
Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from the Seeds of Caesalpinia sappan in Cancer Cells



Abstract
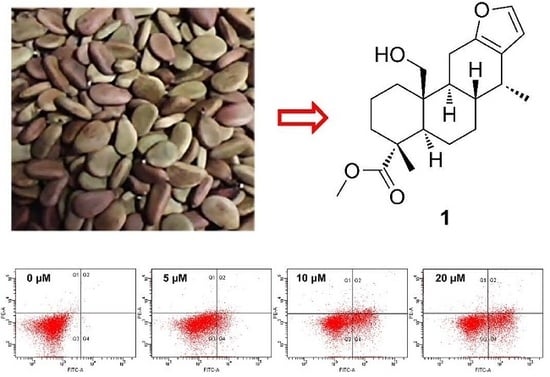
:
1. Introduction
2. Results and Discussion
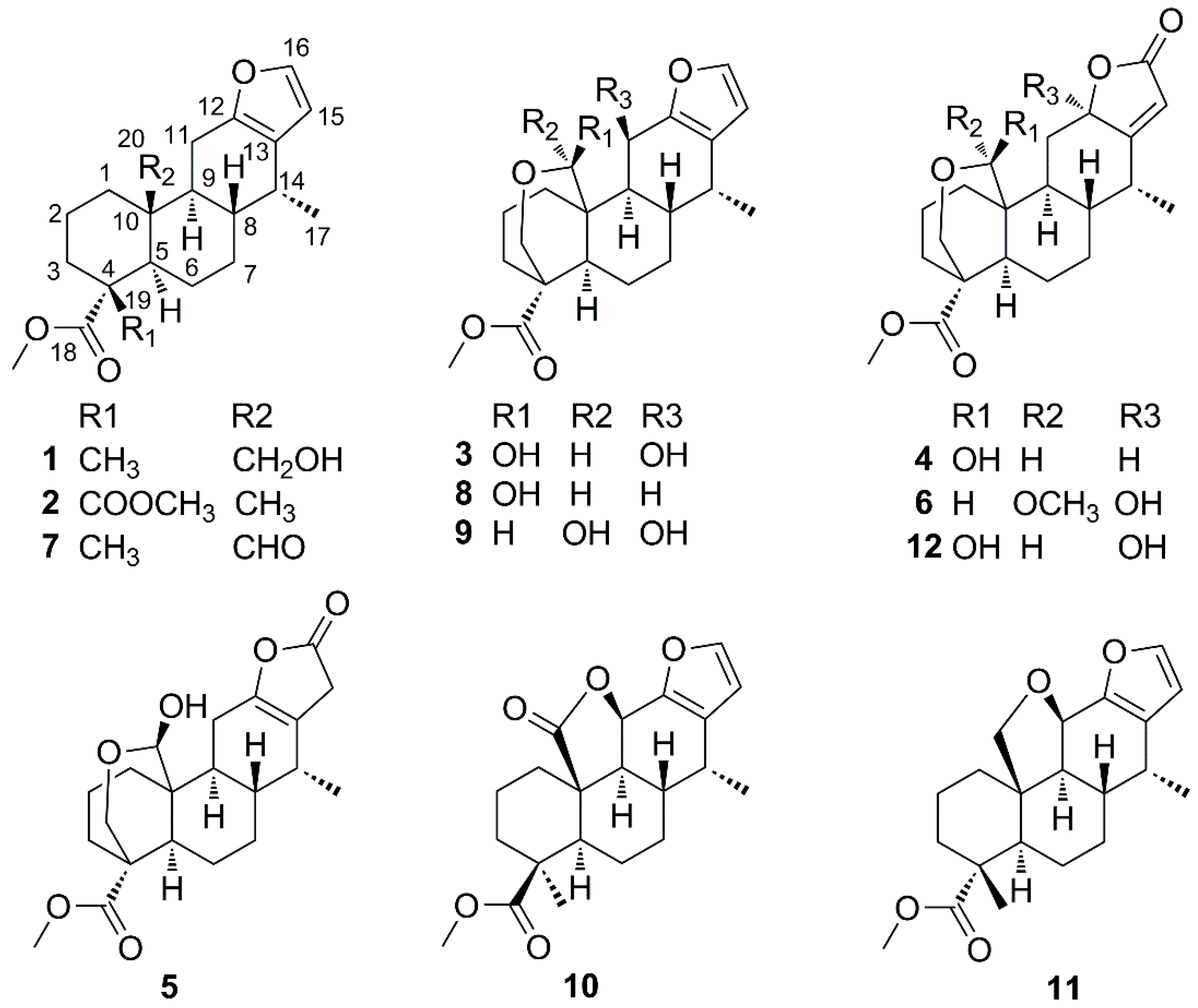
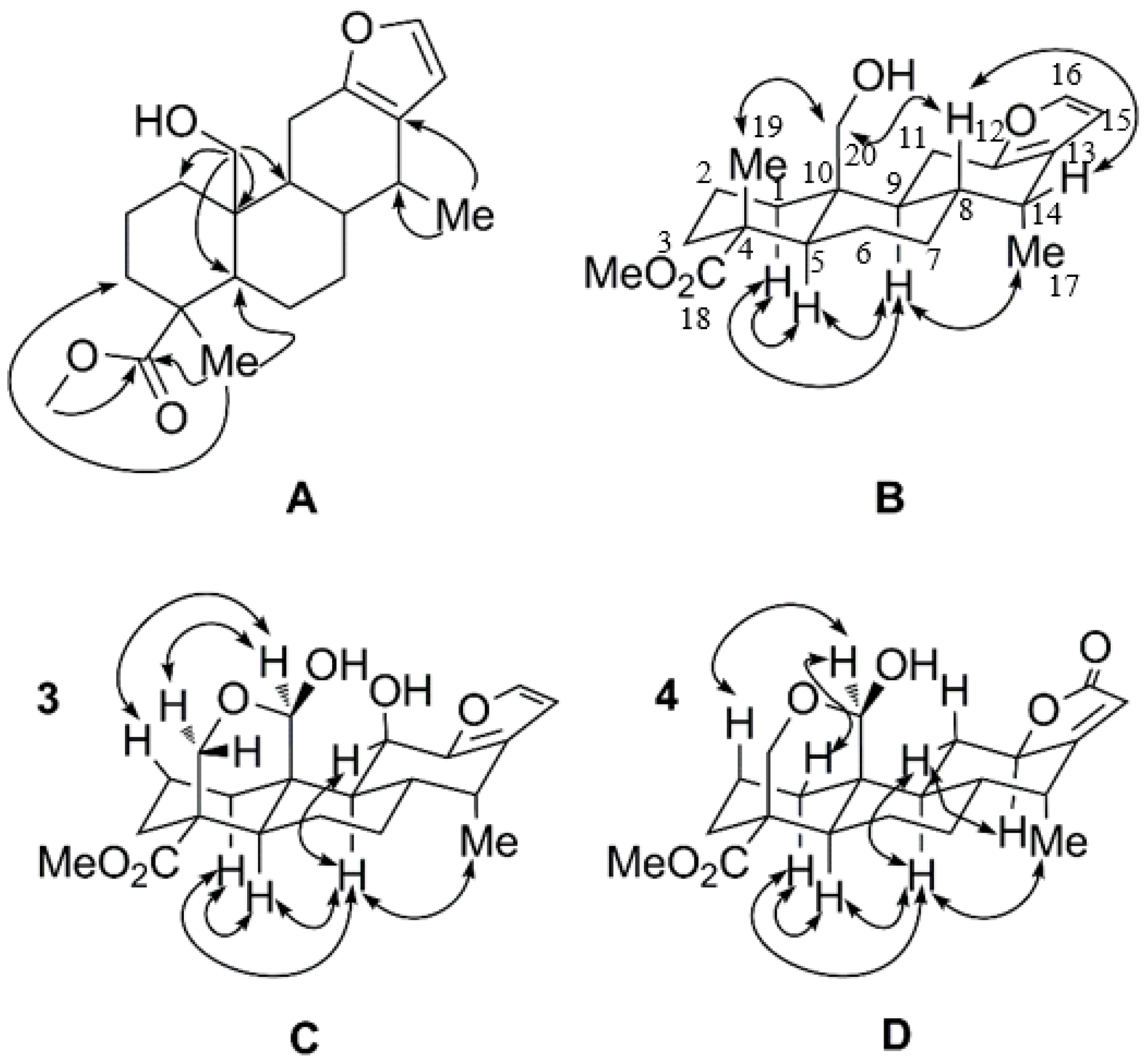
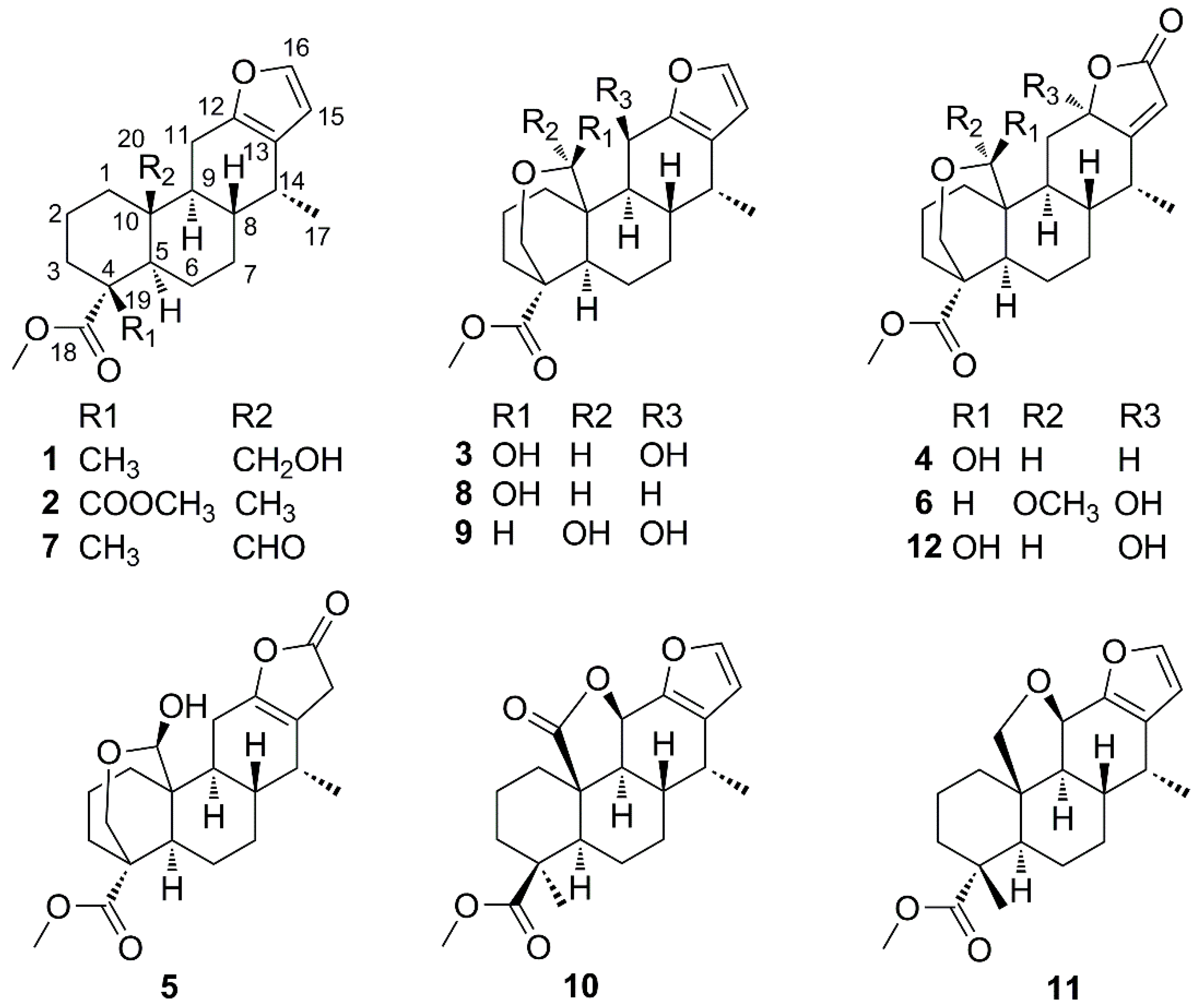
2.1. Identification of New Compounds
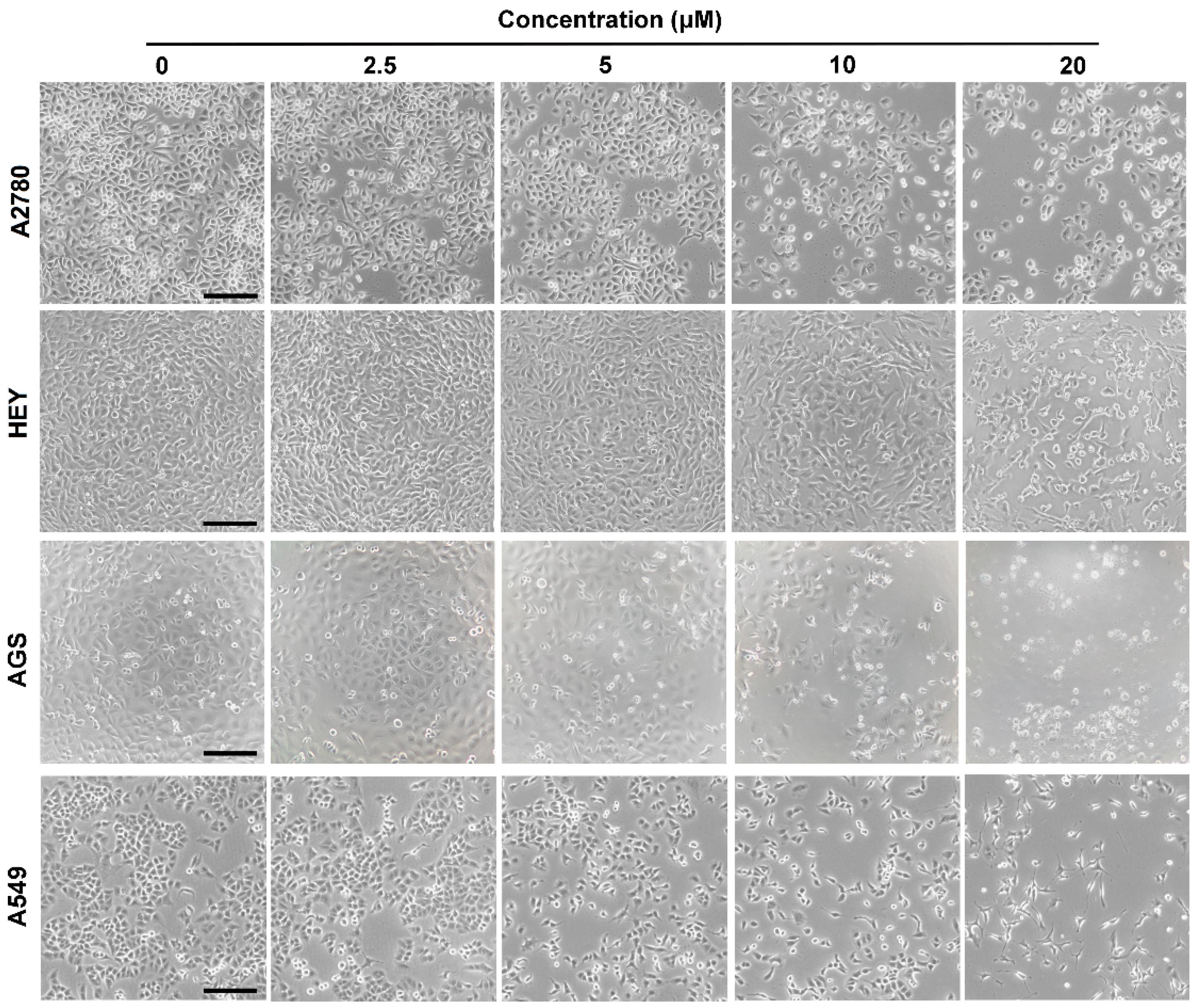
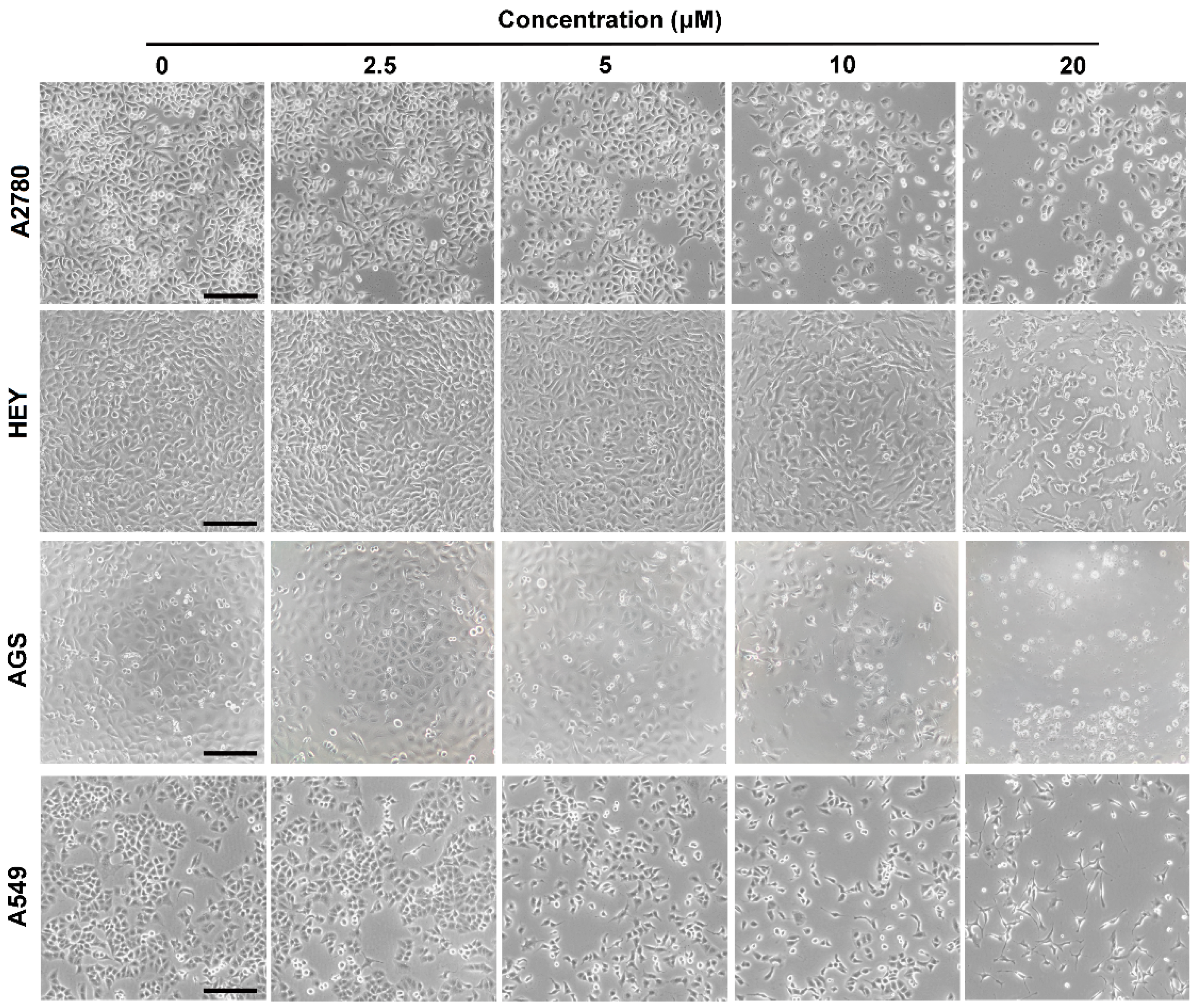
2.2. Cytotoxicity Assay
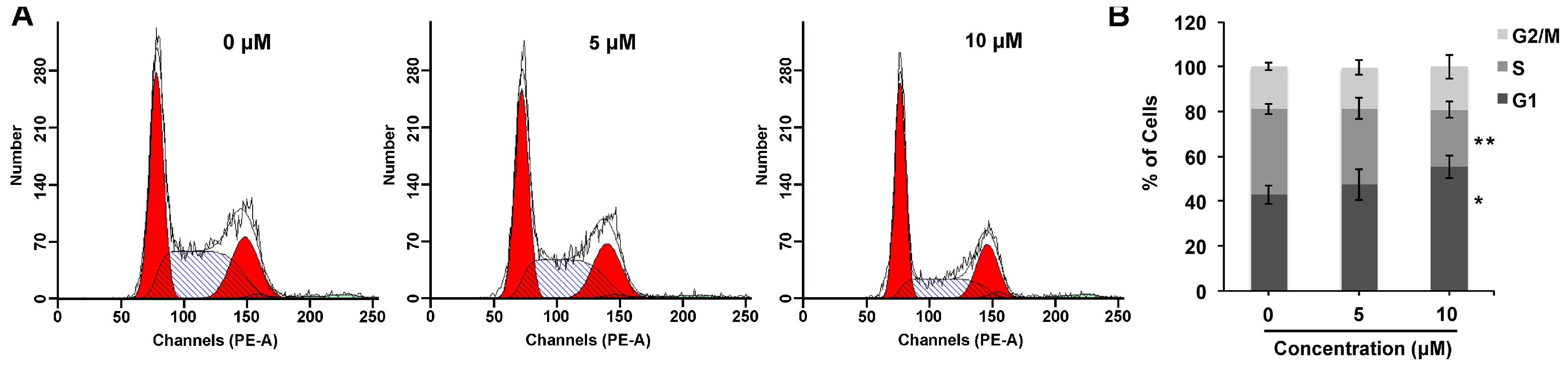
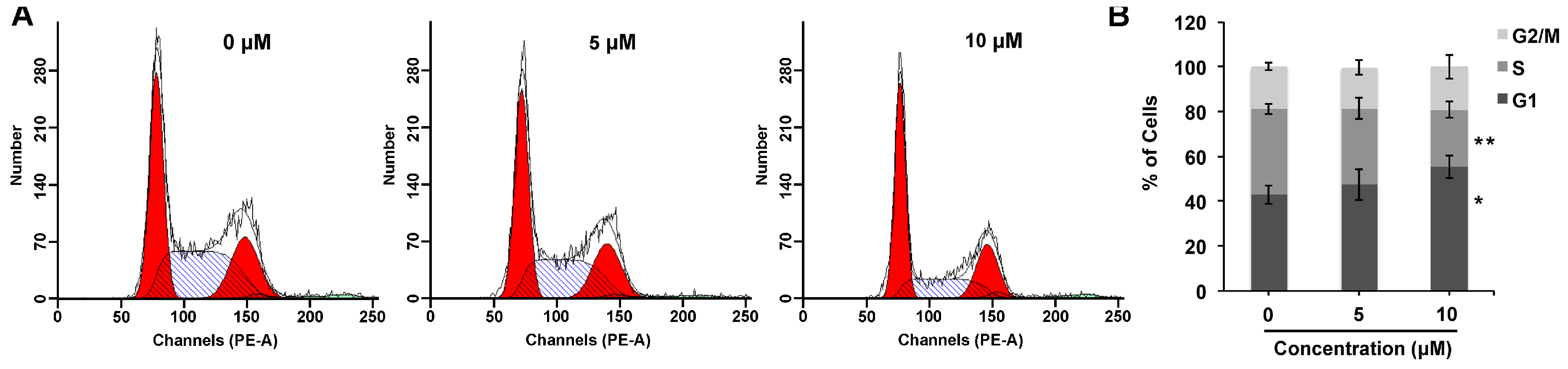
2.3. Compound 1 Mediates G1 Cell Cycle Arrest
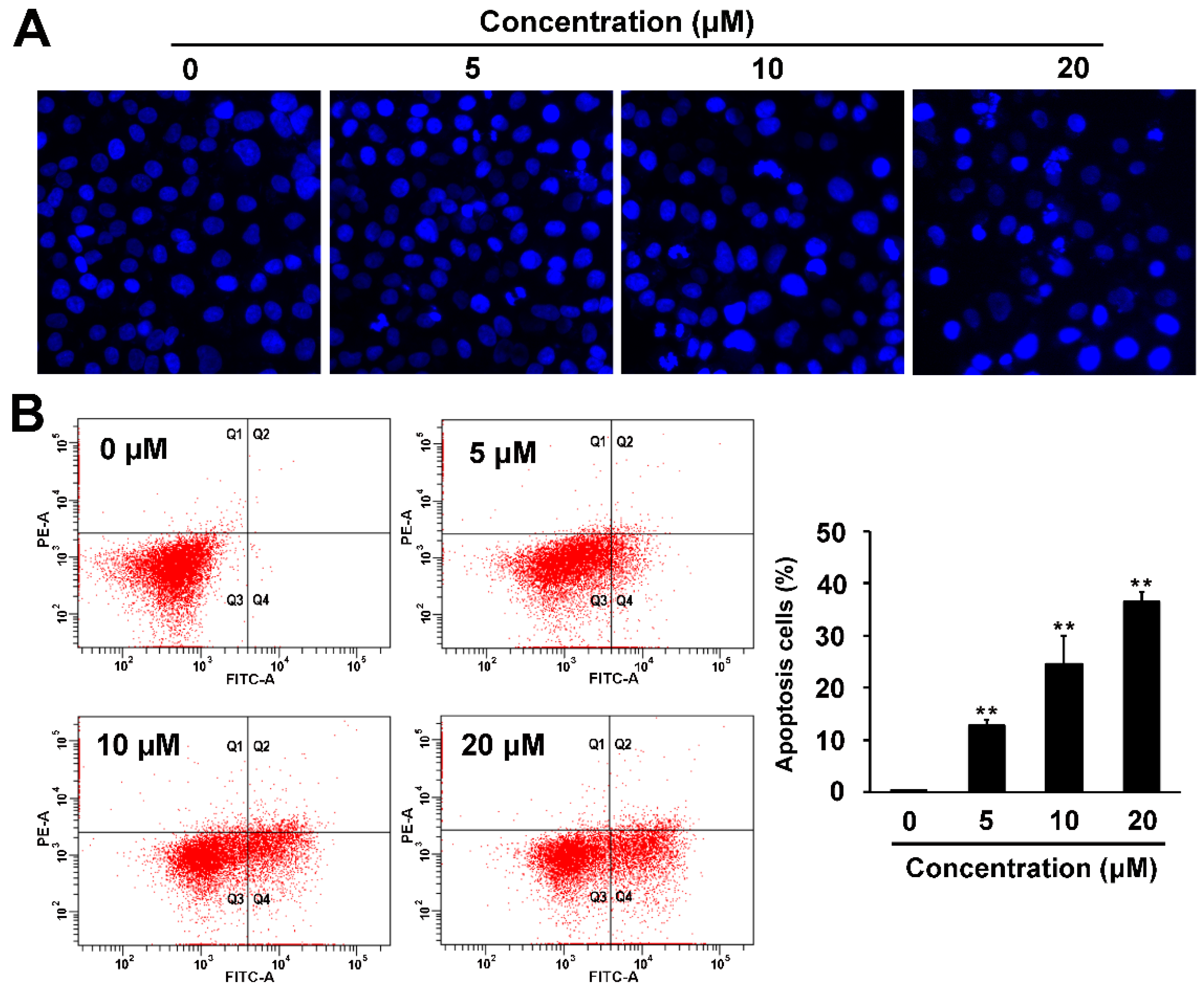
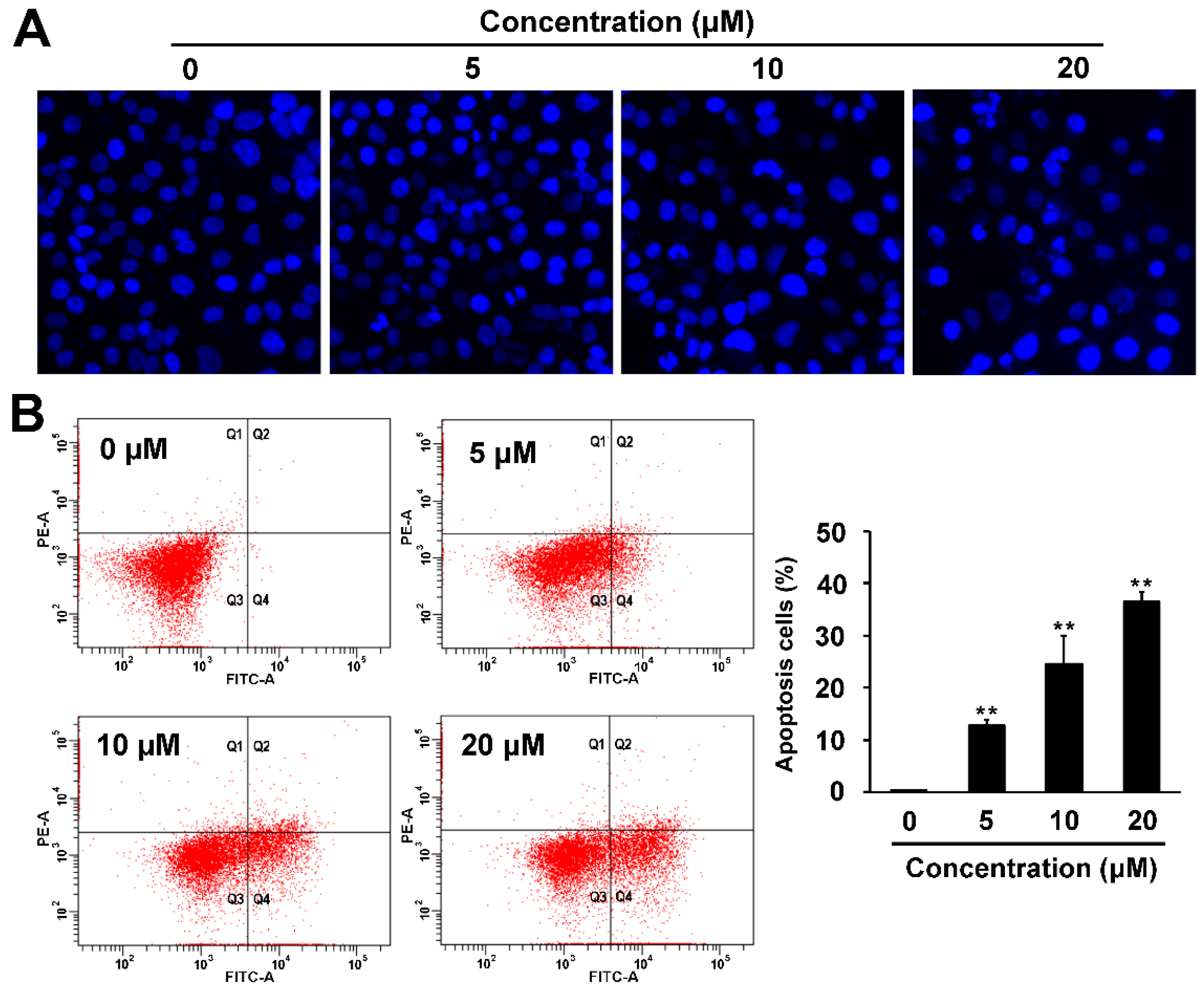
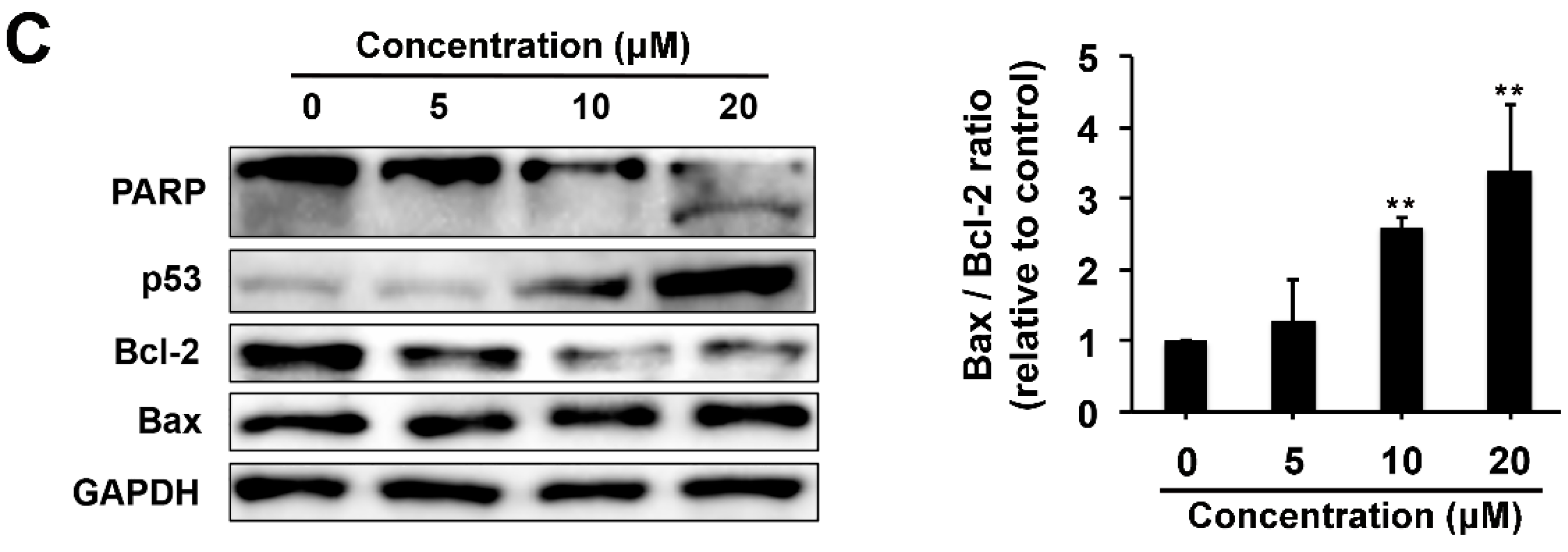
2.4. Pro-apoptotic and p53 Suppressing Activities of Compound 1
3. Materials and Methods
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data
3.5. Reagents
3.6. Cell Culture
3.7. MTT Assay
3.8. Cell Cycle Analysis
3.9. Hoechst 33342 Staining Assay
3.10. Annexin V/PI Staining Assay
3.11. Western Blot Analysis
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CD | Circular Dichroism |
| 13C-NMR | 13C Nuclear Magnetic Resonance |
| 1D-NMR | 1 Dimension Nuclear Magnetic Resonance |
| 2D-NMR | 2 Dimension Nuclear Magnetic Resonance |
| DMSO | Dimethyl Sulphoxide |
| ESIMS | Electrospray Ionization Mass Spectrometry |
| FBS | Fetal Bovine Serum |
| HMBC | Heteronuclear Multiple Bond Correlation |
| 1H-NMR | 1H Nuclear Magnetic Resonance |
| HRESIMS | High-resolution ESIMS |
| HSQC | Heteronuclear Single-quantum Correlation |
| IR | Infrared Spectroscopy |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium Bromide |
| PBS | Phosphate-buffered Saline |
| Preparative HPLC | Preparative High Performance Liquid Chromatography |
| ROESY | Rotating-frame Nuclear Overhauser Enhancement Spectroscopy |
| SDS | Sodium Dodecyl Sulfate |
| TLC | Thin Layer Chromatography |
| UV | Ultraviolet Spectroscopy |
References
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; IARC Nonserial Publication: Lyon, France, 2015. [Google Scholar]
- Park, S.H.; Sohn, T.S.; Lee, J.; Lim, D.H.; Hong, M.E.; Kim, K.M.; Sohn, I.; Jung, S.H.; Choi, M.G.; Lee, J.H. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: Final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J. Clin. Oncol. 2015, 33, 3130–3136. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P. Drug resistance: Challenges to effective therapy. Curr. Cancer Drug Tar. 2011, 11, 613–623. [Google Scholar] [CrossRef]
- Millimouno, F.M.; Dong, J.; Yang, L.; Li, J.; Li, X.M. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev. Res. 2014, 7, 1081–1107. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, H.Y.; Lin, H.; Su, H.; Xing, D.M.; Du, L.J. Brazilein protects the brain against focal cerebral ischemia reperfusion injury correlating to inflammatory response suppression. Eur. J. Pharmacol. 2007, 558, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.L.; Shu, S.H.; Qin, H.L.; Lee, S.M.; Wang, Y.T.; Du, G.H. In vitro anti-influenza viral activities of constituents from Caesalpinia sappan. Planta Med. 2009, 75, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Yodsaoue, O.; Cheenpracha, S.; Karalai, C.; Ponglimanont, C.; Tewtrakul, S. Anti-allergic activity of principles from the roots and heartwood of Caesalpinia sappan on antigen-induced β-hexosaminidase release. Phytother. Res. 2009, 23, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Badami, S.; Moorkoth, S.; Rai, S.R.; Kannan, E.; Bhojraj, S. Antioxidant activity of Caesalpinia sappan heartwood. Biol. Pharm. Bull. 2003, 26, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Yang, K.M.; Jeon, S.D.; Kim, J.H.; Khil, L.Y.; Chang, T.S.; Moon, C.K. Brazilin modulates immune function mainly by augmenting T cell activity in halothane administered mice. Planta Med. 1997, 63, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Yodsaoue, O.; Cheenpracha, S.; Karalai, C.; Ponglimanont, C.; Chantrapromma, S.; Fun, H.K.; Kanjana-Opas, A. Phanginin A–K, diterpenoids from the seeds of Caesalpinia sappan Linn. Phytochemistry 2008, 69, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wu, F.H.; Qu, W.; Liang, J.Y. Two new cassane diterpenoids from the seeds of Caesalpinia sappan Linn. Chin. J. Nat. Med. 2012, 10, 218–221. [Google Scholar] [CrossRef]
- Ma, G.X.; Zhu, Y.D.; Sun, Z.H.; Yuan, J.Q.; Xie, Y.; Zhang, X.P.; Tian, Y.; Yang, J.S.; Wu, H.F.; Xu, X.D. Three new cassane diterpenes from the seeds of Caesalpinia sappan. Phytochem. Lett. 2014, 8, 141–144. [Google Scholar] [CrossRef]
- Tran, M.H.; Nguyen, M.T.; Nguyen, H.D.; Nguyen, T.D.; Phuong, T.T. Cytotoxic constituents from the seeds of Vietnamese Caesalpinia sappan. Pharm. Biol. 2015, 53, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.X.; Wu, H.F.; Chen, D.L.; Zhu, N.L.; Zhu, Y.D.; Sun, Z.H.; Li, P.F.; Yang, J.S.; Yuan, J.Q.; Xu, X.D. Antimalarial and antiproliferative cassane diterpenes of Caesalpinia sappan. J. Nat. Prod. 2015, 78, 2364–2371. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.X.; Nguyen, N.T.; Dang, P.H.; Thi Ho, P.; Nguyen, M.T.; Van Can, M.; Dibwe, D.F.; Ueda, J.Y.; Awale, S. Cassane diterpenes from the seed kernels of Caesalpinia sappan. Phytochemistry 2016, 122, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Yin, Y.H.; Hu, L.H. Five new cassane-type diterpenes from Caesalpinia crista. Helv. Chim. Acta 2009, 92, 121–126. [Google Scholar] [CrossRef]
- Ochieng, C.O.; Owuor, P.O.; Mang’uro, L.A.; Akala, H.; Ishola, I.O. Antinociceptive and antiplasmodial activities of cassane furanoditerpenes from Caesalpinia volkensii H. root bark. Fitoterapia 2012, 83, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Kamikawa, S.; Oshimo, S.; Ohta, E.; Nehira, T.; Omura, H.; Ohta, S. Cassane diterpenoids from the roots of Caesalpinia decapetala var. japonica and structure revision of caesaljapin. Phytochemistry 2016, 121, 50–57. [Google Scholar] [PubMed]
- Wu, J.M.; Chen, G.; Xu, X.T.; Huo, X.L.; Wu, S.L.; Wu, Z.H.; Gao, H.Y. Seven new cassane furanoditerpenes from the seeds of Caesalpinia minax. Fitoterapia 2014, 92, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.X.; Xu, X.D.; Cao, L.; Yuan, J.Q.; Yang, J.S.; Ma, L.Y. Cassane-type diterpenes from the seeds of Caesalpinia minax with their antineoplastic activity. Planta Med. 2012, 78, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.X.; Yuan, J.Q.; Wu, H.F.; Cao, L.; Zhang, X.P.; Xu, L.J.; Wei, H.; Wu, L.Z.; Zheng, Q.X.; Li, L.Y.; et al. Caesalpins A–H, bioactive cassane-type diterpenes from the seeds of Caesalpinia minax. J. Nat. Prod. 2013, 76, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Ishihara, R.; Hayashi, K.; Matsuura, N.; Akashi, H.; Nozaki, H. Cassane-type diterpenoids from Caesalpinia echinata (Leguminosae) and their NF-κB signaling inhibition activities. Phytochemistry 2015, 116, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Ishihara, R.; Hayashi, K.; Sunadome, M.; Matsuura, N.; Nozaki, H. New cassane-type diterpenoids of Caesalpinia echinata (Leguminosae) exhibiting NF-κB inhibitory activities. Chem. Pharm. Bull. 2014, 62, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Zhang, Q.W.; Ye, Y.; Lin, L.G. Naturally occurring furanoditerpenoids: Distribution, chemistry and their pharmacological activities. Phytochem. Rev. 2016. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Chen, D.L.; Shiloh, A.; Luo, J.Y.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Attardi, L.D. Cancer: A piece of the p53 puzzle. Nature 2015, 520, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1–12 are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1α | 1.13, m | 1.14, dd (13.1, 4.8) | 1.48, m | 1.20, m | 1.26, m |
| 1β | 2.27, m | 1.75, m | 1.78, m | 2.05, m | 1.46, m |
| 2α | 1.60, m | 1.87, m | 1.61, m | 1.64, m | 1.62, m |
| 2β | 1.44, m | 1.68, m | 2.01, m | 2.28, m | 2.31, m |
| 3α | 1.77, m | 1.63, m | 1.56, m | 1.93, m | 1.94, m |
| 3β | 1.61, m | 2.42, dd (13.0, 1.3) | 2.01, m | 2.05, m | 2.00, m |
| 5 | 1.81, m | 2.01, dd (12.1, 2.3) | 1.83, m | 1.68, m | 1.65, m |
| 6α | 1.21, m | 1.45, m | 1.57, m | 1.21, m | 1.24, m |
| 6β | 1.41, m | 1.64, m | 1.75, m | 2.10, m | 1.62, m |
| 7α | 1.40, m | 1.57, m | 1.19, m | 1.36, m | 1.36, m |
| 7β | 1.70, m | 1.76, m | 1.76, m | 1.56, m | 1.67, m |
| 8 | 1.88, m | 1.78, m | 2.17, m | 2.32, m | 2.34, m |
| 9 | 1.61, m | 1.56, m | 1.83, m | 1.50, m | 2.24, m |
| 11α | 2.71, dd (16.8, 6.4) | 2.60, dd (16.9, 6.8) | 4.73, d (3.1) | 2.60, m | 2.25, m |
| 11β | 2.34, m | 2.35, dd (16.9, 11.8) | - | 1.74, m | 2.36, m |
| 12 | - | - | - | 4.83, dd (11.6, 6.0) | - |
| 14 | 2.61, m | 2.62, m | 2.66, m | 2.92, m | 2.38, m |
| 15 | 6.18, d (1.7) | 6.18, d (1.7) | 6.23, d (1.7) | 5.67, s | α: 3.18, d (18.6) β: 3.01, d (18.6) |
| 16 | 7.21, d (1.7) | 7.22, d (1.7) | 7.32, d (1.7) | - | - |
| 17 | 0.97, d (7.0) | 0.99, d (7.1) | 0.98, d (7.1) | 1.04, d (7.1) | 0.88, d (7.0) |
| 19α | 1.29, s | - | 3.50, d (12.5) | 3.69, d (12.0) | 3.72, d (11.7) |
| 19β | 4.89, d (12.6) | 4.33, dd (11.9, 2.1) | 4.35, dd (11.8, 2.5) | ||
| 20 | α: 3.86, d (12.1) β: 3.97, d (12.1) | 0.74, s | 4.98, s | 4.83, d (2.1) | 4.98, s |
| 18-OMe | 3.68, s | 3.71, s | 3.72, s | 3.67, s | 3.67, s |
| 19-OMe | - | 3.74, s | - | - | - |
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 31.4 | 39.1 | 34.9 | 37.8 | 37.8 |
| 2 | 17.6 | 19.1 | 17.9 | 21.0 | 20.9 |
| 3 | 35.4 | 34.7 | 35.7 | 35.7 | 35.6 |
| 4 | 49.4 | 57.6 | 47.3 | 45.6 | 45.6 |
| 5 | 51.5 | 50.5 | 46.2 | 45.1 | 45.1 |
| 6 | 23.5 | 25.7 | 27.8 | 23.9 | 23.5 |
| 7 | 30.5 | 31.7 | 28.1 | 29.0 | 21.9 |
| 8 | 36.6 | 36.1 | 38.1 | 41.2 | 36.5 |
| 9 | 45.0 | 45.2 | 45.1 | 41.7 | 41.9 |
| 10 | 40.6 | 37.0 | 43.4 | 38.7 | 38.2 |
| 11 | 23.1 | 22.5 | 70.1 | 33.8 | 29.2 |
| 12 | 149.6 | 149.5 | 146.7 | 79.6 | 149.1 |
| 13 | 122.1 | 122.5 | 129.5 | 175.5 | 115.6 |
| 14 | 31.4 | 31.7 | 32.6 | 36.9 | 32.2 |
| 15 | 109.7 | 109.7 | 109.5 | 110.9 | 34.9 |
| 16 | 140.3 | 140.6 | 143.1 | 174.0 | 176.9 |
| 17 | 16.9 | 17.8 | 14.2 | 13.3 | 14.3 |
| 18 | 179.2 | 173.8 | 176.1 | 177.2 | 175.7 |
| 19 | 19.1 | 172.6 | 67.1 | 61.5 | 61.9 |
| 20 | 61.2 | 13.7 | 106.2 | 96.8 | 97.3 |
| 18-OMe | 52.0 | 52.8 | 52.2 | 51.8 | 51.8 |
| 19-OMe | 52.0 |
| Compounds | A2780 | HEY | AGS | A549 |
|---|---|---|---|---|
| 1 | 10.4% ± 4.7% | 10.2% ± 9.8% | 4.9% ± 1.3% | 32.9% ± 13.0% |
| 2 | 92.0% ± 7.1% | 95.7% ± 6.4% | 80.8% ± 9.7% | 80.0% ± 8.0% |
| 3 | 74.8% ± 8.2% | 79.9% ± 12.7% | 71.7% ± 18.2% | 74.6% ± 5.4% |
| 4 | 97.3% ± 7.4% | 95.2% ± 6.0% | 80.8% ± 15.7% | 79.9% ± 10.0% |
| 5 | 91.7% ± 1.4% | 97.2% ± 2.8% | 83.0% ± 16.6% | 84.9% ± 10.0% |
| 6 | 95.2% ± 1.0% | 91.3% ± 9.6% | 82.3% ± 9.9% | 83.5% ± 7.6% |
| 7 | 37.9% ± 5.6% | 68.2% ± 5.5% | 69.5% ± 8.9% | 73.5% ± 12.0% |
| 8 | 49.5% ± 5.8% | 41.6% ± 9.0% | 14.6% ± 2.3% | 50.9% ± 12.3% |
| 9 | 64.4% ± 4.6% | 74.1% ± 7.0% | 71.1% ± 12.7% | 72.8% ± 4.7% |
| 10 | 57.4% ± 4.5% | 71.4% ± 1.9% | 62.7% ± 13.5% | 75.2% ± 9.8% |
| 11 | 95.4% ± 2.7% | 91.7% ± 6.1% | 85.8% ± 9.4% | 74.8% ± 13.8% |
| 12 | 92.1% ± 6.6% | 97.5% ± 2.0% | 82.1% ± 14.0% | 82.2% ± 10.6% |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, H.; Zhang, L.-L.; Liu, Q.-Y.; Feng, L.; Ye, Y.; Lu, J.-J.; Lin, L.-G. Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from the Seeds of Caesalpinia sappan in Cancer Cells. Molecules 2016, 21, 791. https://doi.org/10.3390/molecules21060791
Bao H, Zhang L-L, Liu Q-Y, Feng L, Ye Y, Lu J-J, Lin L-G. Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from the Seeds of Caesalpinia sappan in Cancer Cells. Molecules. 2016; 21(6):791. https://doi.org/10.3390/molecules21060791
Chicago/Turabian StyleBao, Han, Le-Le Zhang, Qian-Yu Liu, Lu Feng, Yang Ye, Jin-Jian Lu, and Li-Gen Lin. 2016. "Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from the Seeds of Caesalpinia sappan in Cancer Cells" Molecules 21, no. 6: 791. https://doi.org/10.3390/molecules21060791
APA StyleBao, H., Zhang, L.-L., Liu, Q.-Y., Feng, L., Ye, Y., Lu, J.-J., & Lin, L.-G. (2016). Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from the Seeds of Caesalpinia sappan in Cancer Cells. Molecules, 21(6), 791. https://doi.org/10.3390/molecules21060791