Quantitative Prediction of Solvation Free Energy in Octanol of Organic Compounds

Abstract
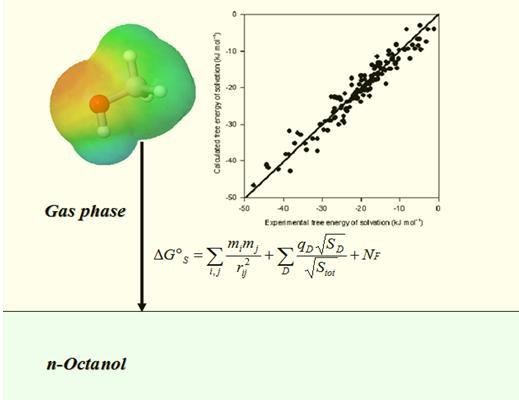
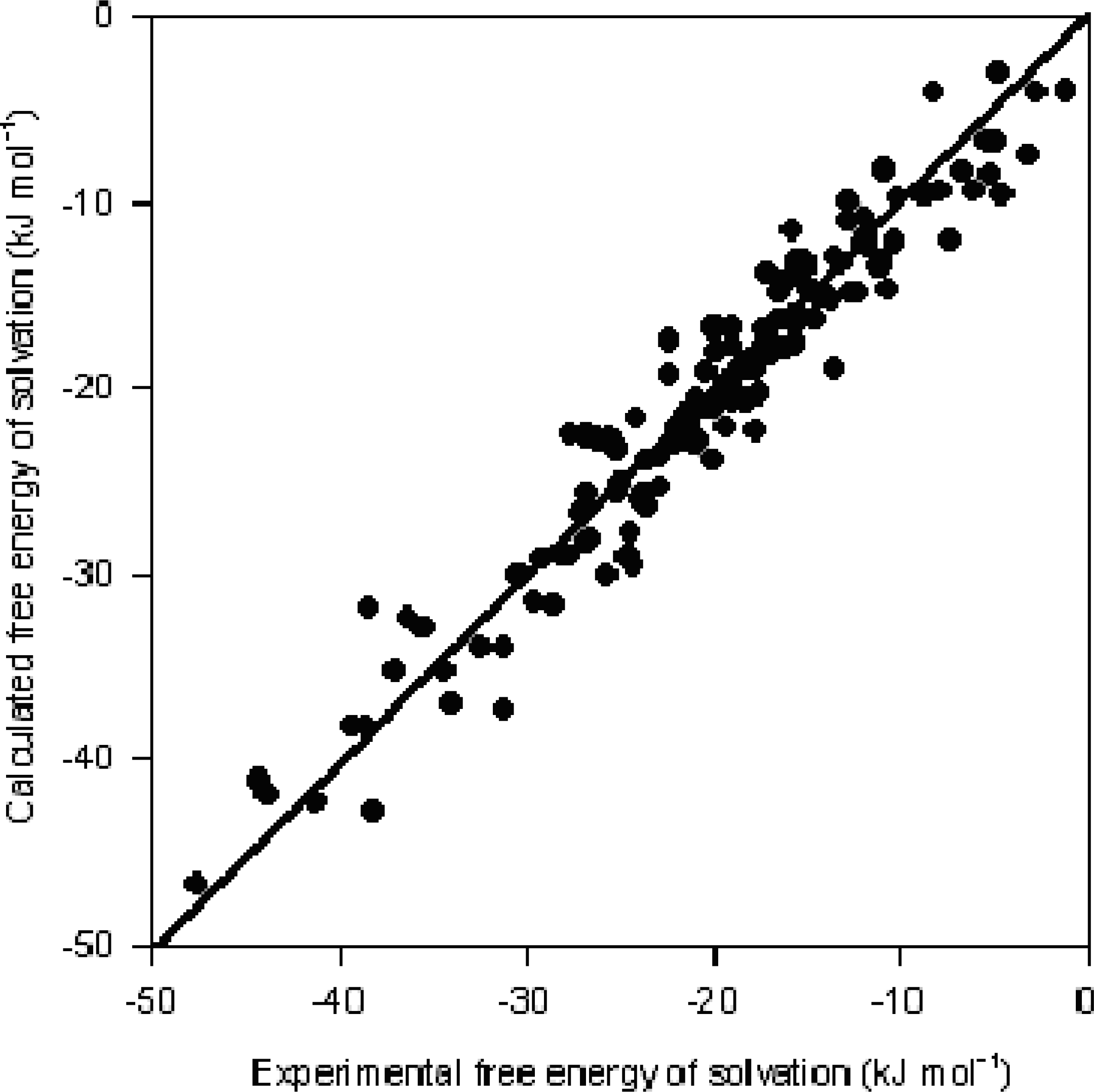
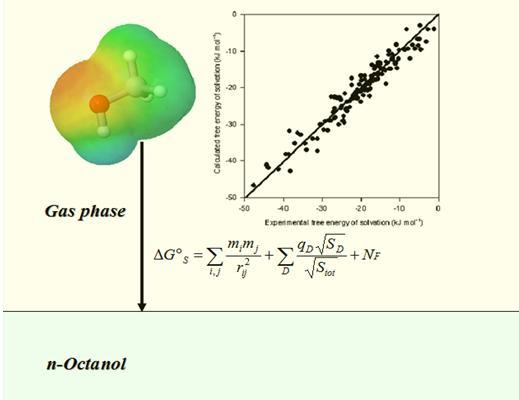
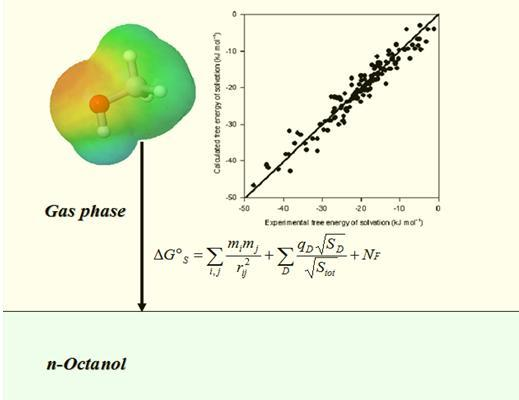
:
1. Introduction
2. Experimental
2.1. Chemical data
2.2. Computational methodology
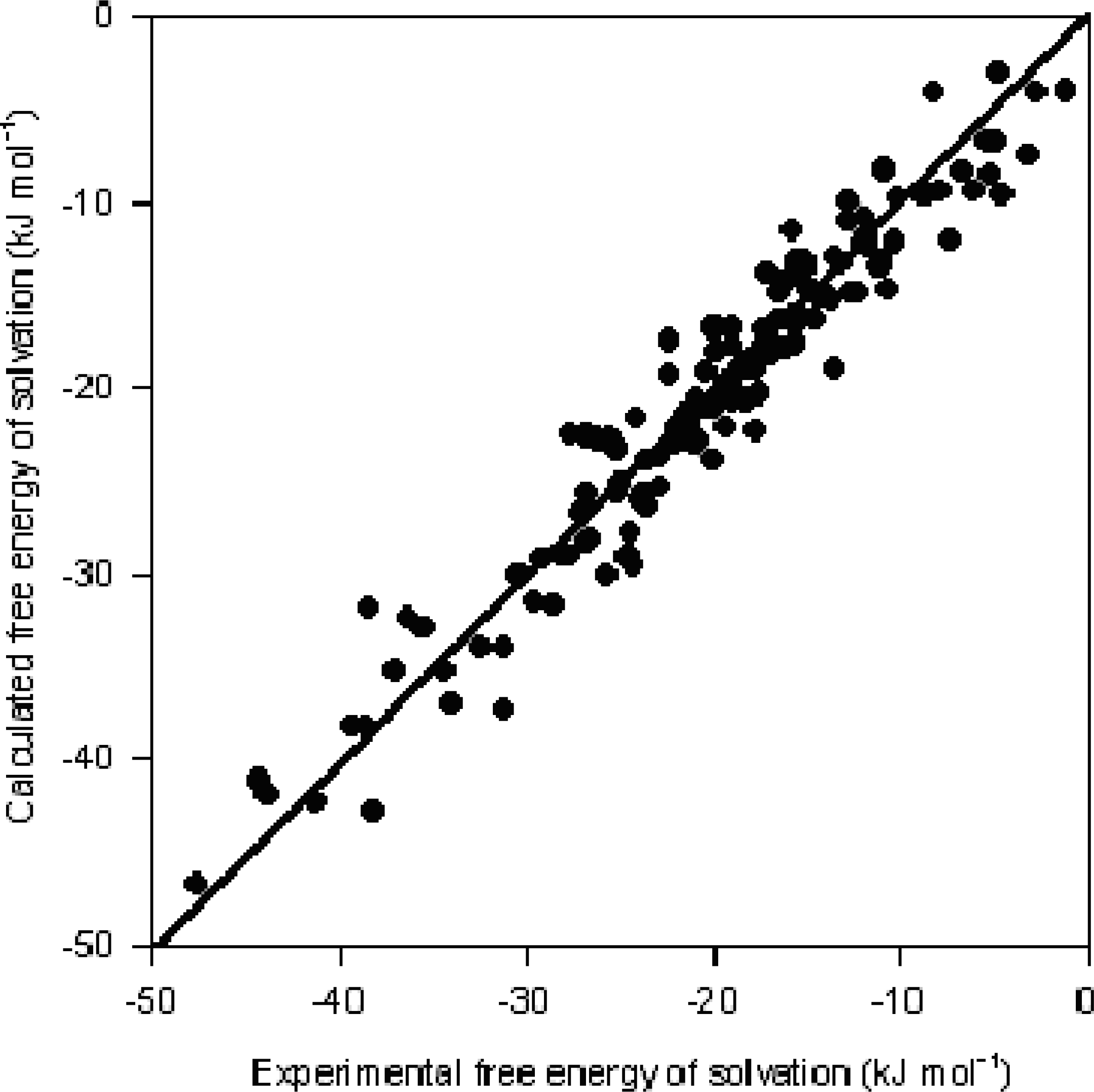
3. Results and Discussion
3.1. Physical meaning of the descriptors involved
4. Conclusions
Acknowledgments
References
- Ruelle, P. The n-octanol and n-hexane/water partition coefficient of environmentally relevant chemicals predicted from the mobile order and disorder (MOD) thermodynamics. Chemosphere 2000, 40, 457–512. [Google Scholar]
- Sandier, SI. Unusual chemical thermodynamics. J. Chem. Thermodynam 1999, 31, 3–25. [Google Scholar]
- Leo, A. Octanol/water partition coefficient. In Handbook of Property Estimation Methods for Chemicals, Environmental and Health Sciences; Boethling, RS, Mackay, D, Eds.; Lewis Publishers: Boca Ratón, FL, USA, 2000; pp. 89–114. [Google Scholar]
- Delgado, EJ; Alderete, JB; Jaña, GA. DFT derived solvation models for organic compounds in alkane solvents. Chem. Phys 2006, 325, 220–224. [Google Scholar]
- Cramer, CJ; Truhlar, DG. Implicit solvation models: Equilibria, structure, spectra, and dynamics. Chem. Rev 1999, 99, 2161–2200. [Google Scholar]
- Duffy, EM; Jorgensen, WL. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc 2000, 122, 2878–2888. [Google Scholar]
- Katritzky, AR; Oliferenko, AA; Oliferenko, PV; Petrukhin, R; Tatham, DB; Maran, U; Lomaka, A; Acree, WE. A general treatment of solubility. 1. The QSPR correlation of solvation free energies of single solutes in series of solvents. J. Chem. Inf. Comput. Sci 2003, 43, 1794–1805. [Google Scholar]
- Wang, J; Wang, W; Huo, S; Lee, M; Kollman, PA. Solvation model on weighted solvent accessible surface area. J. Phys. Chem B 2001, 105, 5055–5067. [Google Scholar]
- Sepassi, K; Yalkowsky, SH. Simplified estimation of the octanol-air partition coefficient. Ind. Eng. Chem. Res 2007, 46, 2220–2223. [Google Scholar]
- Thuens, S; Dreyer, A; Sturm, R; Temme, C; Ebingaus, R. Determination of the octanol-air partition coefficients (KOA) of fluorotelomer alcohols. J. Chem. Eng. Data 2008, 53, 223–227. [Google Scholar]
- Staikova, M; Wania, F; Donaldson, DJ. Molecular polarizability as a single-parameter predictor of vapour pressures and octanol-air partitioning coefficients of non-polar compounds: A priori approach and results. Atmos. Environ 2004, 38, 213–225. [Google Scholar]
- Hyperchem 7.0; Hypercube, Inc.: Gainesville, FL, USA, 2002.
- Ampac 5.0; Semichem, Inc.: Shawnee, KS, USA, 1994.
- Codessa 2.63; Semichem, Inc.: Shawnee, KS, USA, 1999.
- SigmaStat 30; SPSS Inc: Chicago, IL, USA, 2003.
- Li, J; Zhu, T; Hawkins, GD; Winget, P; Liotard, DA; Cramer, CJ; Truhlar, DG. Extension of the platform of applicability of the SM5.42R universal solvation model. Theor. Chem. Ace 1999, 103, 9–63. [Google Scholar]
- Katritzky, AR; Mu, L; Lobanov, VS; Karelson, M. Correlation of boiling points with molecular structure. 1. A training set of 298 diverse organics and a test set of 9 simple organics. J. Phys. Chem 1996, 100, 10400–10407. [Google Scholar]
- Stanton, DT; Jurs, PC. Development and use of charged partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Anal. Chem 1990, 62, 2323–2329. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Coefficien t | Std. Error | P-value | t-test | Std. Coeff. | VIF | |
|---|---|---|---|---|---|---|
| Constant | 0.46 | 0.55 | 0.401 | 0.86 | ||
| GI | −0.033 | 8.96x10−4 | < 0.001 | −36.91 | −0.81 | 1.02 |
| HA | −29.77 | 1.11 | < 0.001 | −26.75 | −0.58 | 1.03 |
| NF | 4.09 | 0.25 | < 0.001 | 16.37 | 0.36 | 1.05 |
| R2 = 0.93, R2CV = 0.93, R2df = 0.93, s = 2.41 | ||||||
| Analysis of Variance
| |||||
|---|---|---|---|---|---|
| DF | SS | MS | F | P | |
| Regression | 3 | 11700 | 3900 | 670 | < 0.001 |
| Residual | 143 | 832 | 5.82 | ||
| Total | 146 | 12533 | 85.84 | ||
| Name | GI | HA dependent HDCA-2 | NF | exp. (kJ·mol−1) | calc. (kJ·mol−1) | Standardized residual |
|---|---|---|---|---|---|---|
| Alkanes | ||||||
| Ethane | 122.30 | 0.00 | 0 | −2.68 | −4.04 | 0.56 |
| Propane | 204.48 | 0.00 | 0 | −5.27 | −6.75 | 0.61 |
| Cyclopropane | 251.65 | 0.00 | 0 | −6.69 | −8.30 | 0.67 |
| 2-Methylpropane | 285.71 | 0.00 | 0 | −6.07 | −9.43 | 1.39 |
| 2,2-Dimethylpropane | 366.02 | 0.00 | 0 | −7.28 | −12.08 | 1.99 |
| n-Butane | 286.70 | 0.00 | 0 | −7.78 | −9.46 | 0.70 |
| Cyclopentane | 407.86 | 0.00 | 0 | −11.09 | −13.46 | 0.98 |
| n-Pentane | 368.90 | 0.00 | 0 | −10.25 | −12.17 | 0.80 |
| n-Hexane | 451.10 | 0.00 | 0 | −12.59 | −14.89 | 0.95 |
| Cyclohexane | 492.57 | 0.00 | 0 | −14.48 | −16.25 | 0.74 |
| Methylcyclohexane | 574.01 | 0.00 | 0 | −13.43 | −18.94 | 2.29 |
| n-Heptane | 533.30 | 0.00 | 0 | −15.65 | −17.60 | 0.81 |
| n-Octane | 615.51 | 0.00 | 0 | −17.49 | −20.31 | 1.17 |
| Chlorotrifluoromethane | 495.97 | 0.00 | 3 | −8.24 | −4.10 | −1.72 |
| Dichlorodifluoromethane | 503.91 | 0.00 | 2 | −5.23 | −8.45 | 1.34 |
| Fluorotrichloromethane | 521.24 | 0.00 | 1 | −11.00 | −13.11 | 0.88 |
| 1,1,2-Trichloro-1,2,2-trifluoroethane | 818.05 | 0.00 | 3 | −10.63 | −14.73 | 1.70 |
| 1-Bromo-1-chloro-2,2,2-trifluoroethane | 832.16 | 0.00 | 3 | −13.68 | −15.19 | 0.63 |
Alkanes | ||||||
| Bromotrifluoromethane | 596.15 | 0.00 | 3 | −3.14 | −7.40 | 1.77 |
| Dichloromethane | 300.40 | 0.00 | 0 | −12.84 | −9.91 | −1.21 |
| Trichloromethane | 427.79 | 0.00 | 0 | −15.94 | −14.12 | −0.76 |
| Chloroethane | 250.24 | 0.00 | 0 | −10.79 | −8.26 | −1.05 |
| 1,1,1 -Trichloroethane | 501.12 | 0.00 | 0 | −15.44 | −16.54 | 0.46 |
| 1,1 -Difluoroethane | 340.12 | 0.00 | 2 | −4.73 | −3.04 | −0.70 |
| 1,1,2-Trichloroethane | 507.54 | 0.00 | 0 | −18.95 | −16.75 | −0.91 |
| 1-Chloropropane | 332.45 | 0.00 | 0 | −12.80 | −10.97 | −0.76 |
| 2-Chloropropane | 329.56 | 0.00 | 0 | −11.88 | −10.88 | −0.42 |
| Bromomethane | 293.94 | 0.00 | 0 | −10.17 | −9.70 | −0.20 |
| Dibromomethane | 550.46 | 0.00 | 0 | −17.49 | −18.17 | 0.28 |
| Tribromomethane | 801.73 | 0.00 | 0 | −23.51 | −26.46 | 1.22 |
| Bromoethane | 370.65 | 0.00 | 0 | −12.13 | −12.23 | 0.04 |
| 2-Bromopropane | 453.05 | 0.00 | 0 | −14.23 | −14.95 | 0.30 |
| 1-Bromobutane | 535.20 | 0.00 | 0 | −17.41 | −17.66 | 0.10 |
| 1-Bromopentane | 617.39 | 0.00 | 0 | −19.58 | −20.37 | 0.33 |
| Nitroethane | 495.74 | 0.00 | 0 | −16.44 | −16.36 | −0.03 |
| 1-Nitropropane | 577.83 | 0.00 | 0 | −18.58 | −19.07 | 0.20 |
| 2-Nitropropane | 575.60 | 0.00 | 0 | −17.70 | −18.99 | 0.54 |
| 1-Nitrobutane | 659.99 | 0.00 | 0 | −21.38 | −21.78 | 0.17 |
Alkenes | ||||||
| Ethylene | 122.21 | 0.00 | 0 | −1.13 | −4.03 | 1.20 |
| Propylene | 206.54 | 0.00 | 0 | −4.77 | −6.82 | 0.85 |
| 2-Methylpropene | 290.01 | 0.00 | 0 | −8.49 | −9.57 | 0.45 |
| 1-Butene | 288.64 | 0.00 | 0 | −4.56 | −9.53 | 2.06 |
| 1-Hexene | 453.03 | 0.00 | 0 | −12.30 | −14.95 | 1.10 |
| 1,3-Butadiene | 290.52 | 0.00 | 0 | −8.79 | −9.59 | 0.33 |
| cis-1,2-Dichloroethylene | 400.65 | 0.00 | 0 | −15.52 | −13.22 | −0.95 |
| trans-1,2-Dichloroethylene | 399.06 | 0.00 | 0 | −15.10 | −13.17 | −0.80 |
| Trichloroethylene | 537.42 | 0.00 | 0 | −15.69 | −17.73 | 0.85 |
| Tetrachloroethylene | 674.93 | 0.00 | 0 | −17.74 | −22.27 | 1.88 |
| 3-Bromopropene | 455.31 | 0.00 | 0 | −13.81 | −15.03 | 0.50 |
Alkynes | ||||||
| 1-Pentyne | 375.21 | 0.00 | 0 | −11.67 | −12.38 | 0.30 |
| 1-Hexyne | 457.40 | 0.00 | 0 | −14.35 | −15.09 | 0.31 |
Aromatics | ||||||
| Benzene | 504.85 | 0.00 | 0 | −15.56 | −16.66 | 0.46 |
| Toluene | 588.93 | 0.00 | 0 | −19.04 | −19.43 | 0.16 |
| Ethylbenzene | 670.93 | 0.00 | 0 | −21.25 | −22.14 | 0.37 |
| m-Xylene | 672.96 | 0.00 | 0 | −21.97 | −22.21 | 0.10 |
| o-Xylene | 672.78 | 0.00 | 0 | −21.21 | −22.20 | 0.41 |
| p-Xylene | 673.07 | 0.00 | 0 | −21.71 | −22.21 | 0.21 |
| Naphtalene | 887.05 | 0.00 | 0 | −29.16 | −29.27 | 0.05 |
| Anthracene | 1269.00 | 0.00 | 0 | −43.81 | −41.88 | −0.80 |
| Bromobenzene | 767.77 | 0.00 | 0 | −22.84 | −25.34 | 1.04 |
| Fluorobenzene | 617.17 | 0.00 | 1 | −16.19 | −16.28 | 0.04 |
| 1,4-Dibromobenzene | 1031.60 | 0.00 | 0 | −31.25 | −34.04 | 1.16 |
| p-Bromotoluene | 852.06 | 0.00 | 0 | −26.61 | −28.12 | 0.63 |
| Chlorobenzene | 641.42 | 0.00 | 0 | −20.92 | −21.17 | 0.10 |
| 1,2-Dichlorobenzene | 779.27 | 0.00 | 0 | −25.15 | −25.72 | 0.23 |
| 1,4-Dichlorobenzene | 778.61 | 0.00 | 0 | −23.72 | −25.69 | 0.82 |
| Nitrobenzene | 879.27 | 0.00 | 0 | −27.74 | −29.02 | 0.53 |
| 2-Nitrotoluene | 962.38 | 0.00 | 0 | −28.45 | −31.76 | 1.37 |
| 2,2′-Dichlorobiphenyl | 1158.20 | 0.00 | 0 | −39.37 | −38.22 | −0.48 |
| 2,3 -Dichlorobiphenyl | 1160.50 | 0.00 | 0 | −38.62 | −38.30 | −0.13 |
| 2,2,3′-Trichlorobiphenyl | 1295.30 | 0.00 | 0 | −38.16 | −42.74 | 1.90 |
Alcohols | ||||||
| Methanol | 142.96 | 0.44 | 0 | −16.19 | −17.82 | 0.67 |
| Ethanol | 224.20 | 0.44 | 0 | −18.24 | −20.50 | 0.94 |
| Ethylene glycol | 326.09 | 0.89 | 0 | −31.13 | −37.26 | 2.54 |
| 1-Propanol | 306.49 | 0.43 | 0 | −21.00 | −22.92 | 0.79 |
| 2-Propanol | 305.16 | 0.40 | 0 | −19.33 | −21.98 | 1.10 |
| 1,1,1 -Trifluoro-2-propanol | 638.10 | 0.43 | 3 | −21.42 | −21.59 | 0.07 |
| Hexafluoro-2-propanol | 972.10 | 0.47 | 6 | −24.10 | −21.53 | −1.07 |
| 1-Butanol | 388.67 | 0.45 | 0 | −23.89 | −26.22 | 0.97 |
| tert-Butyl alcohol | 385.36 | 0.38 | 0 | −20.00 | −24.03 | 1.67 |
| 1-Pentanol | 470.88 | 0.43 | 0 | −26.78 | −28.34 | 0.65 |
| 1-Hexanol | 553.07 | 0.45 | 0 | −29.54 | −31.65 | 0.87 |
| 1-Heptanol | 635.29 | 0.44 | 0 | −32.43 | −34.06 | 0.68 |
| 1-Octanol | 717.49 | 0.45 | 0 | −34.02 | −37.07 | 1.27 |
| 1-Decanol | 881.86 | 0.45 | 0 | −41.34 | −42.50 | 0.48 |
| Allyl alcohol | 308.24 | 0.43 | 0 | −22.05 | −22.97 | 0.38 |
| Phenol | 612.17 | 0.41 | 0 | −36.36 | −32.41 | −1.64 |
| 4-Bromophenol | 875.94 | 0.41 | 0 | −44.31 | −41.11 | −1.33 |
| 2-Cresol | 695.97 | 0.34 | 0 | −35.52 | −33.09 | −1.01 |
| 3-Cresol | 696.13 | 0.41 | 0 | −34.31 | −35.18 | 0.36 |
| 4-Cresol | 696.29 | 0.41 | 0 | −36.99 | −35.18 | −0.75 |
| 2,2,2-Trifluoroethanol | 557.11 | 0.50 | 3 | −20.13 | −21.00 | 0.36 |
| 2-Methoxyethanol | 432.48 | 0.45 | 0 | −24.39 | −27.67 | 1.36 |
Ethers | ||||||
| Methyl propyl ether | 413.02 | 0.00 | 0 | −15.19 | −13.63 | −0.65 |
| Methyl isopropyl ether | 411.69 | 0.00 | 0 | −15.19 | −13.59 | −0.67 |
| Methyl tert-butyl ether | 492.04 | 0.00 | 0 | −15.19 | −16.24 | 0.43 |
| Diethyl ether | 412.05 | 0.00 | 0 | −15.19 | −13.60 | −0.66 |
| Tetrahydrofuran | 451.51 | 0.00 | 0 | −16.44 | −14.90 | −0.64 |
| Anisole | 718.19 | 0.00 | 0 | −22.89 | −23.70 | 0.34 |
| Ethyl phenyl ether | 799.40 | 0.00 | 0 | −23.64 | −26.38 | 1.14 |
| 1,2-Dimethoxyethane | 538.90 | 0.00 | 0 | −19.04 | −17.78 | −0.52 |
| 1,4-Dioxane | 580.19 | 0.00 | 0 | −20.46 | −19.15 | −0.55 |
| l,l-Dichloro-2,2-difluoroethylmethyl ether | 800.57 | 0.00 | 2 | −16.82 | −18.24 | 0.59 |
Aldehydes | ||||||
| Formaldehyde | 147.19 | 0.27 | 0 | −13.51 | −12.90 | −0.26 |
| Propanal | 312.73 | 0.22 | 0 | −17.28 | −16.87 | −0.17 |
| Butanal | 394.91 | 0.22 | 0 | −19.33 | −19.58 | 0.10 |
| Benzaldehyde | 696.53 | 0.24 | 0 | −25.65 | −30.13 | 1.86 |
| m-Hydroxybenzaldehyde | 803.86 | 0.68 | 0 | −47.66 | −46.77 | −0.37 |
| p-Hydroxybenzaldehyde | 804.41 | 0.64 | 0 | −51.71 | −45.60 | −2.54 |
Ketones | ||||||
| Acetone | 312.77 | 0.10 | 0 | −13.18 | −13.30 | 0.05 |
| 2-Butanone | 395.36 | 0.10 | 0 | −15.82 | −16.02 | 0.08 |
| 3,3-Dimethylbutanone | 554.92 | 0.08 | 0 | −18.95 | −20.69 | 0.72 |
| 2-Pentanone | 476.64 | 0.10 | 0 | −18.20 | −18.71 | 0.21 |
| 3-Pentanone | 476.00 | 0.11 | 0 | −18.24 | −18.98 | 0.31 |
| Cyclopentanone | 518.01 | 0.11 | 0 | −20.96 | −20.37 | −0.25 |
| 2-Hexanone | 559.88 | 0.09 | 0 | −21.00 | −21.16 | 0.06 |
Ketones | ||||||
| 2-Heptanone | 642.08 | 0.09 | 0 | −23.64 | −23.87 | 0.09 |
| 2-Octanonne | 724.29 | 0.09 | 0 | −26.69 | −26.58 | −0.05 |
| Acetophenone | 778.46 | 0.11 | 0 | −28.20 | −28.96 | 0.32 |
Esters | ||||||
| Methyl formate | 363.42 | 0.00 | 0 | −11.80 | −11.99 | 0.08 |
| Methyl acetate | 446.71 | 0.00 | 0 | −14.81 | −14.74 | −0.03 |
| Ethyl acetate | 527.89 | 0.00 | 0 | −16.99 | −17.42 | 0.18 |
| Propyl acetate | 610.15 | 0.00 | 0 | −19.04 | −20.13 | 0.45 |
| Butyl acetate | 692.32 | 0.00 | 0 | −20.75 | −22.85 | 0.87 |
| Methyl propionate | 528.62 | 0.00 | 0 | −16.99 | −17.44 | 0.19 |
| Methyl butyrate | 605.56 | 0.00 | 0 | −19.20 | −19.98 | 0.33 |
| Methyl pentanoate | 692.80 | 0.00 | 0 | −21.46 | −22.86 | 0.58 |
| Methyl benzoate | 911.96 | 0.00 | 0 | −30.38 | −30.09 | −0.12 |
Amines | ||||||
| Methylamine | 139.01 | 0.23 | 0 | −15.82 | −11.43 | −1.82 |
| Ethylamine | 219.44 | 0.22 | 0 | −17.11 | −13.79 | −1.38 |
| Propylamine | 301.77 | 0.23 | 0 | −19.96 | −16.81 | −1.31 |
| Butylamine | 383.96 | 0.22 | 0 | −22.38 | −19.22 | −1.31 |
| Diethylamine | 395.38 | 0.17 | 0 | −19.87 | −18.11 | −0.73 |
| Dipropylamine | 559.65 | 0.16 | 0 | −25.19 | −23.23 | −0.81 |
| Trimethylamine | 328.18 | 0.09 | 0 | −15.06 | −13.51 | −0.64 |
Miscellaneous | ||||||
| Piperazine | 544.97 | 0.39 | 0 | −24.27 | −29.59 | 2.21 |
| Aniline | 609.22 | 0.22 | 0 | −27.11 | −26.65 | −0.19 |
| Morpholine | 562.85 | 0.22 | 0 | −25.06 | −25.12 | 0.03 |
| Piperidine | 518.83 | 0.19 | 0 | −26.23 | −22.78 | −1.43 |
| Pyridine | 529.10 | 0.00 | 0 | −22.34 | −17.46 | −2.02 |
| 2-Methylpyridine | 611.55 | 0.09 | 0 | −25.69 | −22.86 | −1.17 |
| 3-Methylpyridine | 613.61 | 0.08 | 0 | −26.78 | −22.63 | −1.72 |
| 4-Methylpyridine | 613.30 | 0.08 | 0 | −27.61 | −22.62 | −2.07 |
| 2-Ethylpyridine | 693.46 | 0.09 | 0 | −26.78 | −25.56 | −0.50 |
| 2-Methylpyrazine | 717.78 | 0.18 | 0 | −24.56 | −29.05 | 1.86 |
| Benzonitrile | 689.18 | 0.00 | 0 | −25.48 | −22.74 | −1.14 |
| 2,6-Dichlorobenzonitrile | 963.97 | 0.00 | 0 | −38.41 | −31.81 | −2.74 |
| Name | GI | HA dependent HDCA-2 | NF | exp. (kJ·nnol−1) | calc. (kJ·mol−1) | Standardized residual |
|---|---|---|---|---|---|---|
| 1,2,3 -Trichlorobenzene | 917.09 | 0.00 | 0 | −30.36 | −30.26 | −0.04 |
| 1,2,4-Trichlorobenzene | 916.84 | 0.00 | 0 | −29.10 | −30.26 | 0.46 |
| 1,2,3,4-Tetrachloro-benzene | 1055.20 | 0.00 | 0 | −33.21 | −34.82 | 0.64 |
| Pentachlorobenzene | 1193.60 | 0.00 | 0 | −37.09 | −39.39 | 0.92 |
| 1,2,3 -Trichloronaphthalene | 1297.90 | 0.00 | 0 | −43.48 | −42.83 | −0.26 |
| Methane | 39.19 | 0.00 | 0 | 2.13 | −1.29 | 1.37 |
| Chloromethane | 169.87 | 0.00 | 0 | −7.42 | −5.61 | −0.73 |
| Tetrachloromethane | 549.75 | 0.00 | 0 | −14.49 | −18.14 | 1.46 |
| Monochloroethane | 250.23 | 0.00 | 0 | −9.87 | −8.26 | −0.64 |
| 1,1 -Dichloroethane | 377.46 | 0.00 | 0 | −14.61 | −12.46 | −0.86 |
| 1,1,1,2-Tetrachloroethane | 632.35 | 0.00 | 0 | −22.65 | −20.87 | −0.71 |
| Chloropropane | 332.47 | 0.00 | 0 | −13.18 | −10.97 | −0.88 |
| 1,2,3-Trichloropropane | 589.65 | 0.00 | 0 | −25.79 | −19.46 | −2.53 |
| 4:2 FTOH | 1537.40 | 0.46 | 9 | −26.08 | −27.62 | 0.62 |
| 6:2 FTOH | 2136.40 | 0.46 | 13 | −27.50 | −31.03 | 1.41 |
| 8:2 FTOH | 2735.40 | 0.46 | 17 | −31.84 | −34.43 | 1.04 |
| 10:12 FTOH | 3334.50 | 0.46 | 21 | −32.58 | −37.84 | 2.11 |
| 12:2 FTOH | 3933.60 | 0.46 | 25 | −35.38 | −41.25 | 2.35 |
| Ethyl formate | 444.76 | 0.00 | 0 | −12.50 | −14.68 | 0.87 |
| Propyl formate | 526.87 | 0.00 | 0 | −15.18 | −17.39 | 0.88 |
| Isopentanol | 469.57 | 0.45 | 0 | −25.79 | −28.89 | 1.24 |
| Cyclohexanol | 593.62 | 0.39 | 0 | −29.56 | −31.20 | 0.66 |
| Hexanal | 559.31 | 0.22 | 0 | −25.16 | −25.01 | −0.06 |
| 4-Methyl-2-pentanone | 558.35 | 0.09 | 0 | −18.83 | −21.10 | 0.91 |
| Ethyl propionate | 610.06 | 0.00 | 0 | −17.97 | −20.13 | 0.86 |
| Isopropyl acetate | 608.75 | 0.00 | 0 | −18.09 | −20.09 | 0.80 |
| Di-isopropyl ether | 573.56 | 0.00 | 0 | −15.18 | −18.93 | 1.50 |
| Ethyl butyrate | 692.21 | 0.00 | 0 | −20.31 | −22.84 | 1.01 |
| Pentafluorobenzene | 1070.60 | 0.00 | 5 | −14.49 | −14.88 | 0.16 |
| Hexafluorobenzene | 1184.70 | 0.00 | 6 | −12.04 | −14.56 | 1.01 |
| 1-Propanethiol | 335.70 | 0.19 | 0 | −14.73 | −16.73 | 0.80 |
| Thiophenol | 646.51 | 0.18 | 0 | −25.06 | −26.69 | 0.65 |
| Thioanisole | 783.48 | 0.10 | 0 | −27.07 | −28.83 | 0.70 |
| Dimethyl sulfide | 309.60 | 0.10 | 0 | −17.74 | −13.19 | −1.82 |
| Diethyl sulfide | 470.87 | 0.09 | 0 | −17.11 | −18.22 | 0.44 |
| Dipropyl sulfide | 635.15 | 0.08 | 0 | −16.28 | −23.34 | 2.82 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delgado, E.J.; Jaña, G.A. Quantitative Prediction of Solvation Free Energy in Octanol of Organic Compounds. Int. J. Mol. Sci. 2009, 10, 1031-1044. https://doi.org/10.3390/i10031031
Delgado EJ, Jaña GA. Quantitative Prediction of Solvation Free Energy in Octanol of Organic Compounds. International Journal of Molecular Sciences. 2009; 10(3):1031-1044. https://doi.org/10.3390/i10031031
Chicago/Turabian StyleDelgado, Eduardo J., and Gonzalo A. Jaña. 2009. "Quantitative Prediction of Solvation Free Energy in Octanol of Organic Compounds" International Journal of Molecular Sciences 10, no. 3: 1031-1044. https://doi.org/10.3390/i10031031