Quantum Chemical and Experimental Studies on the Mechanism of Alkylation of β-Dicarbonyl Compounds. The Synthesis of Five and Six Membered Heterocyclic Spiro Derivatives

Abstract
:Introduction
Results and Discussion
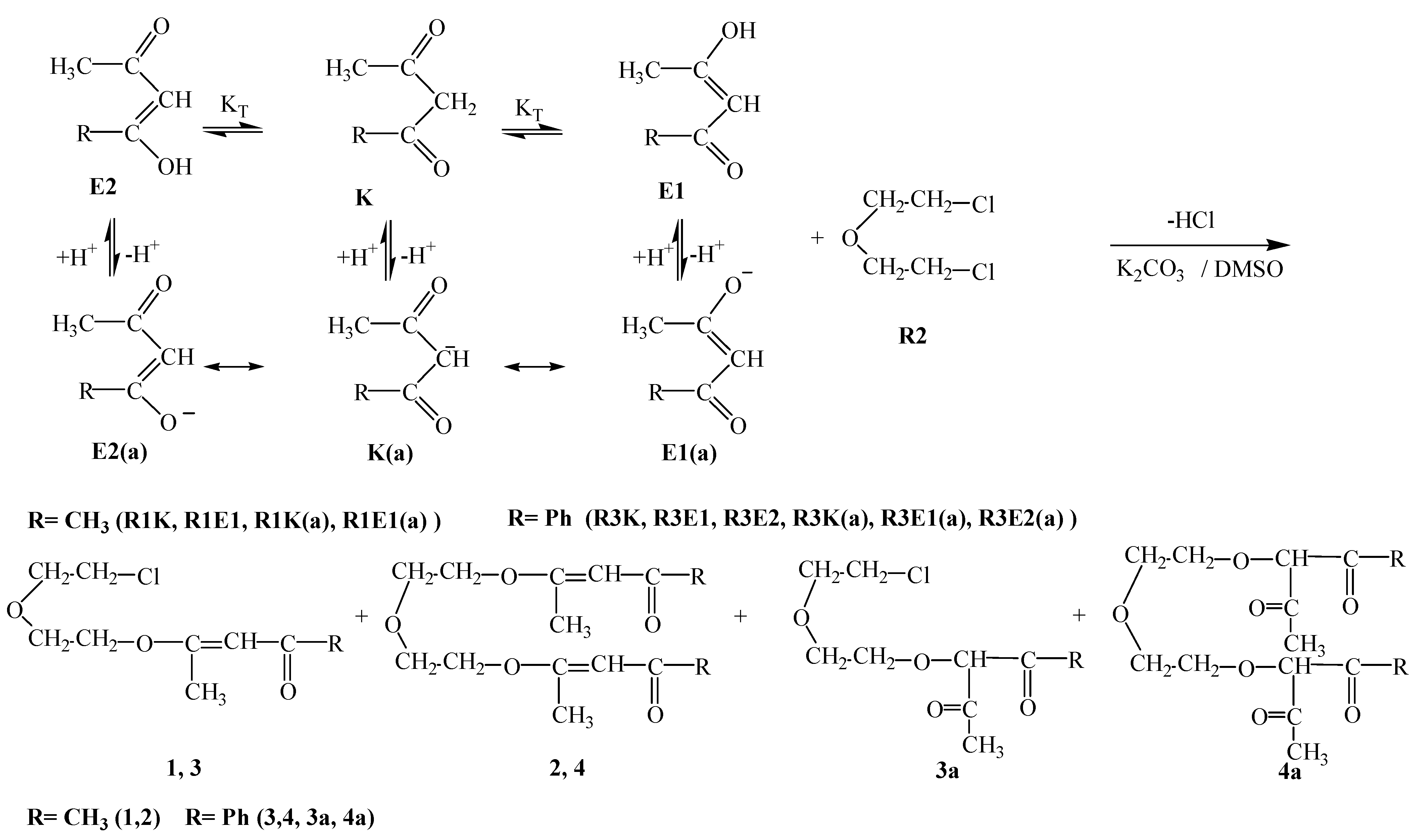
Theoretical Approaches
Discussion of Computational Work
 Enol tautomerism of the main molecules and the obtained data is collected in Table 2.R1E equilibrium suggests the predominance of the keto form (i.e. the R1K form) in aqueous media, it seems that this situation is reversed in basic media and the enolate form R1E1(a) predominates over the carbene form R1K(a) and the reaction proceeds by the nucleophilic attack of R1E1(a) on R2 to first form compound 1 and then it proceeds via a second attack of R1E1(a) on 1 to form compound 2. Further evidence to support this argument is the higher nucleophilicity, η; and the higher basicity (i.e. smaller pKa value for deprotonation) of R1E1(a) compared to the R1K1(a) form (Table 1 and Table 2) .
Enol tautomerism of the main molecules and the obtained data is collected in Table 2.R1E equilibrium suggests the predominance of the keto form (i.e. the R1K form) in aqueous media, it seems that this situation is reversed in basic media and the enolate form R1E1(a) predominates over the carbene form R1K(a) and the reaction proceeds by the nucleophilic attack of R1E1(a) on R2 to first form compound 1 and then it proceeds via a second attack of R1E1(a) on 1 to form compound 2. Further evidence to support this argument is the higher nucleophilicity, η; and the higher basicity (i.e. smaller pKa value for deprotonation) of R1E1(a) compared to the R1K1(a) form (Table 1 and Table 2) .
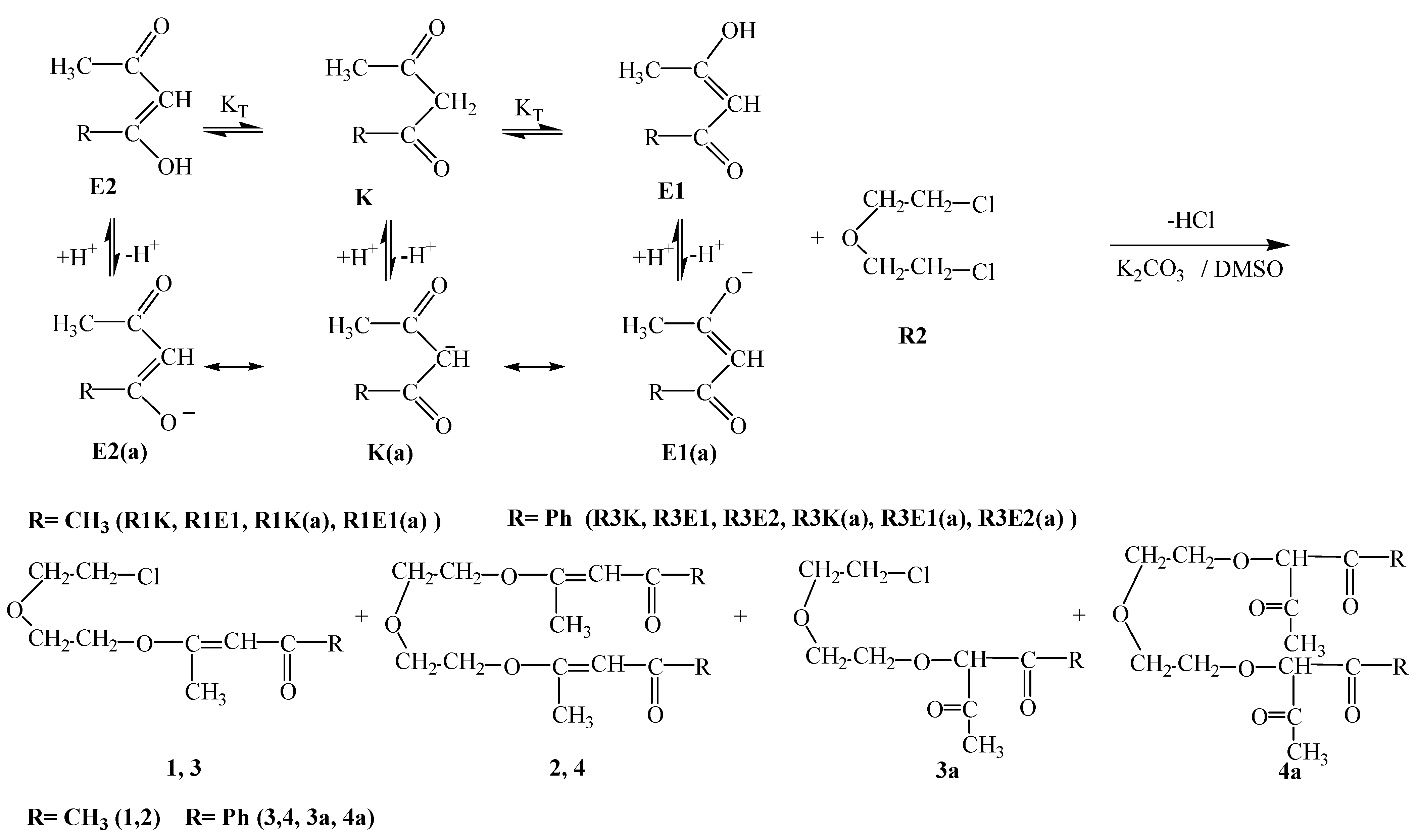{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔH (cal mol-1) | ΔS (cal mol-1) | ΔG (kcal mol-1)a | ΔHf (kcalmol-1) | HOMO | LUMO | Nucleophilicity (η)b | Experimental Yield (% ) |
|---|---|---|---|---|---|---|---|---|
| T = 343 K (ε = 47.24) | ||||||||
| R1K | 6249.087 | 86.069 | -23.273 | -105.766 | -11.329 | 0.018 | -11.347 | |
| R2 | 5604.130 | 85.404 | -23.689 | -75.785 | -10.857 | 0.718 | -11.575 | |
| 1 | 9064.189 | 108.952 | -28.306 | -37.861 | -10.065 | -1.863 | -8.202 | 59 |
| 2 | 12782.249 | 131.221 | -32.227 | -103.808 | -9.921 | -1.859 | -8.062 | 15 |
| 3 | 11268.273 | 120.470 | -30.053 | -99.461 | -9.936 | -0.802 | -9.134 | 57 |
| 4 | 15983.897 | 146.434 | -34.243 | -132.297 | -9.834 | -0.753 | -9.081 | 16 |
| R1K(a) | 12604.066 | 98.677 | -21.239 | -185.011 | -8.685 | 0.625 | -9.310 | |
| R1E1 | 13327.849 | 101.118 | -21.356 | -87.118 | -9.904 | 0.017 | -9.921 | |
| R1E1(a) | 12604.066 | 97.186 | -20.731 | -185.181 | -8.804 | -0.406 | -8.398 | |
| R3K | 7242.087 | 93.873 | -24.956 | -69.146 | -10.145 | -0.739 | -9.406 | |
| R3K(a) | 18143.301 | 120.397 | -23.153 | -147.226 | -8.707 | -0.105 | -8.602 | |
| R3E1 | 18129.246 | 119.193 | -22.754 | -47.677 | -9.913 | -0.538 | -9.375 | |
| R3E1(a) | 18190.937 | 120.608 | -23.178 | -148.021 | -8.847 | 0.098 | -8.945 | |
| R3E2 | 18222.994 | 119.722 | -22.842 | -47.937 | -10.041 | -0.155 | -9.886 | |
| R3E2(a) | 18924.776 | 124.037 | -23.620 | -146.083 | -8.685 | -0.320 | -8.365 | |
| H3O+ | 2750.472 | 47.158 | -13.425 | 61.928 | -15.994 | 1.652 | -17.646 | |
| H20 | 2730.416 | 46.135 | -13.095 | -61.414 | -12.794 | 4.268 | -17.062 | |
| R4K | 8118.436 | 97.785 | -25.422 | -111.147 | -11.392 | 0.065 | -11.457 | |
| 5 | 10389.545 | 114.701 | -28.953 | -142.383 | -10.042 | -0.481 | -9.561 | 46 |
| 6 | 11016.198 | 115.088 | -28.459 | -148.209 | -10.938 | -0.143 | -10.795 | 24 |
| 7 | 15999.285 | 145.441 | -33.887 | -220.448 | -10.033 | -0.509 | -9.524 | 12 |
| R4K(a) | 18370.540 | 119.539 | -22.631 | -193.605 | -8.797 | 0.436 | -9.233 | |
| R4E1 | 19259.580 | 122.297 | -22.688 | -92.314 | -9.970 | -0.437 | -9.533 | |
| R4E1(a) | 18483.670 | 120.297 | -22.823 | -193.418 | -8.779 | 0.444 | -9.223 | |
| R5K | 6741.119 | 90.785 | -24.398 | -149.949 | -11.482 | 0.049 | -11.531 | |
| 8 | 10185.203 | 111.495 | -28.058 | -185.961 | -11.056 | -0.037 | -11.019 | 55 |
| 9 | 10080.245 | 113.231 | -28.768 | -183.145 | -10.103 | -0.223 | -9.880 | 48 |
| 10 | 14245.501 | 137.720 | -32.992 | -292.902 | -9.958 | -0.297 | -9.661 | 10 |
| R5K(a) | 15761.846 | 122.368 | -26.210 | -229.112 | -8.791 | 0.665 | -9.456 | |
| R5E1 | 16477.994 | 114.511 | -22.799 | -129.609 | -10.005 | -0.251 | -9.754 | |
| R5E1(a) | 16726.542 | 117.887 | -23.709 | -228.808 | -8.826 | 0.634 | -9.460 | |
| R5E2 | 16610.422 | 115.076 | -22.861 | -123.669 | -9.608 | -0.196 | -9.412 | |
| R5E2(a) | 15770.958 | 112.325 | -26.187 | -229.079 | -8.789 | 0.666 | -9.455 | |
| R6K | 8091.277 | 100.368 | -26.335 | -195.638 | -11.606 | 0.308 | -11.914 | |
| 11 | 10618.015 | 114.735 | -28.736 | -230.841 | -11.146 | 0.184 | -11.330 | 57 |
| 12 | 10731.533 | 118.688 | -29.978 | -245.052 | -10.955 | 0.144 | -11.099 | 10 |
| 13 | 15969.012 | 148.524 | -34.974 | -423.969 | -11.063 | -0.061 | -11.002 | 14 |
| R6K(a) | 17368.114 | 117.216 | -22.837 | -273.403 | -8.799 | 0.867 | -9.666 | |
| R6E1 | 19569.412 | 126.417 | -23.792 | -168.694 | -10.095 | -0.201 | -9.894 | |
| R6E1(a) | 17334.207 | 117.216 | -22.871 | -273.373 | -8.792 | 0.870 | -9.662 | |
| T = 363 K (ε = 78.40) | ||||||||
| R7K | 11364.453 | 114.981 | -30.374 | -185.028 | -11.063 | -0.041 | -11.022 | |
| R8 | 3563.453 | 59.771 | -18.133 | -17.071 | -10.560 | 2.442 | -13.002 | |
| R9 | 3637.903 | 59.273 | -17.878 | 15.501 | -9.707 | 2.716 | -12.423 | |
| R10 | 6603.077 | 85.778 | -24.534 | 42.036 | -9.330 | -0.082 | -9.248 | |
| 14-OH | 12074.647 | 118.843 | -31.065 | -142.197 | -10.704 | 0.012 | -10.716 | 91 |
| 14-NH2 | 12606.540 | 121.978 | -31.671 | -115.892 | -9.717 | 0.198 | -9.915 | 91 |
| 14-NHPh | 14384.721 | 131.234 | -33.253 | -84.046 | -9.060 | -0.157 | -8.903 | 91 |
| R7K(a) | 24864.521 | 141.158 | -26.376 | -243.164 | -8.464 | 0.652 | -9.116 | |
| R7E1 | 25643.604 | 143.758 | -26.541 | -151.205 | -9.905 | 0.403 | -10.308 | |
| R7E1(a) | 11364.453 | 139.490 | -39.270 | -243.571 | -8.501 | 0.685 | -9.186 | |
| H3O+ | 2919.050 | 47.636 | -14.373 | 62.097 | -15.994 | 1.642 | -17.636 | |
| H2O | 2891.295 | 46.591 | -14.021 | -61.254 | -12.794 | 4.268 | -17.062 | |
| OH- | 2524.759 | 42.407 | -12.869 | -142.332 | -11.201 | 6.455 | -17.656 | |
| T = 473 K (ε = 78.40) | ||||||||
| 14-OH | 20047.920 | 137.927 | -45.192 | -134.917 | -10.704 | 0.012 | -10.716 | |
| 14-NH2 | 20884.456 | 141.853 | -46.212 | -107.621 | -9.717 | 0.200 | -9.917 | |
| 14-NHPh | 24748.689 | 156.029 | -24.120 | -73.683 | -9.060 | -0.157 | -8.903 | |
| 15 | 15544.809 | 116.427 | -39.525 | -81.917 | -11.084 | -0.640 | -10.444 | 95 |
| 16 | 15831.565 | 117.270 | -39.637 | -59.973 | -9.654 | -0.446 | -9.208 | 71 |
| 17 | 20574.229 | 136.912 | -44.185 | -25.753 | -9.800 | -0.461 | -9.339 | 90 |
| CH3CH2OH | 6808.702 | 74.566 | -28.461 | -59.262 | -11.102 | 3.295 | -14.397 | |
| T = 373 K (ε = 25.30) | ||||||||
| R11 | 13085.134 | 123.690 | -33.051 | -228.325 | -11.137 | 0.188 | -11.325 | |
| R12 | 5027.115 | 72.694 | -22.088 | -60.778 | -9.847 | 0.931 | -10.778 | |
| R13 | 5120.630 | 73.307 | -22.223 | -6.014 | -9.697 | -0.457 | -9.240 | |
| 18(a) | 11732.778 | 114.256 | -30.885 | -243.788 | -9.322 | 0.771 | -10.093 | 96 |
| 18E | 12135.123 | 116.007 | -31.136 | -137.535 | -9.974 | -0.335 | -9.639 | |
| 20K | 11753.872 | 113.663 | -30.642 | -147.469 | -10.046 | -0.109 | -9.937 | 92 |
| 19(a) | 11673.276 | 114.631 | -31.084 | -191.810 | -9.579 | -0.376 | -9.203 | 94 |
| 19E | 12750.364 | 120.777 | -32.274 | -79.395 | -9.802 | -0.788 | -9.014 | |
| 21K | 12047.588 | 116.145 | -31.274 | -86.109 | -10.326 | -1.368 | -8.958 | 90 |
| H3O+ | 3004.184 | 47.867 | -14.850 | 62.182 | -15.994 | 1.642 | -17.636 | |
| H2O | 2791.943 | 46.591 | -14.668 | -62.254 | -12.794 | 4.268 | -17.062 | |
| Reaction | δΔG(T)a | pKTb | KTc | δΔG(BH)d | pKae | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| T = 343 K, (ε = 47.24) | ||||||||||
| R1K R1E | 1.917 | 1.221 | 0.060 | - | - | |||||
| R1K R1K(a) | - | - | - | 1.703 | 1.085 | |||||
| R1E R1E(a) | - | - | - | 0.294 | 0.187 | |||||
| R3K R3E1 | 2.202 | 1.403 | 0.040 | - | - | |||||
| R3K R3E2 | 2.114 | 1.347 | 0.045 | - | - | |||||
| R3K R3K(a) | - | - | - | 1.472 | 0.938 | |||||
| R3E1 R3E1(a) | - | - | - | -0.755 | -0.481 | |||||
| R3E2 R3E2(a) | - | - | - | -1.109 | -0.706 | |||||
| R4K R4E | 2.734 | 1.742 | 0.018 | - | - | |||||
| R4K R4K(a) | - | - | - | 2.460 | 1.567 | |||||
| R4E R4E(a) | - | - | - | -0.466 | -0.297 | |||||
| R5K R5E1 | 1.599 | 1.019 | 0.096 | - | - | |||||
| R5K R5E2 | 1.537 | 0.979 | 0.105 | - | - | |||||
| R5K R5K(a) | - | - | - | -2.143 | -1.365 | |||||
| R5E1 R5E1(a) | - | - | - | -1.241 | -0.791 | |||||
| R5E2 R5E2(a) | - | - | - | -3.657 | -2.330 | |||||
| R6K R6E | 2.543 | 1.620 | 0.024 | - | - | |||||
| R6K R6K(a) | - | - | - | 3.167 | 2.017 | |||||
| R6E R6E(a) | - | - | - | 0.590 | 0.376 | |||||
| T = 363 K, (ε = 78.40) | ||||||||||
| R7K R7E | 3.833 | 2.307 | 4.932E-3 | - | - | |||||
| R7K R7K(a) | - | - | - | 3.646 | 2.194 | |||||
| R7E R7E(a) | - | - | - | -13.081 | -7.875 | |||||
| T = 373 K, (ε = 25.30) | ||||||||||
| 20K 18E | -0.494 | -0.298 | 1.986 | - | - | |||||
| 21K 19E | -0.955 | -0.560 | 3.631 | - | - | |||||
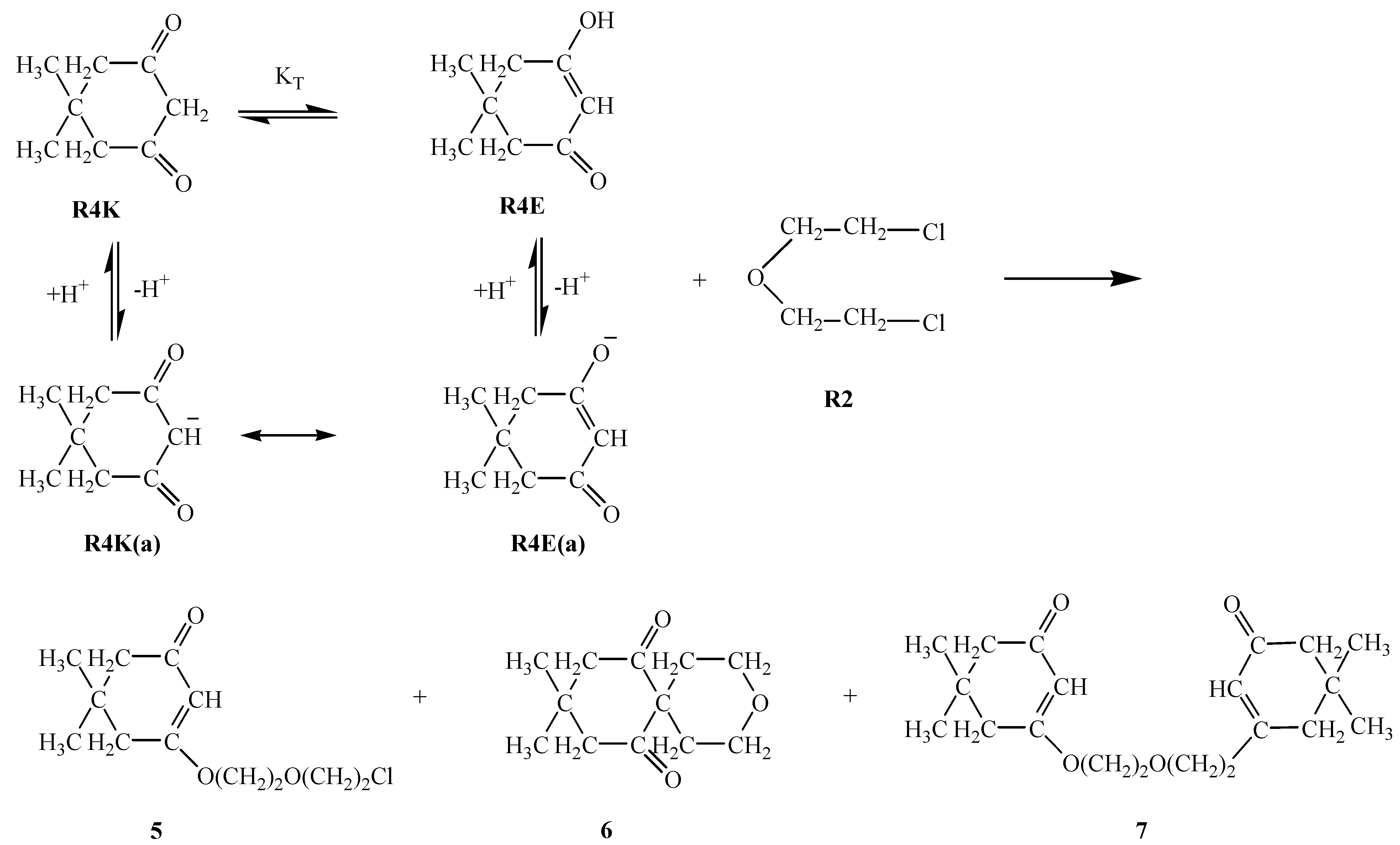
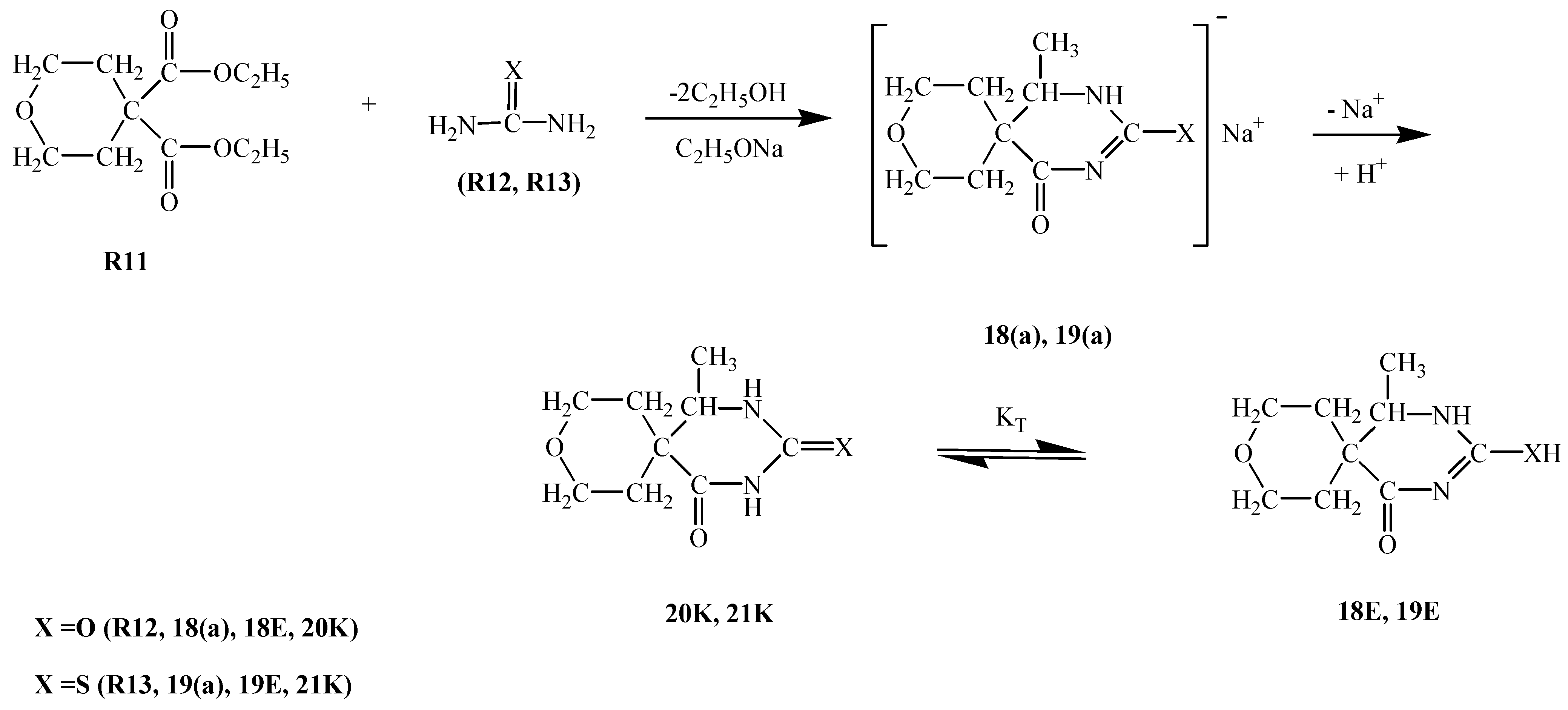
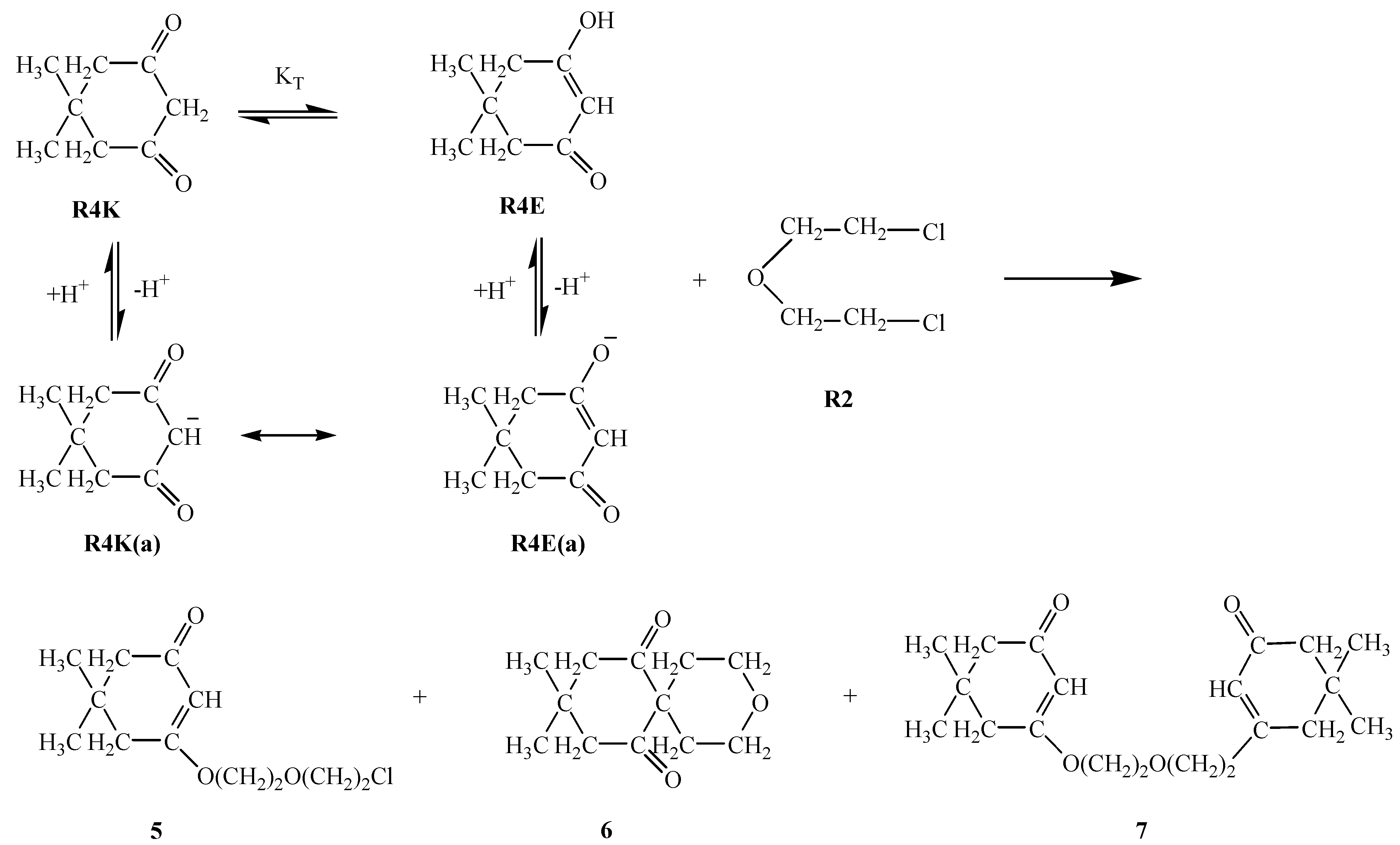
R3E1 and R3K R3E2 equilibria, respectively, suggest the predominance of the keto form (i.e. R3K) in aqueous media, it appears that in basic media a competition among two enolate ions and one carbene ion becomes inevitable. Although the respective nucleophilicity values are ranked in the increasing order R3E1(a) < R3K(a) < R3E2(a), the magnitudes of the differences are not too large (Table 1). The same analogy exists within the pKa values: the basicity increases (or acidity decreases) in the order R3K(a) < R3E1(a) < R3E2(a) and again the magnitudes of the differences might allow for competition. It seems that in this case the competition was won by the R3E1(a) enolate ion which attacks R2 to form compound 3 in 57 % yield. An attack of the second R3E1(a) enolate ion on compound 3 then afforded compound 4 in 16 % yield.R4E equilibrium suggests the predominance of the keto form (i.e. R4K) in aqueous media, but again in basic media it would seem that a competition exists between the enolate ion R4E(a), formed by deprotonation of the enol form R4E, and the carbene ion R4K(a), which forms by deprotonation of the keto form R4K. Since the yield of compound 5 (46%) is the highest, that implies that the R4E(a) enolate ion attacks R2 to form compound this compound. A further attack of enolate R4E(a) ion on compound 5 affords compound 7 in 12 % yield. Alternatively, when carbene ion R4K(a) attacks R2 then compound 6 is formed (in 24 % yield) by an intramolecular ring closure reaction as follows:
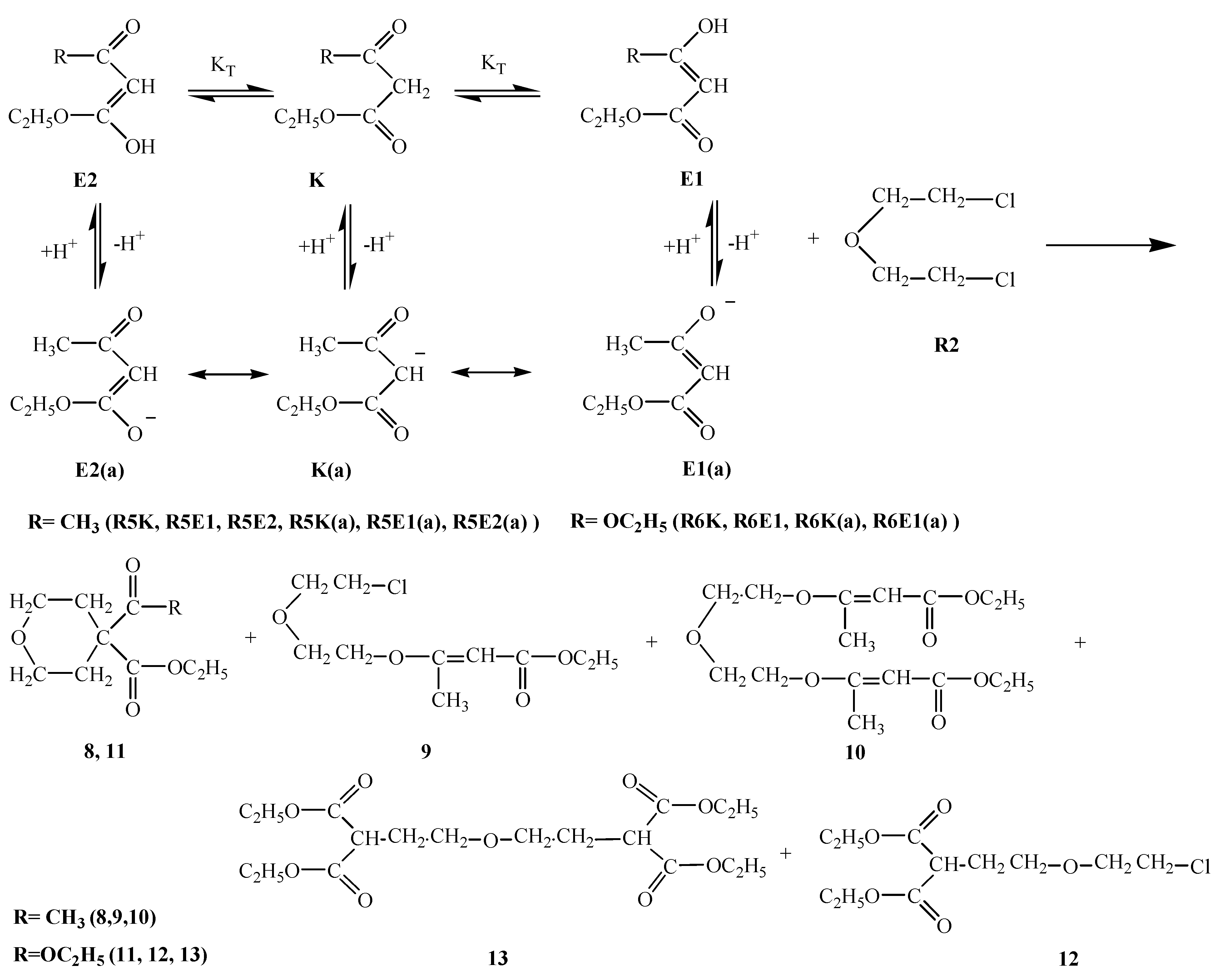
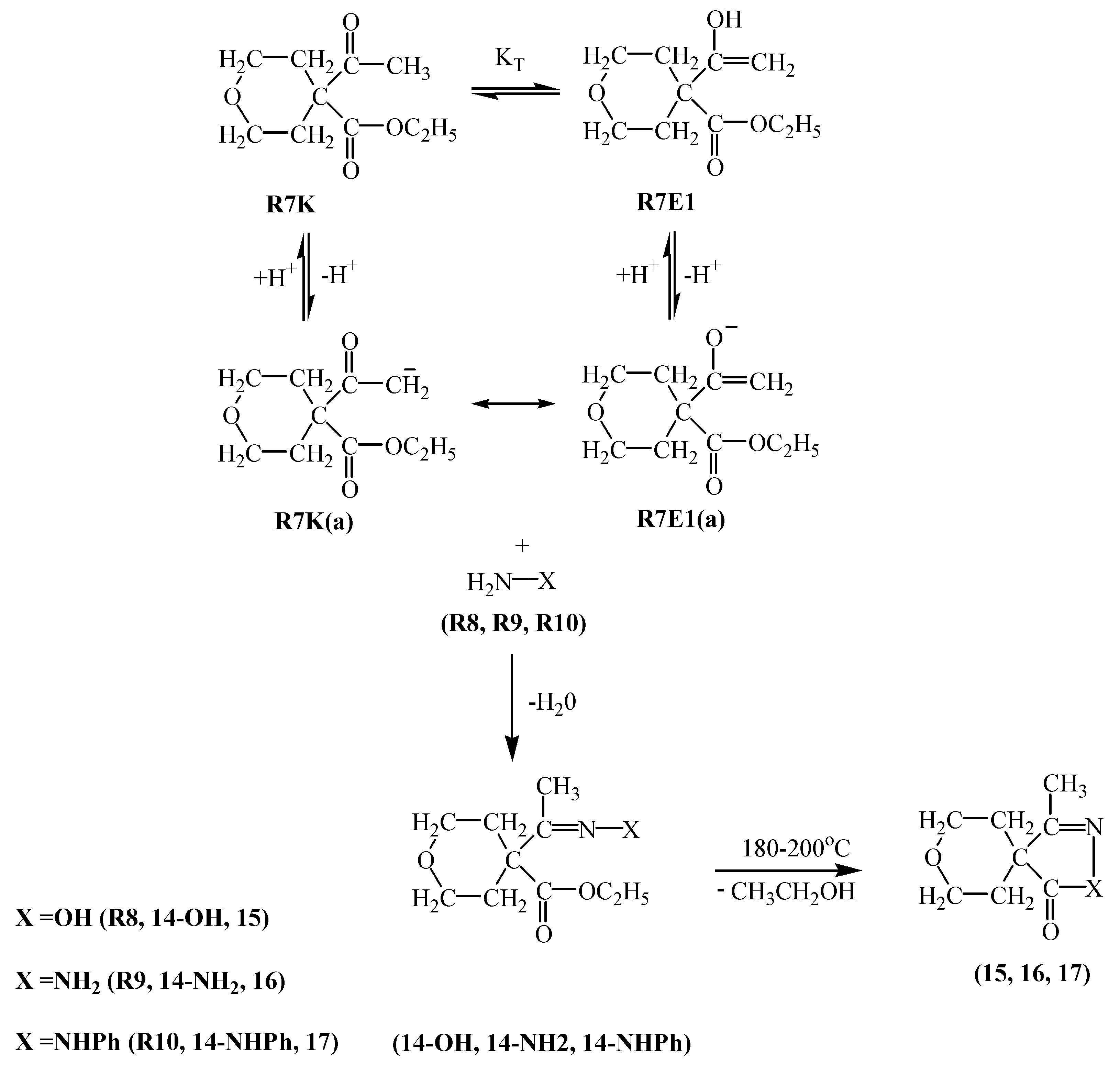
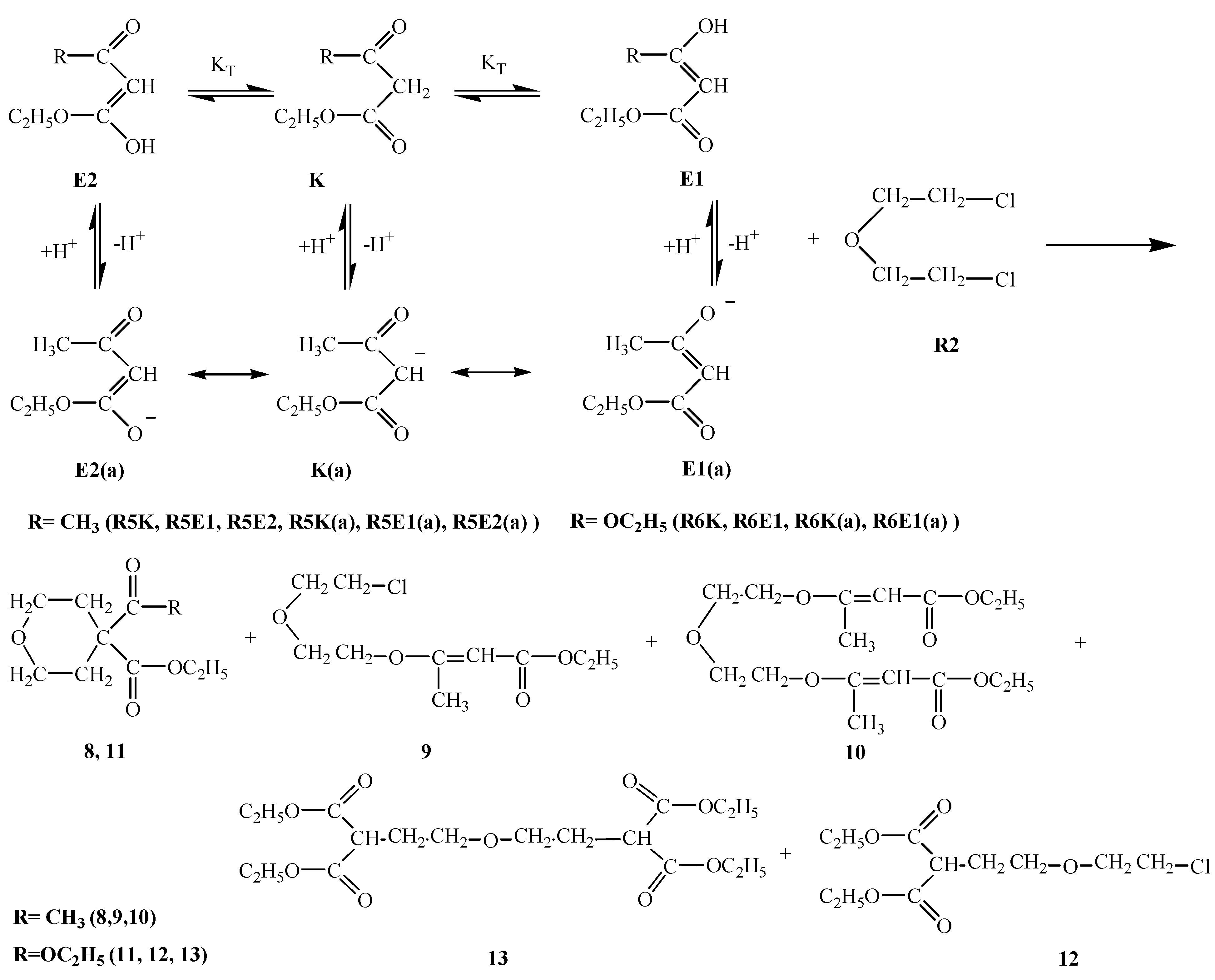
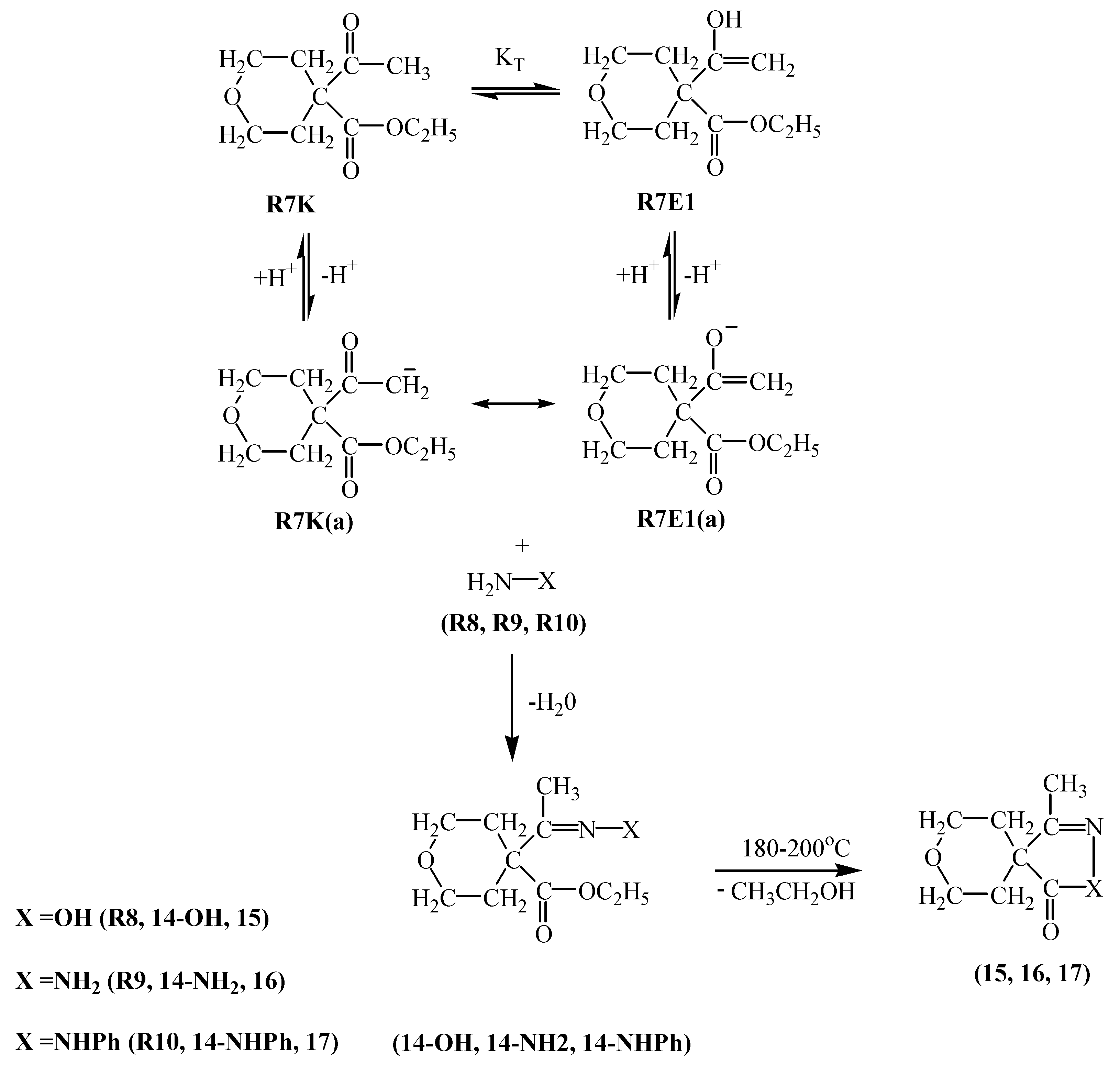
 R5E1 and R5K R5E2 equilibria indicate the favorability of the keto form R5K over the enol forms R5E1 and R5E2 (Table 2). However, in basic media there seems to be a competition among the enolate ions and carbene ion. When we consider the higher yield of compound 8 it seems that this time the enolate ion R5K(a) is favored and this ion attacked R2 to afford compound 8 in 55 % yield. A competitive reaction is the attack of enolate ion R5E1(a) on R2 to afford compound 9 in a yield of 49 %. Attack of the same enolate ion R5E1(a) on compound 9 affords the molecule 10 in 10 % yield. The nucleophilicity of those three species were found to be in the increasing order: R5E1(a) < R5K(a) < R5E2(a), which accounts for the higher yield of the R5E2(a) enolate initiated reaction giving compounds 9 and 10 (total yield is 59 %) (Table 1). The basicity order is found be be in the increasing order R5E1(a) < R5K < R5E2(a) (Table 2). The higher basicity (or lower acidity) of R5E2(a) is further evidence for the higher yield of compounds 9 and 10.R6E equilibrium suggests the ketone form R6K is favored (Table 2). When we take into account the percent yield and the structures of the products 11-13 it seems that only the carbene ion R6K(a), formed by deprotonation of R6K in basic media, acts as nucleophile to attack R2 and give compound 12 in 10 % yield and a subsequent intramolecular rearrangement of compound 12 in basic media produces compound 11 in a 57 % yield. Alternatively, attack of the carbene ion on 12 produces compound 13 in 14 % yield. The nucleophilicities of enolate and carbene ions are almost the same (Table 1) but the basicity of the carbene ion is greater than that of the enolate ions (Table 2) which explains why the enolate ion is inactive in this reaction. For the formation of compounds 14-OH, 14-NH2 and 14-NHPh (Scheme 4) the KT value of 0.01 for the R7K R7E equilibrium suggests the keto form R7K is favored (Table 2). It seems that the formation of compounds 14-OH, 14-NH2 and 14-NHPh occurs by nucleophilic attack of R8, R9 and R10 on R7K, which is more electropositive compared to R7E. These products were found to produced in about 91 % yield. These products rearrange into compounds 15, 16 and 17 respectively. The overall mechanism can be summarized as follows:
R5E1 and R5K R5E2 equilibria indicate the favorability of the keto form R5K over the enol forms R5E1 and R5E2 (Table 2). However, in basic media there seems to be a competition among the enolate ions and carbene ion. When we consider the higher yield of compound 8 it seems that this time the enolate ion R5K(a) is favored and this ion attacked R2 to afford compound 8 in 55 % yield. A competitive reaction is the attack of enolate ion R5E1(a) on R2 to afford compound 9 in a yield of 49 %. Attack of the same enolate ion R5E1(a) on compound 9 affords the molecule 10 in 10 % yield. The nucleophilicity of those three species were found to be in the increasing order: R5E1(a) < R5K(a) < R5E2(a), which accounts for the higher yield of the R5E2(a) enolate initiated reaction giving compounds 9 and 10 (total yield is 59 %) (Table 1). The basicity order is found be be in the increasing order R5E1(a) < R5K < R5E2(a) (Table 2). The higher basicity (or lower acidity) of R5E2(a) is further evidence for the higher yield of compounds 9 and 10.R6E equilibrium suggests the ketone form R6K is favored (Table 2). When we take into account the percent yield and the structures of the products 11-13 it seems that only the carbene ion R6K(a), formed by deprotonation of R6K in basic media, acts as nucleophile to attack R2 and give compound 12 in 10 % yield and a subsequent intramolecular rearrangement of compound 12 in basic media produces compound 11 in a 57 % yield. Alternatively, attack of the carbene ion on 12 produces compound 13 in 14 % yield. The nucleophilicities of enolate and carbene ions are almost the same (Table 1) but the basicity of the carbene ion is greater than that of the enolate ions (Table 2) which explains why the enolate ion is inactive in this reaction. For the formation of compounds 14-OH, 14-NH2 and 14-NHPh (Scheme 4) the KT value of 0.01 for the R7K R7E equilibrium suggests the keto form R7K is favored (Table 2). It seems that the formation of compounds 14-OH, 14-NH2 and 14-NHPh occurs by nucleophilic attack of R8, R9 and R10 on R7K, which is more electropositive compared to R7E. These products were found to produced in about 91 % yield. These products rearrange into compounds 15, 16 and 17 respectively. The overall mechanism can be summarized as follows:

Conclusions
Experimental
General
Syntheses; general method for the alkylation reactions of β-carbonyl derivatives with 2-chloro-1-(2-chloroethoxy)ethane
 :1.1258;
:1.1258;  : 1.4964; 1H-NMR (CCl4): δ(ppm) = 1.94 (s, 3H), 2.12 (s, 3H), 3.37-3.87 (m, 8H), 5.36 (s, 1H); Anal. Calc. for C9H15ClO3: C, 52.30; H, 7.26; Cl, 17.19 %, found: C, 52.33; H, 7.28; Cl, 17.17 %.:1.4997; :1.0935; 1H-NMR (CCl4): δ (ppm) = 1.96 (s, 6H), 2.11 (s, 6H), 3.73 (m, 8H), 5.36 (s, 2H); Anal. Calc. for C14H22O5: C, 62.61; H, 8.17 %, found: C, 62.22; H, 8.15 %.:1.5305; :1.1722; 1H-NMR (CCl4): δ (ppm) = 1.92 (s, 3H), 3.39-3.88 (m, 8H), 7.36-7.96 (m, 5H); Anal. Calc. for C14H17ClO3: C, 62.57; H, 6.33; Cl, 13.22 %, found: C, 62.55; H, 6.32; Cl, 13.24 %.:1.5305; :1.1580; 1H-NMR (CCl4): δ (ppm) = 1.95 (s, 6H), 7.34-7.97 (m, 10H); Anal. Calc. for C24H26O5: C, 73.10; H, 6.60 %, found: C, 73.12; H, 6.58 %.:1.5130; :1.2554; 1H-NMR (CCl4): δ (ppm) = 0.98 (s, 6H), 2.00 (s, 2H), 3.85 (t, 2H), 5.15 (s, 1H); Anal. Calc. for C12H19ClO3: C, 58.42; H, 7.71; Cl, 14.40 %. Found: C, 58.41; H, 7.33; Cl, 14.38 %.:1.4648; :1.1065; 1H-NMR (CCl4): δ (ppm) = 1.25 (s, 3H), 2.06 (m, 4H), 4.12 (q, 2H); Anal. Calc. for C10H16O4: C, 60.00; H, 8.00 %, found: C, 60.07; H, 8.05 %.:1.4755; :1.1342; 1H-NMR (CCl4): δ (ppm) = 1.25 (s, 3H), 2.55 (s, 3H), 3.37-4.85 (m, 10H), 4.88 (s, 1H); Anal. Calc. for C10H14ClO4: C, 50.84; H, 7.19; Cl, 15.01, found: C, 50.72; H, 7.21; Cl, 14.99 %.:1.4554; :1.1081; 1H-NMR (CCl4): δ (ppm) = 1.12 (t, 6H), 1.84 (m, 4H), 4.12 (q, 4H); Anal. Calc. for C11H18O5: C, 57.39; H, 7.83, found: C, 57.37; H, 7.81 %.:1.4542; :1.1346; 1H-NMR (CCl4): δ (ppm) = 1.30 (t, 6H), 2.00 (m, 2H), 3.50 (m, 7H), 4.12 (q, 4H); Anal. Calc. for C11H19ClO5: C, 49.53; H, 7.13; Cl, 13.32, found: C, 49.51; H, 7.11; Cl, 13.30 %.:1.4552; :1.1175; 1H-NMR (CCl4): δ (ppm) = 1.12 (t, 12H), 1.87 (m, 4H), 3.36 (m, 6H) 4.00 (q, 8H); Anal. Calc. for C18H30O9: C, 55.39; H, 7.69, found: C, 57.37; H, 7.70 %.
: 1.4964; 1H-NMR (CCl4): δ(ppm) = 1.94 (s, 3H), 2.12 (s, 3H), 3.37-3.87 (m, 8H), 5.36 (s, 1H); Anal. Calc. for C9H15ClO3: C, 52.30; H, 7.26; Cl, 17.19 %, found: C, 52.33; H, 7.28; Cl, 17.17 %.:1.4997; :1.0935; 1H-NMR (CCl4): δ (ppm) = 1.96 (s, 6H), 2.11 (s, 6H), 3.73 (m, 8H), 5.36 (s, 2H); Anal. Calc. for C14H22O5: C, 62.61; H, 8.17 %, found: C, 62.22; H, 8.15 %.:1.5305; :1.1722; 1H-NMR (CCl4): δ (ppm) = 1.92 (s, 3H), 3.39-3.88 (m, 8H), 7.36-7.96 (m, 5H); Anal. Calc. for C14H17ClO3: C, 62.57; H, 6.33; Cl, 13.22 %, found: C, 62.55; H, 6.32; Cl, 13.24 %.:1.5305; :1.1580; 1H-NMR (CCl4): δ (ppm) = 1.95 (s, 6H), 7.34-7.97 (m, 10H); Anal. Calc. for C24H26O5: C, 73.10; H, 6.60 %, found: C, 73.12; H, 6.58 %.:1.5130; :1.2554; 1H-NMR (CCl4): δ (ppm) = 0.98 (s, 6H), 2.00 (s, 2H), 3.85 (t, 2H), 5.15 (s, 1H); Anal. Calc. for C12H19ClO3: C, 58.42; H, 7.71; Cl, 14.40 %. Found: C, 58.41; H, 7.33; Cl, 14.38 %.:1.4648; :1.1065; 1H-NMR (CCl4): δ (ppm) = 1.25 (s, 3H), 2.06 (m, 4H), 4.12 (q, 2H); Anal. Calc. for C10H16O4: C, 60.00; H, 8.00 %, found: C, 60.07; H, 8.05 %.:1.4755; :1.1342; 1H-NMR (CCl4): δ (ppm) = 1.25 (s, 3H), 2.55 (s, 3H), 3.37-4.85 (m, 10H), 4.88 (s, 1H); Anal. Calc. for C10H14ClO4: C, 50.84; H, 7.19; Cl, 15.01, found: C, 50.72; H, 7.21; Cl, 14.99 %.:1.4554; :1.1081; 1H-NMR (CCl4): δ (ppm) = 1.12 (t, 6H), 1.84 (m, 4H), 4.12 (q, 4H); Anal. Calc. for C11H18O5: C, 57.39; H, 7.83, found: C, 57.37; H, 7.81 %.:1.4542; :1.1346; 1H-NMR (CCl4): δ (ppm) = 1.30 (t, 6H), 2.00 (m, 2H), 3.50 (m, 7H), 4.12 (q, 4H); Anal. Calc. for C11H19ClO5: C, 49.53; H, 7.13; Cl, 13.32, found: C, 49.51; H, 7.11; Cl, 13.30 %.:1.4552; :1.1175; 1H-NMR (CCl4): δ (ppm) = 1.12 (t, 12H), 1.87 (m, 4H), 3.36 (m, 6H) 4.00 (q, 8H); Anal. Calc. for C18H30O9: C, 55.39; H, 7.69, found: C, 57.37; H, 7.70 %.Synthesis of 1,2-azolones
General method for the preparation of barbituric acids.
Computational Details
References
- Zefirov, N. S.; Kuznetsova, T. S.; Kozhushkov, S. I.; Surmina, L. S.; Rashchupkina, Z. A. Cycloalcylation by the α,ω-Dibromides of Compounds Containing an Activated Methylene Group as a Method for the Synthesis of 1,1-Disubstitutedcycloalkanes. J. Org. Chem. USSR 1983, 19, 474–480. [Google Scholar]
- Zefirov, N. S.; Kuznetsova, T. S.; Kozhushkov, S. I. Alkylation of Cyclic 1,3-Diketones by α,ω-Dibromides. J. Org. Chem. USSR 1983, 19, 1412–1415. [Google Scholar]
- Akhmedov, S. T.; Sadykov, N. S.; Ismailov, V. M.; Akhundova, M. A.; Mamedov, M. M.; Kozhushkov, S. I.; Zefirov, N. S. Synthesis of New Pyrazole and Isoxazole Derivatives Based on Products of Dondensation of β-Dicarbonyl Compounds with 1,2,3-Trihalopropanes. Chem. Heterocycl. Comp. 1987, 23, 651–654. [Google Scholar]
- Zefirov, N. S.; Kozhushkov, S. I.; Kuznetsova, T. S.; Lukin, K. A.; Kazimirchik, I. V. Vinylspiropentane. J. Org. Chem. USSR 1988, 24, 605–610. [Google Scholar]
- Zefirov, N. S.; Lukin, K. A.; Kozhushkov, S. I.; Kuznetsova, T. S.; Domarev, A. M.; Sosonkin, I. M. Synthesis of Spirofused Cyclopropanes. J. Org. Chem. USSR 1989, 25, 278–284. [Google Scholar]
- Yufit, D. S.; Lukin, K. A.; Kozhushkov, S. I.; Struchkov, Yu. T.; Zefirov, N. S. Crystal and Molecular Structure of 1,1-Dichlorotetraspiro[2.0.0.2.0.2.0.1]-Undecane. Dokl. Chem. (Engl. Transl.) 1991, 320, 288–292. [Google Scholar]
- Lukin, K. A.; Masunova, A. Yu.; Kozhushkov, S. I.; Kuznetsova, T. S.; Urgak, B. I.; Piven’, V. A.; Zefirov, N. S. Reaction of Allenes with 1,1-Dichloroethane and Butyllithium. J. Org. Chem. USSR 1991, 27, 422–425. [Google Scholar]
- de Meijere, A.; Kozhushkov, S.; Puls, C.; Haumann, T.; Boese, R.; Cooney, M. J.; Scott, L. T. Hexaspiro[2.4.2.4.2.4.2.4.2.4.2.4]Dotetraconta-4,6,11,13,18,20,25,27,32,34,39,41-Dodecain - ein Explodierendes [6]Rotan. Angew. Chem. Int. Ed. Engl. 1994, 33, 869–871. [Google Scholar]
- de Meijere, A.; Becker, H.; Kozhushkov, S. I.; Noltemeyer, M. Intramolecular Pauson-Khand Reactions of Methylenecyclopropane and Bicyclopropylidene Derivatives as a Synthetic Approach to Spiro(Cyclopropanbicyclo-[N.3.0]Alkenones. Eur. J. Org. Chem. 2004. to be submitted. [Google Scholar]
- Kozhushkov, S. I.; Späth, T.; Kosa, M.; Apeloig, Y.; Yufit, D. S.; de Meijere, A. Relative Stabilities of Spirocyclopropanated Cyclopropyl Cations. Eur. J. Org. Chem. 2003, 4234–4242. [Google Scholar]
- Markó, I. E.; Vanherck, J.-C.; Ates, A.; Tinant, B.; Declercq, J.-P. Efficient and Convergent Stereocontrolled Spiroannulation of Ketones. Tetrahedron Lett. 2003, 44, 3333–3336. [Google Scholar]
- Frank, D.; Kozhushkov, S. I.; Labahn, T.; de Meijere, A. Striving for Unusually Strained Oxiranes: Epoxidation of Spirocyclopropanated Methylenecyclopropanes. Tetrahedron 2002, 58, 7007–7007. [Google Scholar]
- Piacente, S.; Bifulco, G.; Pizza, C.; Stochmal, A.; Oleszek, W. A novel Phenolic Spiro Derivative. Tetrahedron Lett. 2002, 43, 9133–9136. [Google Scholar]
- Keglevich, G.; Forintos, H.; Keserű, G. M.; Hegedűs, L.; Tőke, L. Synthesis of the Spiro Derivatives of 1,2-Oxaphosphetes by [2+2] Cycloaddition of Cyclic 1-(2,4,6-Triisopropylphenyl)Phosphine Oxides with Dimethyl Acetylenedicarboxylate. Tetrahedron 2000, 56, 4823–4828. [Google Scholar]
- Mérour, J.-Y.; Mamai, A.; Malapel, B.; Gadonneix, P. Heterodiene Cycloadditions: Synthesis and Oxidation of Pyrano[3,2,b]Indoles into Spiro Derivatives. Tetrahedron 1997, 53, 987–1002. [Google Scholar]
- Gauvin-Hussenet, C.; Séraphin, D.; Cartier, D.; Laronze, J.-Y. ; Lévy, J. Approaches Towards Indolic Analogues of Cephalotaxine Via a Spiro-Cyclohexene Strategy. Tetrahedron Lett. 1993, 34, 465–468. [Google Scholar]
- Hossain, N.; Papchikhin, A.; Plavec, J.; Chattopadhyaya, J. Synthesis of 2′- and 3′-Spiro-isoxazolidine Derivatives of Thymidine & Their Conversions to 2′,3′-Dideoxy-2′,3′-Didehydro-3′-C-Substituted Nucleosides by Radical Promoted Fragmentation. Tetrahedron 1993, 49, 10133–10156. [Google Scholar]
- Desmaële, D.; d'Angelo, J. Enantioselective Synthesis of Oxa-Spiro Compounds. Tetrahedron Lett. 1989, 30, 345–348. [Google Scholar]
- Haack, R. A.; Beck, K, R. Synthesis of Substituted Spiro[4.5]deca-3,6,9-Triene-2,8-Diones: an Expeditious Route to the Spiro[4.5]Decane Terpene Skeleton. Tetrahedron Lett. 1989, 30, 1605–1608. [Google Scholar]
- Cordero, F. M.; Anichini, B.; Goti, A; Brandi, A. Rearrangement of Isoxazoline-5-spiro Derivatives. Part 4. Synthesis of Medium Size Benzofused Azaheterocycles. Tetrahedron 1989, 45, 5917–5924. [Google Scholar]
- Arenal, I.; Bernabé, M.; Cuevas, O.; Alvarez, E. F. Reaction of 5(4h)-Thiazolones with Diazomethane. Tetrahedron 1983, 39, 1387–1393. [Google Scholar]
- Chan, L.; Matlin, S. A. New Spiro Derivatives of Penicillin. Tetrahedron Lett. 1981, 22, 4025–4028. [Google Scholar]
- Belikov, V.G. Farmatsevticheskaia Khimiia; Uchebnik. V 2-kh chastiakh. Chast' 1: Obshchaia Farmatsevticheskaia Khimiia; Vysshaia Shkola Second Edition; Moskva, 1993; p. 432. [Google Scholar]
- Stewart, J.J.P. MOPAC 7.0, Quantum Chemistry Program Exchange; University of Indiana: Bloomington, IN, USA.
- CS ChemOffice; Cambridge Scientific Computing Inc.: Cambridge, MA, USA.
- Sample Availability: Contact the authors.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sadikov, N.; Nasibov, S.; Ogretir, C.; Berber, H.; Hüseyinli, A. Quantum Chemical and Experimental Studies on the Mechanism of Alkylation of β-Dicarbonyl Compounds. The Synthesis of Five and Six Membered Heterocyclic Spiro Derivatives. Molecules 2004, 9, 922-938. https://doi.org/10.3390/91100922
Sadikov N, Nasibov S, Ogretir C, Berber H, Hüseyinli A. Quantum Chemical and Experimental Studies on the Mechanism of Alkylation of β-Dicarbonyl Compounds. The Synthesis of Five and Six Membered Heterocyclic Spiro Derivatives. Molecules. 2004; 9(11):922-938. https://doi.org/10.3390/91100922
Chicago/Turabian StyleSadikov, Nurettin, Sahin Nasibov, Cemil Ogretir, Halil Berber, and Ali Hüseyinli. 2004. "Quantum Chemical and Experimental Studies on the Mechanism of Alkylation of β-Dicarbonyl Compounds. The Synthesis of Five and Six Membered Heterocyclic Spiro Derivatives" Molecules 9, no. 11: 922-938. https://doi.org/10.3390/91100922