
Total Synthesis and Pharmacological Investigation of Cordyheptapeptide A
by
Suresh Kumar
1,†, 
Rajiv Dahiya
2,*,†, 
Sukhbir Lal Khokra
1,
Rita Mourya
3,
Suresh V. Chennupati
4 and
Sandeep Maharaj
2 1
Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra 136119, Haryana, India
2
Laboratory of Peptide Research and Development, School of Pharmacy, Faculty of Medical Sciences, The University of the West Indies, St. Augustine, Trinidad & Tobago, West Indies
3
School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, P.O. Box 196, Gondar 6200, Ethiopia
4
Department of Pharmacy, College of Medical and Health Sciences, Wollega University, P.O. Box 395, Nekemte, Ethiopia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2017, 22(6), 682; https://doi.org/10.3390/molecules22060682
Submission received: 23 March 2017
/
Revised: 17 April 2017
/
Accepted: 18 April 2017
/
Published: 27 May 2017
(This article belongs to the Collection Bioactive Compounds)
Abstract
:The present investigation reports the synthesis of a phenylalanine-rich N-methylated cyclopeptide, cordyheptapeptide A (8), previously isolated from the insect pathogenic fungus Cordyceps sp. BCC 1788, accomplished through the coupling of N-methylated tetrapeptide and tripeptide fragments followed by cyclization of the linear heptapeptide unit. Structure elucidation of the newly synthesized cyclopolypeptide was performed by means of FT-IR, 1H-NMR, 13C-NMR, and fast atom bombardment mass spectrometry (FABMS), and screened for its antibacterial, antidermatophytic, and cytotoxic potential. According to the antimicrobial activity results, the newly synthesized N-Methylated cyclopeptide exhibited potent antibacterial activity against Gram-negative bacteria Pseudomonas aeruginosa and Klebsiella pneumoniae and antifungal activity against dermatophytes Trichophyton mentagrophytes and Microsporum audouinii at a concentration of 6 μg/mL, in comparison to the reference drugs, gatifloxacin and griseofulvin. In addition, cyclopolypeptide 8 displayed suitable levels of cytotoxicity against Dalton’s lymphoma ascites (DLA) and Ehrlich’s ascites carcinoma (EAC) cell lines.
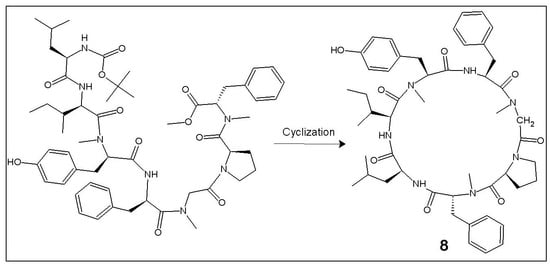
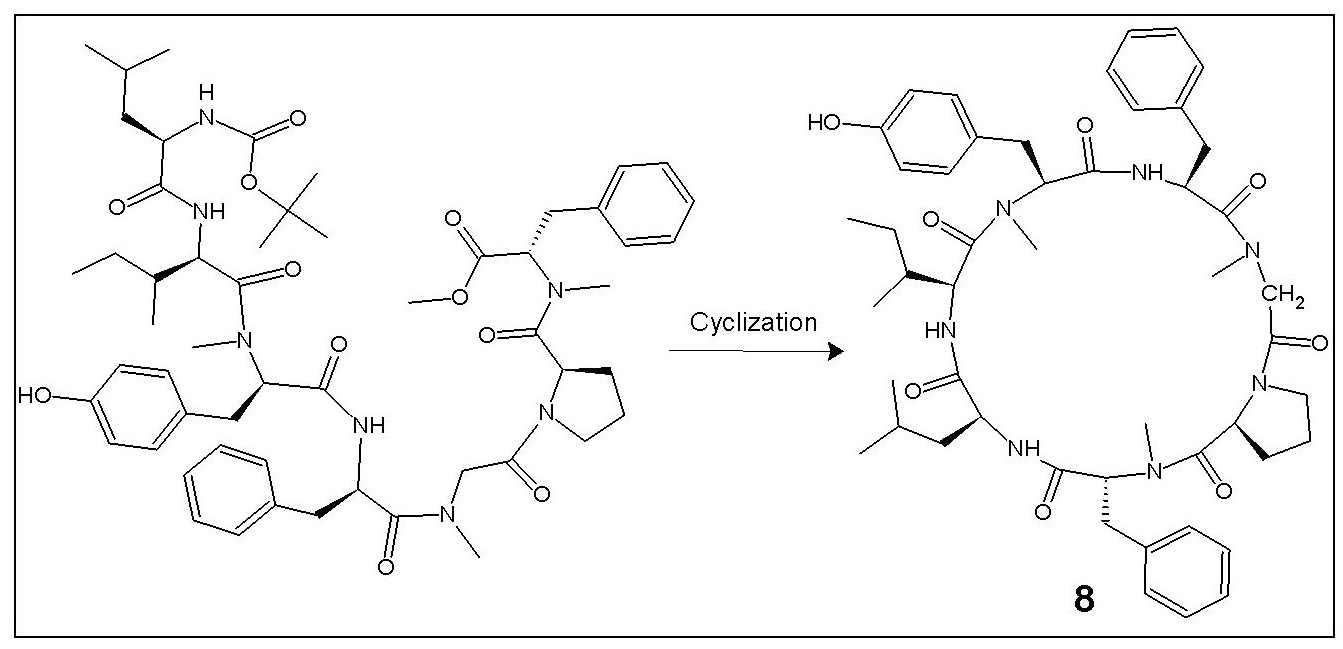
1. Introduction
Nature is an attractive source of new therapeutic compounds. In drug discovery, microorganisms play a tremendous role especially by producing a number of candidates that are effective against microbes [1]. The previous literature is enriched with potential data indicating the ability of microorganisms such as fungi and bacteria, which produce a variety of natural products with diverse bioactivities [2,3]. Currently, microorganism-derived peptides (MdPs) are attracting the attention of scientists [4,5] because of the unique molecular structures and a wide array of associated bioactivities like antimycobacterial and antimalarial activity [6,7], antibacterial activity [8,9], cytotoxicity [10], and antiviral activity [11]. Cyclopeptides produced by microbes including cycloaspeptides, halolitoralins [12], psychrophilins, talaromins, ustiloxins, and unguisins are associated with many biological properties and some of the peptides from fungi have even gained entrance into the pharmaceutical market. e.g., cyclosporins, ergopeptides, etc.
The structure of a homodectic heptapeptide, cordyheptapeptide A, previously isolated from the insect pathogenic fungus Cordyceps sp. BCC 1788, was confirmed by spectroscopic techniques including FT-IR, 1H/13C-NMR, distortionless enhancement of polarisation transfer (DEPT), high-resolution electron ionization mass spectrometry (HREIMS) and the amino acid residues were established on the basis of correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), heteronuclear multiple quantum correlation (HMQC), and heteronuclear multiple bond correlation (HMBC) spectroscopy. In terms of the biological response, natural cordyheptapeptide A exhibited antimalarial activity against Plasmodium falciparum K1 and cytotoxicity to Vero cell lines with 50% inhibitory concentration values of 5.35 and >56.88 μM, respectively [13]. Another peptide of the series, cordyheptapeptide B, was isolated from a fungal strain Cordyceps sp. and the absolute configuration of the cordyheptapeptide B was indicated by chromatographic analysis of the acid hydrolyzate. Cordyheptapeptide B was found to exhibit cytotoxicity against several cell lines including KB, BC, NCI-H187, and Vero cell lines with IC50 values in range of 0.66–3.1 μM [14]. In 2011, the total synthesis of cordyheptapeptide B was reported for the first time utilizing a solution-phase technique [15]. Cordyheptapeptide B differs from cordyheptapeptide A by having an N-methyl-l-phenylalanine residue instead of the N-methyl-l-tyrosine. Three new cyclic peptides, cordyheptapeptides C–E, were isolated from the marine-derived fungus A. persicinum SCSIO 115 and their planar structures were confirmed by extensive mass spectrometry (MS), 1D and 2D NMR spectroscopic analyses. Cordyheptapeptides C and E displayed cytotoxicity against the SF-268, MCF-7, and NCI-H460 tumor cell lines with IC50 values in the range of 2.5 to 12.1 μM [16].
Given the diverse pharmacological activities possessed by MdPs and other cyclooligopeptides [17,18,19] and the continued efforts of our research group [15,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45] for synthesizing N-methylated and other cyclopolypeptides for quantitative yield in the laboratory, the present work was directed toward the synthesis, characterization, and bioactivity screening of a phenylalanine-rich cyclopolypeptide, cordyheptapeptide A for the antimicrobial and cytotoxic properties.
2. Results
2.1. Chemistry
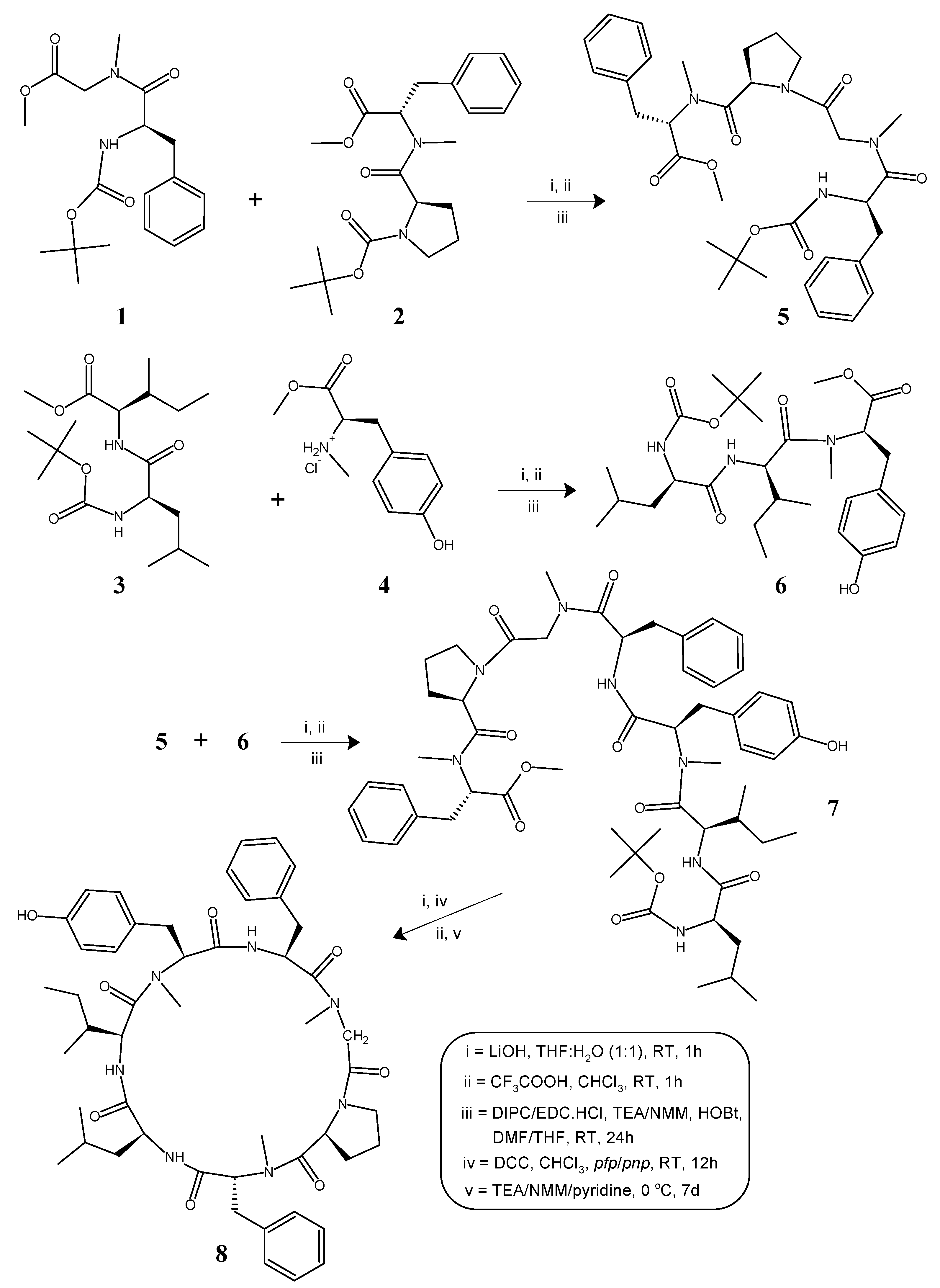
The cycloheptapeptide molecule was split into three dipeptide units viz. Boc-l-Phe-N(Me)Gly-OMe (1), Boc-l-Pro-d-N(Me)Phe-OMe (2), and Boc-l-Leu-l-Ile-OMe (3), and a single amino acid unit l-N(Me)Tyr-OMe·HCl (4). N-methylation of Boc-protected phenylalanine and tyrosine methyl esters was done by treatment with methyliodide and sodium hydride as per the literature [46]. Dipeptide units (1–3) were prepared by the coupling of Boc-amino acids such as Boc-l-Phe, Boc-l-Pro, and Boc-l-Leu with the corresponding amino acid methyl ester hydrochlorides such as N(Me)Gly-OMe·HCl, d-N(Me)Phe-OMe·HCl, and l-Ile-OMe·HCl by following the modified Bodanzsky method [47]. After deprotection at the carboxy terminus, dipeptide 1 was coupled with dipeptide 2, deprotected at the amino terminus, to obtain the tetrapeptide unit Boc-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-OMe (5). The carboxyl group of dipeptide 3 was deprotected by alkaline hydrolysis using lithium hydroxide (LiOH) and the deprotected peptide was coupled with the amino acid unit 4, utilizing different carbodiimides to obtain the tripeptide unit Boc-l-Leu-l-Ile-l-N(Me)Tyr-OMe (6). After removal of the ester group of tripeptide 6 and the Boc-group of tetrapeptide 5, the deprotected units (5a, 6a) were coupled to obtain the linear heptapeptide unit Boc-l-Leu-l-Ile-l-N(Me)Tyr-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-OMe (7). The methyl ester group of the linear peptide fragment was replaced by the pentafluorophenyl (pfp)/p-nitrophenyl (pnp) ester group. The Boc-group of the resulting compound was removed using trifluoroacetic acid (CF3COOH), and the deprotected linear fragment was now cyclized by keeping the entire contents at 0 °C for 7 days in the presence of catalytic amounts of triethylamine (TEA) or N-methylmorpholine (NMM) or pyridine to get the cyclic product 8. The structures of the newly synthesized cycloheptapeptide as well as that of the intermediate tri/tetra/heptapeptides were confirmed by FT-IR, 1H-NMR spectroscopy, and elemental analysis. In addition, the mass spectra and 13C-NMR spectroscopy were recorded for the linear and cyclic heptapeptide only. The synthetic pathway for the newly synthesized N-methylated heptacyclopeptide is shown in Figure 1.
2.2. Pharmacological Activity Studies
The synthesized linear and cycloheptapeptide (7, 8) were subjected to the short term in vitro cytotoxicity study against the Dalton’s lymphoma ascites and Ehrlich’s ascites carcinoma cell lines at concentrations of 62.5–3.91 μg/mL using 5-fluorouracil (5-FU) as the reference compound. The activity was determined by measuring the inhibition (%) of the cell lines [48]. The CTC50 values were determined by the graphical extrapolation method. The results of the cytotoxic activity studies are presented in Table 1.
Moreover, the linear and heptacyclopeptide (7, 8) were further evaluated for their antimicrobial activity against the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus, and Gram-negative bacteria Pseudomonas aeruginosa, Klebsiella pneumoniae, dermatophytes Microsporum audouinii, Trichophyton mentagrophytes, diamorphic fungi Candida albicans, and other fungal strains, including Aspergillus niger at concentrations of 50–6.25 μg/mL by using the modified Kirby-Bauer disc diffusion method [49]. The minimum inhibitory concentration (MIC) values of the test compounds were determined by the tube dilution technique. Gatifloxacin and griseofulvin were used as the reference drugs and DMF/DMSO were used as the control. The results of the antibacterial and antifungal studies are presented in Table 2.
3. Discussion
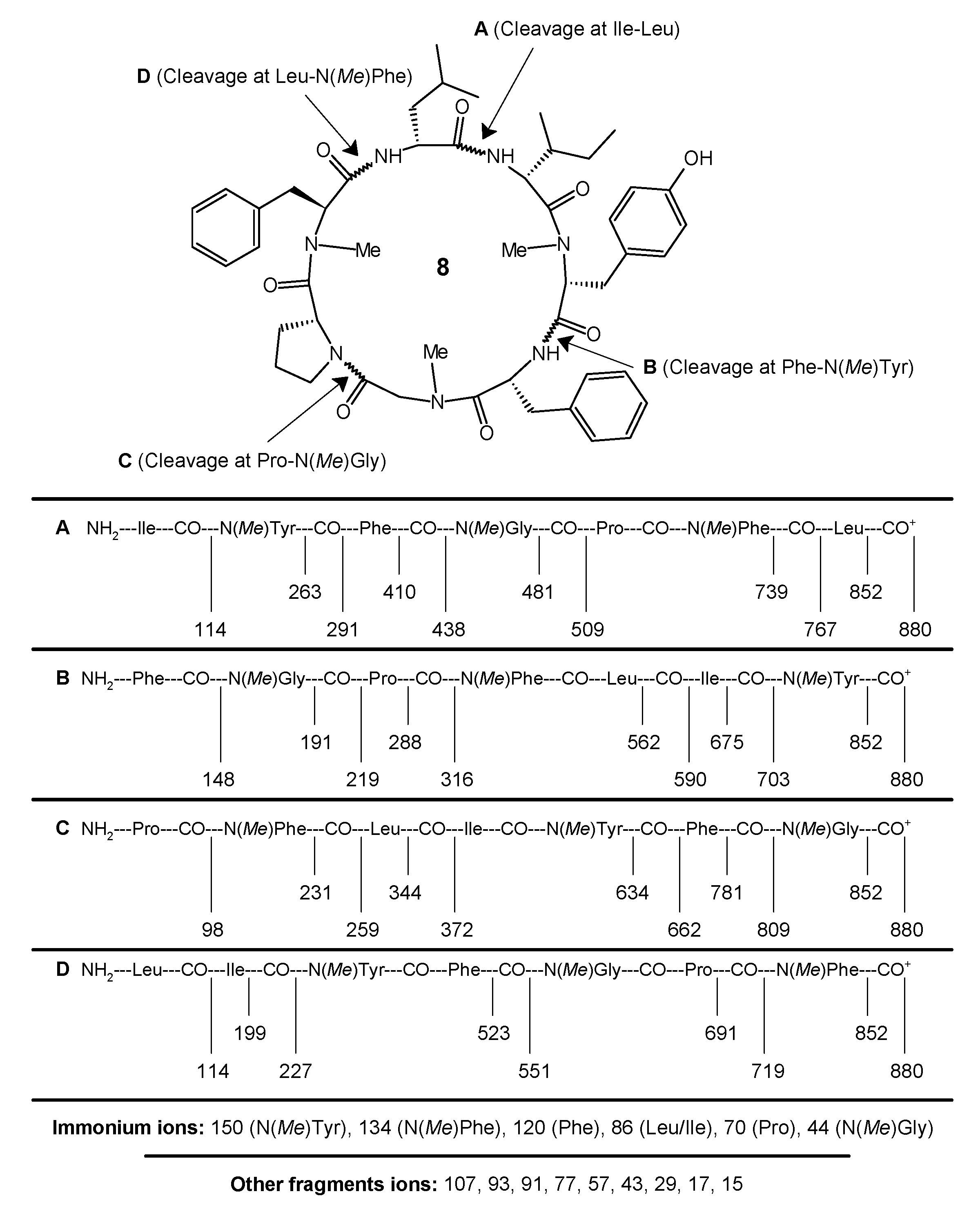
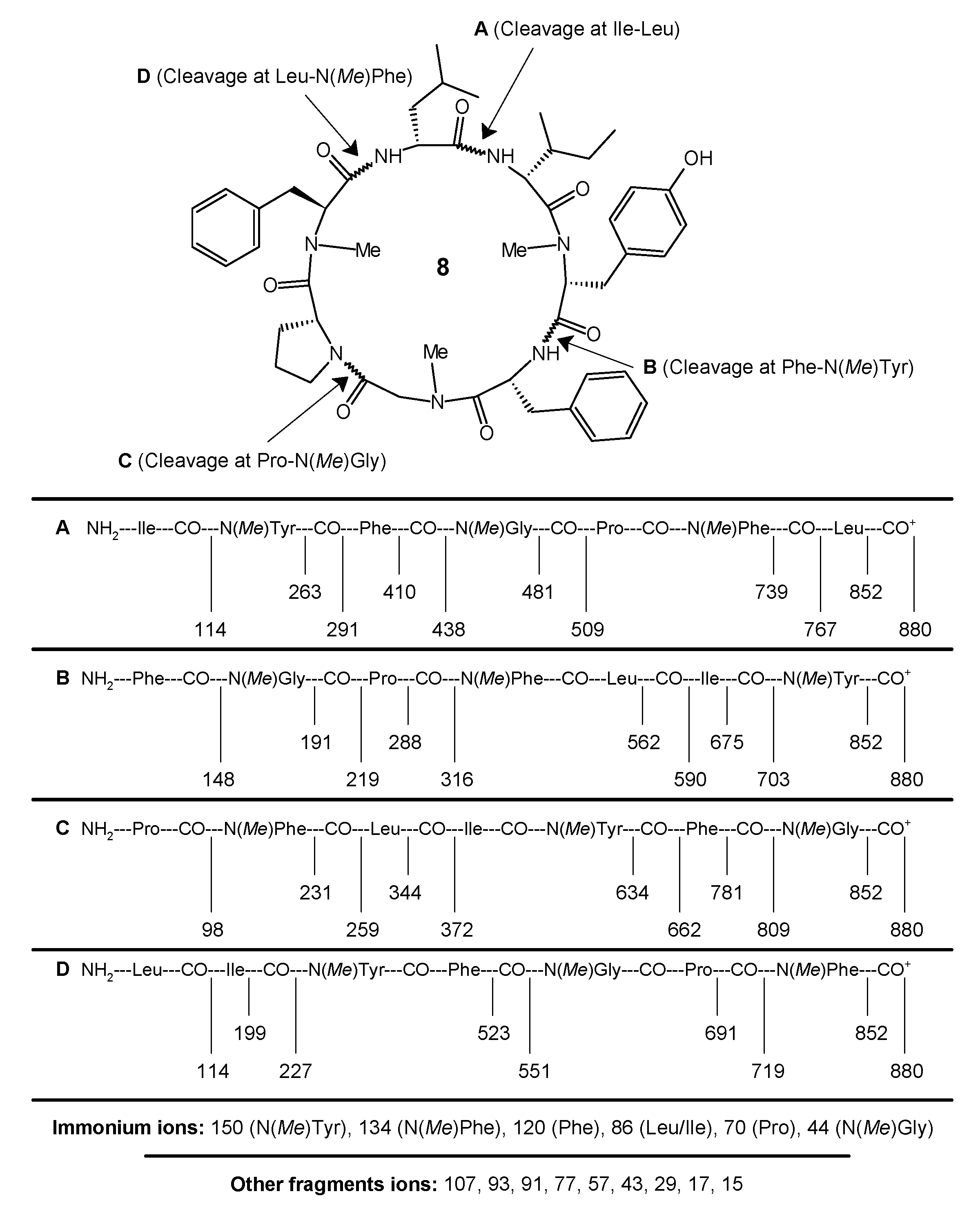
The synthesis of the N-methylated cyclopolypeptide 8 was accomplished with 83% yield, and the N-methylmorpholine proved to be an effective base for the cyclization of the linear heptapeptide unit. The cyclization of the linear peptide fragment was supported by the disappearance of the absorption bands at 1744, 1268, 1393, 1381, and 939 cm−1 (C=Ostr, C–Ostr, ester and C–Hbend, CH3(rock), tert-butyl group) in the IR spectra of 8. The formation of the cyclopeptide was further confirmed by the disappearance of the singlets at 3.56 and 1.51 ppm corresponding to the three protons of the methyl ester group and the nine protons of the tert-butyl group of the Boc in the 1H-NMR spectrum and the disappearance of the singlets at 154.5 and 79.8, and 52.9 and 28.3 ppm corresponding to the carbon atoms of the ester and tert-butyl groups in the 13C-NMR spectrum of 8, respectively. Furthermore, the 1H-NMR and 13C-NMR spectra of the synthesized cyclic heptapeptide showed the characteristic peaks confirming the presence of all of the 65 protons and 49 carbon atoms. A large 13C chemical shift difference in the 13C chemical shifts between the Cβ and Cγ signals of the proline residue (Δδβγ = 32.0 − 23.6 = 8.4) indicates a cis configuration for the proline residue in the synthesized N-methylated cyclopolypeptide 8. The retention times (tR, min) of the observed peaks of the 1-fluoro-2-4-dinitrophenyl-5-l-alanine amide (FDAA) derivatized hydrolysis products of cyclopeptide 8 were 18.27 for the amino acid gly, 27.23 for l-leu, 28.90 for l-ile, 20.54 for l-tyr, 17.44 for l-phe, 22.36 for d-phe, and 17.42 for l-pro, and were all in agreement with the values for standard amino acid derivatives during the HPLC analysis. The appearance of the pseudomolecular ion peak (M + 1)+ at m/z = 880 corresponding to the molecular formula C49H65N7O8 in the mass spectrum of 8, along with other fragment ion peaks resulting from the cleavage at ‘Phe-N(Me)Tyr’, ‘Pro-N(Me)Gly’, ‘Ile-Leu’, and ‘Leu-N(Me)Phe’ amide bonds showed the exact sequence of the attachment of all of the seven amino acid moieties in a chain (Figure 2). In addition, the presence of the immonium ion peaks at m/z 150 [N(Me)Tyr], 134 [N(Me)Phe], 120 [Phe], 86 [Leu/Ile], 70 [Pro], and 44 [N(Me)Gly] further confirmed all the amino acid moieties in the cyclopeptide structure. Furthermore, the elemental analysis of 8 afforded the values with a tolerance of ±0.02 strictly in accordance with the molecular composition.
Comparison of the antibacterial activity data suggested that the cycloheptapeptide 8 possessed potent bioactivity against the Gram-negative bacteria K. pneumonia and P. aeruginosa, in addition to a satisfactory level of activity against the Cutaneous fungi M. audouinii and T. mentagrophytes with the MIC values of 6 μg/mL when compared to the reference drugs, gatifloxacin and griseofulvin. No significant level of bioactivity was observed against the pathogenic fungi C. albicans and A. niger and the Gram-positive bacteria B. subtilis and S. aureus, in comparison to the standard drugs. Moreover, the newly synthesized N-methylated cyclopeptide 8 exhibited a good level of cytotoxic activity against the DLA and EAC cell lines with CTC50 values of 10.6 and 14.6 μM, respectively, in comparison to the standard drug, 5-fluorouracil (5-FU) (CTC50 values of 37.4 and 90.6 μM, respectively). The possible mechanism of the cytotoxic action of cycloheptapeptide 8 might be through apoptosis via induction of the early cell death, nuclear fragmentation, and the internucleosomal DNA scission. In addition, the analysis of the pharmacological activity data revealed that cycloheptapeptide 8 displayed more bioactivity against the pathogenic microbes and cell lines when compared to its linear form 7. The enhanced activity is due to the reduction in the degree of freedom for each constituent within the ring by the cyclization of the peptides. Usually, cyclic peptides show better biological activity compared to their linear counterparts due to the conformational rigidity, which decreases the entropy term of the Gibbs free energy, therefore allowing the enhanced binding toward target molecules, or receptor selectivity. Another advantage from the cyclic structure is the resistance to hydrolysis by the exopeptidases due to the lack of both amino and carboxyl termini. Furthermore, the cyclic peptides can be resistant even to the endopeptidases, as the structure is less flexible than the linear peptides [50]. Cordyheptapeptide A differs from the cordyheptapeptide B in one amino acid moiety i.e., N-methyl-l-tyrosine in place of the N-methyl-l-phenylalanine residue. This structural change results in the enhanced bioactivity against the Gram-negative bacteria and variation in the cytotoxic properties against the DLA and EAC cell lines [14].
4. Materials and Methods
Amino acids, trifluoroacetic acid, pentafluorophenol (pfp), p-nitrophenol (pnp), pyridine (C5H5N), N-methylmorpholine (NMM), triethylamine (TEA), Diisopropylcarbodiimide (DIPC), Dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), and di-tert-butyl-pyrocarbonate (Boc2O) were purchased from Spectrochem Limited (Mumbai, India). The IR spectra were recorded on a Shimadzu 8700 FTIR spectrophotometer (Shimadzu, Japan) using a thin film supported on KBr pellets for the synthesized cyclic heptapeptide and CHCl3 as the solvent for the intermediate semisolids. The 1H-NMR and 13C-NMR spectra were recorded on a Bruker AC-NMR spectrometer (Brucker, Billerica, MA, USA) at 300 MHz using CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. The mass spectrum was recorded on a JMS-DX 303 Mass spectrometer (Jeol, Tokyo, Japan) operating at 70 eV using the fast atom bombardment technique.
4.1. General Method for the Synthesis of Linear N-methylated Tri/Tetrapeptide Units (5, 6)
The N-methyl amino acid methyl ester hydrochloride or dipeptide methyl ester (0.01 mol) was dissolved in DMF (25 mL). To this, TEA (0.021 mol) was added at 0 °C and the reaction mixture was stirred for 15 min. The Boc-dipeptide (0.01 mol) in the DMF (25 mL), DIPC (1.26 g, 0.01 mol), and HOBt (1.34 g, 0.01 mol) were added with stirring. The stirring was first done for 1 h at 0–5 °C and then for a further 24 h at room temperature (RT). After the completion of the reaction, the reaction mixture was diluted with an equal amount of water. The precipitated solid/semisolid was then filtered/separated and washed with water, and purified from the mixture of CHCl3-petroleum ether (b.p. 60–80 °C) followed by cooling at 0 °C to obtain the title compounds.
4.1.1. Tert-Butyloxycarbonyl-l-phenylalanyl-N(Me)glycyl-l-prolyl-d-N(Me)phenylalanine Methyl Ester (5)
Semisolid mass; Yield 79%; [α]D = −52.3° (c = 0.25, CHCl3); Rf = 0.48 (CHCl3·MeOH—8:2); IR (CHCl3): v 3136 (m, -NH str, amide), 2999, 2996 (m, -CH str, cyclic CH2, Pro), 2927, 2622, 2854–2851 (m, -CH str, asym and sym, CH2), 2870, 2866 (m, -CH str, sym, CH3), 1742 (s, -C=O str, ester), 1664–1659, 1642 (s, -C=O str, 3° and 2° amide), 1587, 1479 (m, skeletal bands, arom. rings), 1532 (m, -NH bend, 2° amide), 1391, 1376 (s, -CH bend, tert-Butyl group), 1270 (s, C-O str, ester), 933 (w, CH3 rocking, tert-Butyl group), 726–722, 690, 687 (s, -CH bend, out-of-plane, arom. ring) cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.52–7.48 (2H, tt, J = 6.8, 4.45 Hz, m-H’s, Phe-1), 7.15–7.12 (1H, t, J = 6.2 Hz, p-H, Phe-2), 7.05–7.01 (2H, tt, J = 6.75, 4.5 Hz, m-H’s, Phe-2), 6.93–6.90 (1H, t, J = 6.25 Hz, p-H, Phe-1), 6.86–6.84 (2H, dd, J = 8.8, 4.15 Hz, o-H’s, Phe-1), 6.75–6.73 (2H, dd, J = 8.75, 4.2 Hz, o-H’s, Phe-2), 6.51 (1H, br. s, -NH, Phe-1), 5.39–5.36 (1H, t, J = 5.2 Hz, α-H, Phe-2), 4.87–4.83 (1H, q, J = 5.55 Hz, α-H, Phe-1), 4.45–4.42 (1H, t, J = 6.9 Hz, α-H, Pro), 3.89 (2H, s, α-H, Gly), 3.59–3.56 (2H, t, J = 7.15 Hz, δ-H’s, Pro), 3.54 (3H, s, OCH3), 3.17–3.11 (4H, m, β-H’s, Phe-1 and Phe-2), 2.99 (3H, s, NCH3, Phe), 2.94 (3H, s, NCH3, Gly), 2.69–2.64 (2H, q, β-H’s, Pro), 1.96–1.89 (2H, m, γ-H’s, Pro), 1.52 (9H, s, tert-Butyl group) ppm; C33H44N4O7 (608): calcd. C 65.11, H 7.29, N 9.20; found C 65.10, H 7.32, N 9.22.
4.1.2. Tert-Butyloxycarbonyl-l-leucyl-l-isoleucyl-l-N(Me)tyrosine Methyl Ester (6)
Semisolid mass; Yield 83%; [α]D = +81.3° (c = 0.25, CHCl3); Rf = 0.71 (CHCl3·MeOH - 9:1); IR (CHCl3): v 3372 (m, -OH str, arom. ring), 3136, 3132 (m, -NH str, amide), 2929, 2854 (m, -CH str, asym and sym, CH2), 2967–2963, 2869, 2863 (m, -CH str, asym and sym, CH3), 1745 (s, -C=O str, ester), 1667, 1644–1641 (s, -C=O str, 3° and 2° amide), 1588, 1471 (m, skeletal bands, arom. ring), 1539, 1535 (m, -NH bend, 2° amide), 1466 (m, -CH bend (scissoring), CH2), 1393, 1375 (s, -CH bend, tert-Butyl group), 1379, 1364 (s, -CH bend, iso-propyl group), 1274 (s, C-O str, ester), 933, 920 (w, CH3 rocking, tert-Butyl and iso-propyl groups), 729, 685 (s, -CH bend, oop, arom. ring) cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.09 (1H, br. s, -NH, Ile), 6.87–6.83 (2H, dd, J = 8.6, 4.85 Hz, m-H’s, Tyr), 6.79–6.75 (2H, dd, J = 8.55, 5.25 Hz, o-H’s, Tyr), 6.06 (1H, br. s, -NH, Leu), 5.95 (1H, br. s, -OH, Tyr), 4.46–4.43 (1H, t, J = 5.8 Hz, α-H, Tyr), 4.37–4.34 (1H, t, J = 8.6 Hz, α-H, Ile), 4.22–4.18 (1H, q, J = 6.85 Hz, α-H, Leu), 3.57 (3H, s, OCH3), 3.11–3.09 (2H, d, J = 5.8 Hz, β-H’s, Tyr), 3.04 (3H, s, NCH3), 2.06–1.95 (3H, m, β-H’s, Leu and Ile), 1.65–1.59 (2H, m, γ-H’s, Ile), 1.58–1.53 (1H, m, γ-H’s, Leu), 1.50 (9H, s, tert-Butyl group), 1.04–1.02 (3H, d, J = 5.9 Hz, γ′-H’s, Ile), 1.01–0.99 (6H, d, J = 6.25 Hz, δ-H’s, Leu), 0.95–0.93 (3H, d, J = 7.75 Hz, δ-H’s, Ile) ppm; C28H45N3O7 (535): calcd. C 62.78, H 8.47, N 7.84; found C 62.75, H 8.49, N 7.85.
4.2. Deprotection of the Tetrapeptide Unit (5) at the Amino End and Tripeptide Unit (6) at the Carboxyl End
The Boc-protected tetrapeptide 5 (6.08 g, 0.01 mol) was dissolved in CHCl3 (15 mL) and treated with CF3COOH (2.28 g, 0.02 mol). The resulting solution was stirred at room temperature for 1 h, and then washed with a saturated NaHCO3 solution (25 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by the crystallization from CHCl3 and petroleum ether (b.p. 40–60 °C) to obtain the pure deprotected compound 5a.
For deprotection at the carboxyl end, LiOH (0.36 g, 0.015 mol) was added to a solution of tripeptide 6 (5.35 g, 0.01 mol) in THF·H2O (1:1, 36 mL) at 0 °C. The mixture was stirred at room temperature for 1 h and then acidified to pH 3.5 with 1 M H2SO4. The aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was finally crystallized from methanol and ether to obtain the pure deprotected compound 6a.
4.2.1. l-Phenylalanyl-N(Me)glycyl-l-prolyl-d-N(Me)phenylalanine Methyl Ester (5a)
Semisolid mass; Yield 83%; [α]D = –84.1° (c = 0.25, CHCl3); Rf = 0.61 (CHCl3·MeOH - 8:2); IR (CHCl3): v 3496, 3389 (w, -NH str, amine), 2997, 2995 (m, -CH str, cyclic CH2, Pro), 2926–2622, 2855–2849 (m, -CH str, asym and sym, CH2), 2868, 2865 (m, -CH str, sym, CH3), 1739 (s, -C=O str, ester), 1667–1662, 1643 (s, -C=O str, 3° and 2° amide), 1616 (m, -NH bend, amine), 1589, 1477 (m, skeletal bands, arom. rings), 1534 (m, -NH bend, 2° amide), 1334 (m, -CN str, amine), 1269 (s, C-O str, ester), 725–722, 689, 685 (s, -CH bend, out-of-plane, arom. ring) cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.29–7.25 (2H, tt, J = 6.75, 4.5 Hz, m-H’s, Phe-1), 7.17–7.14 (1H, t, J = 6.15 Hz, p-H, Phe-2), 7.06–7.02 (2H, tt, J = 6.8, 4.45 Hz, m-H’s, Phe-2), 6.97–6.94 (1H, t, J = 6.2 Hz, p-H, Phe-1), 6.77–6.69 (4H, m, o-H’s, Phe-1 and Phe-2), 5.38–5.35 (1H, t, J = 5.15 Hz, α-H, Phe-2), 4.46–4.43 (1H, t, J = 6.85 Hz, α-H, Pro), 3.98–3.93 (1H, m, α-H, Phe-1), 3.82 (2H, s, α-H, Gly), 3.61–3.58 (2H, t, J = 7.2 Hz, δ-H’s, Pro), 3.52 (3H, s, OCH3), 3.07–3.05 (2H, d, J = 5.6 Hz, β-H’s, Phe-2), 2.97 (3H, s, NCH3, Phe-2), 2.92 (3H, s, NCH3, Gly), 2.75–2.73 (2H, d, J = 5.65 Hz, β-H’s, Phe-1), 2.68–2.63 (2H, q, β-H’s, Pro), 2.25 (2H, s, NH2, Phe-1), 1.95–1.89 (2H, m, γ-H’s, Pro) ppm; C28H36N4O5 (508): calcd. C 66.12, H 7.13, N 11.02; found C 66.10, H 7.15, N 11.05.
4.2.2. Tert-Butyloxycarbonyl-l-leucyl-l-isoleucyl-l-N(Me)tyrosine (6a)
Semisolid mass; Yield 79%; [α]D = +67.7° (c = 0.25, CHCl3); Rf = 0.82 (CHCl3·MeOH—9:1); IR (CHCl3): v 3315–2530 (m/br, -OH str, -COOH), 3375 (m, -OH str, Tyr), 3133, 3129 (m, -NH str, amide), 2927, 2856 (m, -CH str, asym and sym, CH2), 2966–2962, 2868–2863 (m, -CH str, asym and sym, CH3), 1710 (m, -C=O str, -COOH), 1668, 1644–1639 (s, -C=O str, 3° and 2° amide), 1589, 1469 (m, skeletal bands, arom. ring), 1536–1532 (m, -NH bend, 2° amide), 1462 (m, -CH bend (scissoring), CH2), 1423 (m, C-O-H bend, -COOH), 1392, 1377 (s, -CH bend, tert-Butyl group), 1378, 1362 (s, -CH bend, iso-propyl group), 936, 919 (w, CH3 rocking, tert-Butyl and iso-propyl groups), 727, 683 (s, -CH bend, oop, arom. ring) cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.19 (2H, br. s, -OH, Tyr and -COOH), 7.08 (1H, br. s, -NH, Ile), 6.82–6.78 (2H, dd, J = 8.55, 4.9 Hz, m-H’s, Tyr), 6.93–6.89 (2H, dd, J = 8.6, 5.3 Hz, o-H’s, Tyr), 6.02 (1H, br. s, -NH, Leu), 5.35–5.32 (1H, t, J = 8.55 Hz, α-H, Ile), 4.52–4.49 (1H, t, J = 5.75 Hz, α-H, Tyr), 4.19–4.15 (1H, q, J = 6.9 Hz, α-H, Leu), 3.17 (3H, s, NCH3), 3.13–3.11 (2H, d, J = 5.75 Hz, β-H’s, Tyr), 2.05–1.96 (3H, m, β-H’s, Leu and Ile), 1.67–1.61 (2H, m, γ-H’s, Ile), 1.59–1.54 (1H, m, γ-H’s, Leu), 1.52 (9H, s, tert-Butyl group), 1.03–1.01 (3H, d, J = 5.85 Hz, γ′-H’s, Ile), 0.99–0.97 (6H, d, J = 6.3 Hz, δ-H’s, Leu), 0.93–0.91 (3H, d, J = 7.8 Hz, δ-H’s, Ile) ppm; C27H43N3O7 (521): calcd. C 62.17, H 8.31, N 8.06; found C 62.19, H 8.32, N 8.09.
4.3. Procedure for the Preparation of the Linear Heptapeptide Unit and Its Cyclized Form (7, 8)
The tetrapeptide methyl ester, l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-OMe (5a, 5.08 g, 0.01 mol) was dissolved in 30 mL of THF, 0.021 mol of NMM was added at 0 °C, and the resulting mixture was stirred for 15 min. The Boc-protected tripeptide, Boc-l-Leu-l-Ile-l-N(Me)Tyr-OH (6a, 5.21 g, 0.01 mol) was dissolved in 30 mL of THF and DIPC/EDC·HCl (1.26 g/1.92 g, 0.01 mol) and HOBt (1.34 g, 0.01 mol) were added to the above mixture with stirring. The stirring was continued for 24 h, after which the reaction mixture was filtered and the filtrate was washed with 25 mL each of the 5% NaHCO3 and saturated NaCl solutions. The organic layer was dried over the anhydrous Na2SO4, filtered, and evaporated in vacuum. Finally, the recrystallization of the crude product was carried out from a mixture of the CHCl3-petroleum ether (b.p. 40–60 °C) followed by cooling at 0 °C to obtain the Boc-l-Leu-l-Ile-l-N(Me)Tyr-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-OMe (7) as the pale-yellow semisolid mass. The linear heptapeptide unit (7, 5.06 g, 0.005 mol) was then deprotected at the carboxyl terminal using the lithium hydroxide (LiOH, 0.18 g, 0.0075 mol) to obtain Boc-l-Leu-l-Ile-l-N(Me)Tyr-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-OH. To a solution of the deprotected heptapeptide (4.99 g, 0.005 mol) in CHCl3 (50 mL), pentafluorophenol (pfp, 1.23 g, 0.0067 mol) and DCC (1.06 g, 0.005 mol) were added followed by stirring at room temperature for 12 h. The filtrate of the above reaction mixture was washed with 10% NaHCO3 (3 × 20 mL) and 5% HCl (2 × 20 mL) solutions to obtain the corresponding pentafluorophenyl ester Boc-l-Leu-l-Ile-l-N(Me)Tyr-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-Opfp. The Boc-group of the resulting unit (4.66 g, 0.004 mol) was removed using CF3COOH (0.91 g, 0.008 mol) to obtain the deprotected product, l-Leu-l-Ile-l-N(Me)Tyr-l-Phe-N(Me)Gly-l-Pro-d-N(Me)Phe-Opfp, which was dissolved in CHCl3 (25 mL) and TEA/NMM/pyridine (2.8 mL/2.21 mL/1.61 mL, 0.021 mol) was added. Then, the entire contents were kept at 0 °C for 7 days. Then the reaction mixture was washed with 10% NaHCO3 (3 × 25 mL) and 5% HCl (2 × 25 mL) solutions. The organic layer was dried over anhydrous Na2SO4 and the crude cyclized compound was recrystallized from the CH2Cl2/n-hexane to obtain the cyclic product cyclo(l-leucyl-l-isoleucyl-l-N(Me)tyrosyl-l-phenylalanyl-N(Me)glycyl-l-prolyl-d-N(Me)phenylalanyl) (8).
4.3.1. Tert-Butyloxycarbonyl-l-leucyl-l-isoleucyl-l-N(Me)tyrosyl-l-phenylalanyl-N(Me)glycyl-l-prolyl-d-N(Me)phenylalanine Methyl Ester (7)
Semisolid mass; Yield 78%; [α]D = −103.2° (c = 0.25, MeOH); Rf = 0.73 (CHCl3·MeOH—9:1); IR (CHCl3): v 3369 (m, -OH str, arom. ring), 3135–3129 (m, -NH str, amide), 2998–2994 (m, -CH str, cyclic CH2, Pro), 2928, 2625, 2853–2849 (m, -CH str, asym and sym, CH2), 2968, 2962, 2872, 2866 (m, -CH str, asym and sym, CH3), 1744 (s, -C=O str, ester), 1666–1659, 1643–1639 (s, -C=O str, 3° and 2° amide), 1589–1584, 1477–1473 (m, skeletal bands, arom. rings), 1537–1533 (m, -NH bend, 2° amide), 1393, 1381 (s, -CH bend, tert-Butyl group), 1375, 1364 (s, -CH bend, iso-propyl group), 1268 (s, C-O str, ester), 939, 921 (w, CH3 rocking, tert-Butyl and iso-propyl groups), 728–723, 689, 685 (s, -CH bend, out-of-plane, arom. rings) cm−1; 1H-NMR (300 MHz, CDCl3): δ 9.22 (1H, br. s, -NH, Phe-1), 7.19–7.16 (2H, tt, J = 6.75, 4.45 Hz, m-H’s, Phe-1), 7.14–7.11 (1H, t, J = 6.15 Hz, p-H, Phe-2), 7.08 (1H, br. s, -NH, Ile), 7.06–7.03 (2H, tt, J = 6.8, 4.5 Hz, m-H’s, Phe-2), 7.02–6.97 (2H, dd, J = 8.55, 4.9 Hz, m-H’s, Tyr), 6.92–6.89 (1H, t, J = 6.3 Hz, p-H, Phe-1), 6.85–6.83 (2H, dd, J = 8.75, 4.15 Hz, o-H’s, Phe-1), 6.74–6.69 (2H, dd, J = 8.6, 5.25 Hz, o-H’s, Tyr), 6.75–6.73 (2H, dd, J = 8.8, 4.15 Hz, o-H’s, Phe-2), 6.05 (1H, br. s, -NH, Leu), 5.97 (1H, br. s, -OH, Tyr), 5.38–5.35 (1H, t, J = 5.15 Hz, α-H, Phe-2), 4.89–4.85 (1H, q, J = 5.6 Hz, α-H, Phe-1), 4.45–4.42 (1H, t, J = 8.55 Hz, α-H, Ile), 4.40–4.37 (1H, t, J = 6.85 Hz, α-H, Pro), 4.35–4.32 (1H, t, J = 5.75 Hz, α-H, Tyr), 4.20–4.16 (1H, q, J = 6.9 Hz, α-H, Leu), 3.91 (2H, s, α-H, Gly), 3.61–3.58 (2H, t, J = 7.2 Hz, δ-H’s, Pro), 3.56 (3H, s, OCH3), 3.11–3.07 (4H, m, β-H’s, Phe-1 and Phe-2), 3.04 (3H, s, NCH3, Tyr), 2.99 (3H, s, NCH3, Phe-2), 2.95 (3H, s, NCH3, Gly), 2.89–2.87 (2H, d, J = 5.75 Hz, β-H’s, Tyr), 2.70–2.65 (2H, q, β-H’s, Pro), 2.05–1.93 (3H, m, β-H’s, Leu and Ile), 1.91–1.86 (2H, m, γ-H’s, Pro), 1.67–1.61 (2H, m, γ-H’s, Ile), 1.58–1.53 (1H, m, γ-H’s, Leu), 1.51 (9H, s, tert-Butyl group), 1.07–1.05 (3H, d, J = 5.85 Hz, γ′-H’s, Ile), 1.03–1.01 (6H, d, J = 6.3 Hz, δ-H’s, Leu), 0.99–0.97 (3H, d, J = 7.8 Hz, δ-H’s, Ile) ppm; 13C-NMR (CDCl3): δ = 175.3 (C=O, Tyr), 172.6 (C=O, Leu), 170.5, 169.5, 167.3 (3 C, C=O, Phe-1, Ile and Phe-2), 166.9, 165.1 (2 C, C=O, Gly and Pro), 156.2 (p-C, Tyr), 154.5 (C=O, Boc), 139.2 (γ-C, Phe-2), 136.6 (γ-C, Phe-1), 130.8 (2 C, o-C’s, Tyr), 129.6 (2 C, o-C’s, Phe-2), 128.5 (2 C, m-C’s, Tyr), 128.1 (2 C, o-C’s, Phe-1), 127.4 (2 C, m-C’s, Phe-1), 126.9 (2 C, m-C’s, Phe-2), 126.5 (γ-C, Tyr), 126.1, 125.6 (2 C, γ-C’s, Phe-1 and Phe-2), 79.8 (α-C, Boc), 60.1, 57.7, 54.5 (3 C, α-C’s, Tyr, Phe-2 and Pro), 52.9 (OCH3), 51.3, 48.2 (2 C, α-C’s, Leu and Ile), 46.8, 45.3 (2 C, α-C, Gly and Phe-1), 39.9 (δ-C, Pro), 38.0, 37.2 (2 C, β-C’s, Leu and Phe-1), 36.2 (NCH3, Gly), 34.4, 33.6, 32.3 (3 C, β-C’s, Ile, Tyr and Phe-2), 31.5, 29.7 (2 C, NCH3, Phe-2 and Tyr), 29.2 (β-C, Pro), 28.3 (3 C, β-C’s, Boc), 26.6, 25.2 (2 C, γ-C’s, Pro and Ile), 24.4 (2 C, δ-C’s, Leu), 22.1 (γ-C, Leu), 17.4 (γ′-C, Leu), 10.1 (δ-C, Ile); C55H77N7O11 (1012): calcd. C 65.26, H 7.67, N 9.69; found C 65.25, H 7.65, N 9.72.
4.3.2. Cyclo(l-leucyl-l-isoleucyl-l-N(Me)tyrosyl-l-phenylalanyl-N(Me)glycyl-l-prolyl-d-N(Me)phenylalanyl) (8)
Pale yellow crystals; m.p. 178–180 °C (179.6–179.8 °C for natural cordyheptapeptide A [13]); Yield 83% (NMM), 79% (C5H5N), 68% (TEA); [α]D = −68.6° (c = 0.56, CHCl3) (–68.5° for natural cordyheptapeptide A [12]); Rf = 0.59 (CHCl3·MeOH—9:1); IR (KBr): v 3365 (m, -OH str, arom. ring), 3133, 3129–3126 (m, -NH str, amide), 2999–2993 (m, -CH str, cyclic CH2, Pro), 2929–2624, 2855, 2849 (m, -CH str, asym and sym, CH2), 2969–2963, 2874, 2867 (m, -CH str, asym and sym, CH3), 1668–1662, 1644, 1632 (s, -C=O str, 3° and 2° amide), 1588–1582, 1479–1474 (m, skeletal bands, arom. rings), 1539–1534 (m, -NH bend, 2° amide), 1376, 1364 (s, -CH bend, iso-propyl group), 923 (w, CH3 rocking, iso-propyl group), 729–722, 686, 682 (s, -CH bend, out-of-plane, arom. rings) cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.64 (1H, br. s, -NH, Phe), 8.23 (1H, br. s, -NH, Leu), 7.65 (2H, tt, J = 6.75, 4.5 Hz, m-H’s, Phe-2), 7.44 (2H, tt, J = 6.8, 4.45 Hz, m-H’s, Phe-1), 7.23 (1H, t, J = 6.15 Hz, p-H, Phe-2), 7.01 (2H, dd, J = 8.6, 4.85 Hz, m-H’s, Tyr), 6.85 (1H, t, J = 6.25 Hz, p-H, Phe-1), 6.71 (2H, dd, J = 8.8, 4.15 Hz, o-H’s, Phe-1), 6.46 (2H, dd, J = 8.55, 5.3 Hz, o-H’s, Tyr), 6.37 (2H, dd, J = 8.8, 4.15 Hz, o-H’s, Phe-2), 5.95 (1H, br. s, -OH, Tyr), 5.89 (1H, br. s, -NH, Ile), 5.59 (1H, t, J = 5.2 Hz, α-H, Phe-2), 5.44 (2H, s, α-H, Gly), 5.37 (1H, q, J = 5.55 Hz, α-H, Phe-1), 4.95 (1H, q, J = 6.85 Hz, α-H, Leu), 4.46 (1H, t, J = 8.6 Hz, α-H, Ile), 4.35 (1H, t, J = 6.85 Hz, α-H, Pro), 3.64 (2H, t, J = 7.15 Hz, δ-H’s, Pro), 3.45 (1H, t, J = 5.8 Hz, α-H, Tyr), 3.07 (3H, s, NCH3, Phe), 2.94 (3H, s, NCH3, Gly), 2.74 (2H, q, β-H’s, Pro), 2.62 (3H, s, NCH3, Tyr), 2.46 (2H, d, J = 5.8 Hz, β-H’s, Tyr), 2.13 (4H, m, β-H’s, Phe-1 and Phe-2), 1.75 (2H, m, γ-H’s, Pro), 1.61 (2H, m, γ-H’s, Ile), 1.47 (3H, m, β-H’s, Leu and Ile), 1.25 (6H, d, J = 6.25 Hz, δ-H’s, Leu), 1.13 (3H, d, J = 5.9 Hz, γ′-H’s, Ile), 0.97 (3H, d, J = 7.75 Hz, δ-H’s, Ile), 0.85 (1H, m, γ-H’s, Leu) ppm; 13C-NMR (CDCl3): δ = 174.4 (C=O, Leu), 172.5 (C=O, Pro), 170.7 (C=O, Ile), 170.4 (C=O, Tyr), 170.1, 168.7 (2 C, C=O, Phe and NMePhe), 168.2 (C=O, Gly), 155.9 (p-C, Tyr), 139.4 (γ-C, Phe-2), 137.3 (γ-C, Phe-1), 133.1 (γ-C, Tyr), 132.3 (2 C, m-C’s, Phe-1), 130.2 (2 C, o-C’s, Tyr), 129.8 (2 C, m-C’s, Tyr), 128.9 (2 C, o-C’s, Phe-1), 128.1 (2 C, m-C’s, Phe-2), 127.2 (2 C, m-C’s, Phe-1), 122.9, 122.2 (2 C, γ-C’s, Phe-2 and Phe-1), 68.9, 58.6, 57.9, 54.8 (4 C, α-C’s, Tyr, Ile, Pro and Phe-2), 50.6, 49.9, 47.3 (3 C, α-C’s, Gly, Phe-1 and Leu), 43.5 (δ-C, Pro), 42.9, 42.3 (2 C, β-C’s, Leu and Phe-1), 40.1 (NCH3, Tyr), 39.8, 36.9 (2 C, β-C’s, Tyr and Phe-2), 36.6 (β-C, Ile), 35.7 (NCH3, Phe), 32.0 (β-C, Pro), 30.3 (NCH3, Gly), 29.7, 24.9, 23.6 (3 C, γ-C’s, Leu, Ile and Pro), 22.0 (2 C, δ-C’s, Leu), 17.1 (γ′-C, Leu), 10.3 (δ-C, Ile); MS (FAB, 70 eV): m/z (%) = 880 (100) [M + 1]+, 852 (13) [880-CO]+, 809 (77) [Pro-N(Me)Phe-Leu-Ile-N(Me)Tyr-Phe]+, 781 (29) [809-CO]+, 767 (49) [Ile-N(Me)Tyr-Phe-N(Me)Gly-Pro-N(Me)Phe]+, 739 (21) [767-CO]+, 719 (59) [Leu-Ile-N(Me)Tyr-Phe-N(Me)Gly-Pro]+, 703 (39) [Phe-N(Me)Gly-Pro-N(Me)Phe-Leu-Ile]+, 691 (13) [719-CO]+, 675 (26) [703-CO]+, 662 (52) [Pro-N(Me)Phe-Leu-Ile-N(Me)Tyr]+, 634 (32) [662-CO]+, 606 (69) [Ile-N(Me)Tyr-Phe-N(Me)Gly-Pro]+, 590 (78) [Phe-N(Me)Gly-Pro-N(Me)Phe-Leu]+, 578 (17) [606-CO]+, 562 (24) [590-CO]+, 551 (54) [Leu-Ile-N(Me)Tyr-Phe]+, 523 (25) [551-CO]+, 509 (44) [Ile-N(Me)Tyr-Phe-N(Me)Gly]+, 481 (22) [509-CO]+, 438 (52) [Ile-N(Me)Tyr-Phe]+, 410 (19) [438-CO]+, 372 (44) [Pro-N(Me)Phe-Leu]+, 344 (25) [662-CO]+, 316 (66) [Phe-N(Me)Gly-Pro]+, 291 (41) [Ile-N(Me)Tyr]+, 288 (22) [316-CO]+, 263 (20) [291-CO]+, 259 (39) [Pro-N(Me)Phe]+, 231 (15) [662-CO]+, 227 (37) [Leu-Ile]+, 219 (46) [Phe-N(Me)Gly]+, 199 (21) [227-CO]+, 191 (17) [219-CO]+, 150 (22) [N(Me)Tyr immonium ion, C9H12NO]+, 148 (16) [Phe]+, 134 (26) [N(Me)Phe immonium ion, C9H12N]+, 120 (16) [Phe immonium ion, C8H10N]+, 114 (20) [Ile/Leu]+, 107 (27) [C7H7O]+, 98 (14) [Pro]+, 93 (19) [C6H5O]+, 91 (23) [C7H7]+, 86 (29) [Leu/Ile immonium ion, C5H12N]+, 77 (20) [C6H5]+, 70 (24) [Pro immonium ion, C4H8N]+, 57 (18) [C4H9]+, 44 (9) [N(Me)Gly immonium ion, C2H6N]+, 43 (13) [C3H7]+, 29 (11) [C2H5]+, 17 (10) [OH]+, 15 (16) [CH3]+; C49H65N7O8 (879): calcd. C 66.87, H 7.44, N 11.14; found C 61.85, H 7.47, N 11.15.
4.4. Preparation and Analysis of the Marfey Derivatives
About 0.5 mg of the synthesized cycloheptapeptide 8 was hydrolyzed by heating in 1 mL of 6 M HCl at 110 °C for 24 h. After cooling, the solution was evaporated to dryness and redissolved in 50 μL of water. Then 100 μL of a 1% w/v solution of FDAA (Marfey’s reagent) in acetone was added to the acid hydrolyzate solution (or to 50 μL of a 50 mM solution of the respective amino acid). After addition of 20 μL of the 1 M NaHCO3 solution, the mixture was incubated for 1 h at 40 °C. The reaction was stopped by the addition of 10 μL of 2 M HCl. Finally, the solvents were evaporated to dryness and the residue was dissolved in 1 mL of MeOH. An aliquot of this solution (20 μL for the cyclopeptide 8 and 10 μL for the standards) was analyzed by HPLC (Phenomenex Luna C18, 4.6 × 250 mm, 5 μm, solvents: (A) water + 0.05% TFA, (B) MeCN, linear gradient: 0 min 35% B, 30 min 45% B, 1 mL min−1, 25 °C). The retention times (min) of the FDAA amino acid derivatives used as the standards were as follows: gly (18.24), l-ile (28.92), d-ile (33.32), l-tyr (20.51), d-tyr (33.08), l-leu (27.25), d-leu (25.05), l-phe (17.41), d-phe (22.32), l-pro (17.45), and d-pro (18.39). Retention times (min) and relative peak area (%) of the observed peaks of the FDAA derivatized hydrolysis products of cyclopeptide 8 were as follows: gly (18.27, 2.39%), l-leu (27.23, 2.48%), l-ile (28.90, 3.67%), l-tyr (20.54, 5.24%), l-phe (17.44, 7.33%), d-phe (22.36, 6.26%), and l-pro (17.42, 7.82%).
4.5. Biological Evaluation Procedures
4.5.1. Cytotoxic Screening
The linear and cyclic heptapeptides (7, 8) were subjected to the short term in vitro cytotoxicity study at 62.5–3.91 μg/mL against the DLA and EAC cell lines using 5-fluorouracil (5-FU) as the reference compound. The different dilutions of both compounds ranging from 62.5–3.91 μg/mL were prepared in Dulbecco’s minimum essential medium and 0.1 mL of each diluted test compound was added to 0.1 mL of the DLA cells (1 × 106 cells/mL) and EAC cells (1 × 106 cells/mL). The resulting suspensions were incubated at 37 °C for 3 h followed by performing the tryphan blue dye test and the calculation of the growth inhibition (%). The CTC50 values were determined by the graphical extrapolation method. The controls were also tested at 62.5–3.91 μg/mL against both cell lines. The results of the cytotoxicity studies are listed in Table 1.
4.5.2. Antimicrobial Screening
The newly synthesized linear and cyclic heptapeptides (7, 8) were evaluated for their antibacterial and antifungal potential against two Gram-positive bacteria, B. subtilis and S. aureus, two Gram-negative bacteria, P. aeruginosa and K. pneumoniae, and the diamorphic fungal strain C. albicans and three other fungal strains, including A. niger and two Cutaneous fungal strains M. audouinii and T. mentagrophytes at the concentrations of 50–6.25 μg/mL. The MIC values of the test compounds were determined by the tube dilution technique. Both the linear and cyclic heptapeptides were dissolved separately to prepare a stock solution of 1 mg/mL using the DMF or DMSO. The stock solution was aseptically transferred and suitably diluted with the sterile broth medium to contain seven different concentrations of each test compound ranging from 200–3.1 μg/mL in the different test tubes. The inoculation of all the tubes was carried out with one of the test microbes. The process was repeated with different test bacteria/fungi and the different samples. The tubes inoculated with the bacterial cultures were incubated at 37 °C for 18 h and the fungal cultures were incubated at 37 °C for 48 h. Finally, the presence/absence of growth of the bacteria/fungi was observed. From these results, the MIC of each test compound was determined against each test bacterium/fungus. Gatifloxacin and griseofulvin were used as the reference drugs with the pure solvents (DMF and DMSO) as the negative controls for the antibacterial and antifungal studies, respectively. The Petri plates inoculated with the bacterial cultures were incubated at 37 °C for 18 h and those inoculated with the fungal cultures were incubated at 37 °C for 48 h. The diameters of the inhibition zones (in mm) were measured and the average diameters for the test samples were calculated in triplicate. The diameters obtained for the test samples were compared with that produced by the standard drug. The results of the antimicrobial studies are presented in Table 2.
The experimental details of the biological activity studies are described in our previously published reports [51,52,53,54,55]. As per the International Union of Pure and Applied Chemistry (IUPAC) rules, the N-methylated cycloheptapeptide 8 can be named as “3,15-Dibenzyl-9-(sec-butyl)-12-(4-hydroxybenzyl)-6-isobutyl-2,11,17-trimethylperhydropyrrolo [1,2-a][1,4,7,10,13,16,19]heptaazacyclohenicosine-1,4,7,10,13,16,19-heptaone”.
5. Conclusions
The first successful synthesis of a N-methylated cyclopeptide, cordyheptapeptide A (8), was accomplished in the present study, with reasonable yield via the coupling reactions utilizing different carbodiimides. The EDC·HCl/TEA coupling method proved to be yield-effective, in comparison to the method utilizing the DIPC/TEA or NMM, providing 9–10% extra yield. The pentafluorophenyl ester was shown to be better than the p-nitrophenyl ester, for the activation of the acid functionality of the linear heptapeptide unit. The NMM was found to be a good base for the intramolecular cyclization of the linear peptide fragment in comparison to the TEA or pyridine. The synthesized heptacyclopeptide displayed potent antibacterial activity especially against the Gram-negative bacteria, and effectiveness against the pathogenic dermatophtytes and the significant level of cytotoxicity. The Gram-negative bacteria were found to be more sensitive than the Gram-positive bacteria towards the newly synthesized peptide. On passing the toxicity tests, the N-methylated cyclopeptide 8 may prove to be a good candidate for clinical studies and can be a new antibacterial, anti-dermatophyte, and the anticancer drug of the future.
Supplementary Materials
The following are available online. Figure S1: Mass spectrum for the N-methylated cyclic heptapeptide, cordyheptapeptide A (8), Figure S2: 13C-NMR spectrum for the N-methylated cyclic heptapeptide, cordyheptapeptide A (8), Table S1: Various steric and lipophilicity parameters for the N-methylated linear and cyclic heptapeptide (7, 8).
Acknowledgments
The authors are thankful to the Faculty of Pharmacy, Jamia Hamdard University, Delhi, India for the spectral analysis. Also, the authors wish to thank the J.S.S. College of Pharmacy, Ooty, Tamilnadu, India for the cytotoxic activity studies.
Author Contributions
S.K., S.L.K., and R.D. conceived and designed the experiments; S.K. performed the experiments; S.L.K., S.M., R.M., S.K., and S.V.C. analyzed the data; S.M. and R.M. contributed reagents/materials/analysis tools; S.K. and R.D. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Amedei, A.; D’Elios, M.M. New therapeutic approaches by using microorganism-derived compounds. Curr. Med. Chem. 2012, 19, 3822–3840. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, H.B. Natural products from microorganisms. Science 1980, 208, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Leslie Gunatilaka, A.A. Natural products from plant-associated microorganisms: Distribution, structural diversity, bioactivity, and implications of their occurrence. J. Nat. Prod. 2006, 69, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, P.W.; Blunt, J.W.; Munro, M.H.; Larsen, T.O.; Christophersen, C. Psychrophilin B and C: Cyclic nitropeptides from the psychrotolerant fungus Penicillium rivulum. J. Nat. Prod. 2004, 67, 1950–1952. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Lang, G.; Steffens, S.; Schaumann, K. Petrosifungins A and B, novel cyclodepsipeptides from a sponge-derived strain of Penicillium brevicompactum. J. Nat. Prod. 2004, 67, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, P.W.; Larsen, T.O.; Christophersen, C. Bioactive cyclic peptides from the psychrotolerant fungus Penicillium algidum. J. Antibiot. 2005, 58, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Vongvanich, N.; Kittakoop, P.; Isaka, M.; Trakulnaleamsai, S.; Vimuttipong, S.; Tanticharoen, M.; Thebtaranonth, Y. Hirsutellide A, a new antimycobacterial cyclohexadepsipeptide from the entomopathogenic fungus Hirsutella kobayasii. J. Nat. Prod. 2002, 65, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Chen, A.J.; Lan, J.; Zhang, H.; Chen, X.; Liu, X.; Zou, Z. Sesquiterpenes and cyclopeptides from the endophytic fungus Trichoderma asperellum Samuels, Lieckf. & Nirenberg. Chem. Biodivers. 2012, 9, 1205–1212. [Google Scholar] [PubMed]
- Zou, X.; Niu, S.; Ren, J.; Li, E.; Liu, X.; Che, Y. Verrucamides A-D, antibacterial cyclopeptides from Myrothecium verrucaria. J. Nat. Prod. 2011, 74, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Liu, H.; Liu, X.; Che, Y. Cycloaspeptides F and G, cyclic pentapeptides from a Cordyceps-colonizing isolate of Isaria farinosa. J. Nat. Prod. 2009, 72, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Bao, J.; Zhang, X.Y.; Tu, Z.C.; Shi, Y.M.; Qi, S.H. Asperterrestide A, a cytotoxic cyclic tetrapeptide from the marine-derived fungus Aspergillus terreus SCSGAF0162. J. Nat. Prod. 2013, 76, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Pathak, D. Synthesis, characterization and biological evaluation of halolitoralin B—A natural cyclic peptide. Asian J. Chem. 2007, 19, 1499–1505. [Google Scholar]
- Rukachaisirikul, V.; Chantaruk, S.; Tansakul, C.; Saithong, S.; Chaicharernwimonkoon, L.; Pakawatchai, C.; Isaka, M.; Intereya, K. A cyclopeptide from the insect pathogenic fungus Cordyceps sp. BCC 1788. J. Nat. Prod. 2006, 69, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Isaka, I.; Srisanoh, U.; Lartpornmatulee, N.; Boonruangprapa, T. ES-242 derivatives and cycloheptapeptides from Cordyceps sp. strains BCC 16173 and BCC 16176. J. Nat. Prod. 2007, 70, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Gautam, H. Solution phase synthesis and bioevaluation of cordyheptapeptide B. Bull. Pharm. Res. 2011, 1, 1–10. [Google Scholar]
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C-E, from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Y.; Dahiya, R.; Qin, H.L.; Mourya, R.; Maharaj, S. Natural proline-rich cyclopolypeptides from marine organisms: Chemistry, synthetic methodologies and biological status. Mar. Drugs 2016, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Dahiya, R. Cyclic peptides as novel antineoplastic agents: A review. J. Sci. Pharm. 2003, 4, 125–131. [Google Scholar]
- Dahiya, R. Cyclopolypeptides with antifungal interest. Coll. Pharm. Commun. 2013, 1, 1–15. [Google Scholar]
- Dahiya, R.; Pathak, D. Synthetic studies on a natural cyclic tetrapeptide-halolitoralin C. J. Pharm. Res. 2006, 5, 69–73. [Google Scholar]
- Dahiya, R.; Pathak, D.; Himaja, M.; Bhatt, S. First total synthesis and biological screening of hymenamide E. Acta Pharm. 2006, 56, 399–415. [Google Scholar] [PubMed]
- Dahiya, R.; Pathak, D. First total synthesis and biological evaluation of halolitoralin A. J. Serbian Chem. Soc. 2007, 72, 101–107. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis, characterization and biological evaluation of a glycine-rich peptide—Cherimolacyclopeptide E. J. Chil. Chem. Soc. 2007, 52, 1224–1229. [Google Scholar] [CrossRef]
- Dahiya, R.; Kaur, K. Synthetic and biological studies on natural cyclic heptapeptide: Segetalin E. Arch. Pharm. Res. 2007, 30, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R. Synthesis of a phenylalanine-rich peptide as potential anthelmintic and cytotoxic agent. Acta Pol. Pharm. 2007, 64, 509–516. [Google Scholar] [PubMed]
- Dahiya, R. Synthetic and pharmacological studies on longicalycinin A. Pak. J. Pharm. Sci. 2007, 20, 317–323. [Google Scholar] [PubMed]
- Dahiya, R.; Kumar, A. Synthesis and biological activity of a potent analog of natural cyclopeptide. Int. J. Nat. Appl. Sci. 2007, 3, 433–440. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis, spectroscopic and biological investigation of cyclic octapeptide: Cherimolacyclopeptide G. Turk. J. Chem. 2008, 32, 205–215. [Google Scholar]
- Dahiya, R. Total synthesis and biological potential of psammosilenin A. Arch. Pharm. Chem. Life Sci. 2008, 341, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R. Synthetic studies on a cyclic hexapeptide from Dianthus superbus. Chem. Pap. 2008, 62, 527–535. [Google Scholar] [CrossRef]
- Dahiya, R. Synthesis and in vitro cytotoxic activity of a natural peptide of plant origin. J. Iran. Chem. Soc. 2008, 5, 445–452. [Google Scholar] [CrossRef]
- Dahiya, R.; Sharma, R.D. Synthesis and bioactivity of a novel cyclic hexapeptide from Stellaria delavayi. Eur. J. Sci. Res. 2008, 21, 277–287. [Google Scholar]
- Dahiya, R.; Kumar, A. Synthetic and biological studies on a cyclopolypeptide of plant origin. J. Zhejiang Univ. Sci. B 2008, 9, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Kaur, K. Synthetic and Pharmacological Investigation of Segetalin C as a Novel Antifungal and Cytotoxic Agent. Arzneim. Forsch. 2008, 58, 29–34. [Google Scholar]
- Dahiya, R.; Maheshwari, M.; Kumar, A. Toward the synthesis and biological evaluation of hirsutide. Monatsh. Chem. 2009, 140, 121–127. [Google Scholar] [CrossRef]
- Dahiya, R.; Kumar, A.; Gupta, R. Synthesis, cytotoxic and antimicrobial screening of a proline-rich cyclopolypeptide. Chem. Pharm. Bull. 2009, 57, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Maheshwari, M.; Yadav, R. Synthetic, cytotoxic and antimicrobial activity studies on annomuricatin B. Z. Naturforsch. 2009, 64b, 237–244. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Total synthesis and antimicrobial activity of a natural cycloheptapeptide of marine-origin. Mar. Drugs 2010, 8, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Gautam, H. Synthetic and pharmacological studies on a natural cyclopeptide from Gypsophila arabica. J. Med. Plants Res. 2010, 4, 1960–1966. [Google Scholar]
- Dahiya, R.; Gautam, H. Toward the first total synthesis of gypsin D: A natural cyclopolypeptide from Gypsophila arabica. Am. J. Sci. Res. 2010, 11, 150–158. [Google Scholar]
- Dahiya, R.; Gautam, H. Synthesis, characterization and biological evaluation of cyclomontanin D. Afr. J. Pharm. Pharmacol. 2011, 5, 447–453. [Google Scholar] [CrossRef]
- Dahiya, R.; Gautam, H. Toward the synthesis and biological screening of a cyclotetrapeptide from marine bacteria. Mar. Drugs 2011, 9, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S. First total synthesis and biological potential of a heptacyclopeptide of plant origin. Chin. J. Chem. 2016, 34, 1158–1164. [Google Scholar] [CrossRef]
- Dahiya, R.; Singh, S. Synthesis, characterization and biological screening of diandrine A. Acta Pol. Pharm. 2017, 3. accepted. [Google Scholar]
- Dahiya, R.; Singh, S. Synthesis, characterization, and biological activity studies on fanlizhicyclopeptide A. Iran. J. Pharm. Res. 2017. accepted. [Google Scholar]
- Das, P.; Himaja, M. Design and synthesis of 4-[2′-(5′-nitro)imidazolyl]benzoyl (N-methyl) amino acids and peptides. Int. J. Drug Dev. Res. 2010, 2, 364–370. [Google Scholar]
- Bodanzsky, M.; Bodanzsky, A. The Practice of Peptide Synthesis; Springer: New York, NY, USA, 1984; pp. 78–143. [Google Scholar]
- Kuttan, R.; Bhanumathy, P.; Nirmala, K.; George, M.C. Potential anticancer activity of turmeric (Curcuma longa). Cancer Lett. 1985, 29, 197–202. [Google Scholar] [CrossRef]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1996, 45, 493–496. [Google Scholar]
- Joo, S.N. Cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. (Seoul) 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S. Toward the synthesis and pharmacological screening of a natural cycloheptapeptide of plant origin. Nat. Prod. Commun. 2017, 12, 379–383. [Google Scholar]
- Dahiya, R.; Pathak, D.; Bhatt, S. Synthesis and biological evaluation of a novel series of 2-(4-chloro-3-methylphenoxy) acetyl amino acids and peptides. J. Saudi Chem. Soc. 2006, 10, 165–176. [Google Scholar]
- Dahiya, R.; Pathak, D. Synthetic studies on novel benzimidazolopeptides with antimicrobial, cytotoxic and anthelmintic potential. Eur. J. Med. Chem. 2007, 42, 772–798. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Singh, S.; Sharma, A.; Chennupati, S.V.; Maharaj, S. First total synthesis and biological screening of a proline-rich cyclopeptide from a Caribbean marine sponge. Mar. Drugs 2016, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, R.; Kumar, A.; Yadav, R. Synthesis and biological activity of peptide derivatives of iodoquinazolinones/nitroimidazoles. Molecules 2008, 13, 958–976. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |
Figure 1.
Synthetic route of the N-methylated cyclopeptide, cordyheptapeptide A (8).
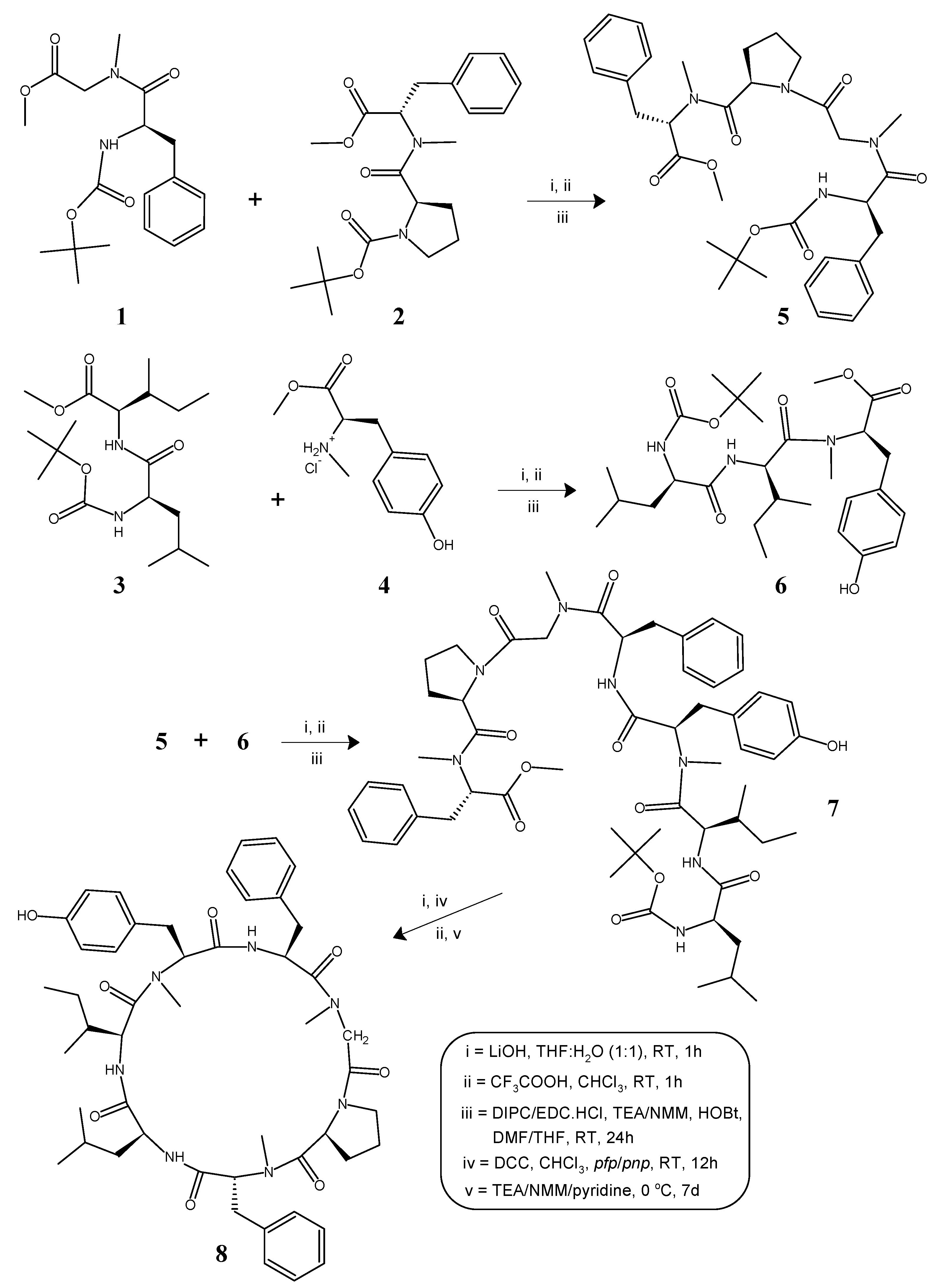
Figure 2.
Mass fragmentation pattern for the newly synthesized N-methylated cyclopeptide, cordyheptapeptide A (8).
Figure 2.
Mass fragmentation pattern for the newly synthesized N-methylated cyclopeptide, cordyheptapeptide A (8).

{kind=link}
{kind=link}
{kind=link}
Table 1.
Cytotoxic activity data for the linear and cycloheptapeptide (7, 8).
| Compd. | Conc. (μg/mL) | Dalton’s Lymphoma Ascites (DLA) Cells | Ehrlich’s Ascites Carcinoma (EAC) Cells | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Live Cells Counted | No. of Dead Cells | % GI a | CTC50 (μM) b | Live Cells Counted | No. of Dead Cells | % GI | CTC50 (μM) | ||
| 7 | 62.50 | 0 | 38 | 100.0 | 0 | 28 | 100.0 | ||
| 31.25 | 11 | 27 | 71.05 | 8 | 20 | 71.43 | |||
| 15.63 | 21 | 17 | 44.74 | 19.9 | 17 | 11 | 39.29 | 22.6 | |
| 7.81 | 25 | 12 | 34.21 | 21 | 7 | 25.00 | |||
| 3.91 | 34 | 4 | 10.53 | 24 | 4 | 14.29 | |||
| 8 | 62.50 | 0 | 38 | 100.0 | 0 | 28 | 100.0 | ||
| 31.25 | 7 | 31 | 81.58 | 4 | 24 | 85.71 | |||
| 15.63 | 15 | 23 | 60.53 | 10.6 | 11 | 17 | 60.71 | 14.6 | |
| 7.81 | 22 | 16 | 42.11 | 18 | 10 | 35.71 | |||
| 3.91 | 27 | 10 | 28.95 | 22 | 6 | 21.43 | |||
| Control | 62.50 | 38 | 0 | – | 28 | 0 | – | ||
| 31.25 | 38 | 0 | – | 28 | 0 | – | |||
| 15.63 | 38 | 0 | – | – | 28 | 0 | – | – | |
| 7.81 | 38 | 0 | – | 28 | 0 | – | |||
| 3.91 | 38 | 0 | – | 28 | 0 | – | |||
| Std. | 62.50 | 0 | 38 | 100.0 | 0 | 28 | 100.0 | ||
| (5-FU) | 31.25 | 0 | 38 | 100.0 | 0 | 28 | 100.0 | ||
| 15.63 | 10 | 28 | 73.68 | 37.4 | 11 | 17 | 60.71 | 90.6 | |
| 7.81 | 13 | 25 | 65.79 | 19 | 9 | 32.14 | |||
| 3.91 | 22 | 16 | 42.11 | 23 | 5 | 17.86 | |||
a % growth inhibition (GI) = 100 − [{(Celltotal – Celldead) × 100}/Celltotal]; b CTC50 = cytotoxic concentration inhibiting 50% of growth.
Table 2.
Antimicrobial evaluation data for the linear and cycloheptapeptide (7, 8).
| Compound | Diameter of the Zone of Inhibition (mm) | |||||||
|---|---|---|---|---|---|---|---|---|
| Bacterial Strains | Fungal Strains | |||||||
| B. sub. | S. aur. | P. aeru. | K. pneu. | C. alb. | M. audo. | A. niger | T. menta. | |
| 7 | 10(50) | – | 21(6) | 23(6) | 12(25) | 18(6) | – | 16(6) |
| 8 | 12(50) | 10(25) | 25(6) | 26(6) | 15(25) | 22(6) | 9(25) | 20(6) |
| DMF † | – | – | – | – | ||||
| DMSO ‡ | – | – | – | – | ||||
| Gatifloxacin | 18(12.5) * | 27(6) | 23(6) | 25(6) | – | – | – | – |
| Griseofulvin | – | – | – | – | 20(6) | 18(6) | 20(12.5) | 19(6) |
B. sub.: Bacillus subtilis, S. aur.: Staphylococcus aureus, P. aeru.: Pseudomonas aeruginosa, K. pneu.: Klebsiella pneumoniae, C. alb.: Candida albicans, M. audo.: Microsporum audouinii, A. niger: Aspergillus niger, T. menta.: Trichophyon mentagrophytes; * Values in the bracket are the MIC values (μg/mL); † dimethylformamide—negative control for the antibacterial studies; ‡ dimethylsulfoxide—negative control for the antifungal studies.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kumar, S.; Dahiya, R.; Khokra, S.L.; Mourya, R.; Chennupati, S.V.; Maharaj, S. Total Synthesis and Pharmacological Investigation of Cordyheptapeptide A. Molecules 2017, 22, 682. https://doi.org/10.3390/molecules22060682
AMA Style
Kumar S, Dahiya R, Khokra SL, Mourya R, Chennupati SV, Maharaj S. Total Synthesis and Pharmacological Investigation of Cordyheptapeptide A. Molecules. 2017; 22(6):682. https://doi.org/10.3390/molecules22060682
Chicago/Turabian StyleKumar, Suresh, Rajiv Dahiya, Sukhbir Lal Khokra, Rita Mourya, Suresh V. Chennupati, and Sandeep Maharaj. 2017. "Total Synthesis and Pharmacological Investigation of Cordyheptapeptide A" Molecules 22, no. 6: 682. https://doi.org/10.3390/molecules22060682