Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum

1
State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau 999078, China
2
Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization, Jiangsu Key Laboratory for High Technology Research of TCM Formulae, and National and Local Collaborative Engineering Center of Chinese Medicinal Resources Industrialization and Formulae Innovative Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China
*
Authors to whom correspondence should be addressed.
Molecules 2017, 22(7), 1073; https://doi.org/10.3390/molecules22071073
Submission received: 2 June 2017
/
Revised: 21 June 2017
/
Accepted: 22 June 2017
/
Published: 28 June 2017
(This article belongs to the Section Natural Products Chemistry)
Abstract
:Ganoderma resinaceum is usually used as ethnomedicine for immune-regulation, hyperglycemia, and liver disease. To date, only a few chemical constituents have been reported from G. resinaceum. In this study, fifteen nortriterpenoids including six new nortriterpenoids (1–6) and nine known analogs (7–15), were separated and purified from the fruiting bodies of G. resinaceum. New compounds were identified as lucidone I (1), lucidone J (2), lucidone K (3), lucidone I (4), ganosineniol B (5), and ganosineniol C (6), based on analysis of extensive spectroscopic data (high resolution mass spectrometry (HRMS), nuclear magnetic resonance (NMR), infrared (IR), and ultraviolet (UV)). The known compounds were assigned as lucidone A (7), lucidone B (8), lucidone H (9), lucidone E (10), lucidone F (11), lucidone D (12), lucidone C (13), ganoderense F (14), and ganosineniol A (15), by comparing their spectroscopic data with those reported in the literature. Compounds 3, 4, and 7–13 were examined for α-glucosidase inhibitory activity and display no significant activity, but the finding may support that the side chain of ganoderma triterpenoids played an important role in α-glucosidase inhibitory activity.
1. Introduction
Nortriterpenoids, derived from lanostane-type triterpenoids due to degradation of side chain, are a class of secondary metabolites in Ganoderma [1,2]. Although substantial triterpenoids have been reported from Ganoderma, nortriterpenoid is rare. The common nortriterpenoids possess 24 or 27 carbons skeleton in Ganoderma, for example, lucidones A–H, and lucidenic acids A–N. However, two novel nortriterpenoids, methyl ganosinensate A and ganosinensic acid A, contain an unusual four-membered ring skeleton produced by a bond across C-1 to C-11 [3]. Nortriterpenoids isolated from Ganoderma showed a wide range of biological activities, such as anti-tumor [4,5,6], anti-inflammatory [7], neurotrophic [8], hepatoprotective [9], and anti-HIV-1 protease activities [10]. Hence, nortriterpenoids deserve our close attention due to its structure diversity and good model for the pharmaceutical field.
As a member of the genus Ganoderma, G. resinaceum has been used for immune-regulation, hyperglycemia, and liver disease [11]. The extract of G. resinaceum exhibited antimicrobial, antioxidant, and inhibitory activities against acetyl cholinesterase, tyrosinase, α-amylase and α-glucosidase [12]. At present, only 17 triterpenoids had been obtained from the fruiting bodies of G. resinaceum, whose biological properties included cytotoxicity [13] and hepatoprotective activities [9]. Obviously, the chemical constituents isolated from G. resinaceum and their biological activities have not been thoroughly characterized yet. Aiming to elucidate bioactive constituents from G. resinaceum, our team carried out phytochemical investigation on G. resinaceum. In this study, six new compounds (1–6) and nine known compounds (7–15) (Figure 1) were obtained from the fruiting bodies of G. resinaceum. The structures of the new compounds were elucidated by extensive spectroscopic data (HRMS, NMR, IR, and UV). The known compounds were identified by comparison of MS and 1 D NMR spectroscopic data with those reported in the literature. Compounds 3, 4, and 7–15 were evaluated for inhibitory activity against α-glucosidase and display no significant activity, however this finding may support that the side chain of ganoderma triterpenoids is critical for α-glucosidase inhibitory activity.
2. Results and Discussion
The 95% ethanol extract of G. resinaceum was partitioned with petroleum ether, EtOAc, and n-BuOH, successively. The triterpene-containing EtOAc and n-BuOH fractions were repeatedly purified by column chromatography over silica gel, MCI gel, ODS gel, Sephadex LH-20, and preparative HPLC to afford 20 nortriterpenoids, including six new compounds (1–6) and nine known compounds (7–15). By comparison of spectroscopic data with those reported in literature, the known compounds were identified as lucidone A (7) [14], lucidone B (8) [14,15], lucidone H (9) [16], lucidone E (10) [9], lucidone F (11) [9], lucidone D (12) [9], lucidone C (13) [15], ganoderense F (14) [17], and ganosineniol A (15) [18]. Structural elucidation of new compounds was as follows.
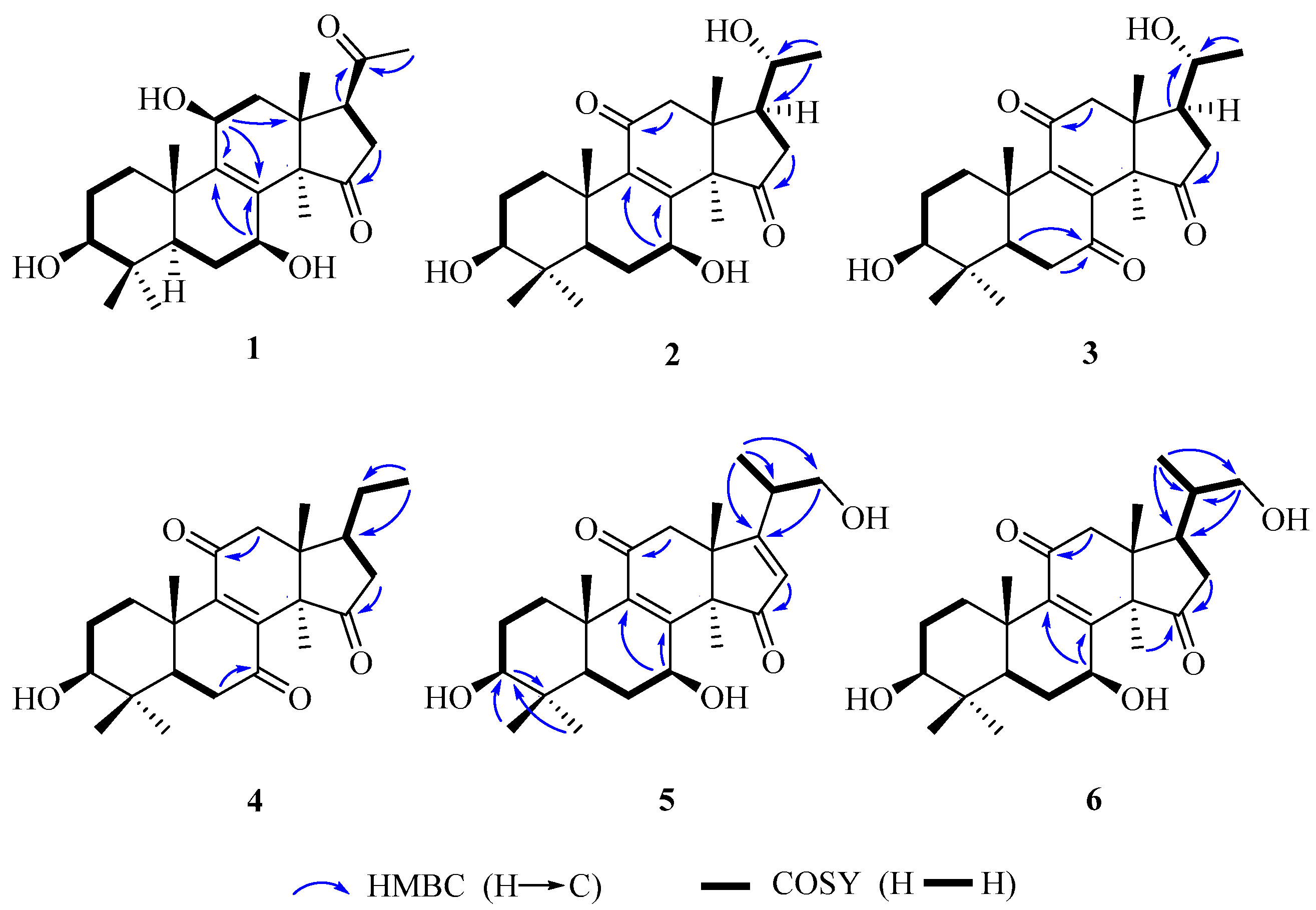
Compound 1 was obtained as white amorphous powder. Its molecular formula was established as C24H36O5 by electrospray ionization (ESI)-HRMS at m/z 403.2484 [M − H]− (calcd. for C24H35O5, 403.2484). Its IR spectrum showed the presence of hydroxy group (3429 cm−1) and carbonyl group (1717 cm−1). 1H-NMR spectrum showed the presences of six singlet methyl signals [δH 0.77, 0.79, 0.90, 1.08, 1.21, and 2.13 (each 3H, s)]. Heteronuclear single quantum coherence (HSQC) and 13C-NMR spectra displayed 24 carbon resonances, including six methyls, five methylenes, five methines including three oxygenated carbon resonances at δC 79.5, 66.9 and 65.3, and eight quaternary carbons including a tetrasubstituted olefinic carbon δC 136.8 and 146.2, and two ketone carbons δC 220.2 and 209.3. The above spectroscopic data suggested 1 to be nortriterpenoid similar to lucidone A (7). However, a comparison of their 13C-NMR data showed that 1 possesses an additional oxygenated methine at δC 65.3 but absent of one ketone group signal at δC 196.7 in 7. Moreover, the chemical sifts of tetrasubstituted olefinic carbons of 1 in 13C-NMR spectrum were significantly different from those of 7 due to the disappearance of α,β-unsaturated ketone conjugated system at C-8/C-9/C-11. Accordingly, it was assumed that 1 was the 11-OH derivative of 7. The 11-OH was further confirmed on the basis of the heteronuclear multiple bond correlation (HMBC) correlations from H-11 (δH 4.48) to C-8 (δH 136.8), C-9 (δC 146.2) and C-13 (δC 43.8), and H-12β (δH 2.00) to C-11 (δH 65.3), and the hydrogen-hydrogen correlation spectroscopy (1H–1H COSY) correlation of H-11 (δH 4.48) with H-12α (δH 2.51). Finally, the planar structure of 1 was established by 1D and 2D NMR spectra.
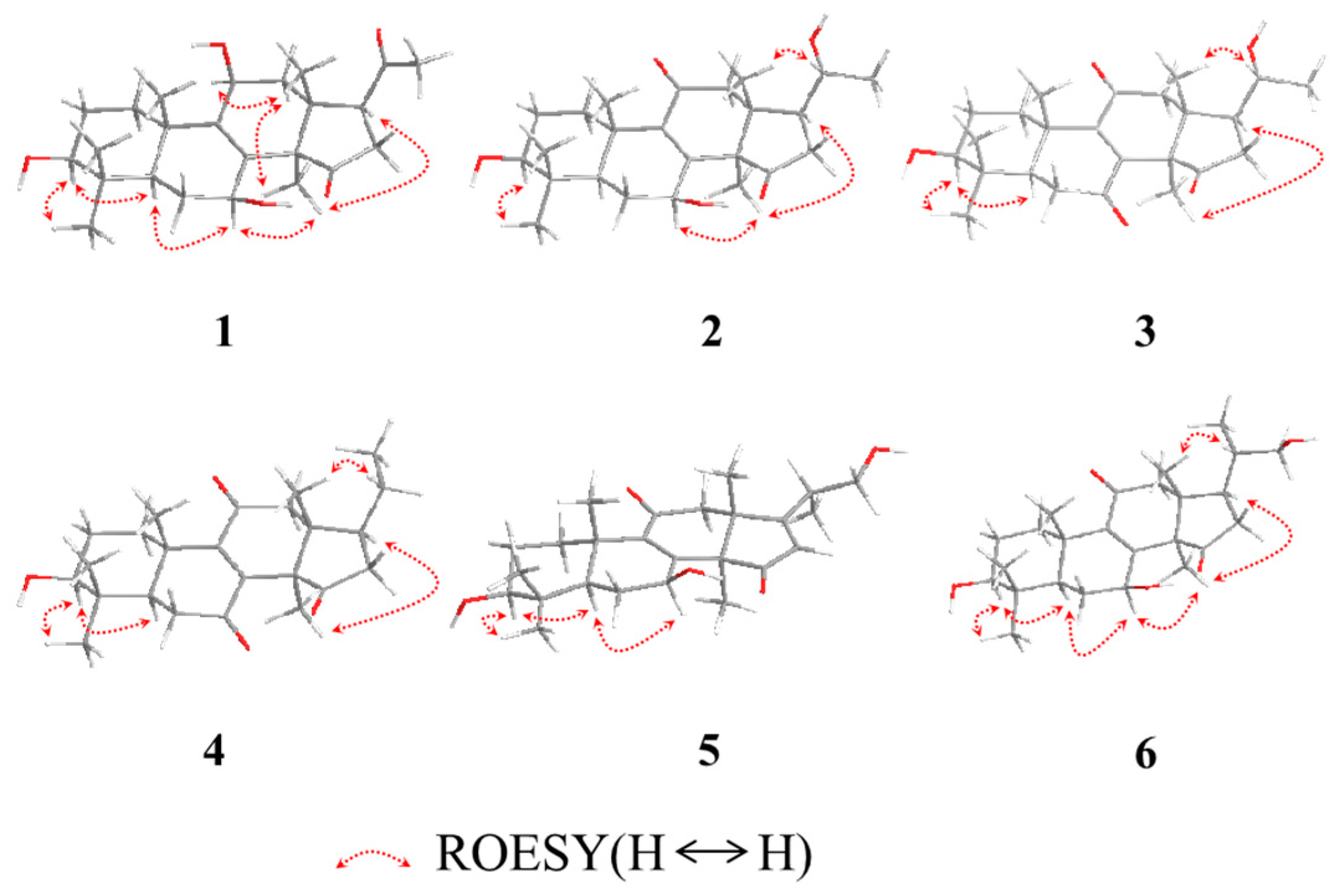
The relative configuration of 1 was established by analyses of rotating-frame nuclear overhauser effect correlation spectroscopy (ROESY) experiment and coupling constant. The ROESY correlations (Figure 2) of H-30 (δH 1.08) with H-12α (δH 2.51) and H-7 (δH 4.54), H-12α with H-11 (δH 4.48), and H-3 (δH 3.08) with H-28 (δH 0.90) and H-5 (δH 0.93) suggested 3-, 7- and 11-OH to be β-orientation, which were also supported by the larger coupling constant (Table 1). Thus, compound 1 was elucidated as 3β,7β,11β-trihydroxy-4,4,14α-trimethyl-15,20-dioxo-5α-pregn-8-ene, named lucidone I.
Compound 2 was isolated as white powder. Its molecular formula was determined to be C24H35O5 due to HRESIMS at m/z 403.2481 [M − H]− (calcd. for C24H35O5, 403.2484). Detail analyses of 13C-NMR (Table 1) and HSQC spectra of 2 showed that the structure of 2 was highly similar as that of 7, except for the presence of an additional oxymethine and the absence of one ketone group in 2. Moreover, 1H-NMR spectrum exhibited that 2 had one doublet methyl signal, whereas 7 did not, suggesting the hydroxy group located at C-20 in 2, which was supported by the HMBC correlations from H-21 (δH 1.20) to C-20 (δC 69.9) and C-17 (δC 48.9), as well as the 1H–1H COSY correlations of H-20 (δH 3.79) with H-21 (δH 1.20) and H-17 (δH 2.28). The 3- and 7-hydroxy groups were assigned as β- and β-orientation, respectively, on the basis of ROESY correlations of H-3 with H-28 and H-5, H-7 with H-5 and H-30. The ROESY correlations of H-17 with H-30, and H-20 with H-18 (Figure 3) assigned the relative configuration of hydroxy attached to C-20 as α-orientation. Consequently, compound 2 was established as 3β,7β,20α-trihydroxy-4,4,14α-trimethyl-11,15-dioxo-5α-pregn-8-ene, named lucidone J.
Compound 3 was obtained as pale yellow powder. It had the molecular formula of C24H34O5 deduced from HRESIMS at m/z 401.2352 [M − H]− (calcd. for C24H33O5, 401.2333). The IR and UV spectra revealed the presence of hydroxy group (3484 cm−1), carbonyl group (1722 cm−1) and α,β-unsaturated carbonyl group (1689 cm−1 and 259 nm). Comparison of spectroscopic data of 3 with that of 2 displayed their structural similarities except for the presence of ketone group to replace oxygenated methine. The ketone group at C-7 was confirmed by the key HMBC correlations from H-5 (δH 1.50) and H-6 (δH 2.60 and 2.40) to C-7 (δC 202.3). The 20α-OH was confirmed on the basis of ROESY correlations of H-17 (δH 2.28) with H-30 (δH 1.52), and H-18 (δH 0.80) with H-20 (δH 3.65) (Figure 3). Accordingly, compound 3 was established as 3β,20α-dihydroxy-4,4,14α-trimethyl-7,11,15-trioxo-5α-pregn-8-ene, named lucidone K.
Compound 4 was obtained as pale yellow powder. Its molecular formula was assigned as C24H34O4 by HRESIMS at m/z 445.2583 [M + CH3COOH − H]− (calcd. for C26H37O6, 445.2590). The hydroxy group (3441 cm−1), carbonyl group (1743 cm−1) and α,β-unsaturated carbonyl group (1680 cm−1) were observed in IR spectrum. The UV absorption at λmax 260 nm showed the presence of α,β-unsaturated carbonyl. A comparison of NMR data of 4 with those of 3 revealed their structural similarities. The major differences were the existence of triplet methyl signal but absent of an oxygenated methine at δC 69.9, suggesting the disappearance of 20-OH in 4. The HMBC correlations from H-21 (δH 0.99) to C-20 (δC 32.9) and C-17 (δC 46.2), combined with the 1H–1H COSY correlations of H-20 (δH 2.13) with H-17 (δH 2.34) and H-21 (δH 0.99), confirmed that the ethyl group was attached to C-17. The 3-OH was assigned as β-orientation by the ROESY correlations of H-3 with H-28 and H-5. Therefore, compound 4 was determined as 3β-hydroxy-4,4,14α-trimethyl-7,11,15-trioxo-5α-pregn-8-ene, named lucidone L.
Compound 5 was isolated as white powder. Its molecular formula was established as C25H36O5 based on HRESIMS at m/z 415.2488 [M − H]− (calcd. for C25H35O5, 415.2484). The IR spectrum showed the presence of hydroxy group (3436 cm−1) and α,β-unsaturated carbonyl group (1664 cm−1). The UV absorption brand λmax 244 nm also supported the presence of α,β-unsaturated carbonyl group. Detail comparison of 1 D NMR spectroscopic data with those of ganoderense F (14) suggested their structural similarities except that the hydroxy group at C-3 replaced the ketone group in 5. The 3-OH was confirmed by the HMBC correlations from H-28 (δH 1.07) and H-29 (δH 0.88) to C-3 (δC 78.2), and H-3 (δH 3.26) to C-28 (δC 28.2) and C-29 (δC 15.6), as well as the 1H–1H COSY correlation of H-3 (δH 3.26) with H-2 (δH 1.70). The hydroxy groups at C-3 and C-7 were all assigned as β-orientation on the basis of the ROESY correlations (Figure 3) of H-3 (δH 3.26) with H-28 (δH 1.07) and H-5 (δH 0.98), and H-7 (δH 4.84) with H-5 (δH 0.98) and H-30 (δH 1.58), Thus, compound 5 was elucidated as 20-carbinol-3β,7β-dihydroxy-4,4,14α-trimethyl-11,15-trioxo-5α-pregn-8,16-dien, named ganosineniol B.
Compound 6 was obtained as white powder. Its molecular formula was determined as C25H38O5 by HRESIMS at m/z 417.2629 [M − H]− (calcd. for C25H37O5, 417.2641). The presence of hydroxy group (3432 cm−1), carbonyl group (1722 cm−1) and α,β-unsaturation group (1655 cm−1 and 254 nm) were confirmed by IR and UV spectra. Detail analyses of 1H, 13C-NMR and HSQC spectra revealed that the structure of 6 was highly similar to that of ganosineniol A (15). The difference between those two constituents was that one oxymethine was converted into ketone in 6. The 13C-NMR spectrum of 6 did not show any signal at around δC 72.6, but revealed an additional downfield shift at δC 218.9. Hence, it was inferred that the ketone was located at C-15. The hypothesis was proved by the key HMBC correlations from H-30 (δH 1.30) and H-16 (δH 2.68 and 2.10) to C-15 (δC 218.9). The ROESY correlations of H-3 (δH 3.08) with H-28 (δH 0.95) and H-5 (δH 0.87), and H-7 (δH 4.78) with H-5 (δH 0.87) and H-30 (δH 1.30) assigned 3-OH and 7-OH as β-orientation. Accordingly, compound 6 was determined as 20-carbinol-3β,7β-dihydroxy-4,4,14α-trimethyl-11,15-dioxo-5α-pregn-8-dien, named ganosineniol C.
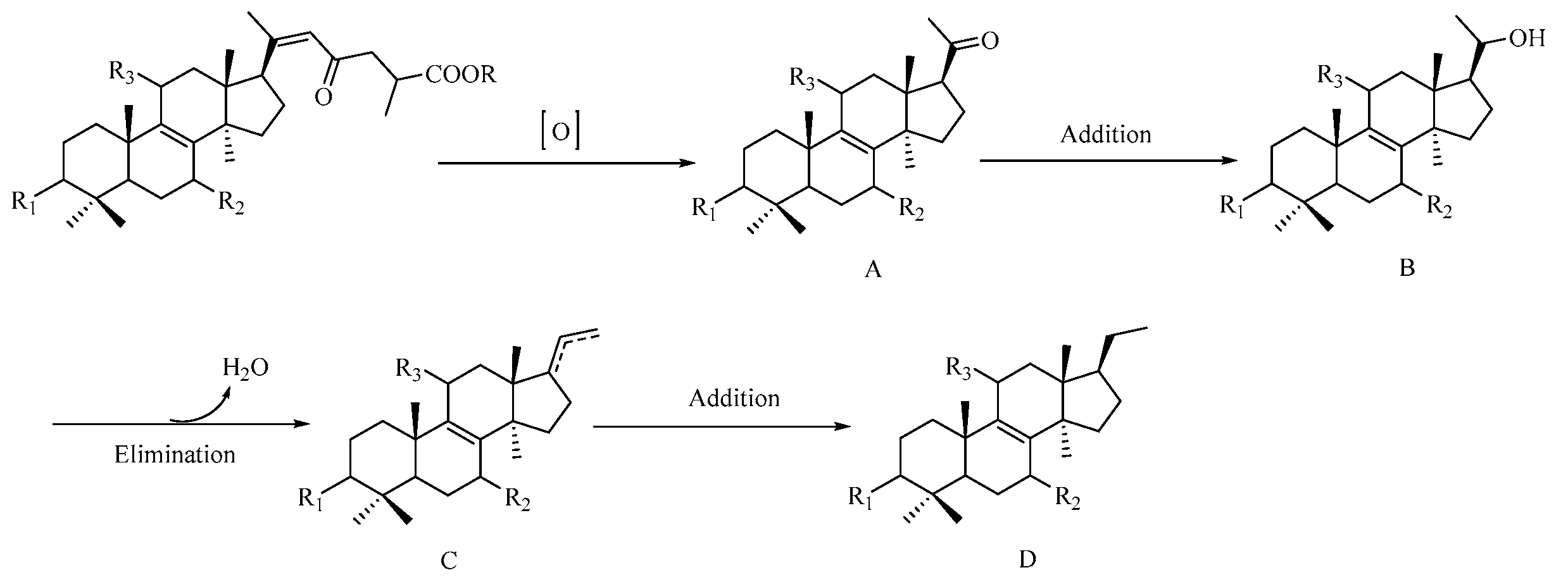
Triterpenoids, biogenetically derived via mevalonic acid pathway, are the main secondary metabolites of Ganoderma. The above-mentioned nortriterpenoids are considered to originate from triterpenoid due to the degradation of its side chain. A possible biogenetic pathway for C24 nortriterpenoids is proposed as shown in Figure 4. The precursor, Ganoderenic acid and its esterified derivatives, undergoes oxidation to yield intermediate A (1, and 7–13). Compounds 7 and 12 were further transformed into 2 and 3, respectively, through an addition reaction. Finally, compound 3 (intermediate B) generated 4 (D) via elimination reaction and addition reaction.
G. resinaceum has been used as ethnomedicine for lowering blood sugar in Nigeria [19]. However, it is unknown whether chemical constituents from G. resinaceum contribute to the traditional medicinal efficacy. Therefore, we considered carrying out α-glucosidase inhibitory assay of isolates from G. resinaceum. Triterpenoids from Ganoderma exhibited significant α-glucosidase inhibitory activity [20,21,22]. Nortriterpenoids from G. resinaceum are considered to originate from triterpenoids due to the degradation of its side chain on basis of analysis of possible biogenetic pathway. Aiming to explore biological active constituents and structure–activity relationship of terpenoids from Ganoderma, compounds 3, 4, and 7–13 were evaluated for α-glucosidase inhibitory activity. Their inhibitions were less than 50% at the concentration of 3 mM (see Supplementary Materials, Table S1). All isolates measured showed lower inhibitory activity compared with positive drug acarbose (IC50 value 2.76 mM), hence, their IC50 value were not measured. Our bioactivity studies may support previous research results that the side chain played an important role in α-glucosidase inhibitory activity of ganoderma triterpenoids, especially the presence of carboxyl acid group [22]. For example, the only difference between ganoderic acid A and 11 was that ganoderic acid A had aliphatic side chain, whereas 11 did not. Ganoderic acid A displayed the same inhibitory activity as positive drug acarbose [22], however, Compound 11 was almost inactive (16% inhibition at the concentration of 3 mM).
3. Materials and Methods
3.1. General Experimental Procedures
NMR spectra data were recorded on a Bruker ascend 600 spectrometer (Bruker, Karlsruhe, Germany) with TMS used as a reference. Optical rotations were measured on PerkinElmer Model 341 polarimeter (PerkinElmer, Waltham, MA, USA). UV spectrum data were acquired using HACH DR6000 UV-visible spectrophotometer (Hach, Loveland, CO, USA). IR spectra were recorded as KBr disks on PerkinElmer Spectrum 100 Series FT-IR spectrometers (PerkinElmer, Waltham, MA, USA). HRESIMS data were obtained on a LTQ Orbitrap XL™ Hybrid Ion Trap-Orbitrap FT-MS spectrometer (Thermo, Waltham, MA, USA). TLC was carried out on silica gel GF254 plates (Yantai Institute of Chemical industry, Yantai, China) and spots were visualized by UV light (254 and/or 365 nm) and spraying with 10% H2SO4 followed by heating. Column chromatography was carried out using silica gel (Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), MCI gel (CHP-20P, 75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan), ODS (35–70 μm, Grace, Maryland, MD, USA), and Sephadex LH-20 (GE Healthcare Bio-Science AB, Uppsala, Sweden) as packing materials. Semi-preparative HPLC was performed on a Shimadzu instrument (Shimadzu, Tokyo, Japan) coupled to CBM-20A system controller, LC-20AP pump, SPD-M20A Photodiode Array Detector and SIL-10AP autosampler and equipped with a Shimadzu PRC-ODS column (250 mm × 20 mm i.d., 15 μm).
3.2. Fungal Material
Fruiting bodies of G. resinaceum were purchased in December 2014 from the Haikou Ruizhitang Wild Lingzhi Co., Ltd. in Hainan province, China, and identified as G. resinaceum Boud by one of author (Prof. S. P. Li). A voucher specimen (No. ICMS-SQC-20141201) has been deposited at Institute of Chinese Medical Sciences, University of Macau.
3.3. Extraction and Isolation
The dried fruiting bodies of G. resinaceum (48 kg) were powdered and extracted with 95% EtOH (600 L) twice under reflux for 2 h. The combined extracts were concentrated under vacuum to afford the crude extract (2.6 kg). After remove of solvent, the extract was dispersed in water and partitioned with petroleum ether, EtOAc and n-BuOH, successively. The HPLC and TLC profiling of EtOAc and n-BuOH extract showed that their constituents were similarities, hence, the EtOAc extract was merged with the n-BuOH extract. The mixture of EtOAc and n-BuOH extract was subjected to silica gel column chromatography (CC) eluted with a gradient of CHCl3–MeOH (100:0–0:100, v/v) to obtain three fractions (E1–E3). E1 (358 g) was separated on silica gel CC eluted with petroleum ether–acetone (100:0–0:100, v/v) to obtain four subfractions (E11–E14).
E12 (135.0 g) was chromatographed on silica gel CC eluted with petroleum ether–EtOAc (10:1–0:1, v/v) to yield fractions E12A and E12B. E12B (55 g) was subjected to MCI gel CC eluted with MeOH–H2O (70:30–100:0, v/v) to yield seven fractions (E12B1–E12B7). E12B3 was separated over silica gel CC eluted with CHCl3–MeOH (30:1, v/v) and then purified over semi-preparative HPLC using MeCN–H2O (30:70, v/v) as mobile phase to afford 7 (151.0 mg) and 10 (118.0 mg). E12B4 was subjected to silica gel CC eluted with CHCl3–MeOH (30:1, v/v) to obtain two fractions (E12B41 and E12B42). The fraction E12B42 was chromatographed over Sephadex LH-20 gel CC eluted with MeOH to yield two fractions (E12B42A and E12B42B). E12B42B was fractionated on semi-preparative HPLC eluted with MeCN–H2O (31:69, v/v) to afford 8 (40.0 mg), 9 (23.0 mg), 11 (110.0 mg), and 14 (35.0 mg).
E13 (103.5 g) was chromatographed over silica gel CC eluted with CHCl3–acetone (10:1–0:1) to yield three fractions (E13A–E13C). E13A was fractionated on MCI gel CC eluted with MeOH–H2O (50:50–100:0, v/v) to obtain three fractions (E13A1–E13A3). E13A1 was chromatograph over silica gel CC eluted with CHCl3–acetone (10:1–8:1) to yield two fractions (E13A11 and E13A12). Compound 12 (2.1 g) was obtained from E13A11 which was separated on Sephadex LH-20 gel CC. E13A12 was subjected to Sephadex LH-20 gel CC eluted with CHCl3–MeOH (1:1, v/v) and further purified over semi-preparative HPLC eluted with MeCN–H2O (35:65, v/v) to obtain 5 (6.0 mg). E13B was fractionated into four fractions (E13B1–E13B4) by ODS CC eluted with MeOH–H2O (50:50–80:20, v/v). E13B1 was purified over Sephadex LH-20 gel CC eluted with CHCl3–MeOH (1:1, v/v) and then semi-preparative HPLC with MeCN–H2O (32:68, v/v) to afford 13 (115.0 mg) and 3 (80.1 mg). E13C was chromatographed over MCI gel CC eluted with MeOH–H2O (40:60–80:20, v/v) to yield E13C1 and E13C2. E13C1 was fractionated on ODS CC eluted with a gradient of MeOH–H2O (50:50–80:20, v/v) to afford five fractions (E13C11–E13C15). Compound 1 (3.6 mg) was obtained from E13C13 which was subjected to Sephadex LH-20 gel CC and further semi-preparative HPLC eluted with MeCN–H2O (32:68, v/v). E13C14 was purified over ODS CC eluted with MeOH–H2O (50:50, v/v) and then semi-preparative HPLC with a MeCN–H2O (32:68, v/v) to afford 2 (5.1 mg) and 6 (35.0 mg).
E2 (102.0 g) was chromatographed over silica gel CC eluted with CHCl3–acetone (5:1–0:1, v/v) to obtain E21 and E22. E21 (75.0 g) was separated on ODS CC eluted with MeOH–H2O (40:60–70:30, v/v) to afford four fractions (E21A–E21D). E21C was separated on ODS CC eluted with MeOH–H2O (40:60, v/v) to afford three fractions (E21C1–E21C3). E21C1 was subjected to Sephadex LH-20 gel CC using MeOH as mobile phase and further semi-preparative HPLC eluted with MeCN–H2O (31:69, v/v) to obtain 4 (40.0 mg). E22 (15.0 g) was fractionated on MCI gel CC eluted with MeOH–H2O (50:50–80:20, v/v) to yield E22A and E22B. E22A was separated on silica gel CC with an isocratic CHCl3–acetone (2:1, v/v) to obtain three fractions (E22A1–E22A3). Compound 15 (18.1 mg) was obtained from E22A2 by semi-preparative HPLC eluted with MeCN–H2O (30:70, v/v).
3.3.1. Lucidone I (1)
White powder; +67.4 (c 0.27, MeOH); UV (MeOH) λmax (log ε) 204 (3.78), 253 (3.41) nm; IR (KBr) vmax 3429, 2691, 2929, 2871, 1717, 1639, 1453, 1383, 1261, 1181 cm−1; 1H- and 13C-NMR spectroscopic data see Table 1; HRESIMS m/z 403.2484 [M − H]− (calcd. for C24H35O5, 403.2484).
3.3.2. Lucidone J (2)
White powder; +80.7 (c 0.38, MeOH); UV (MeOH) λmax (log ε) 202 (3.45), 253 (3.76) nm; IR (KBr) vmax 3432, 2964, 2925, 2866, 1731, 1654, 1457, 1383, 1306, 1172, 1058, 1035 cm−1; 1H- and 13C-NMR spectroscopic data see Table 1; HRESIMS m/z 403.2481 [M − H]− (calcd. for C24H35O5, 403.2484).
3.3.3. Lucidone K (3)
Pale yellow powder; +105.8 (c 0.17, MeOH); UV (MeOH) λmax (log ε) 201 (3.53), 259 (3.68) nm; IR (KBr) vmax 3484, 2982, 2932, 2862, 1742, 1689, 1457, 1426, 1379, 1338, 1300, 1232, 1200, 1163, 1082, 1034, 976 cm−1; 1H- and 13C-NMR spectroscopic data see Table 1; HRESIMS m/z 401.2352 [M − H]− (calcd. for C24H33O5, 401.2333).
3.3.4. Lucidone L (4)
Pale yellow powder; +88.3 (c 0.74, MeOH); UV (MeOH) λmax (log ε) 202 (3.49), 260 (3.58) nm; IR (KBr) vmax 3441, 2974, 2935, 2875, 1743, 1680, 1461, 1383, 1199, 1088, 1035, 928 cm−1; 1H- and 13C-NMR spectroscopic data see Table 1; HRESIMS m/z 445.2583 [M + CH3COOH − H]− (calcd. for C26H37O6, 445.2590).
3.3.5. Ganosineniol B (5)
White powder; +104.6 (c 0.26, MeOH); UV (MeOH) λmax (log ε) 201 (3.62), 244 (3.83), 341(2.05) nm; IR (KBr) vmax 3436, 2967, 2928, 2872, 1664, 1599, 1457, 1379, 1247, 1171, 1102, 1037 cm−1; 1H- and 13C-NMR spectroscopic data see Table 2; HRESIMS m/z 415.2488 [M − H]− (calcd. for C25H35O5, 415.2484).
3.3.6. Ganosineniol C (6)
White powder; +19.9 (c 1.44, MeOH); UV (MeOH) λmax (log ε) 202 (3.44), 254 (3.66), 341(2.04) nm; IR (KBr) vmax 3432, 2968, 2928, 2866, 1722, 1655, 1459, 1380, 1270, 1176, 1034 cm−1; 1H and 13C-NMR spectroscopic data see Table 2; HRESIMS m/z 417.2629 [M − H]− (calcd. for C25H37O5, 417.2641).
3.4. Activity Assay
α-Glucosidase inhibitory activity was examined by the method described by Dengqiang Li et al. [23]. Acarbose, a definite α-glucosidase inhibition, was used as positive drug.
4. Conclusions
In summary, six new nortriterpenoids (1–6), together with nine known analogs (7–15), were isolated and identified from the fruiting bodies of Ganoderma resinaceum. The possible biogenetic pathway for C24 nortriterpenoids was proposed Compounds 3, 4, and 7–15 displayed no significant α-glucosidase inhibitory activity, however, this finding may support that the side chain of ganoderma triterpenoids is critical for α-glucosidase inhibitory activity, especially the presence of carboxyl acid group.
Supplementary Materials
The following are available online: Spectrum copies of Mass, NMR, and IR.
Acknowledgments
This research was financially supported by grants from the Science and Technology Development Fund of Macao (FDCT040/2016/A), and the University of Macau (MYRG2015-00122) to J. Zhao. We thank Shen-Fei Wang who is a master student in Institute of Chinese Medical Sciences, University of Macau, for her assistance in testing α-glucosidase inhibitory activity.
Author Contributions
Xian-Qiang Chen performed the isolation and structure elucidation of the chemicals, activity assay, and the preparation of the manuscript. Ling-Xiao Chen takes part in analysis of ESI-MS. Shao-Ping Li and Jing Zhao planned organized the whole research of this study. Yu-Ping Tang participated in design of this study. All authors approved the final version manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhao, J.; Zhang, X.-Q.; Li, S.-P.; Yang, F.-Q.; Wang, Y.-T.; Ye, W.-C. Quality evaluation of Ganoderma through simultaneous determination of nine triterpenes and sterols using pressurized liquid extraction and high performance liquid chromatography. J. Sep. Sci. 2006, 29, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-W.; Gao, J.-L.; Guan, J.; Qian, Z.-M.; Feng, K.; Li, S.-P. Evaluation of Antiproliferative Activities and Action Mechanisms of Extracts from Two Species of Ganoderma on Tumor Cell Lines. J. Agric. Food Chem. 2009, 57, 3087–3093. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-F.; Liu, J.-Q.; Yan, Y.-X.; Chen, J.-C.; Lu, Y.; Guo, Y.-H.; Qiu, M.-H. Three new triterpenoids containing four-membered ring from the fruiting body of Ganoderma sinense. Org. Lett. 2010, 12, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-L.; Yu, Y.-S.; Yen, G.-C. Lucidenic acid B induces apoptosis in human leukemia cells via a mitochondria-mediated pathway. J. Agric. Food Chem. 2008, 56, 3973–3980. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, K.; Akihisa, T.; Tokuda, H.; Ukiya, M.; Oshikubo, M.; Kimura, Y.; Asano, T.; Nomura, A.; Nishino, H. Lucidenic acids P and Q, methyl lucidenate P, and other triterpenoids from the fungus Ganoderma lucidum and their inhibitory effects on Epstein-Barr virus activation. J. Nat. Prod. 2003, 66, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.S.; Shi, L.S.; Kuo, S.C. Cytotoxicity of Ganoderma lucidum triterpenes. J. Nat. Prod. 2001, 64, 1121–1122. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Nakamura, Y.; Tagata, M.; Tokuda, H.; Yasukawa, K.; Uchiyama, E.; Suzuki, T.; Kimura, Y. Anti-inflammatory and anti-tumor-promoting effects of triterpene acids and sterols from the fungus Ganoderma lucidum. Chem. Biodivers. 2007, 4, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Q.; Ip, F.C.F.; Zhang, D.-M.; Chen, L.-X.; Zhang, W.; Li, Y.-L.; Ip, N.Y.; Ye, W.-C. Triterpenoids with neurotrophic activity from Ganoderma lucidum. Nat. Prod. Res. 2011, 25, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-R.; Liu, J.-Q.; Han, Z.-H.; Yuan, X.-X.; Luo, H.-R.; Qiu, M.-H. Protective effects of triterpenoids from Ganoderma resinaceum on H2O2-induced toxicity in HepG2 cells. Food Chem. 2013, 141, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Zhang, Q.; Ma, C.-M.; Hattori, M. Anti-human immunodeficiency virus-1 protease activity of new lanostane-type triterpenoids from Ganoderma sinense. Chem. Pharm. Bull. 2009, 57, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Ayissi, B.M.K.; Mossebo, D.C. Some noteworthy taxonomic variations in the complex wood-decayer Ganoderma resinaceum (Basidiomycota) with reference to collections from tropical Africa. Kew Bull. 2014, 69, 9542. [Google Scholar] [CrossRef]
- Zengin, G.; Sarikurkcu, C.; Gunes, E.; Uysal, A.; Ceylan, R.; Uysal, S.; Gungor, H.; Aktumsek, A. Two Ganoderma species: Profiling of phenolic compounds by HPLC-DAD, antioxidant, antimicrobial and inhibitory activities on key enzymes linked to diabetes mellitus, Alzheimer’s disease and skin disorders. Food Funct. 2015, 6, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.-M.; Li, S.-H.; Xiao, W.-L.; Sun, H.-D.; Che, C.-T. Two new lanostanoids from Ganoderma resinaceum. J. Asian Nat. Prod. Res. 2007, 9, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Nishitoba, T.; Sato, H.; Sakamura, S. New terpenoids from Ganoderma lucidum and their bitterness. Agric. Biol. Chem. 1985, 49, 1547–1549. [Google Scholar] [CrossRef]
- Nishitoba, T.; Sato, H.; Sakamura, S. New terpenoids, ganolucidic acid D, ganoderic acid L, lucidone C and lucidenic acid G, from the fungus Ganoderma lucidum. Agric. Biol. Chem. 1986, 50, 809–811. [Google Scholar] [CrossRef]
- Nishitoba, T.; Sato, H.; Sakamura, S. Triterpenoids from the fungus Ganoderma lucidum. Phytochemistry 1987, 26, 1777–1784. [Google Scholar] [CrossRef]
- Li, W.; Lou, L.-L.; Zhu, J.-Y.; Zhang, J.-S.; Liang, A.-A.; Bao, J.-M.; Tang, G.-H.; Yin, S. New lanostane-type triterpenoids from the fruiting body of Ganoderma hainanense. Fitoterapia 2016, 115, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Q.; Wang, C.-F.; Li, Y.; Luo, H.-R.; Qiu, M.-H. Isolation and bioactivity evaluation of terpenoids from the medicinal fungus Ganoderma sinense. Planta Med. 2012, 78, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Oyetayo, O.V. Medicinal Uses of Mushrooms in Nigeria: Towards Full and Sustainable Exploitation. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-R.; Huo, X.-K.; Dong, P.-P.; Wang, C.; Huang, S.-S.; Zhang, B.-J.; Zhang, H.-L.; Deng, S.; Liu, K.-X.; Ma, X.-C.; et al. Inhibitory Effects of Highly Oxygenated Lanostane Derivatives from the Fungus Ganoderma lucidum on P-Glycoprotein and α-Glucosidase. J. Nat. Prod. 2015, 78, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bao, L.; Xiong, W.; Ma, K.; Han, J.; Wang, W.; Yin, W.; Liu, H. Lanostane triterpenes from the Tibetan medicinal mushroom Ganoderma leucocontextum and their inhibitory effects on HMG-CoA reductase and α-glucosidase. J. Nat. Prod. 2015, 78, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Fatmawati, S.; Kondo, R.; Shimizu, K. Structure-activity relationships of lanostane-type triterpenoids from Ganoderma lingzhi as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5900–5903. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Q.; Qian, Z.-M.; Li, S.-P. Inhibition of Three Selected Beverage Extracts on α-Glucosidase and Rapid Identification of Their Active Compounds Using HPLC-DAD-MS/MS and Biochemical Detection. J. Agric. Food Chem. 2010, 58, 6608–6613. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–15 are available from corresponding authors. |
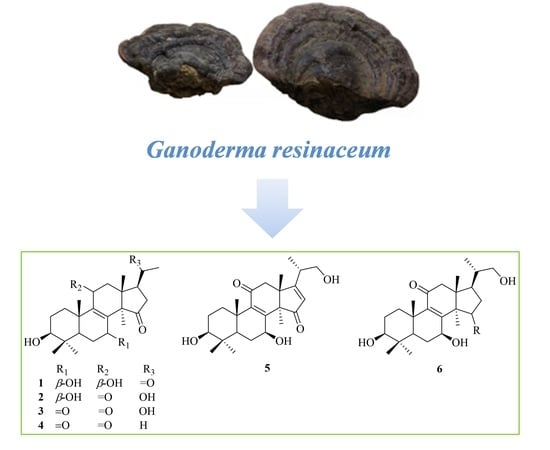
Figure 1.
Structures of compounds 1–15.
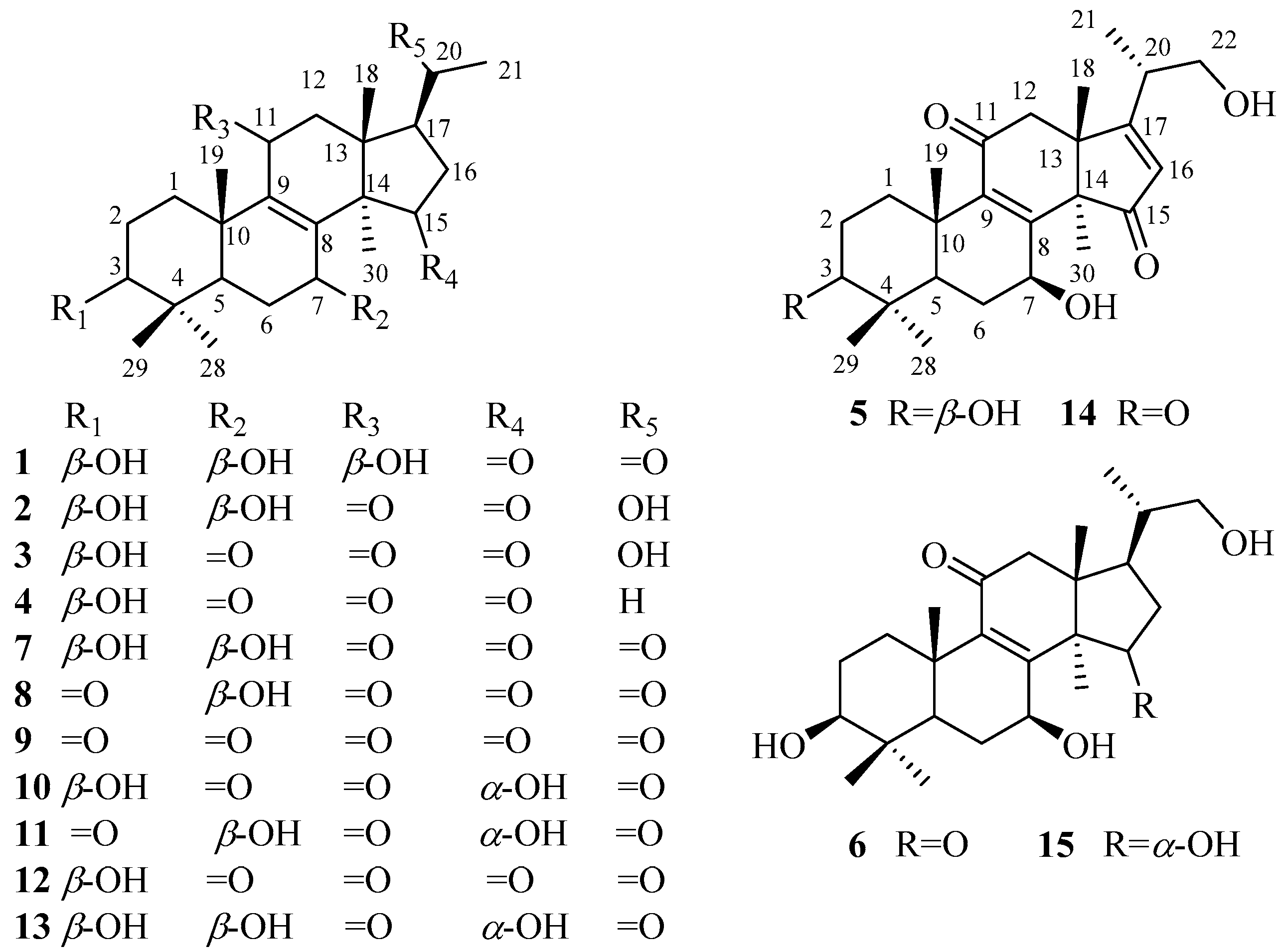
Figure 2.
The key HMBC, and 1H–1H COSY correlations of 1–6.
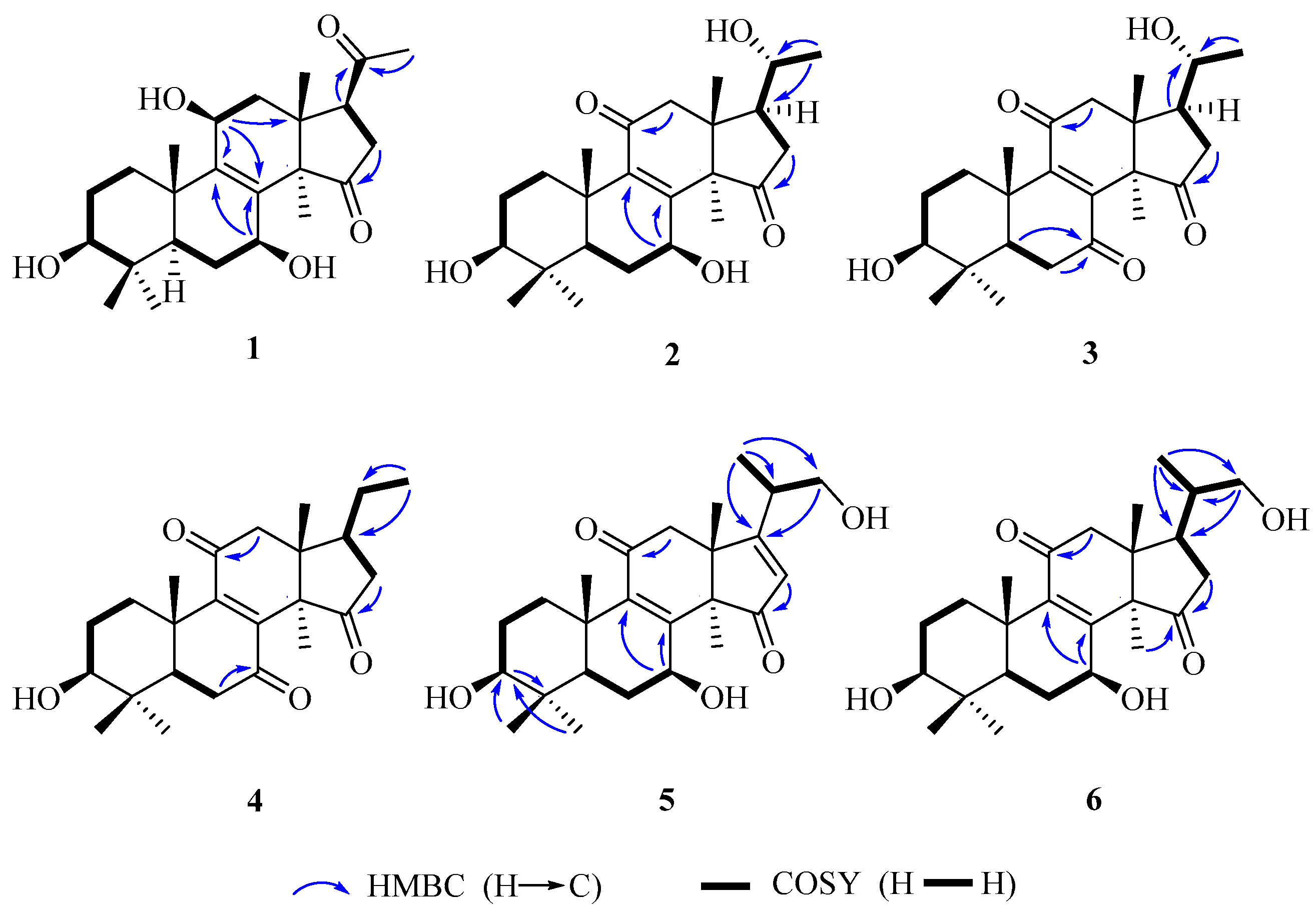
Figure 3.
The key ROESY correlations of 1−6.
Figure 4.
Plausible biogenetic pathway for C24 nortriterpenoids.


{kind=link}
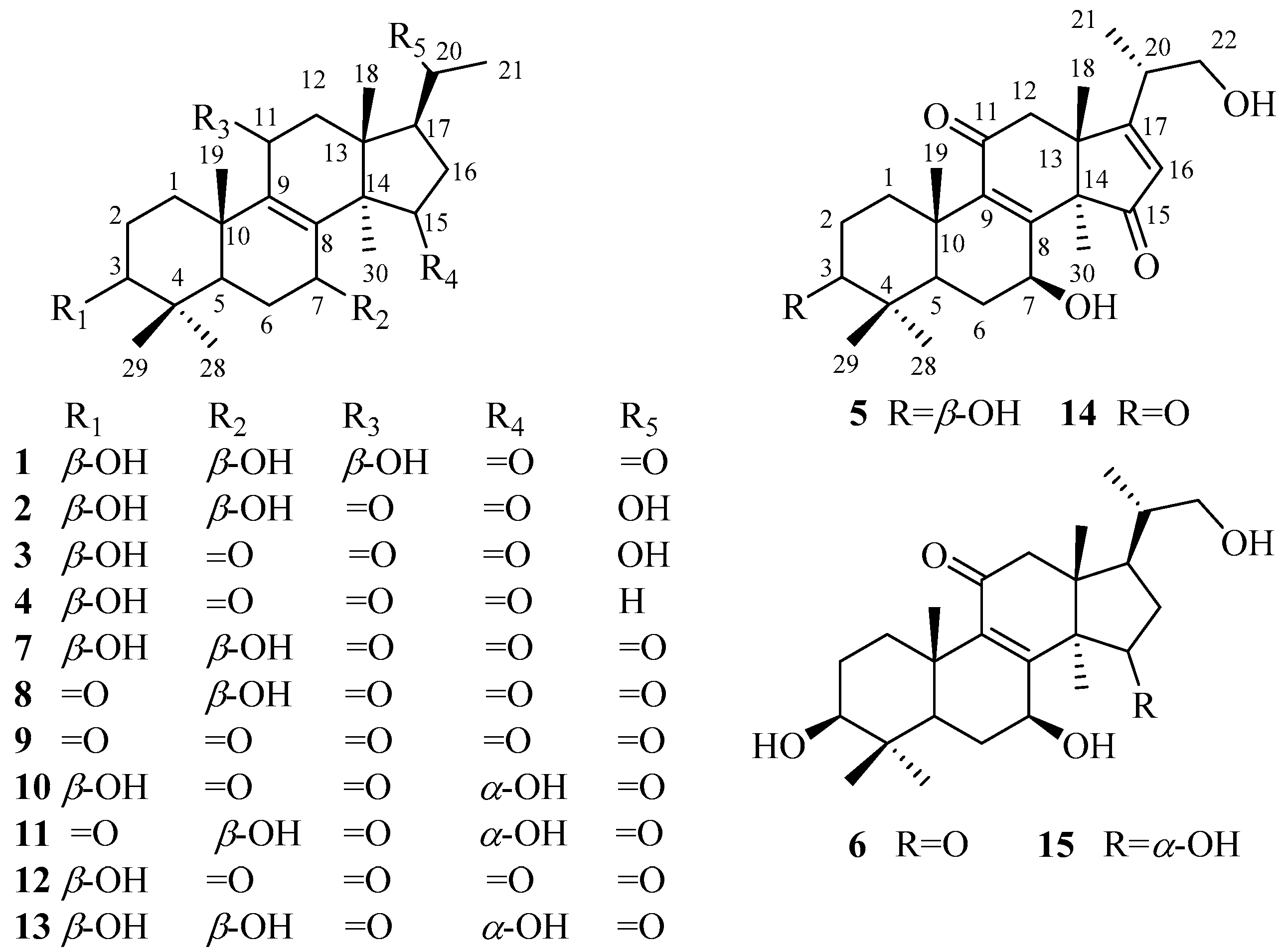{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H-NMR (600 MHz, CD3OD) and 13C-NMR (150 MHz, CD3OD) spectroscopic data of 1–4.
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | |
| 1 | 36.0, CH2 | 2.20, dt (12.6, 3.0); 1.36, m | 36.0, CH2 | 2.78, dt (13.8, 3.0); 1.02, m | 34.9, CH2 | 2.75, dt (13.8, 3.6); 1.16, dt (13.8, 3.6) | 34.9, CH2 | 2.80, m; 1.26, dt (13.5, 3.0) |
| 2 | 28.3, CH2 | 1.60, m; 1.53, m | 28.4, CH2 | 1.60, m; 1.68, m | 28.5, CH2 | 1.66, m; 1.60, m | 28.2, CH2 | 1.72, brd (13.2); 1.66, m |
| 3 | 79.5, CH | 3.08, dd (12.0, 4.2) | 79.1, CH | 3.17, dd (12.0, 4.8) | 78.3, CH | 3.14, dd (11.4, 4.8) | 78.3, CH | 3.21, dd (12.0, 4.2) |
| 4 | 40.1, C | 39.4, C | 40.4, C | 40.3, C | ||||
| 5 | 51.7, CH | 0.93, m | 50.4, CH | 0.96, d (13.2) | 52.8, CH | 1.50, d(1.8) | 52.6, CH | 1.60, d (14.4) |
| 6 | 28.94, CH2 | 2.10, m; 1.61, m | 28.0, CH2 | 2.20, dd (13.2, 9.0); 1.59, m | 37.5, CH2 | 2.60, d (14.4); 2.40, dd (14.4, 1.8) | 37.5, CH2 | 2.68, t (14.4); 2.49, d (14.4) |
| 7 | 66.9, CH | 4.54, t (9.6) | 68.2, CH | 4.85, t (9.0) | 202.3, C | 202.0, C | ||
| 8 | 136.8, C | 158.2, C | 147.3, C | 147.8, C | ||||
| 9 | 146.2, C | 143.3, C | 154.1, C | 153.6, C | ||||
| 10 | 40.2, C | 39.8, C | 41.9, C | 41.9, C | ||||
| 11 | 65.3, CH | 4.48, d (7.2) | 201.5, C | 202.3, C | 201.5, C | |||
| 12 | 42.0, CH2 | 2.51, dd (14.4, 7.2); 2.00, d (14.4) | 51.3, CH2 | 2.89, d (16.2); 2.82, d (16.2) | 50.2, CH2 | 2.90, d (15.6); 2.78, d (15.6) | 50.5, CH2 | 3.05, d (15.6); 2.65, d (15.6) |
| 13 | 43.8 C | 46.6, C | 45.3, C | 45.7, C | ||||
| 14 | 61.0, C | 60.3, C | 58.1, C | 58.7, C | ||||
| 15 | 220.2, C | 218.6, C | 210.2, C | 210.8, C | ||||
| 16 | 37.3, CH2 | 2.82, dd (19.8, 8.4); 2.45, dd (19.8, 9.0) | 40.0, CH2 | 2.63, dd (19.8, 9.0); 2.06, dd (19.8, 9.6) | 38.5, CH2 | 2.60, dd (18.6, 9.0); 1.69, dd (18.6, 8.4) | 41.3, CH2 | 2.82, m; 1.87, dd (18.0, 7.8) |
| 17 | 54.8, CH | 3.22, overlapped | 48.9, CH | 2.28, dd (18.6, 9.0) | 47.9, CH | 2.28, dd (18.6, 9.6) | 46.2, CH | 2.34, m |
| 18 | 19.7, CH3 | 0.79, s | 18.1, CH3 | 1.03, s | 16.9, CH3 | 0.80, s | 16.7, CH3 | 0.83, s |
| 19 | 22.2, CH3 | 1.21, s | 19.0, CH3 | 1.25, s | 18.2, CH3 | 1.26, s | 18.3, CH3 | 1.30, s |
| 20 | 209.3, C | 69.9, CH | 3.79, m | 69.9, CH | 3.65, m | 32.9, CH2 | 2.13, m | |
| 21 | 31.8, CH3 | 2.13, s | 23.8, CH3 | 1.20, d (6.0) | 23.7, CH3 | 1.10, d (6.0) | 20.5, CH3 | 0.99, t (6.0) |
| 28 | 28.89, CH3 | 0.90, s | 28.8, CH3 | 1.04, s | 28.2, CH3 | 0.94, s | 28.5, CH3 | 1.00, s |
| 29 | 16.2, CH3 | 0.77, s | 16.4, CH3 | 0.86, s | 16.8, CH3 | 1.13, s | 16.3, CH3 | 0.88, s |
| 30 | 23.5, CH3 | 1.08, s | 24.6, CH3 | 1.42, s | 16.3, CH3 | 0.82, s | 22.0, CH3 | 1.57, s |
Table 2.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectroscopic data of 5 and 6.
| No. | 5a | 6b | ||
|---|---|---|---|---|
| δC mult. | δH mult. (J in Hz) | δC mult. | δH mult. (J in Hz) | |
| 1 | 34.7, CH2 | 2.94, dt (13.2, 3.6); 1.07, m | 36.0, CH2 | 2.71, dt (13.2, 3.6); 0.95, m |
| 2 | 27.7, CH2 | 1.70, m | 28.4, CH2 | 1.58, m; 1.53, m |
| 3 | 78.2, CH | 3.26, dd (12.0, 4.8) | 79.0, CH | 3.08, dd (12.0, 4.8) |
| 4 | 38.7, C | 39.7, C | ||
| 5 | 49.3, CH | 0.98, d (13.2) | 50.4, CH | 0.87, d (13.8) |
| 6 | 26.1, CH2 | 2.20, dd (13.2, 7.2); 1.64, m | 28.1, CH2 | 2.11, m; 1.49, m |
| 7 | 67.1, CH | 4.84, d (4.2) | 68.1, CH | 4.78, t (3.0) |
| 8 | 158.4, C | 159.0, C | ||
| 9 | 142.2, C | 144.2, C | ||
| 10 | 39.2, C | 40.0, C | ||
| 11 | 197.6, C | 200.6, C | ||
| 12 | 44.2, CH2 | 2.98, d (16.2); 2.58, d (16.2) | 51.6, CH2 | 2.85, d (16.8); 2.55, d (16.8) |
| 13 | 51.3, C | 46.7, C | ||
| 14 | 58.2, C | 60.2, C | ||
| 15 | 211.0, C | 218.9, C | ||
| 16 | 123.5, CH | 5.84, s | 41.6, CH2 | 2.68, d (13.2); 2.10, m |
| 17 | 185.6, C | 43.7, CH | 2.15, m | |
| 18 | 30.6, CH3 | 1.24, s | 16.4, CH3 | 0.77, s |
| 19 | 18.6, CH3 | 1.23, s | 19.0, CH3 | 1.15, s |
| 20 | 36.3, CH | 2.65, m | 39.8, CH | 1.58, m |
| 21 | 16.5, CH3 | 1.18, d (7.2) | 17.5, CH3 | 1.00, d (6.6) |
| 22 | 65.6, CH2 | 3.79, m | 67.6, CH2 | 3.45, dd (11.4, 3.3); 3.32, dd (11.4, 6.0) |
| 28 | 28.2, CH3 | 1.07, s | 28.8, CH3 | 0.95, s |
| 29 | 15.6, CH3 | 0.88, s | 18.0, CH3 | 0.91, s |
| 30 | 33.5, CH3 | 1.58, s | 25.0, CH3 | 1.30, s |
a measured in CDCl3; b measured in CD3OD.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, X.-Q.; Chen, L.-X.; Zhao, J.; Tang, Y.-P.; Li, S.-P. Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules 2017, 22, 1073. https://doi.org/10.3390/molecules22071073
AMA Style
Chen X-Q, Chen L-X, Zhao J, Tang Y-P, Li S-P. Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules. 2017; 22(7):1073. https://doi.org/10.3390/molecules22071073
Chicago/Turabian StyleChen, Xian-Qiang, Ling-Xiao Chen, Jing Zhao, Yu-Ping Tang, and Shao-Ping Li. 2017. "Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum" Molecules 22, no. 7: 1073. https://doi.org/10.3390/molecules22071073