor E, Kotarek JA, Moss MA, Hestekin CN. Monitoring Insulin Aggregation via Capillary Electrophoresis. International Journal of Molecular Sciences. 2011; 12(12):9369-9388. https://doi.org/10.3390/ijms12129369
Chicago/Turabian StylePryor, Elizabeth, Joseph A. Kotarek, Melissa A. Moss, and Christa N. Hestekin. 2011. "Monitoring Insulin Aggregation via Capillary Electrophoresis" International Journal of Molecular Sciences 12, no. 12: 9369-9388. https://doi.org/10.3390/ijms12129369