
Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects

Abstract
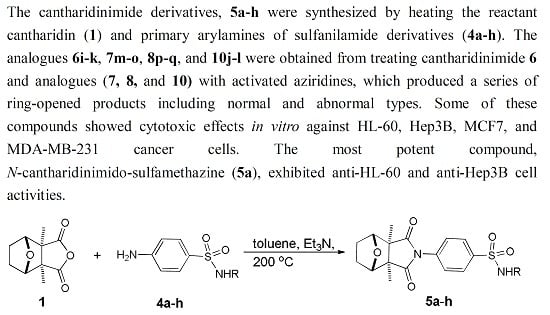
:
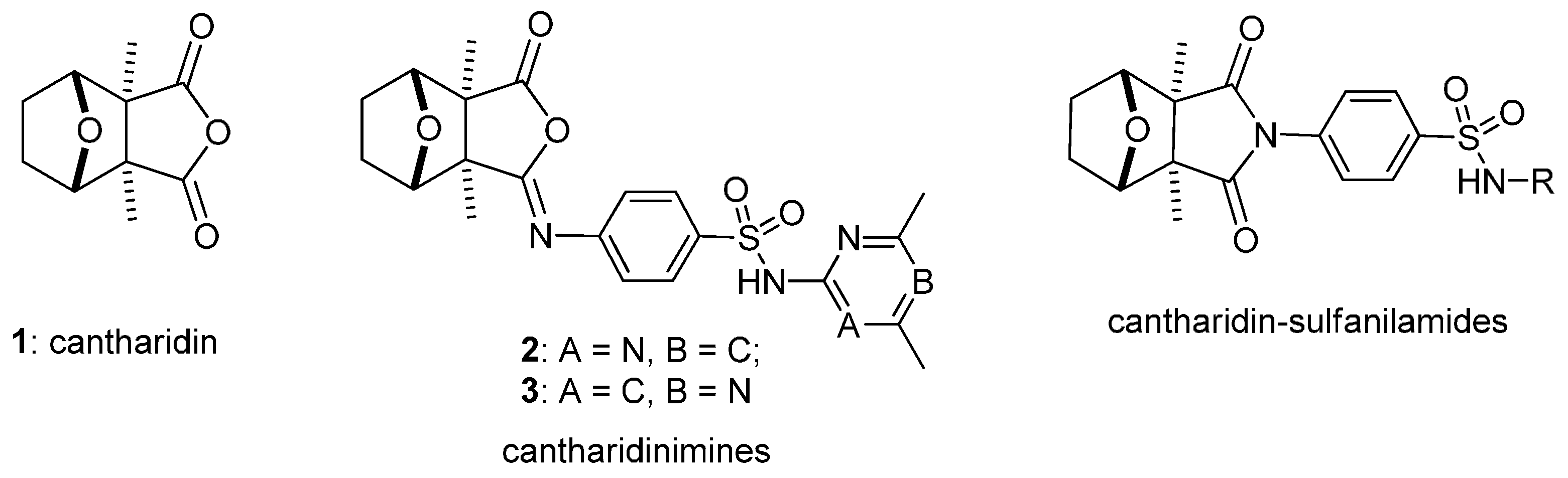
1. Introduction

2. Results and Discussion
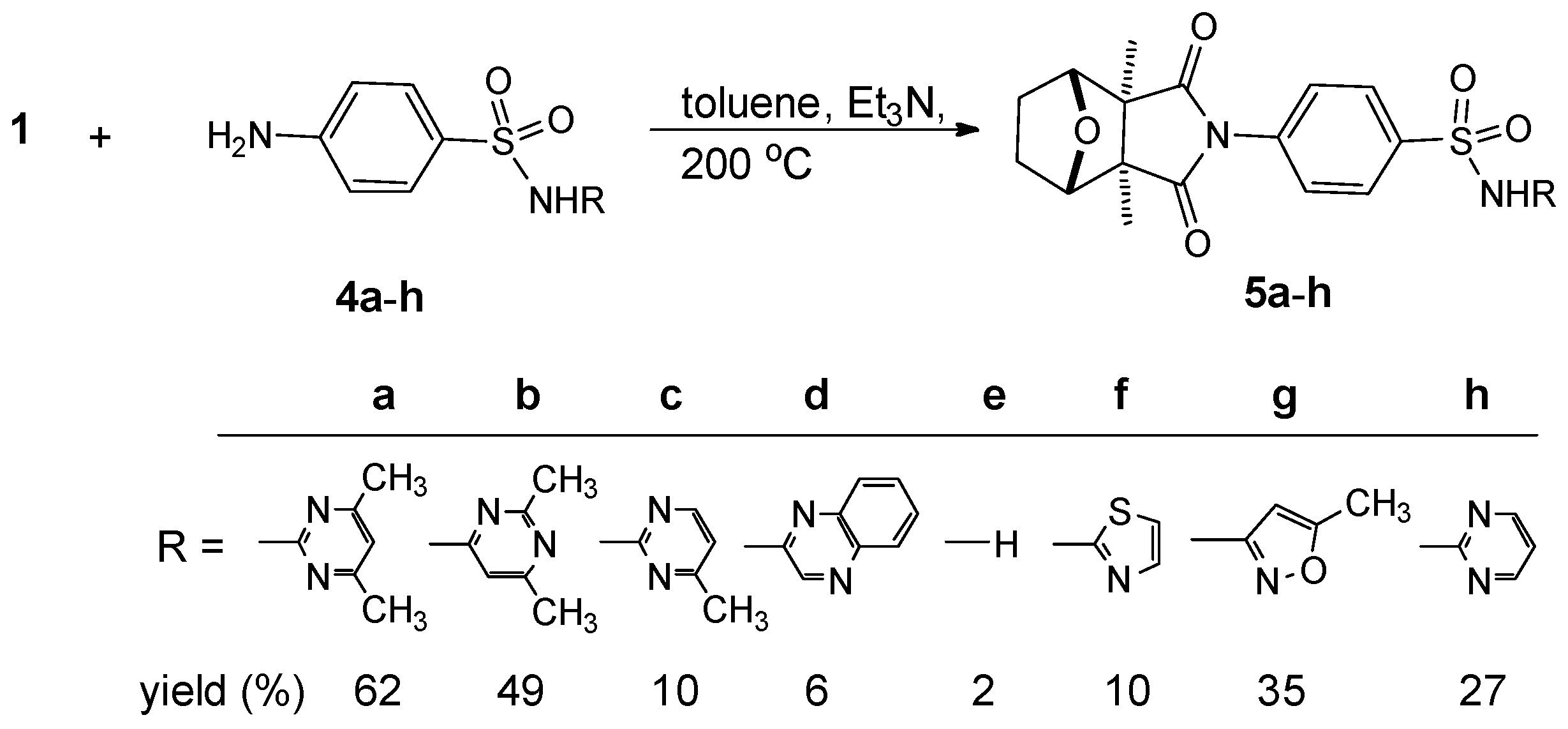
2.1. Chemistry
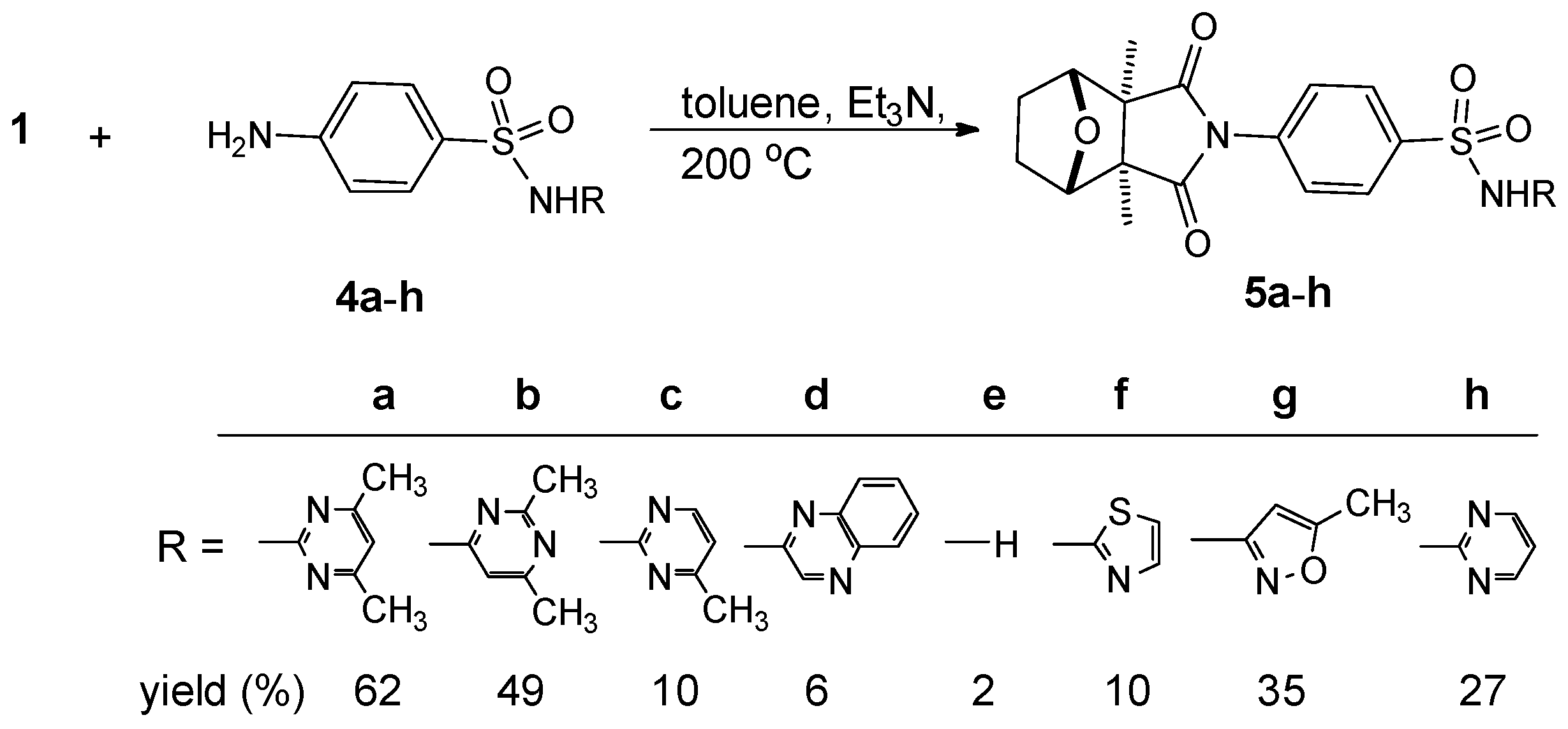

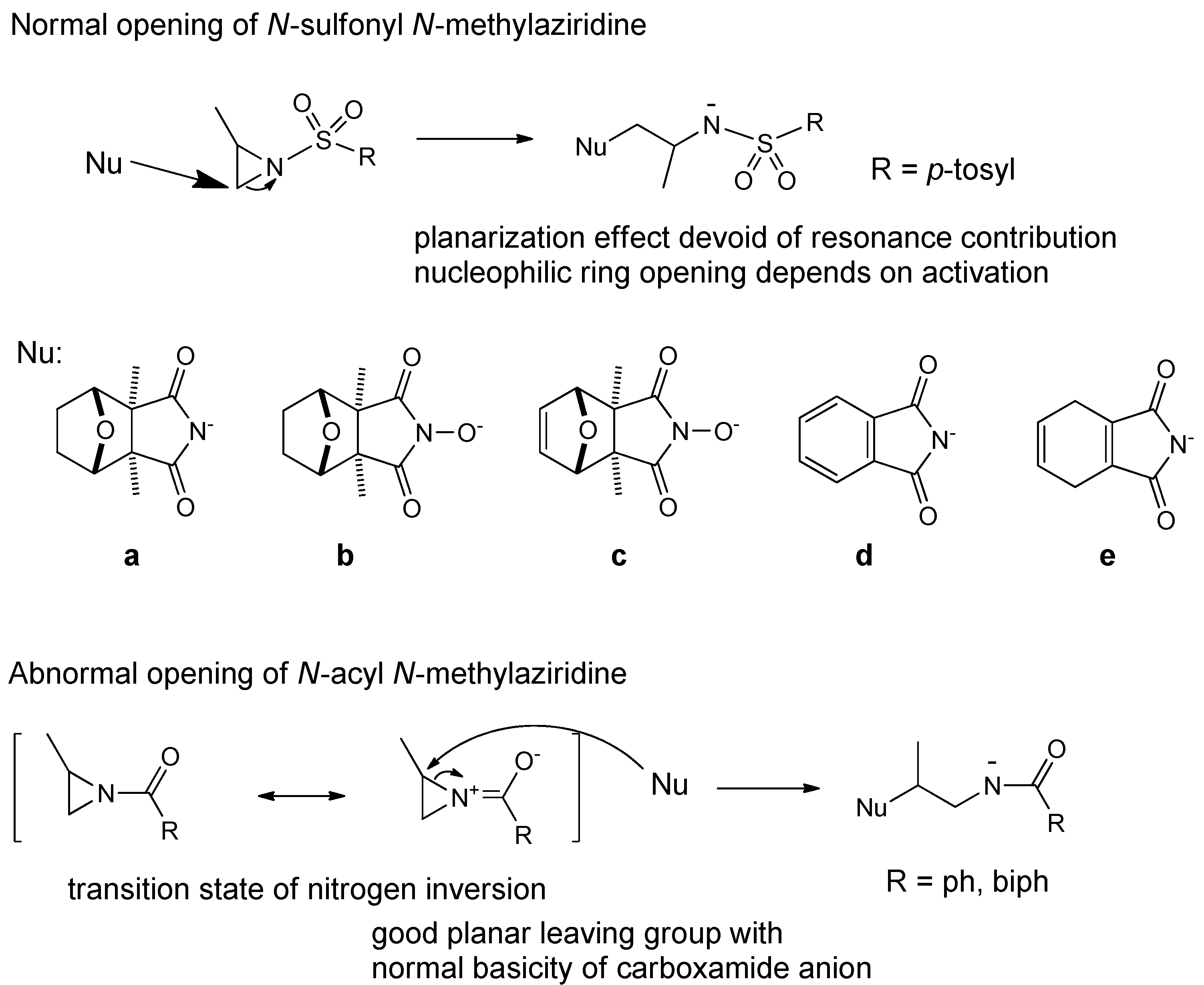

{kind=link}
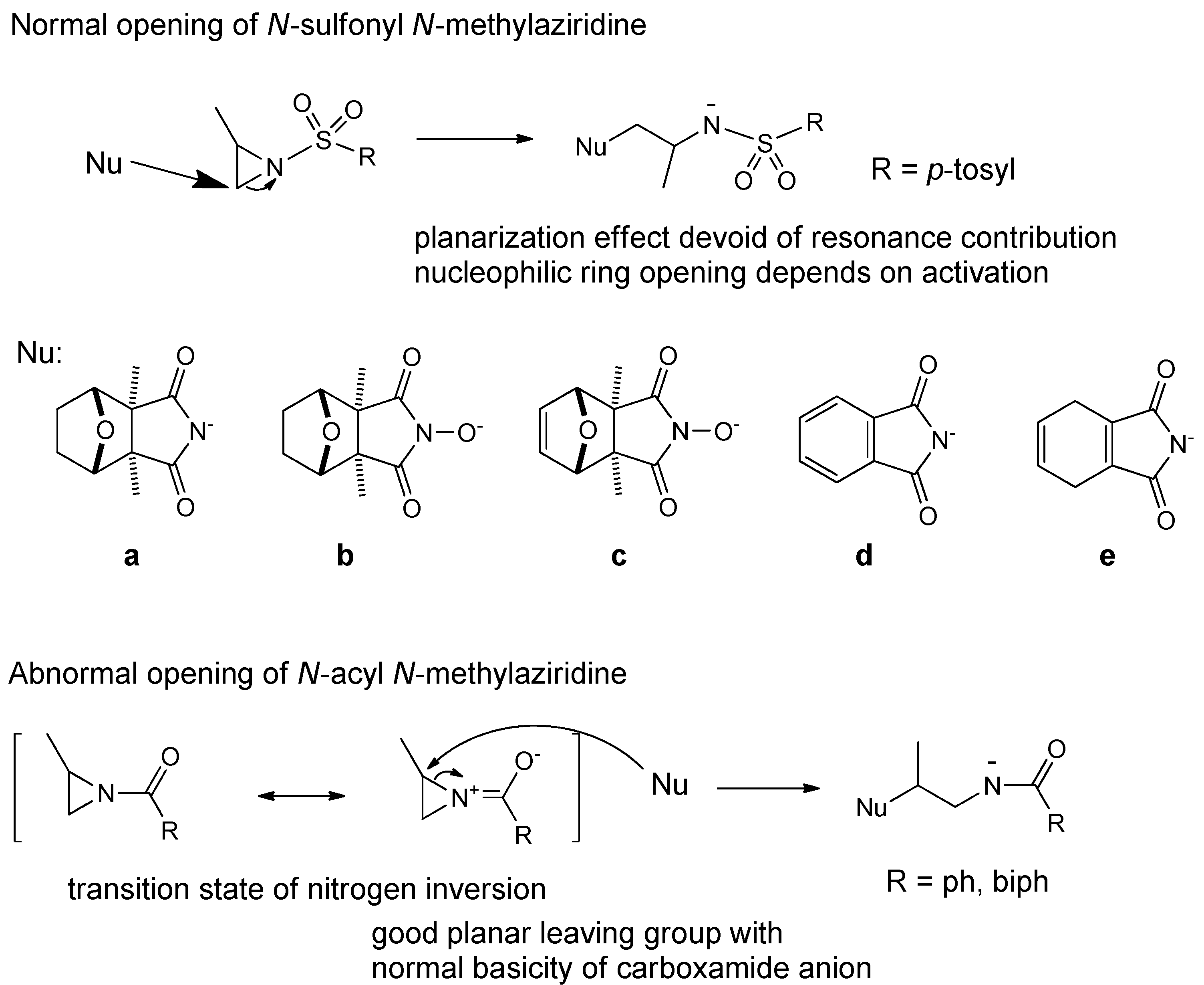{kind=link}
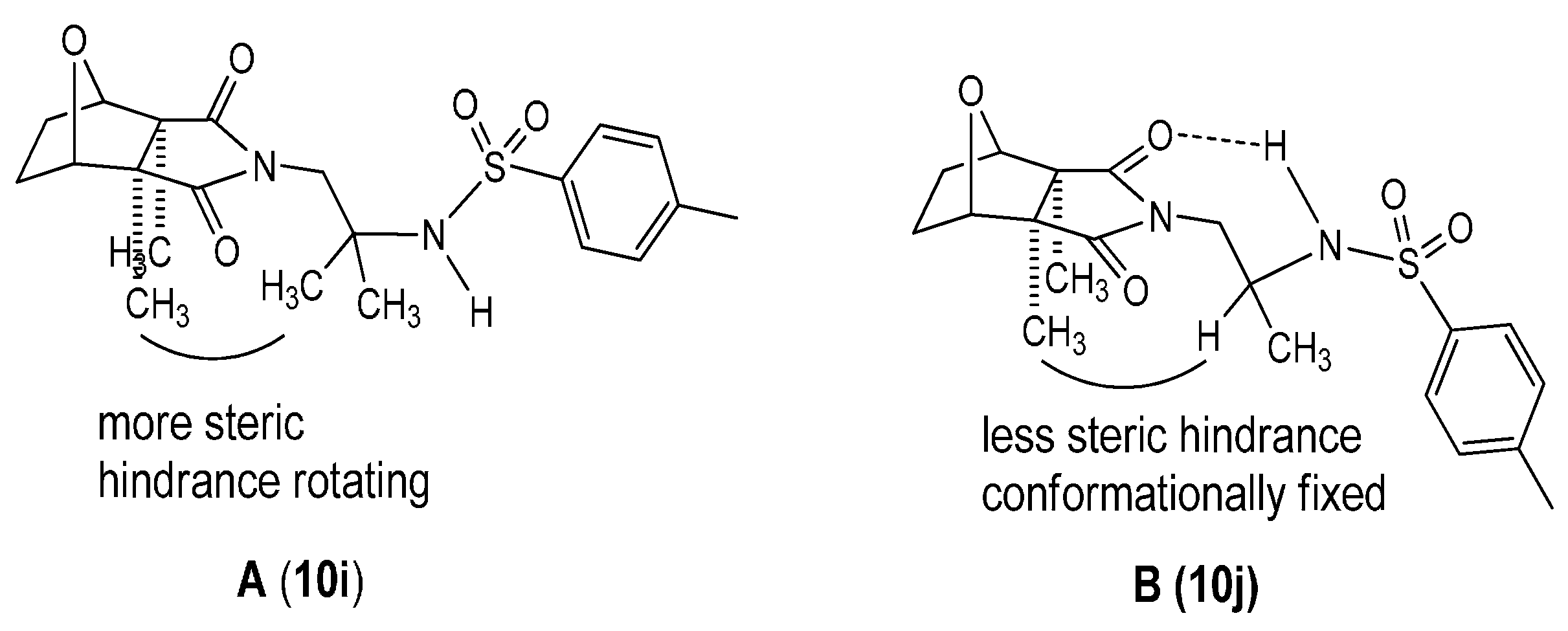{kind=link}
{kind=link}
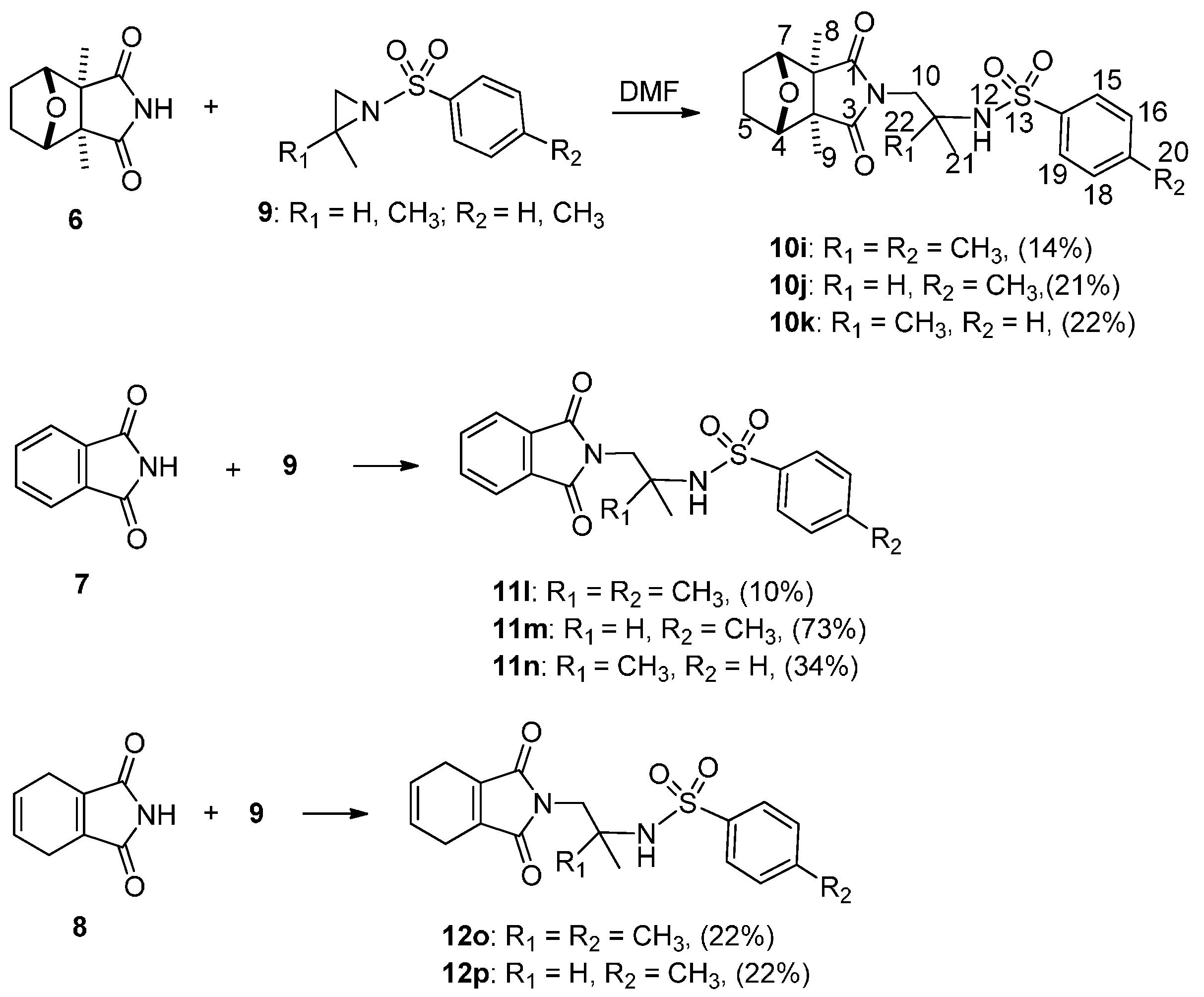{kind=link}
{kind=link}
{kind=link}
| Position | 10i | 10j |
|---|---|---|
| δH (J in Hz) | δH (J in Hz) | |
| 4, 7 | 4.54 (s) | 4.55 (s) |
| 5, 6 | 1.68–1.70 (m); 1.79–1.81 (m) | 1.67-1.70 (m); 1.79–1.83 (m) |
| 8, 9 | 1.22 (s) | 1.21 (s) |
| 10 | 3.51 (s) | 3.38 (dd, 3.8, 3.8); 3.61–3.64 (m) |
| 11 | - | 3.52 (dd, 9.7, 9.7) |
| 12 | 5.77 (s) | 4.68 (d, 9) |
| 15, 19 | 7.72 (d, 8) | 7.70 (d, 8) |
| 16, 18 | 7.23 (d, 8) | 7.27 (d, 8) |
| 20 | 2.39 (s) | 2.41 (s) |
| 21 | 1.14 (s) | 0.93 (d, 6.7) |
| 22 | 1.14 (s) | - |

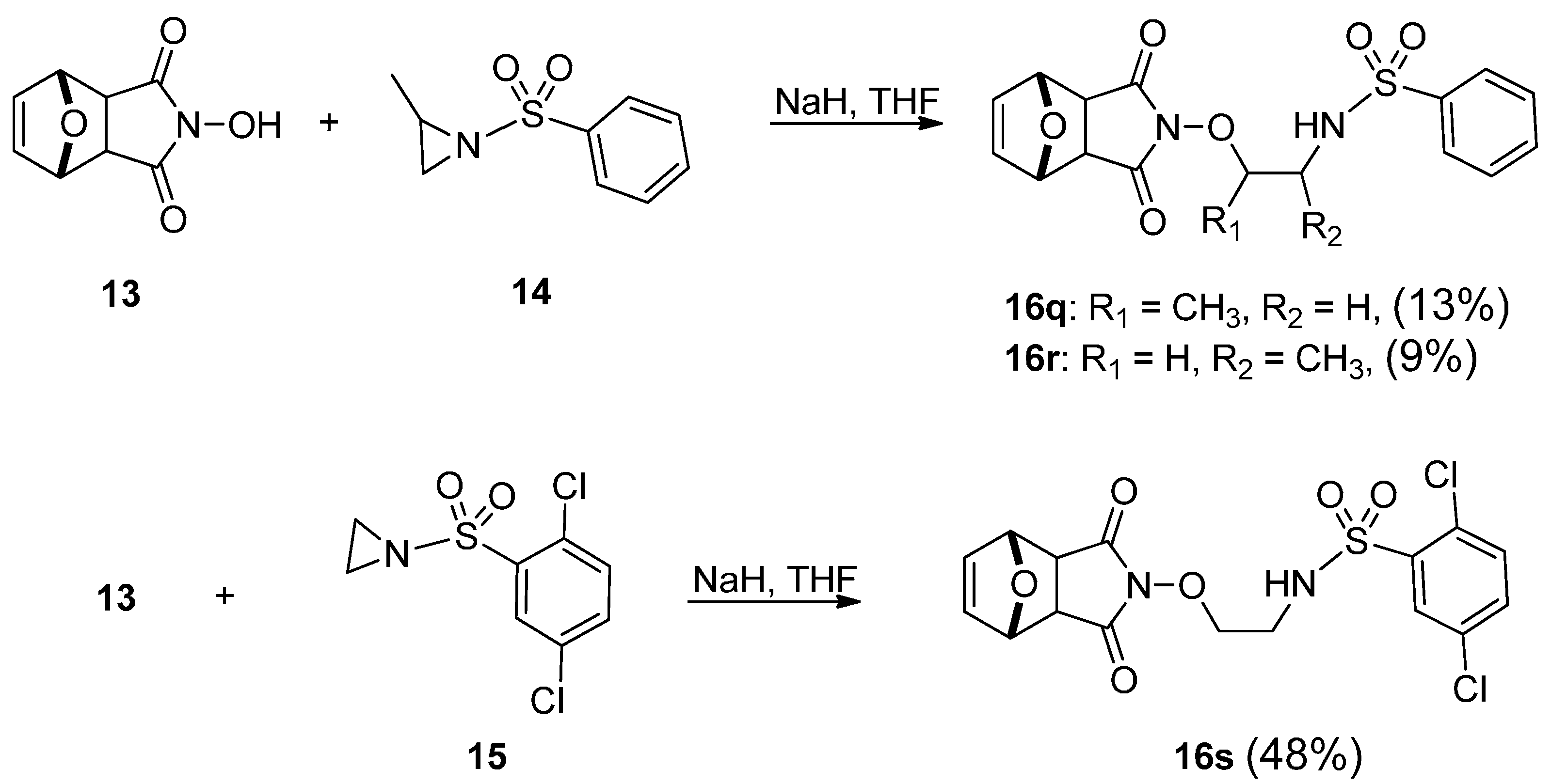
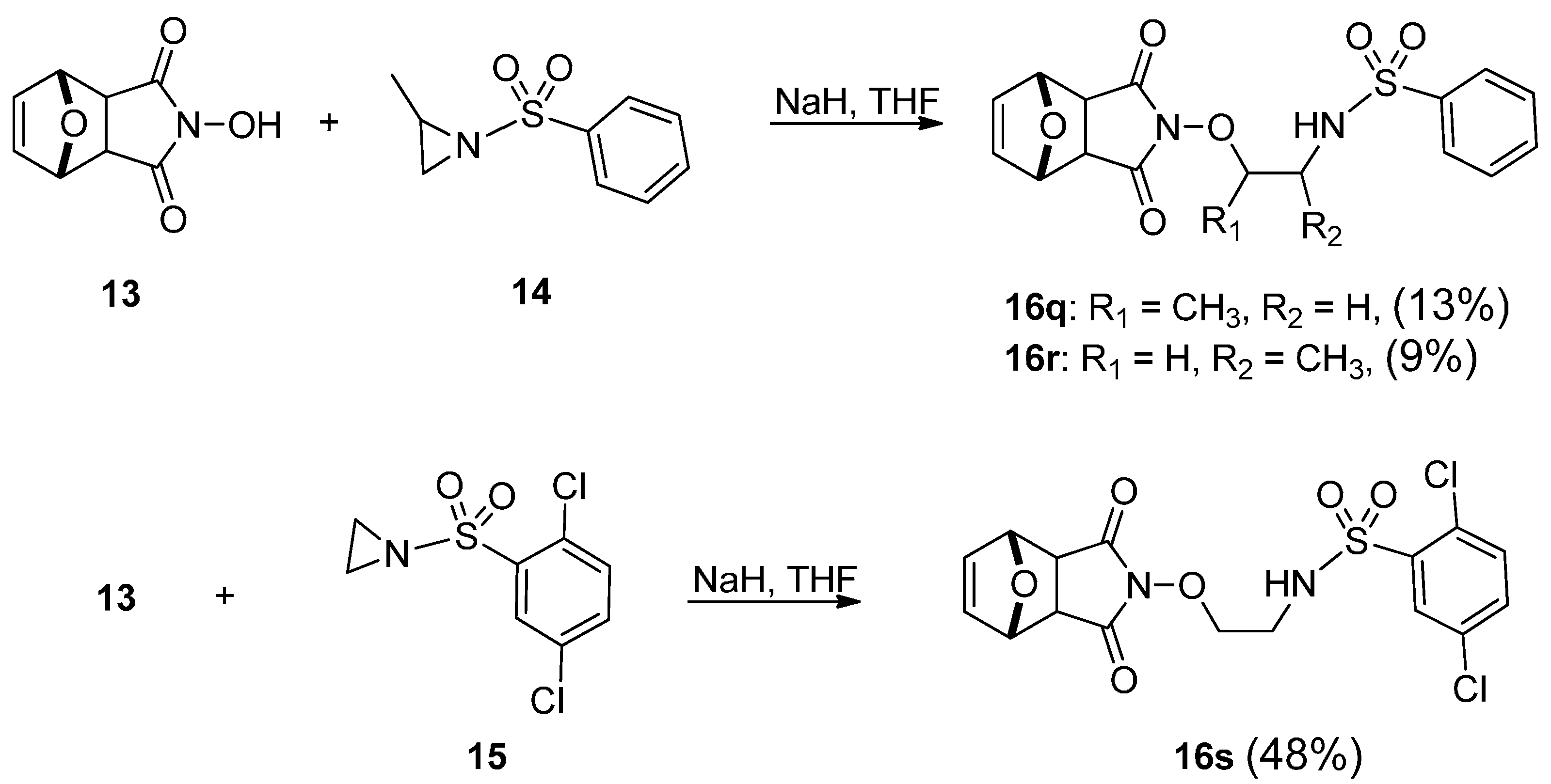
2.2. Pharmacology
| Cpd No. | IC50 (μM) a | |||
|---|---|---|---|---|
| HL-60 | Hep3B | MCF7 | MDA-MB-231 | |
| 1 | 5.6 | 3.4 | - | - |
| 5a | 38.2 | 9 | >100 | >100 |
| 5b | >100 | >100 | >100 | >100 |
| 5c | >100 | >100 | >100 | >100 |
| 5d | >100 | >100 | >100 | >100 |
| 5e | >100 | >100 | >100 | >100 |
| 5f | >100 | >100 | >100 | >100 |
| 5g | >100 | 17 | >100 | >100 |
| 5h | >100 | 80 | >100 | >100 |
| 10i | >100 | >100 | >100 | >100 |
| 10j | >100 | >100 | >100 | >100 |
| 10k | >100 | >100 | >100 | >100 |
| 11l | >100 | >100 | >100 | >100 |
| 11m | >100 | >100 | >100 | >100 |
| 11n | >100 | >100 | >100 | >100 |
| 12o | >100 | >100 | >100 | >100 |
| 12p | >100 | >100 | >100 | >100 |
| 16q | >100 | >100 | >100 | >100 |
| 16r | >100 | >100 | >100 | >100 |
| 16s | >100 | >100 | >100 | >100 |
3. Experimental Section
3.1 Synthesis
3.1.1. General Experimental Procedures
3.1.2. Test Samples
3.1.3. Physical Data of Cantharidinimide Derivatives and Analogues
3.2. Antineoplastic Bioassays
3.2.1. Cell Culture
3.2.2. MTT Assay for Cellular Viability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moed, L.; Shwayder, T.A.; Chang, M.W. Cantharidin revisited: A blistering defense of an ancient medicine. Arch. Dermatol. 2001, 137, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S. Medical uses of Mylabris in ancient china and recent studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef]
- Decker, R.H. Mechanism of acantholysis: The effect of cantharidin on oxidative phosphorylation. J. Investig. Dermatol. 1963, 42, 465–469. [Google Scholar] [CrossRef]
- Li, Y.M.; Casida, J.E. Cantharidin-binding protein: Identification as protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1992, 89, 11867–11870. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.C.; Post, L.O.; Reagor, J.C. GC/MS confirmation of cantharidin toxicosis due to ingestion of blister beetles. Vet. Hum. Toxicol. 1980, 22, 398–399. [Google Scholar] [PubMed]
- Sanchez-Barbudo, I.S.; Camarero, P.R.; Garcia-Montijano, M.; Mateo, R. Possible cantharidin poisoning of a great bustard (Otis tarda). Toxicon 2012, 59, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, R.E. Cantharidin another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 1993, 330, 283–286. [Google Scholar] [CrossRef]
- Efferth, T.; Rauh, R.; Kahl, S.; Tomicic, M.; Bochzelt, H.; Tome, M.E.; Briehl, M.M.; Bauer, R.; Kaina, B. Molecular modes of action of cantharidin in tumor cells. Biochem. Pharmacol. 2005, 69, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xie, L.; Chen, Z.; Zhu, Y.; Sun, Y.; Miao, Y.; Xu, Z.; Han, X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010, 101, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.W.; Ko, S.W.; Tsai, H.Y.; Chung, J.G.; Chiang, J.H.; Chen, K.T.; Chen, Y.C.; Chen, H.Y.; Chen, Y.F.; Yang, J.S. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int. J. Oncol. 2011, 38, 1067–1073. [Google Scholar] [PubMed]
- Hsia, T.C.; Yu, C.C.; Hsu, S.C.; Tang, N.Y.; Lu, H.F.; Huang, Y.P.; Wu, S.H.; Lin, J.G.; Chung, J.G. Cantharidin induces apoptosis of H460 human lung cancer cells through mitochondria-dependent pathways. Int. J. Oncol. 2014, 45, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.P.; Tsai, C.H.; Wu, P.P.; Hsu, S.C.; Liu, H.C.; Huang, Y.P.; Yang, J.H.; Chung, J.G. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and Cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria‑dependent pathways of A375.S2 human melanoma cells. Int. J. Oncol. 2014, 45, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Liu, S.H.; Lee, K.I.; Huang, K.T.; Lu, T.H.; Fang, K.M.; Wu, C.C.; Yen, C.C.; Lai, C.H.; Su, Y.C.; et al. Cantharidin induces apoptosis through the Calcium/PKC-regulated endoplasmic reticulum stress pathway in human bladder cancer cells. Am. J. Chin. Med. 2015, 43, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Prasad, S.B. Antitumor effect of blister beetles: An ethno-medicinal practice in Karbi community and its experimental evaluation against a murine malignant tumor model. J. Ethnopharmacol. 2013, 148, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Garshott, D.M.; Brownell, A.L.; Yoo, G.H.; Lin, H.S.; Freeburg, T.L.; Yoo, N.G.; Kaufman, R.J.; Callaghan, M.U.; Fribley, A.M. Cantharidins induce ER stress and a terminal unfolded protein response in OSCC. J. Dent. Res. 2015, 94, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q. A study on antitumor chemotherapeutic agent-synthesis of N-cantharidine derivatives. Acta Pharmacol. Sin. 1981, 16, 784–786. [Google Scholar]
- Walter, W.G. Antitumor imide derivatives of 7-oxabicyclo[2.2.1]heptane-2,3-dimethyl-2,3-dicarboxylic acid. J. Pharmacol. Sci. 1989, 78, 66–67. [Google Scholar] [CrossRef]
- Kok, S.H.L.; Chui, C.H.; Lam, W.S.; Chen, J.; Tan, J.C.O.; Lau, F.Y.; Cheng, G.Y.M.; Wong, R.S.M.; Chan, A.S.C. Induction of apoptosis on carcinoma cells by two synthetic cantharidin analogues. Int. J. Mol. Med. 2006, 17, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fogh, J.; Fogh, J.M.; Orfeo, T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar] [PubMed]
- Tsauer, W.; Lin, J.G.; Lin, P.Y.; Hsu, F.L.; Chiang, H.C. The effects of cantharidin analogues on xanthine oxidase. Anticancer Res. 1997, 17, 2095–2098. [Google Scholar] [PubMed]
- Essers, M.; Wibbeling, B.; Haufe, G. Synthesis of the first fluorinated cantharidin analogues. Tetrahedron Lett. 2001, 42, 5429–5433. [Google Scholar] [CrossRef]
- McCluskey, A.; Keane, M.A.; Sim, A.T.R.; Walkom, C.C.; Young, D.J.; Bowyer, M.C.; Sakoff, J.A. The first two cantharidin analogues displaying PP1 selectivity. Bioorg. Med. Chem. Lett. 2002, 12, 391–393. [Google Scholar] [CrossRef]
- Hart, M.E.; Chamberlin, R.; Walkom, C.; Sakoff, J.A.; McCluskey, A. Modified norcantharidins: Synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg. Med. Chem. Lett. 2004, 14, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Tatlock, J.H.; Linton, M.A.; Hou, X.J.; Kissinger, C.R.; Pelletier, L.A.; Showalter, R.E.; Tempczyk, A.; Villafranca, J.E. Structure-based design of novel calcineurin (PP2B) inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 1007–1012. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Synthesis and anticancer activity of a series of norcantharidin analogues. Eur. J. Med. Chem. 2012, 54, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Puerto Galvis, C.E.; Vargas Mendez, L.Y.; Kouznetsov, V.V. Cantharidin-based small molecules as potential therapeutic agents. Chem. Biol. Drug Des. 2013, 82, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Luzina, E.L.; Popov, A.V. Synthesis and anticancer activity evaluation of 3,4-mono- and bicyclosubstituted N-(het)aryl trifluoromethyl succinimides. J. Fluorine Chem. 2014, 168, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Shi, S.J.; Shu, H.L.; Chen, H.C.; Lin, C.C.; Liu, P.C.; Wang, L.F. A simple procedure for preparation of N-thiazolyl and N-thiadiazolylcantharidinimides and evalution of their cytotoxicities against human hepatocellular carcinoma cell. Bioorg. Chem. 2000, 28, 266–272. [Google Scholar]
- Lin, L.H.; Huang, H.S.; Lin, C.C.; Lee, L.W.; Lin, P.Y. Effects of cantharidins on human carcinoma cells. Chem. Pharm. Bull. 2004, 52, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Tseng, I.J.; Sheu, S.Y.; Chen, Y.T.; Huang, C.Y.; Lin, C.T.; Lin, P.Y. A simple procedure for preparation of N-thiazol, thiadiazol, pyridyl and sulfanylamidocantharidinimimes analogues and evaluation of their cytotoxicities against human HL-60, MCF7, Neuro-2a and A549 carcinoma cell. Chem. Pharm. Bull. 2012, 60, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Tseng, I.J.; Sheu, S.Y.; Lin, P.Y.; Lee, J.A.; Ou, K.L.; Lee, L.W. Synthesis and evaluation of cantharidinimides on human cancer cells. J. Exp. Clin. Med. 2012, 4, 280–283. [Google Scholar] [CrossRef]
- Yej, C.B.; Lin, P.Y.; Huang, J.M.; Su, C.J.; Yeh, Y.T.; Yang, S.F.; Chou, M.C. Study on synthesis of thalidomide analogues and their bioactivities: Inhibition on iNOS pathway cytotoxic effects. Med. Chem. Res. 2012, 21, 953–963. [Google Scholar]
- Tseng, I.J.; Lin, P.Y.; Sheu, S.Y.; Tung, W.N.; Lin, C.T.; Lin, M.H. Characterization of novel aminobenzylcantharidinimides and their effects on NO induction. J. Chin. Chem. Soc. 2015, 62, 59–63. [Google Scholar] [CrossRef]
- Lin, P.Y.; Bellos, K.; Stamm, H.; Onistschenko, A. Acylated 2,2-dimethylaziridin: Regioselectivity of ring opening thiophenolate; borderline SN2 sue to planarization of nitrogen pyramid. Tetrahedron 1992, 48, 2359–2372. [Google Scholar] [CrossRef]
- Bellos, K.; Stamm, H.; Speth, D. N-S Cleavage is faster than homolytic ring opening in single-electron transfer to some N-sulfonylaziridines. Competition between SN2 and SET. J. Org. Chem. 1991, 56, 6846–6849. [Google Scholar] [CrossRef]
- Lin, P.Y.; Weiee, R.; Werry, J.; Falkenstein, R.; Stamm, H. Neglected aspects of anthracenide (anthracenidyl) chemistry reactions of two N-benzoylaziridines. J. Prakt. Chem. 2000, 342, 153–161. [Google Scholar] [CrossRef]
- Kadioglu, O.; Kermani, N.S.; Kelter, G.; Schumacher, U.; Fiebig, H.H.; Greten, H.J.; Efferth, T. Pharmacogenomics of cantharidin in tumor cells. Biochem. Pharmacol. 2014, 87, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Wosikowski, K.; Schuurhuis, D.; Johnson, K.; Paull, K.D.; Myers, T.G.; Weinstein, J.N.; Bates, S.E. Identification of epidermal growth factor receptor and c-erbB2 pathway inhibitors by correlation with gene expression patterns. J. Natl. Cancer Inst. 1997, 89, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Luzina, E.L.; Popov, A.V. Synthesis and anticancer activity of N-bis(trifluoromethyl)alkyl-N′-thiazolyl and N-bis(trifluoromethyl)alkyl-N′-benzothiazolyl ureas. Eur. J. Med. Chem. 2009, 44, 4944–4953. [Google Scholar] [CrossRef] [PubMed]
- Luzina, E.L.; Popov, A.V. Anticancer activity of N-bis(trifluoromethyl)alkyl-N′-(polychlorophenyl) and N′-(1,2,4-triazolyl) ureas. Eur. J. Med. Chem. 2010, 45, 5507–5512. [Google Scholar] [CrossRef] [PubMed]
- Luzina, E.L.; Popov, A.V. Synthesis, evaluation of anticancer activity and COMPARE analysis of N-bis(trifluoromethyl)alkyl-N′-substituted ureas with pharmacophoric moieties. Eur. J. Med. Chem. 2012, 53, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Luzina, E.L.; Popov, A.V. Synthesis of 3,3,3-trifluoroethyl isocyanate, carbamate and ureas. Anticancer activity evaluation of N-(3,3,3-trifluoroethyl)-N′-substituted ureas. J. Fluorine Chem. 2015, 176, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 10i–k, 11l–n, 12o–p, and 16q–s are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, L.-L.; Tseng, I.-J.; Lin, P.-Y.; Sheu, S.-Y.; Lin, C.-T.; Hsieh, Y.-H.; Lin, Y.-J.; Chen, H.-L.; Lin, M.-H. Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects. Molecules 2016, 21, 100. https://doi.org/10.3390/molecules21010100
Chiang L-L, Tseng I-J, Lin P-Y, Sheu S-Y, Lin C-T, Hsieh Y-H, Lin Y-J, Chen H-L, Lin M-H. Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects. Molecules. 2016; 21(1):100. https://doi.org/10.3390/molecules21010100
Chicago/Turabian StyleChiang, Ling-Ling, Ing-Jy Tseng, Pen-Yuan Lin, Shiow-Yunn Sheu, Ching-Tung Lin, Yun-Han Hsieh, Yi-Jing Lin, Hsiao-Ling Chen, and Mei-Hsiang Lin. 2016. "Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects" Molecules 21, no. 1: 100. https://doi.org/10.3390/molecules21010100
APA StyleChiang, L.-L., Tseng, I.-J., Lin, P.-Y., Sheu, S.-Y., Lin, C.-T., Hsieh, Y.-H., Lin, Y.-J., Chen, H.-L., & Lin, M.-H. (2016). Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects. Molecules, 21(1), 100. https://doi.org/10.3390/molecules21010100