Introduction
The electric moments of a molecule are quantities of fundamental importance in structural chemistry. When a molecule with permanent electric dipole moment
pe is subject to an external constant electrostatic field
E, the change in the dipole moment can be written [
1]
Here pe,j is the jth Cartesian component of the dipole moment, pe(0) is the dipole in the absence of a field and pe(E) is the dipole moment in the presence of the field. The six quantities αij define the dipole polarizability tensor, the quantities βijk define the first dipole hyperpolarizability and so on.
Equation (1) is often written in tensor notation as
Hyperpolarizabilities are generally small and their effect is minimal for weak electric fields. They are important when the applied electric field is large. There has recently been an intense search for molecules with large non-zero hyperpolarizabilities [
2], since these substances have potential as the constituents of non-linear optical materials.
Equations similar to (1) can be written for the higher electric moments, and the quantities of interest are (for example) the quadrupole polarizability and octupole hyperpolarizabilities. Such quantities are rarely encountered in Chemistry.
The energy U of the charge distribution also changes according to the equation
which can again be written more compactly in tensor notation
The experimental determination of a molecular polarizability is far from straightforward, especially if the molecule has little or no symmetry. The mean polarizability
can be determined from the refractive index n of a gas according to the equation
where p is the pressure, k
B the Boltzmann constant, T the thermodynamic temperature and ∈
0 the permittivity of free space. A key assumption in the derivation of equation (5) is that the individual mole-cules are sufficiently far apart on average that they do not interact with each other.
In a condensed phase, the problem is more complicated because the separation between molecules is of the order of molecular dimensions and their interactions can no longer be ignored. The result is that each molecule is polarized not only by the external field but also by the field due to the surrounding molecules. The resultant field is known as the local field
F, and it is usually written in terms of the dielectric polarization
P as
where L is the dimensionless Lorentz factor, which depends on the structure of the phase. L is strictly a tensor, and it can be shown that for cubic and isotropic phases the three principal values are equal to 1/3. This gives the Lorentz local field
The Lorenz-Lorentz equation
gives a molecular expression for the polarizability, and it can be easily derived from equation (7). Here, N is the number of molecules in volume V.
In the case of molecules with a permanent dipole moment, it is necessary to take account of the orientation polarization. The resulting Debye equation
permits polarizabilities and dipole moments to be determined from measurements of the relative per-mittivity ∈
r and the density ρ as a function of temperature. M is the molar mass and N
A the Avogadro constant. Reliable results can only be obtained from dilute solutions [
1].
The experimental techniques described above deal with infinitely dilute gases or make assumptions about the interactions between molecules in condensed phases. Laser Stark spectroscopy is a powerful experimental approach to the basic electric properties of isolated molecules in the gas phase. This type of spectroscopy utilises the interaction between a molecule and an external field. This interaction gives rise to shifts and splittings of individual rotational transitions, and an analysis of these shifts and splittings yields the tensor components of the dipole polarizability of the ground and vibronically excited states. Such a method requires a narrow band tuneable laser together with a molecular beam apparatus to produce isolated molecules, and a capacitor capable of generating an electric field of the order of 100 kV cm
-1. Okruss et. al [
3] have recently exploited this technique to study benzene, and their paper reports the electric polarizabilities of the S
0(
1A
1g) ground electronic and the vibrationally excited 6
1 S
1(
1B
2u) state of benzene. Such accurate experimental data is hard to come by, because of the exacting experimental requirements.
An alternative route to these properties is afforded by molecular modeling. Most dipole polarizability calculations for large molecules have been done at the empirical or semi-empirical level of theory. Indeed some semi-empirical packages such as MOPAC [
4] have polarizability calculations built in as optional properties to be determined once the Hartree-Fock (HF) wavefunction has been calculated. There are a number of semi-empirical schemes in the literature and we will report later the results of AM1 calculations for the molecules under study.
Several authors have used
Ab Initio techniques to study molecular polarizabilities. It is usually possible to obtain respectable agreement with experiment at the HF level of theory for the dipole polarizability tensor
α provided that a careful choice of atomic orbital basis set is made. It is common knowledge that polarizabilities can only be calculated accurately from calculations employing extended basis sets. In particular, these basis sets have to include diffuse functions that can accurately describe the response of a molecular charge distribution to an external electric field. These diffuse (s and p) functions are needed in addition to the normal polarization functions; they are denoted by + and ++ in packages such as GAUSSIAN98 [
5].
Once near the Hartree Fock limit, it is necessary to concern oneself with the correlation contribution to such properties. Until fairly recently, the most usual method of treating electron correlation in such molecules was the Muller Plesset perturbation technique. Such calculations are labelled MPn where n is the order of perturbation. Most post-HF techniques have the common feature that they are extremely expensive in computer resource; MPn calculations usually involve the semi-transformation of integrals from the atomic orbital basis set to the molecular orbital basis set, and this single step can be prohibitive in disk space.
In recent years, density functional techniques have received a great deal of attention in the literature.
The idea is to start from the HF electronic energy expression [
6]
which relates the electronic energy for a one-determinant closed shell ε
el to the electron density matrix
P, the matrix of one-electron integrals
h1, the coulomb matrix
J and the exchange matrix
K.
Density functional theory (DFT) seeks to write the energy expression as
where ε
X is the exchange functional and ε
C the correlation functional, which is of course zero for HF wavefunctions. In order to calculate ε
X and ε
C it is necessary to assume some functional form to the two potentials and then calculate the contribution to the electronic energy as an integral over the electron density (and occasionally the gradient of the electron density). These calculations are performed numerically and tend to consume less computer resource than traditional MPn calculations. There are many variants on the form of the exchange and the correlation functional, most of which are based on the free-electron gas model.
The application of density functional methods to the study of molecular properties is a recent devel-opment [
7], and there is no great pool of expertise to suggest that one formulation is better than any other for the calculation of a given property. In traditional HF theory, one can increase the accuracy of a calculation by systematically extending the atomic orbital basis set, but in DFT the only way forward is to improve the basic model.
Polycyclic aromatic hydrocarbons have been the subject of very many theoretical and experimental studies. They have carcinogenic activities and they form various complexes [
8].
In an earlier paper in this series [
9] we reported corresponding Semi-empirical,
Ab Initio HF and DFT studies on benzene. In the
Ab Initio studies, we used a high quality basis set for geometry optimization and then calculated the polarizabilities at the same geometry but with diffuse functions added to the basis set. To use a common notation, we performed HF/6-311G(3d,2p) // HF/6-311++G(3d,2p) and HF/6-311G(3d,2p) // BLYP/6-311++G(3d,2p) polarizability calculations. We found that the effect of the BLYP density functional procedure was to increase the polarizability components in the molecular plane by some 5% but to leave the polarizability component normal to the molecular plane unchanged.
We reached similar conclusions for naphthalene [
10].
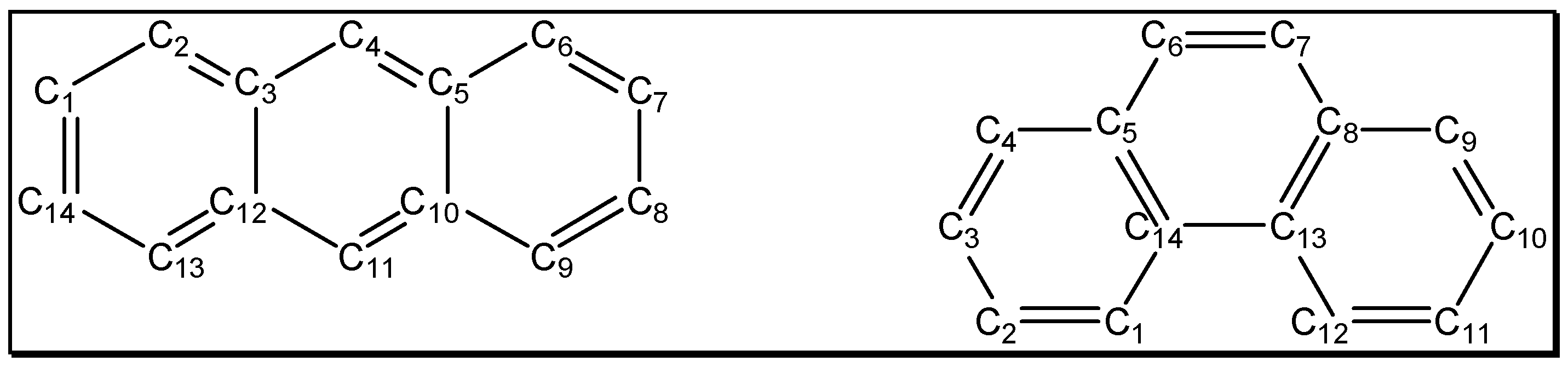
The aim of this paper is to extend our studies to anthracene and phenanthrene.


{kind=link}