
Methods to Increase the Metabolic Stability of 18F-Radiotracers


Helmholtz-Zentrum Dresden-Rossendorf, Institut für Radiopharmazeutische Krebsforschung, Bautzner Landstraße 400, Dresden D-01328, Germany
*
Author to whom correspondence should be addressed.
Molecules 2015, 20(9), 16186-16220; https://doi.org/10.3390/molecules200916186
Submission received: 16 June 2015
/
Revised: 20 August 2015
/
Accepted: 26 August 2015
/
Published: 3 September 2015
(This article belongs to the Special Issue Preparation of Radiopharmaceuticals and Their Use in Drug Development)
Abstract
:The majority of pharmaceuticals and other organic compounds incorporating radiotracers that are considered foreign to the body undergo metabolic changes in vivo. Metabolic degradation of these drugs is commonly caused by a system of enzymes of low substrate specificity requirement, which is present mainly in the liver, but drug metabolism may also take place in the kidneys or other organs. Thus, radiotracers and all other pharmaceuticals are faced with enormous challenges to maintain their stability in vivo highlighting the importance of their structure. Often in practice, such biologically active molecules exhibit these properties in vitro, but fail during in vivo studies due to obtaining an increased metabolism within minutes. Many pharmacologically and biologically interesting compounds never see application due to their lack of stability. One of the most important issues of radiotracers development based on fluorine-18 is the stability in vitro and in vivo. Sometimes, the metabolism of 18F-radiotracers goes along with the cleavage of the C-F bond and with the rejection of [18F]fluoride mostly combined with high background and accumulation in the skeleton. This review deals with the impact of radiodefluorination and with approaches to stabilize the C-F bond to avoid the cleavage between fluorine and carbon.
1. Introduction
Positron emission tomography (PET) and the combined techniques PET/MRT and PET/CT are outstanding imaging instruments and allow for the quantification and localization of physiological as well as pathophysiological processes in vivo, which were analyzed by tracing the appropriate biochemical fundamentals [1]. The basics of PET originate in the coincidental detection of annihilation photons emitted 180° apart, which originate from the radiotracer emitting positron, which again collides with electrons in the surrounding tissue. Measurement and quantification of the tracer distribution were obtained noninvasively in living organisms [2]. Fluorine-18 is an ideal radionuclide due to its favorable nuclear decay properties. It has a half-life of 109.8 min, which provides sufficient time to radiolabel the molecule of interest and localize it in vivo. Additionally, it emits a positron of low kinetic energy, which only travels a short range in tissue leading to high image resolution. However, tracers for PET imaging are always restricted by the kind of molecules that researchers can prepare and label. A summary of commonly used PET radionuclides is found in Table 1.

Table 1.
Most commonly used PET radionuclides with their radiochemical details [3].
| Nuclide | t½ (min) | Production Route | Average Range in H2O (mm) | Eav. (β+) (keV) |
|---|---|---|---|---|
| 11C | 20.4 | 14N(p,α)11C | 1 | 385 |
| 13N | 10 | 16O(p,α)13N | 1.5 | 491 |
| 15O | 2 | 15N(d,n)15O | 2.7 | 735 |
| 18F | 109.8 | 20Ne(d,α)18F 18O(p,n)18F | 0.3 | 242 |
| 68Ga | 67.6 | 68Ge-68Ga generator | 3.7 | 740 |
| 124I | 250.6 | 124Te(p,n)124I | 3 | 188 |
Fluorine-18 is a unique radionuclide for PET imaging. In contrast to other β+ emitting organic radionuclides like 11C, 13N, and 15O, which are inclined to isotopic labeling, fluorine-18 is most commonly incorporated leading to an alteration of the original compound [4,5]. Due to the absence of fluorine in nearly all naturally occurring biomolecules [6], radiolabeling is often accomplished by a formal replacement of a proton or an OH group with 18F (isosteric and isopolar) which is known as bioisosteric labeling (Table 2) [7]. However, in medicinal chemistry, the role of fluorine in drug design and development is expanding rapidly and a wide variety of small compounds/drugs were developed in the past with pharmacological relevance still having one or more fluorine atoms inside [8]. These molecules can serve as brilliant lead structures for 18F-radiotracers. The other variant deals with the connection of small 18F-building blocks or 18F-prosthetic groups like [18F]SFB or [18F]FBAM [9], but this is mostly used with biomacromolecules like peptides, proteins, or antibodies. Both methods come along with changes of biological and/or pharmacological properties of the tracer molecule compared to the original compound. In general, smaller molecules exhibit a larger change in their properties by the introduction of a radionuclide to the considered molecule.
1.1. Nature of the C-F Bond
The similarity in size of fluorine (147 pm), hydrogen (120 pm) and oxygen (152 pm) makes fluorine-18 an appropriate candidate for the preparation of radiotracers, due to its longer half-life time compared to 11C, 13N or 15O (Table 1) [10]. The substitution of single hydrogen or a hydroxyl group by fluorine induces only a slight steric perturbation [11]. The similarity of the C-F to the C-O bond length (Table 2) and the similar electronic properties like the induced dipole due to the inductive effect allows the isoelectronic replacement of an OH group by fluorine [12]. However, fluorine is only a (poor) hydrogen bond acceptor, while an OH group is both a hydrogen donor and an acceptor.
Table 2.
Van der Waals radii [13], electronegativity and aliphatic C-X bond lengths of selected atoms.
| Element X | Van der Waals Radius (pm) | Electronegativity (Pauling Scale) | Bond Length of C-X (pm) |
|---|---|---|---|
| H | 120 | 2.1 | 109 |
| C | 170 | 2.5 | 154 |
| O | 152 | 3.5 | 143 |
| F | 147 | 4.0 | 135 |
Of all the atoms, the fluorine atom possesses the highest electronegativity; therefore, biological aspects have to be considered and can be of advantage in pharmaceutical as well as in radiotracer design. A favorable feature of fluorine is the strong but highly polarized σ bond to carbon [11]; this should make the fluorine a perfect leaving group in case of nucleophilic displacement reactions. However, the fluorine unexpectedly does not show good donor ability despite the high polarization of the C-F bond. This fact can be explained by the strong interaction of the partially positively charged carbon (residue) and the partially negatively charged fluorine which results in the strongest known σ bond in organic chemistry. Additionally, the highest bond dissociation energy (BDE) of approx. 441.3 kJ/mol is found for an aliphatic C-F bond compared to other carbon single bonds [14].
Interestingly, the average BDE differs with the number of covalently bound fluorine. More fluorine atoms bound to carbon increases the BDE and diminishes the C-F bond length [15]. The series of fluoromethane compounds in Scheme 1 demonstrate this trend, which can be explained by each of the C-F bonds pulling p-orbital electron density from the sp3 carbon to the low lying sp2 orbitals of fluorine (Bent’s rule [16]), making the carbon more sp2 in character [17].
Scheme 1.
Comparison of fluoromethanes with increasing number of bound fluorine and their associated bond length and bond dissociation energy.
Scheme 1.
Comparison of fluoromethanes with increasing number of bound fluorine and their associated bond length and bond dissociation energy.
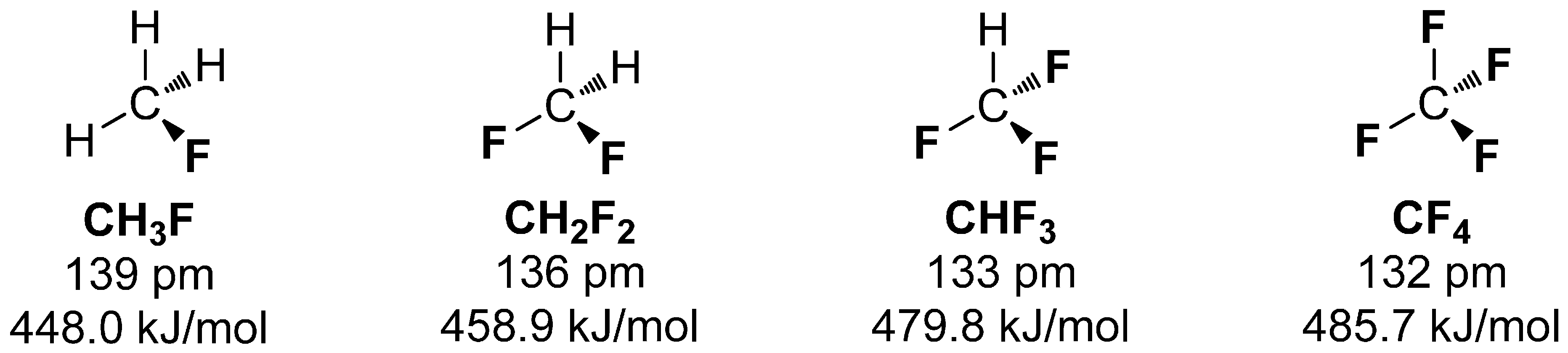
Particular attention has to be made for fluorine in the benzylic position. It was found that the bond enthalpies (DH298) are lower for benzyl fluorides (413.2 kJ/mol) compared to aliphatic compounds (439.2 kJ/mol for methyl fluoride) or aromatic fluorine derivatives (472.7 kJ/mol for fluorobenzene) [11]. Furthermore, the C-F bond length is also extended (138 pm compared to 135 pm average) [18]. The remaining substituents on the aromatic ring also have crucial influence on the stability of the benzylic fluorine especially concerning steric and inductive effects [19]. The introduction of [18F]fluoride into the benzylic position of precursors occurs as previously described for aliphatic compounds, under milder reaction conditions in most of the cases due to the comparatively higher reactivity of the benzylic position. In this regard, the metabolic stability of benzyl [18F]fluorides is also decreased [20]. Additionally, similar observations were made for allyl fluorides [21].
Aromatic CAr(sp2)-F bonds are stronger than aliphatic C(sp3)-F bonds [22] resulting in their bond lengths being shorter: 140 pm (aliphatic) vs. 136 pm (aromatic) [23]. This finding can be explained by the high polarization of the σ-C-F bond, the possible delocalization of the (partial) positive charge of the carbon in the aromatic moiety, and that fluorine possibly acts as a π-electron donor [24,25], which strengthens the C-F bond additionally. Thus, the 18F radiolabeling of aromatic systems should be favored over aliphatic systems.
Bonding of fluorine to a sp-hybridized carbon is also possible, but this results in a highly reactive species due to the repulsion of the free electron pairs of the fluorine and the π-electron system of the triple bond [17]. This trend strongly follows Bent’s rule, which states that the s-character of an atom concentrates in orbitals directed toward electropositive substituents [16]. No 18F-radiotracer with a direct connection of 18F to a triple bond has been developed to date.
To sum up, to produce a stabilized C-F bond, the most important criterion is the hybridization of the carbon. Moreover, inductive and steric effects of further substituents and organic residues influence the (metabolic) stability of the C-F bond as well.
1.2. Possibilities to Introduce Fluorine-18—Short Overview
The radionuclide 18F is produced by a cyclotron using the nuclear reactions shown in Table 1. Once the radionuclide is produced, it must quickly be incorporated in the molecule of interest. Normally, the introduction of 18F into aliphatic molecules (sp3-hybridized carbon) is accomplished using no-carrier-added (n.c.a.) [18F]fluoride and a precursor with a good leaving group (Br, I, OMs, OTs, ONs, OTf, NR3+) in a SN2 reaction. This method has the advantage of preparing radiotracers with high specific activity (AS). A challenging aspect of this labeling procedure is to eliminate traces of water to remove the hydration shell around the fluoride. Polar organic solvents (ACN, DMF, DMSO) were used with a cryptand (Krypofix K2.2.2.) to function as a phase transfer catalyst and to further separate the charge of the cation and fluoride (producing what is called naked fluoride) [26].
Introduction of fluorine-18 into aromatic systems can be performed by several reaction pathways. The classical Balz-Schiemann reaction is only rarely used for this purpose [27]. Commonly, two ways are applied: the nucleophilic aromatic substitution (SNAr) and the electrophilic aromatic substitution (SEAr). The major drawback when using the first variant is the necessity to activate the respective aromatic precursor with electron withdrawing groups (CN, halogens, NO2, C=O) as well as good leaving groups. An isotopic exchange of 19F by 18F is also possible, but this results in a low AS. This is reasoned by the disability to separate the 19F-precursor from the 18F-radiotracer. This is because the AS is always influenced by the applied amount of the 19F-compound. Other appropriate leaving groups for the nucleophilic aromatic displacement are halogens, NO2 or Me3N+. Newer developments are based on iodonium [28] or sulfonium salts [29,30] as precursors and can be used for non-activated aromatic systems as well [31,32].
Using SEAr, [18F]F2 was applied consisting of both 18F and 19F (carrier added, c.a.), thus, the labeling will proceed in an “electrophilic” manner. As a consequence, a minimum of 50% of the elemental fluorine is 19F and therefore not β+-decaying. This pathway leads to a reduced AS of the radiotracers due to the incorporation of 19F. Usually, stannylated precursors, in which the carbon has the partial negative charge, are required for the labeling with [18F]F2 [33].
2. Radiodefluorination
Today’s arsenal of radiotracers comprises more and more complex molecules ranging from small organic and pharmacologically active derivatives such as carbohydrates, amino acids or steroids to high molecular weight compounds like peptides, proteins or oligonucleotides. The development of new radiotracers for molecular imaging has to address important questions on target selection and radiobiological validation. These special requirements are encountered in radiotracer synthesis such as choice of the appropriate radionuclide and suitable labeling position. In this regard, a radiotracer has to meet different criteria to be delivered to the target area of interest such as an adequate lipophilicity, high selectivity to the biological target and a high metabolic stability in vivo [34]. Hence, special attention should be paid to implement fast and highly selective labeling reactions for radiotracers which tolerate other functional groups. One of the most important aspects in the design of new radiopharmaceuticals is the development of metabolically stable tracers to meet the desired requirements and characteristics as mentioned above [35]. The radiolytic decomposition of 18F-radiotracers is also an important issue, especially during isolation and formulation of the tracer. This drawback can be avoided using additives like anti-oxidant stabilizers [36].
Drug metabolism, also known as xenobiotic metabolism, involves the biochemical modification of substances (pharmaceuticals, drugs, poisons, radiotracers). Drugs often are foreign compounds to the organism’s normal biochemistry. This metabolism usually occurs through specialized enzymatic systems by living organisms. Because of this mechanism, lipophilic substances are often converted into more readily hydrophilic derivatives, which are then excreted. The rate of metabolism determines the duration and efficacy of a drug, also known as the biological half-life [37]. In the case of radiopharmaceuticals, the physical half-life of the appendant radionuclide influences this mechanism supplementary.
The reactions in these biochemical pathways are of particular interest in medicine as part of drug metabolism and as a factor contributing to multidrug resistance in infectious diseases, cancer chemotherapy or radiopharmacy. The speed of the homing process of a radioactive drug has to be relatively fast compared to the biological and physical half live of the drug to be able to obtain good signal to background ratio.
Drug metabolism in general is divided into three phases. In phase I, enzymes such as cytochrome P450 oxidase (oxidative metabolism: CYP, FMO, MAO, Mo-CO, aldehyde oxidase, peroxidases, xanthine oxidase; hydrolytic metabolism: esterase, amidases, epoxide hydrolases) introduce reactive or polar groups into the xenobiotics. Afterwards, these modified compounds are conjugated to yield more polar compounds in phase II reactions. These reactions are catalyzed by transferase enzymes (UGT, ST, NAT, GST, MT) [38]. Finally, in phase III, the conjugated xenobiotics may be further processed, before being recognized by efflux transporters and eliminated from the cells. Radiotracers follow this method of degradation as well with one large difference; they are administered in concentrations, which are significantly lower than “normal” pharmaceuticals.
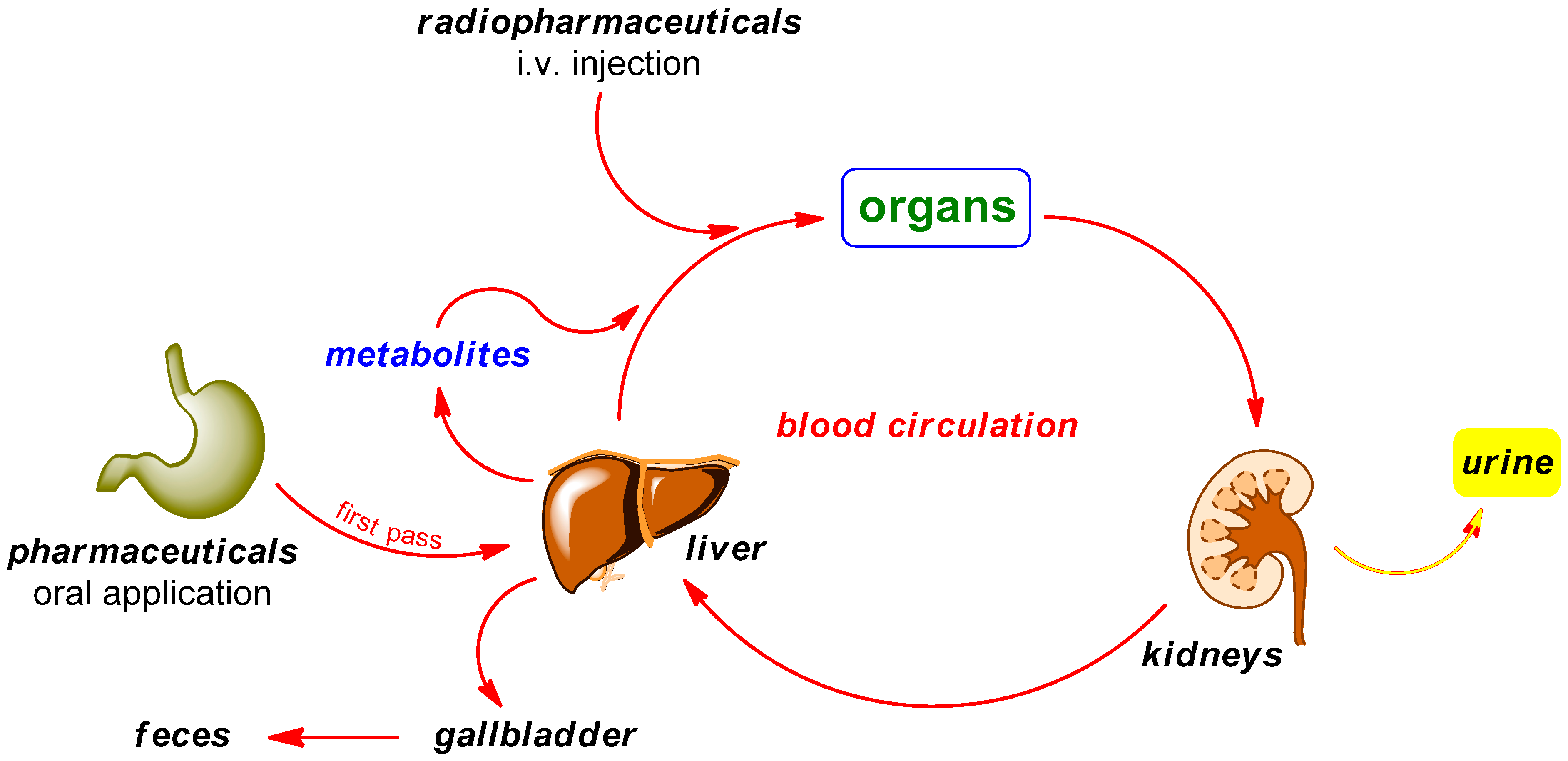
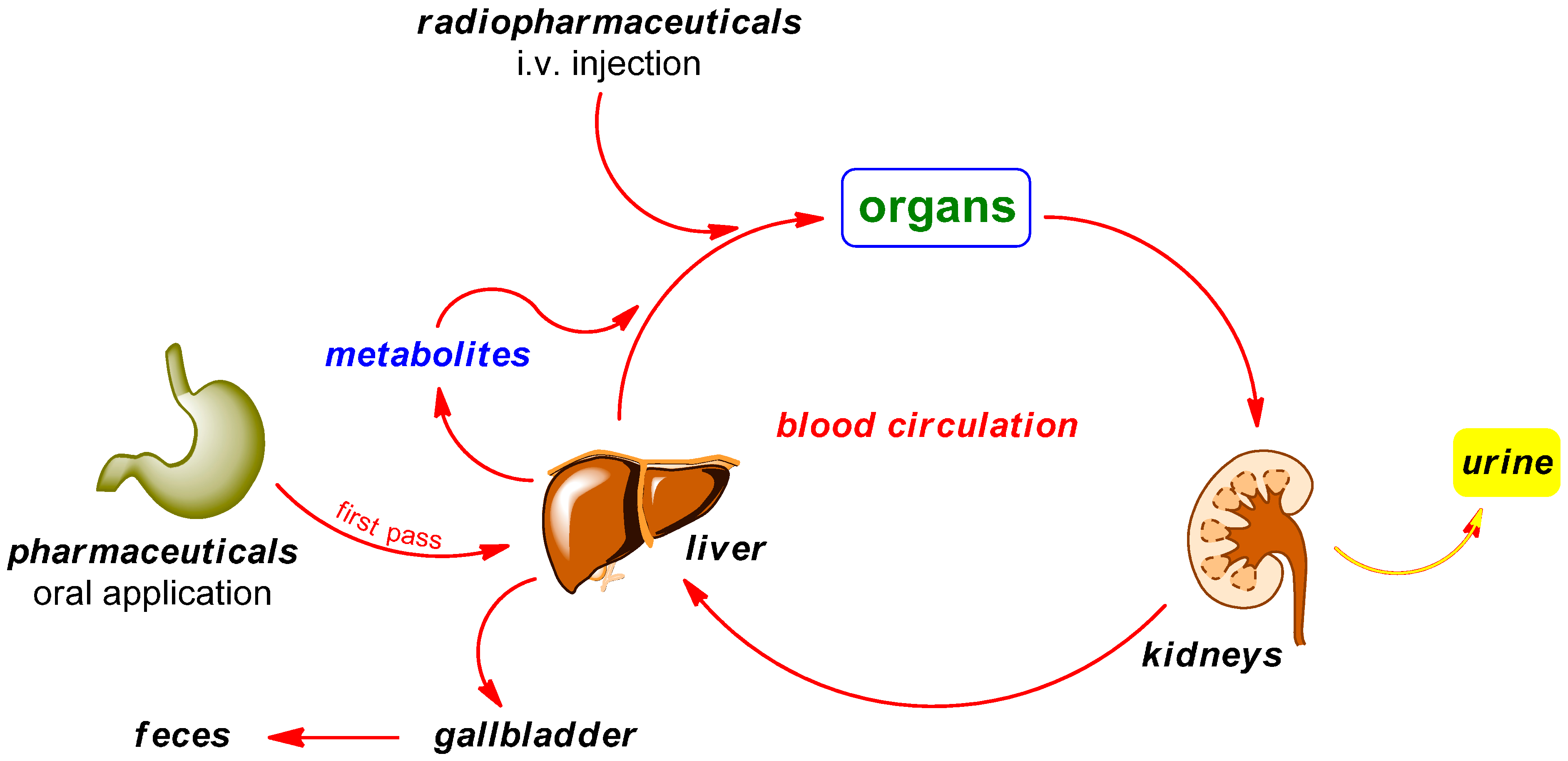
PET radiotracers are typically injected intravenously in contrast to the orally administered “normal” pharmaceuticals. While circulating in the blood and tissues prior to localizing at the target site, a portion of the drug may be metabolized. The major organs involved in this metabolism process are the kidneys and liver. The biotransformation may happen within minutes of administration and the resulting radiometabolites are generally less lipophilic than the original radiotracer. Possible metabolic degradation pathways are illustrated in Scheme 2. Radiodefluorination is known to be a phase I reaction occurring primarily through the action of cytochrome P450 2E1 (CYP2E1) isozyme in liver microsomes [39,40,41].
Scheme 2.
Possible pathways of radiopharmaceuticals in contrast to pharmaceuticals in the body.
Mechanisms of Radiodefluorination
Radiotracers, independent on their corresponding radionuclide, that resist extensive metabolism in vivo over the period of time of a PET scanning session are seldom [42]. Metabolic paths that derivatize rather than disintegrate the respective tracer into small, more polar fragments, can produce unpleasant radiometabolites. Alongside the formation of more polar radiometabolites, the cleavage of [18F]fluoride from the tracer, also known as radiodefluorination, is a major way for several 18F-radiotracers to degrade despite the high strength of the C-F bond [43]. Afterwards, [18F]fluoride as the main metabolite binds primarily to bone and skull. Especially while imaging the central nervous system, [18F]fluoride binding to the skull is problematic [42].
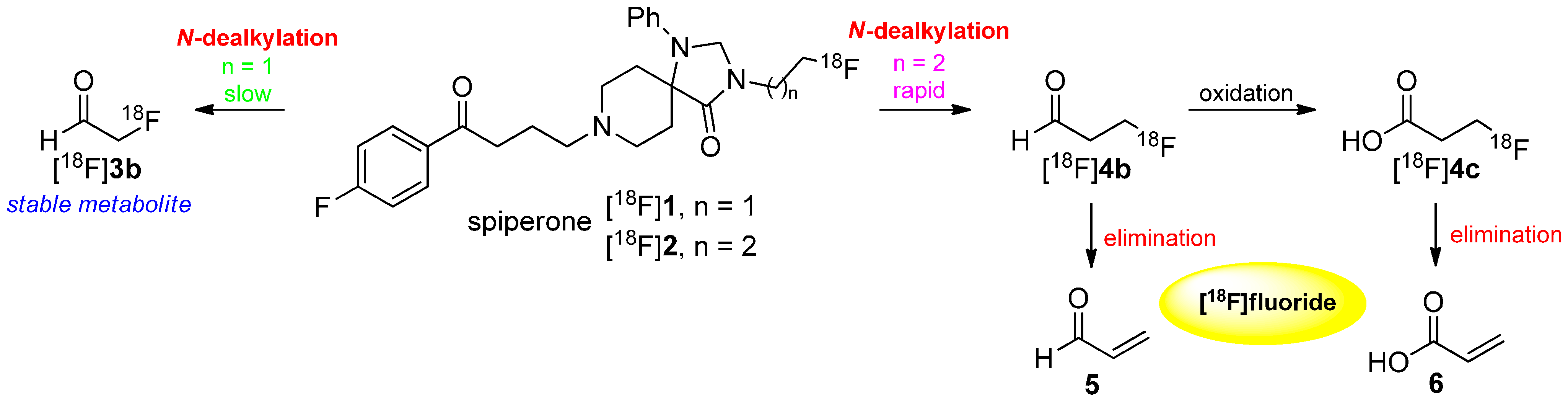
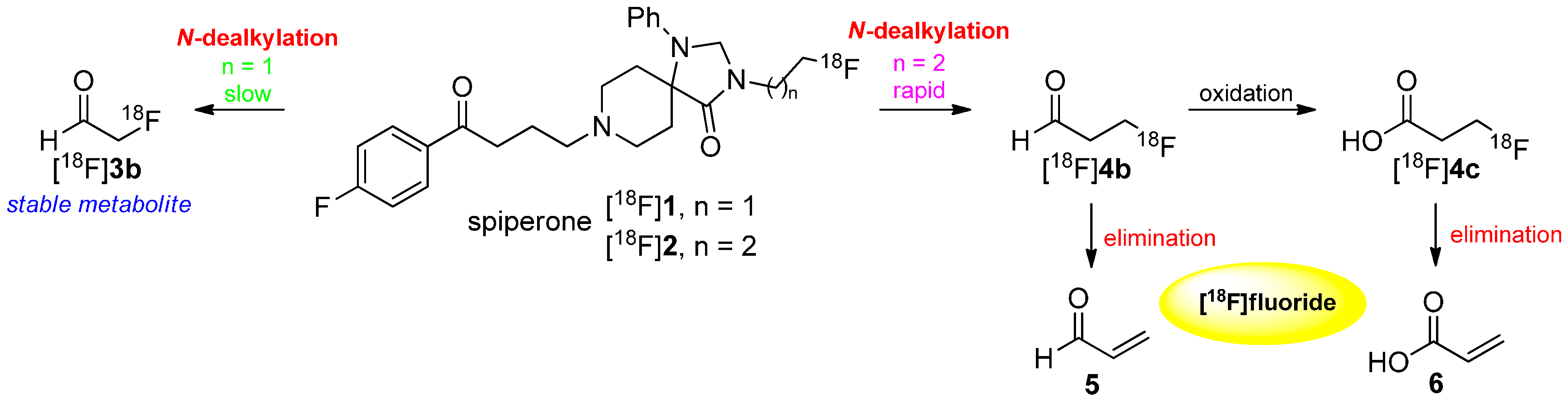
There are several metabolic pathways discussed and proposed for the degradation of [18F]fluoroalkyl chains. Two major factors affect the method of degradation: the location of the fluoroalkyl chain in the molecules and their length. In 1988, Welch and co-workers showed the difference in the metabolism between N-[18F]fluoroethylated and N-[18F]fluoropropylated spiperones [18F]1 and [18F]2 [44]. They proposed a metabolization by N-dealkylation followed by oxidation to the respective [18F]fluoroaldehydes [18F]3b and [18F]4b. As a result, 2-[18F]fluoroacetaldehyde ([18F]3b) is a stable lipophilic metabolite whereas 3-[18F]fluoropropanal ([18F]4b) is unstable towards elimination of [18F]fluoride (retro Michael addition). Further oxidation led to 3-[18F]fluoropropionate ([18F]4c), which eliminated subsequently to [18F]fluoride, too. In general, this kind of metabolism occurs when [18/19F]fluoroalkyl chains are bound to heteroatoms like oxygen, nitrogen or sulfur. Both pathways are shown in Scheme 3.
Scheme 3.
Mechanism of degradation of [18F]1 and [18F]2 leading to N-dealkylation of the radiotracer.
Scheme 3.
Mechanism of degradation of [18F]1 and [18F]2 leading to N-dealkylation of the radiotracer.
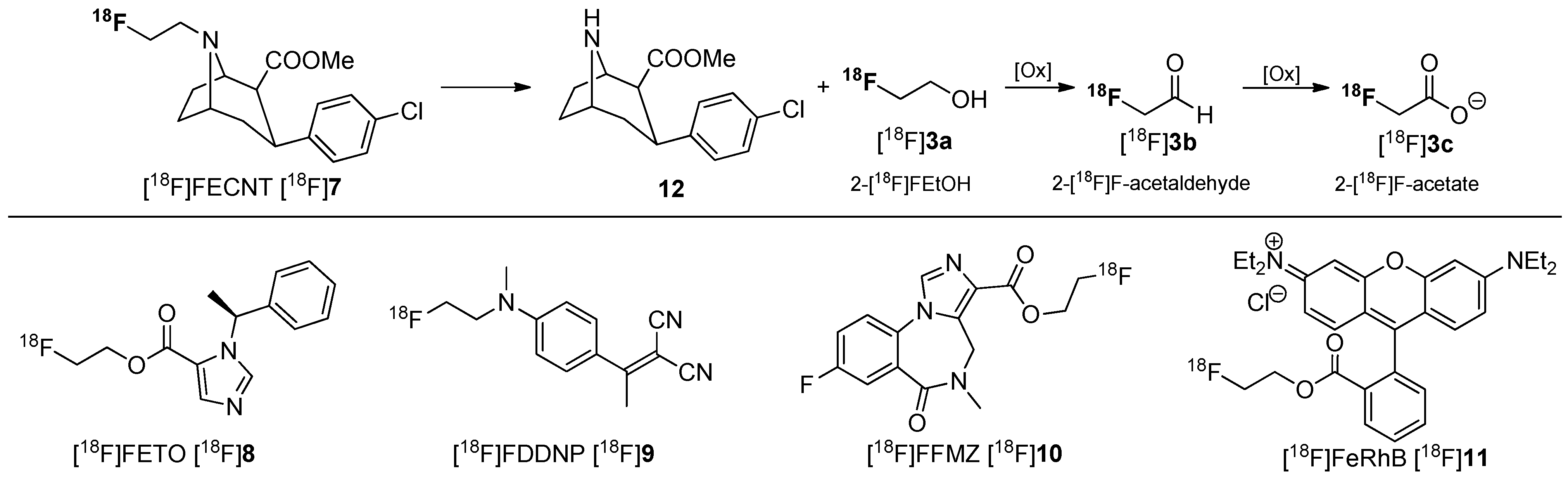
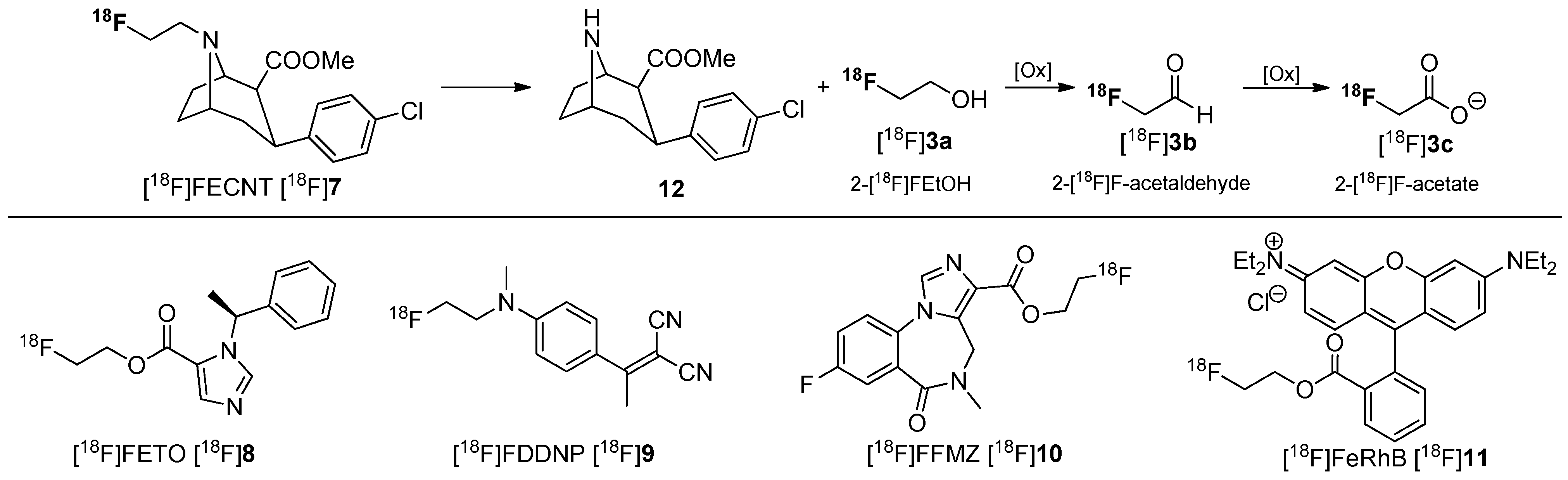
In many cases, 2-[18F]fluoroethanol [18F]3a or its metabolites 2-[18F]fluoroacetaldehyde [18F]3b and 2-[18F]fluoroacetate [18F]3c have been observed during metabolic degradation of several PET tracers containing a 2-[18F]fluoroethyl group such as [18F]FECNT [18F]7 [45], [18F]FETO [18F]8 [46], [18F]FDDNP [18F]9 [47], [18F]FFMZ [18F]10 [48], and [18F]FERhB [18F]11 [49]. The metabolites (presumably 2-[18F]fluoroacetaldehyde [18F]3b and 2-[18F]fluoroacetate [18F]3c) from N-defluoroethylation of [18F]FECNT [18F]7 and [18F]FDDNP [18F]9 have been shown to distribute evenly in the brain, confounding data analysis [46,47]. The metabolic behavior of [18F]FECNT [18F]7 is shown in Scheme 4.
Scheme 4.
Selected [18F]fluoroethylated tracers and assumed metabolic pathway for degradation of [18F]FECNT [18F]7.
Scheme 4.
Selected [18F]fluoroethylated tracers and assumed metabolic pathway for degradation of [18F]FECNT [18F]7.
The behavior of 2-[18F]fluoroethanol ([18F]3a) and 3-[18F]fluoropropanol ([18F]4a) as possible radiometabolites was further investigated during the process of radiodefluorination [50]. The basis of these experiments was the assumption that fluoroalkyl ethers and ester were cleaved to give both aforementioned radiometabolites. Further, it was suggested that 2-[18F]fluoroethanol ([18F]3a) is converted to 2-[18F]fluoroacetaldehyde ([18F]3b), which is then metabolized to 2-[18F]fluoroacetate ([18F]3c). After formation of 2-[18F]fluoroacetyl-CoA, it remains trapped inside the cell [51]. No activity was found in the bone, but it was stated that 2-[18F]fluoroethanol ([18F]3a) behaves like H2[15O]O.
In contrast, when investigating 3-[18F]fluoropropanol ([18F]4a) in vivo as a potential radiometabolite of 3-[18F]fluoropropylated PET tracers a rapid accumulation in the skeleton was observed. This can be explained by free [18F]fluoride being generated from 3-[18F]fluoropropionaldehyde ([18F]4b) or 2-[18F]fluoropropionate ([18F]4c). Both metabolites leading to further β-elimination under release of [18F]fluoride (vide supra).
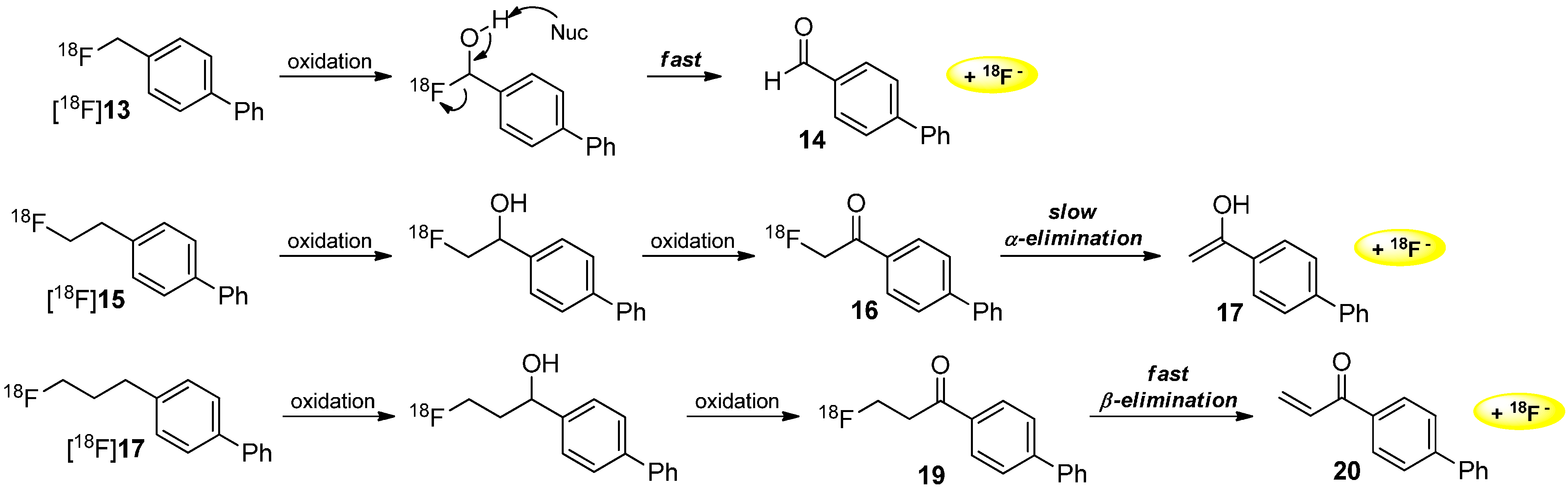
Furthermore, Lee and co-workers demonstrated the radiodefluorination of [18F]fluoroalkyl groups bound to an aromatic system [52]. Several biphenyl derivatives ([18F]13, [18F]15, [18F]17) and their degradation behavior were investigated. In all evaluated reactions, the first step consists of the oxidation of the carbon next to the aromatic ring. In the case of the fluoromethyl group of [18F]13, a fast elimination step followed to give the respective aldehyde and [18F]fluoride. In the case of the fluoroethyl residue of [18F]15, a slow α-elimination occurred to give [18F]fluoride and the remaining enol (ketone). In the third case, β-elimination took place after oxidation of [18F]17 to an α,β-unsaturated system 20 and elimination of [18F]fluoride. The different metabolic degradation of these compounds is investigated (Scheme 5).
Scheme 5.
Different metabolic behavior of [18F]fluoroalkylated aromatic compounds.
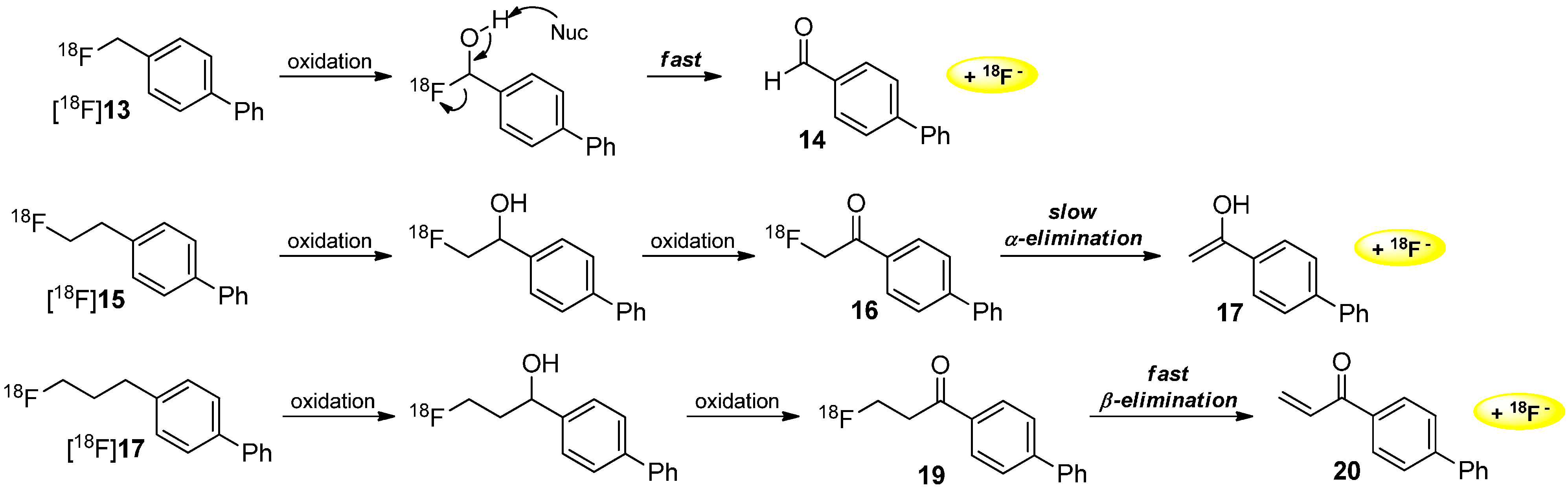
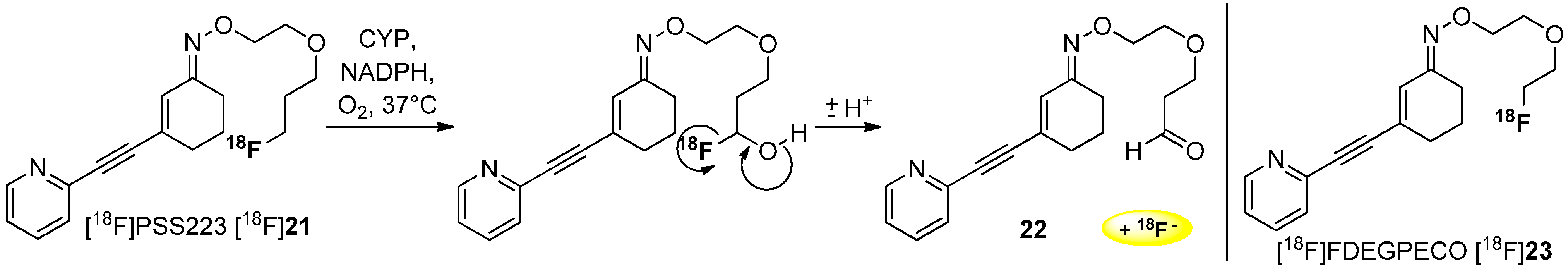
Schibli and co-workers gave an alternative explanation for the mechanism of radiodefluorination. They assumed an oxidation of the carbon next to the [18F]fluorine of tracer [18F]PSS223 [18F]21 involved by cytochrome P450 enzyme (CYP) leading to a separation of [18F]fluoride (Scheme 6). Experiments performed in vivo showed an accumulation of [18F]fluoride in the bone [53] as reported above.
Scheme 6.
Release of [18F]fluoride from [18F]PSS223 [18F]21 during degradation with cytochrome P450 (CYP).
Scheme 6.
Release of [18F]fluoride from [18F]PSS223 [18F]21 during degradation with cytochrome P450 (CYP).
Stability determinations with [18F]PSS223 [18F]21 using rat and human liver microsomal enzymes were executed and pointed out two more polar radiometabolites as demonstrated by radio-UPLC measurements. The degradation process is shown to be NADPH-dependent, which implied the involvement of oxidoreductases. Amongst others, the fluorine-containing carbon atom was oxygenated leading to the release of [18F]fluoride.
The difference in the in vivo behavior between [18F]PSS223 [18F]21 and [18F]FDEGPECO [18F]23 could be explained by the β-heteroatom effect [54,55], by which primary aliphatic bound [18F]fluorine in β-position to heteroatoms (e.g., ROCH2CH2[18F]F) is found to be metabolized at a slower rate. This rationale supports the absence of defluorination for [18F]FDEGPECO [18F]23 containing only a [18F]fluoroethyl group.
3. Methods to Avoid Radiodefluorination
The probably best alternative to avoid radiodefluorination consists of the direct connection of fluorine-18 to a phenyl moiety instead of aliphatic residues wherever applicable [56]. This is consistent with the higher stability of a CAr-F bond compared to a C(sp3)-F bond as previously described. The fluoroaryl groups are stable to metabolism and do not lead to a considerable radiodefluorination. Otherwise, the [18F]fluoroalkyl moiety has to be modified to reduce or avoid rapid metabolic degradation by the following methods.
Deuteration in Direct Neighborhood of Fluorine-18
A fundamental approach in medical chemistry is the application of deuterium to increase the stability of active pharmaceutical ingredients [57], which is useable to raise the metabolic stability of 18F-radiotracers by means of the deuterium-proton exchange at carbon atoms close to the 18F-atom. This procedure can sometimes suppress but not completely prevent the process of radiodefluorination.
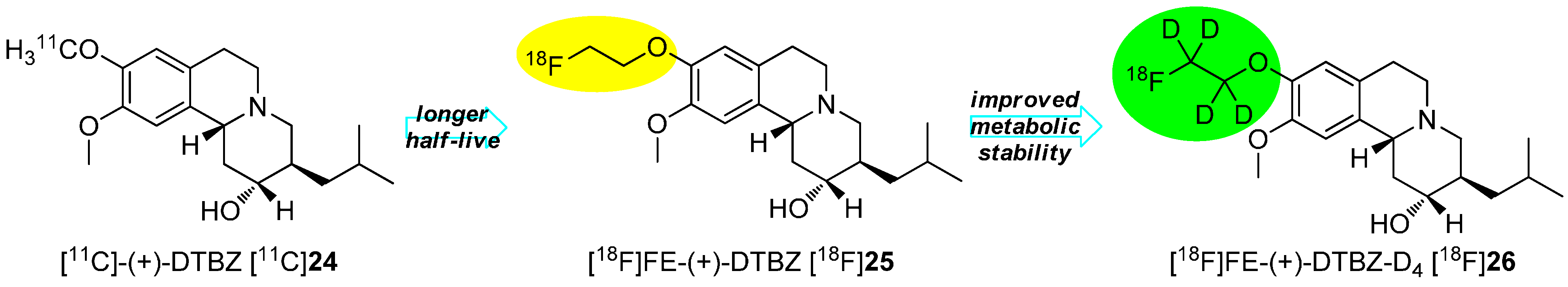
The method of action of this effect is explained by the kinetic isotope effect that reduces the rate of metabolic degradation. The deuterium is not only twice as heavy as hydrogen, but also the zero-point energy is significantly lower than the energy of hydrogen. Due to these differences, the activation energy of the C-D-bond in chemical or biochemical reactions is significantly higher than for C-H-bond. Therefore, reactions on the C-D-bond will proceed considerably slower than the same reactions with a C-H-bond at the same position. In general, the cleavage rate of a C-H bond is 6.7 times faster compared to a C-D bond at 25 °C and it is postulated that the break of the C-H bond is the rate-determining step in this kind of defluorination [58,59]. A successful example of the stabilization by means of deuteration consists of the preparation of [18F]FE-DTBZ-D4 [18F]26, which is pointed out in Scheme 7.
Scheme 7.
Enhancement of the half-life as well as of the metabolic stability of DTBZ 24.
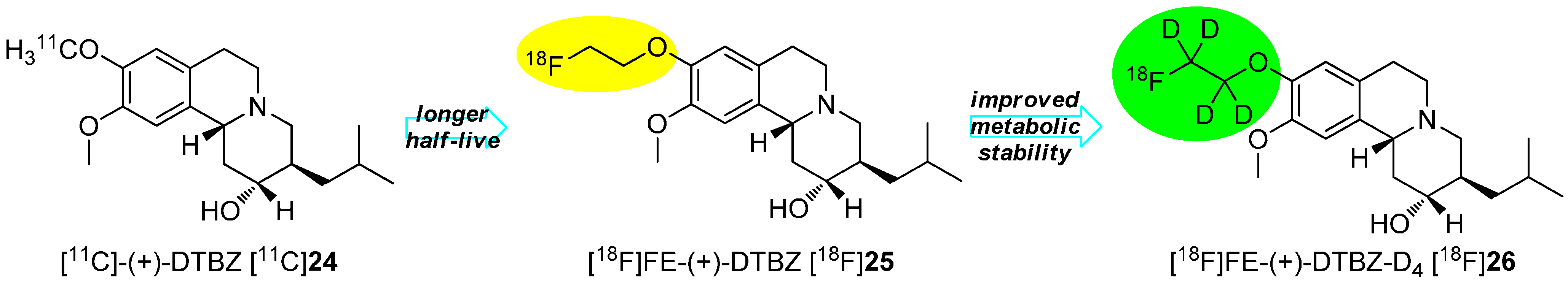
[11C]-(+)-DTBZ [11C]24 was initially used to studied dementia and Parkinson in the clinic [60,61,62]. Further improvements were necessary including the change of the radionuclide to elongate the half-live, which led to the development of [18F]FE-(+)-DTBZ [18F]25 [63,64]. Successful in vitro studies with this 18F-tracer were accomplished followed by in vivo studies showing a high accumulation of radioactivity in joints and bones. To improve the metabolic stability, [18F]FE-(+)-DTBZ-D4 [18F]26 was developed and showed enhanced properties. The main improvement resulted in the considerably reduced bone uptake when comparing both tracers. The defluorination rate (kdefluorination) was determined for both tracers to be 0.012 for [18F]25 and 0.0016 for [18F]26 resulting in an elongated plasma-t1/2 from 46.2 min to 438.7 min [65].
The example in Scheme 7 exhibits the introduction of a deuterated [18F]fluoroalkyl residue via a building block strategy using a fluorine-18 containing deuterated building block. The second general method consists of the gradual introduction of the deuterium followed by 18F-labeling as the last step. In this case, the precursor already possesses the deuterium.
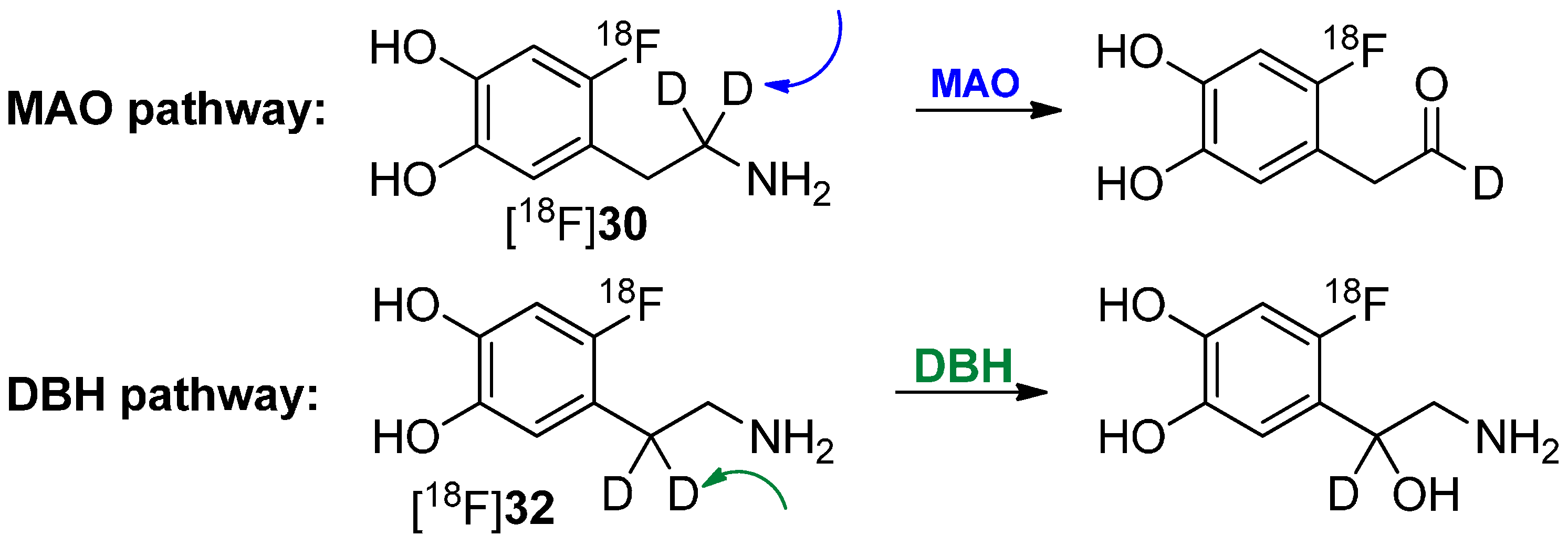
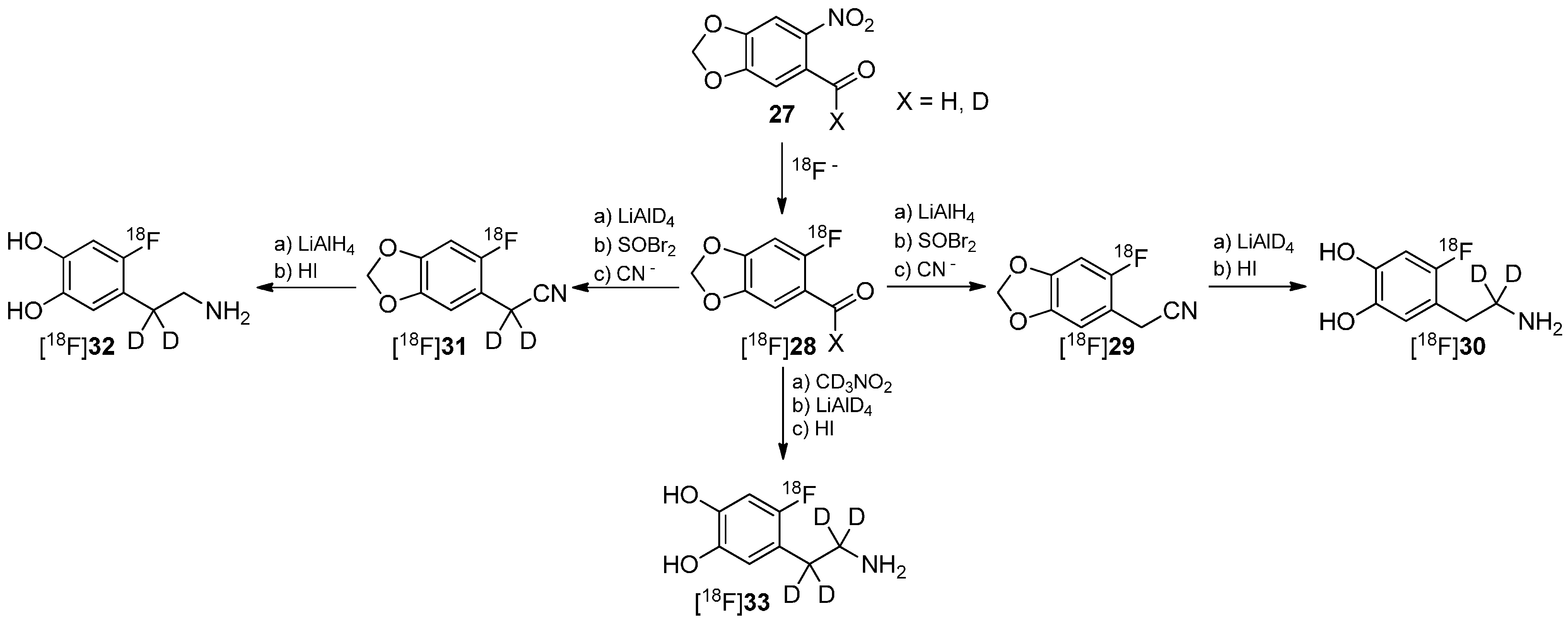
One of the first reports regarding the introduction of deuterium into precursors to prepare 18F-radiotracers was presented by Ding, Fowler and Wolf in 1993 [66]. They introduced deuterium in different positions of the alkyl chain of 6-[18F]fluorodopamine (6-[18F]FDA) regioselectively to execute mechanistic studies regarding the degradation of these derivatives ([18F]30, [18F]32, [18F]33) by monoamine oxidase B (MAO B) and dopamine β-hydrolase (DBH) via PET. The reaction path to precursors and resulting radiotracers is shown in Scheme 8.
Scheme 8.
Synthesis of different regioselectively deuterated [18F]fluorodopamine derivatives.
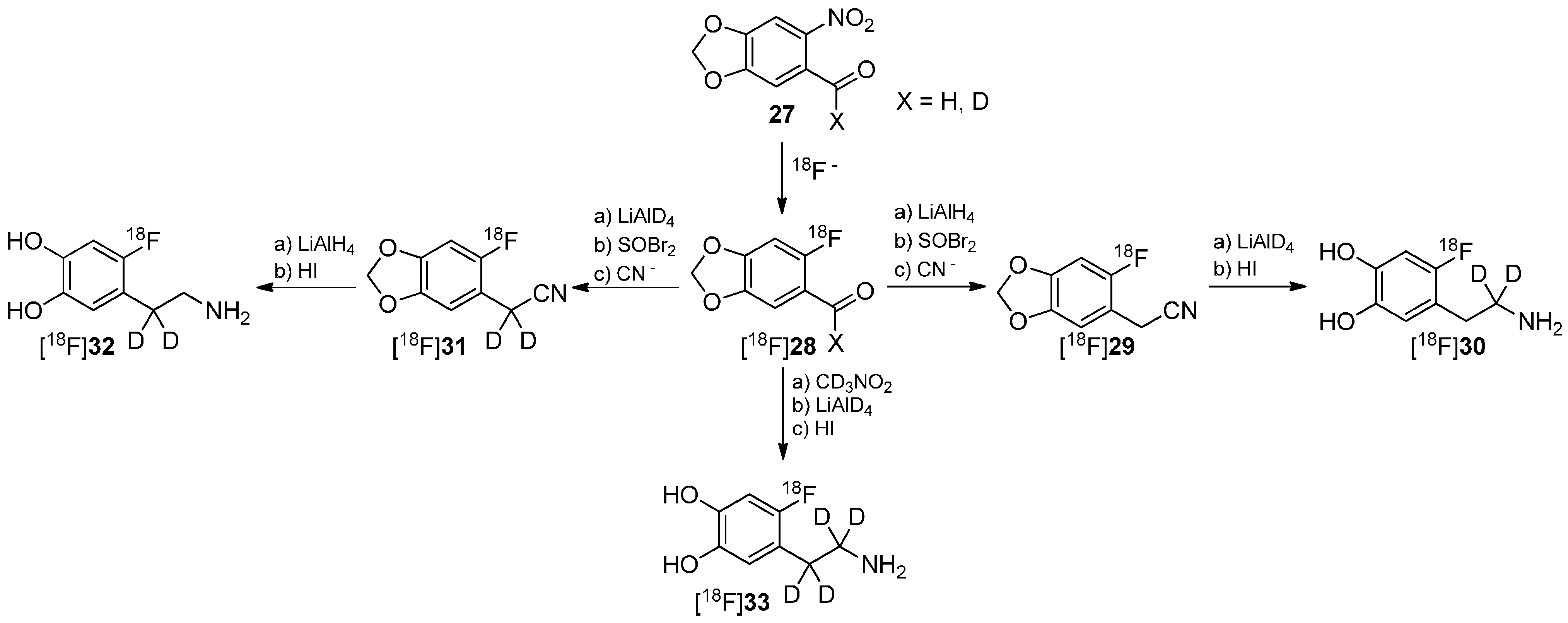
It has recently been shown that MAO B and DBH stereoselectively remove only the pro-R hydrogen of the non-deuterated compounds [67,68,69,70]. Both dopamine compounds [18F]30 and [18F]32 with two deuterium atoms on one carbon were prepared to further verify this finding. Such specifically deuterated derivatives are therefore the most appropriate candidates for unambiguously assessing the contribution of metabolism by MAO and DBH on the kinetics of 6-[18F]fluorodopamine.
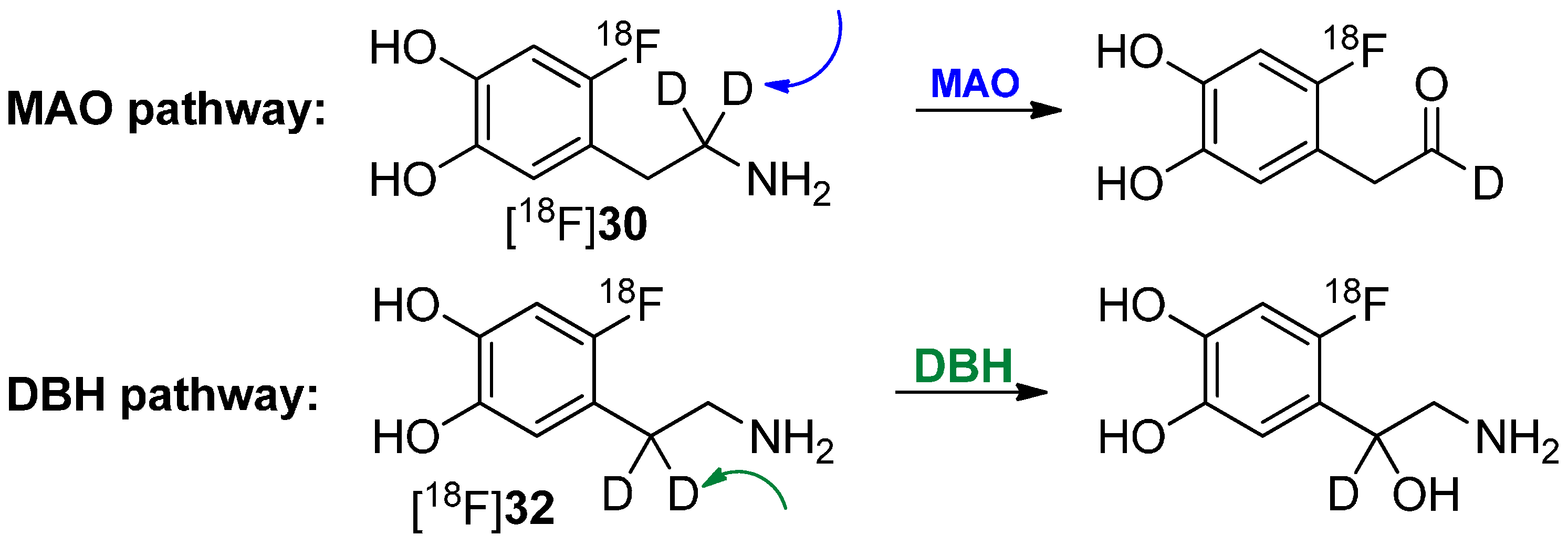
In a following paper, it was shown that [18F]30 has a reduced rate of clearance, consistent with MAO-catalyzed cleavage of the α-C-D bond, whereas [18F]32 showed no change, indicating that cleavage of the β-C-D bond (DBH) is not rate limiting [71]. Both pathways of degradation are shown in Scheme 9. Furthermore, the rate of metabolism was also significantly reduced by pretreatment with pargyline (MAO inhibitor).
Scheme 9.
Metabolic conversion of [18F]30 and [18F]32 by DBH and MAO, respectively.
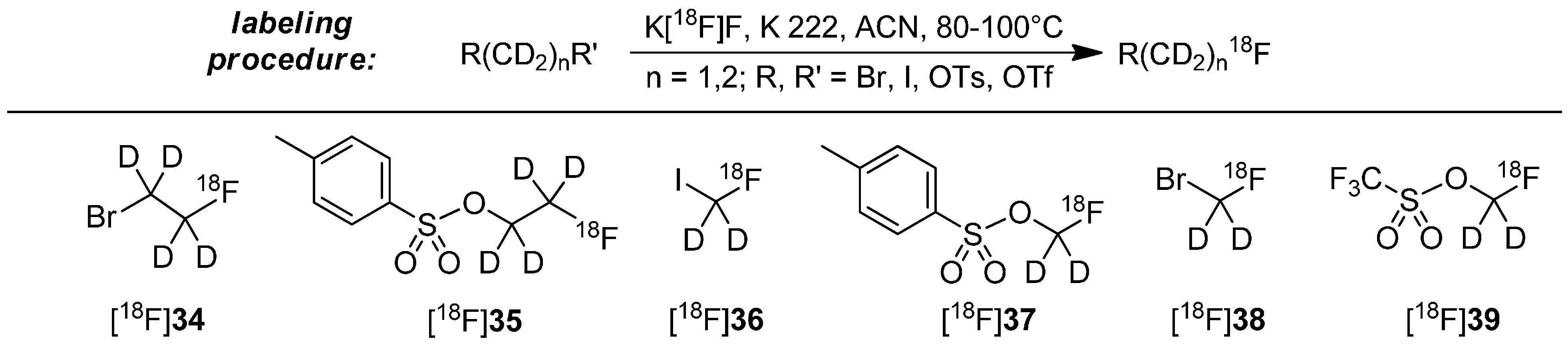
The most often applied approach of the stabilization with deuterium consists of the use of deuterated [18F]fluoroalkyl building blocks. For preparation, dihalogens, disulfonates or derivatives with mixed functions were used as starting material with deuterated methylene (-CD2-) or ethylene (-CD2CD2-) groups. The introduction of 18F follows standard labeling conditions (K222, anhydrous acetonitrile, 80–100 °C, 15–30 min). Examples for building blocks and most common labeling conditions to prepare these building blocks are shown in Scheme 10 [55,65,72,73,74,75,76,77,78,79,80].
Scheme 10.
General labeling procedure to create the deuterated building blocks and known building blocks [18F]34–[18F]39.
Scheme 10.
General labeling procedure to create the deuterated building blocks and known building blocks [18F]34–[18F]39.
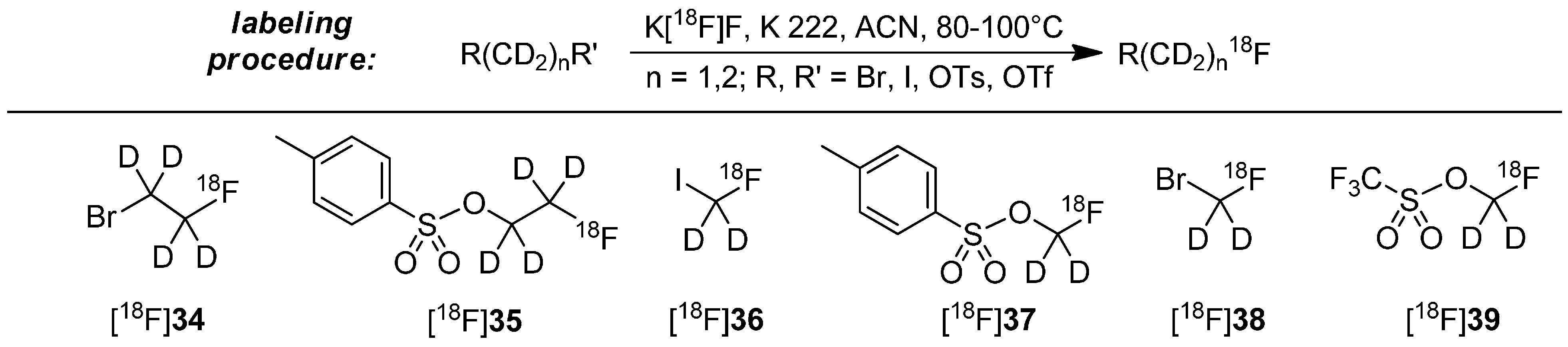
The subsequent labeling procedure with the above mentioned building blocks [18F]34–[18F]39 represents a nucleophilic displacement at the carbon of the building block. Normally, sulfonate leaving groups are superior to halogens, but Schou and co-workers demonstrated that the type of the leaving group has only a small influence on the radiochemical yield (RCY) of the resulting radiotracer [72]. Mostly, the final alkylation reaction of building block with precursor proceeds rapidly (approx. 5 min reaction time) [65,72].
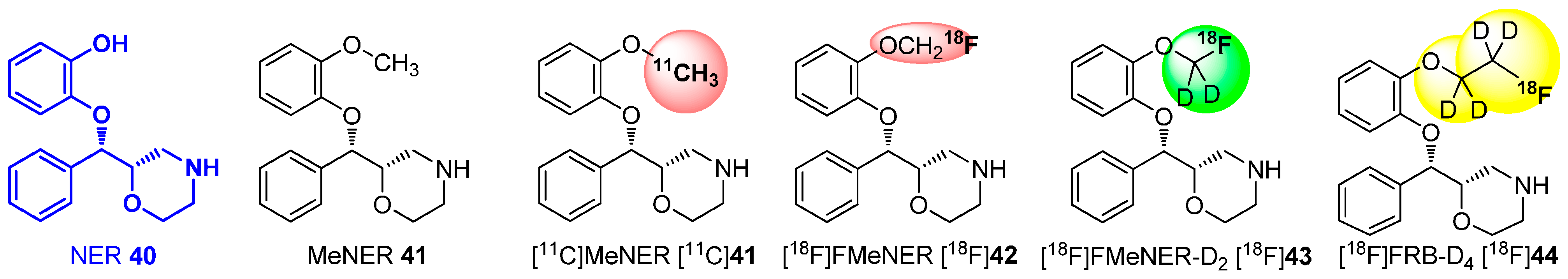
Amongst others, this building block strategy was extensively investigated for MeNER 41 (Scheme 11), which was identified as high-affinity ligand (IC50: 2.5 nM in vitro) for the norepinephrine transporter (NET). Imaging of the NET moved into focus of research to investigate several neuropsychiatric and neurodegenerative disorders. The first successful PET images were obtained with carbon-11 labeled derivative [11C]MeNER [11C]41, which was synthesized by the use of NER 40 as precursor and radiolabeled with [11C]MeOTf. Unfortunately, the binding of this MeNER derivate [11C]41 to the NET proceeded within a range of about 90 min in vivo, which was too long for a carbon-11 labeled tracer (t½ = 20.4 min) [81]. This, led to the development of [18F]FMeNER [18F]42, an improved tracer with fluorine-18 (t½ = 109.77 min) on the methyl group. [18F]42 still bound to the receptor with high affinity, while providing a sufficient half-life for imaging. This tracer was synthesized from the same precursor using bromo[18F]fluoromethane and [18F]fluoromethyl triflate with similar results. In contrast, the signal-to-background ratio and the bone uptake was increased compared to PET images from [11C]MeNER [11C]41. Fortunately, this effect was nearly completely suppressed by the use of the deuterated derivative [18F]FMeNER-D2 [18F]43, which shows the impact of the isotope effect for the development of radiotracers [72].
Scheme 11.
Carbon-11, fluorine-18 and deuterated derivatives of NER 40 to increase metabolic stability.
Scheme 11.
Carbon-11, fluorine-18 and deuterated derivatives of NER 40 to increase metabolic stability.
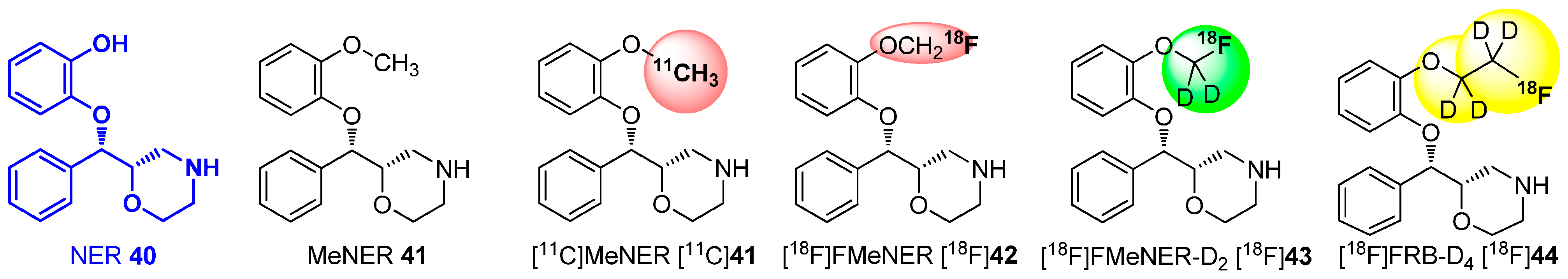
This successful procedure was also applied for the preparation of [18F]FRB, the ethoxy derivative of MeNER 41 based on Reboxetine (IC50(NET): 8.23 nM). For this purpose, the precursor NER 40 was successful labeled with [18F]fluoroethyl bromide and [18F]fluoroethyl bromide-D4 ([18F]34), to give [18F]FRB and [18F]FRB-D4 [18F]44, respectively. Due to the better pharmacological properties of [18F]FMeNER-D2 [18F]43 compared to [18F]FRB-D4 [18F]44, a fully automated synthesis was developed for [18F]FMeNER-D2 [18F]43 in 2013 [80].
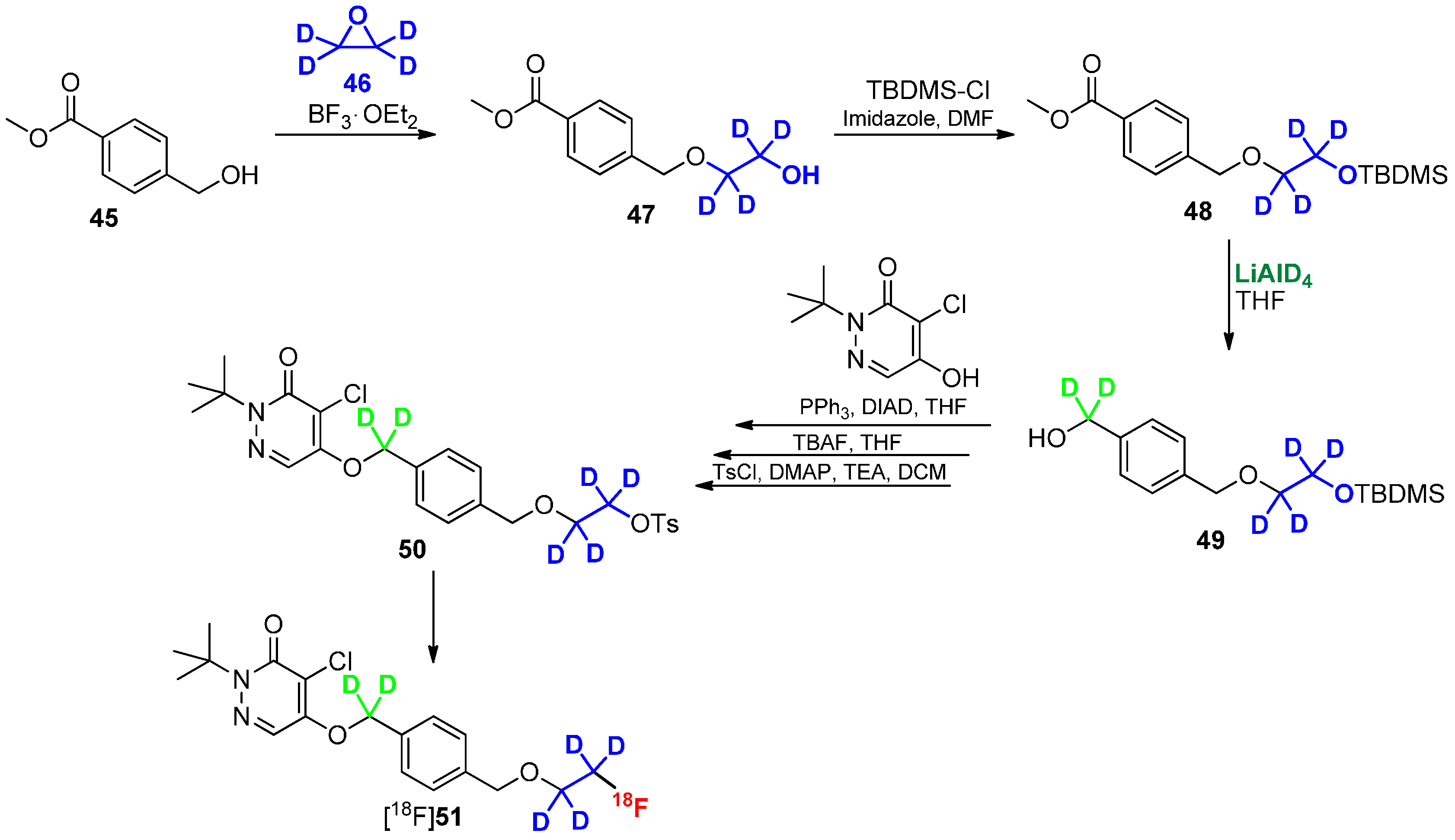
Scheme 12.
Direct approach to introduce deuterium and fluorine-18 into radiotracer [18F]51.
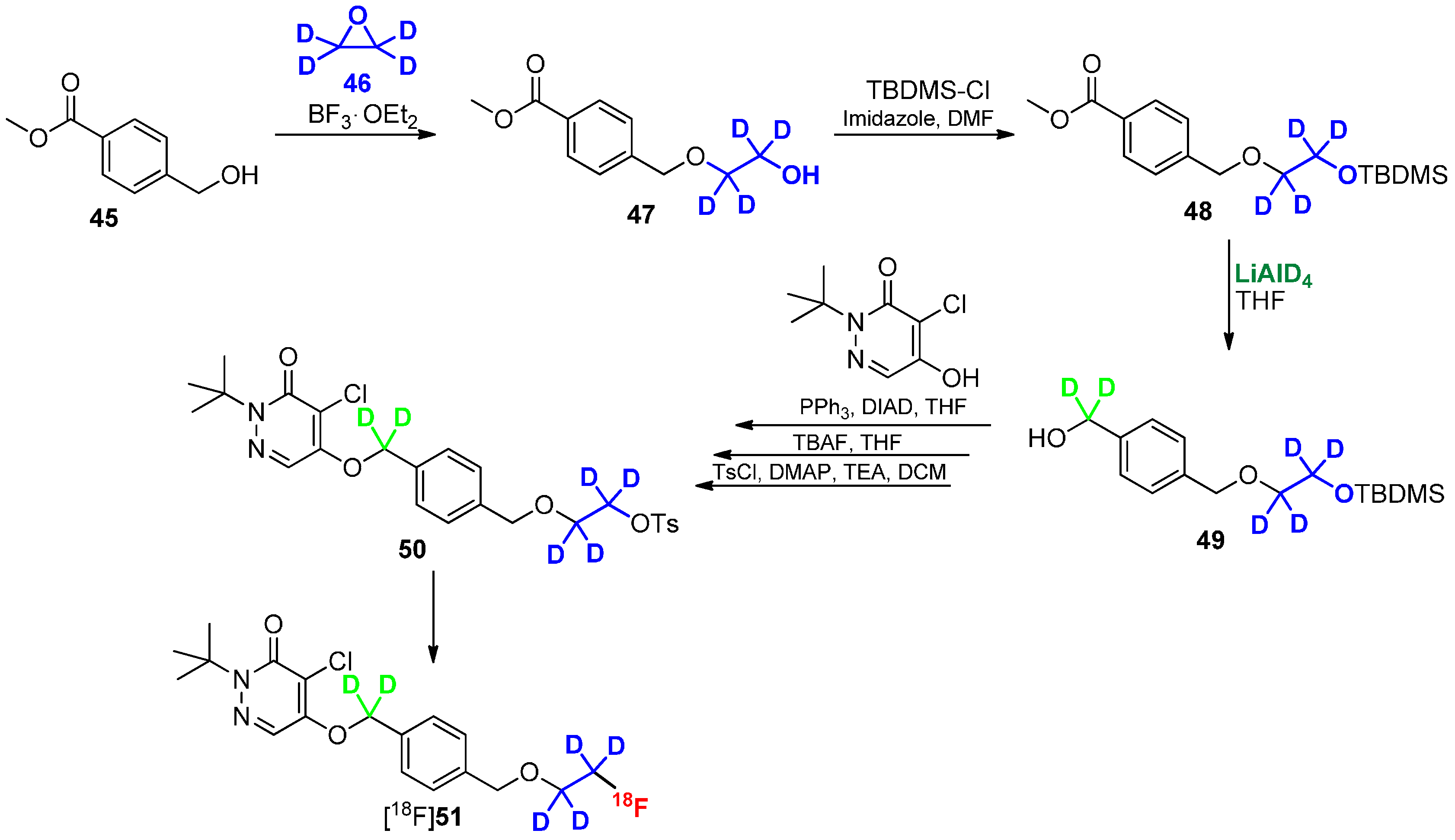
Another promising approach to use deuterated building blocks was shown by Casebier and colleagues [82]. In contrast to the previously discussed approaches, the deuterium containing residue was directly connected to the precursor molecule prior to radiolabeling to avoid a two-step-synthesis of radiotracer. The interesting task of this work was the use of fully deuterated ethylene oxide as building block (Scheme 12), which was introduced via ring-opening reaction. The next steps required the protection of the OH group with TBDMS-Cl followed by reduction of the methyl ester with LiAlD4. The obtained deuterated methylene group is mandatory for a further stabilization of the tracer in terms of metabolic degradation. Upon completion of the basic structure of the molecule, the hydroxyl group was selectively deprotected using TBAF and functionalized with p-tosylchloride for labeling with fluorine-18.
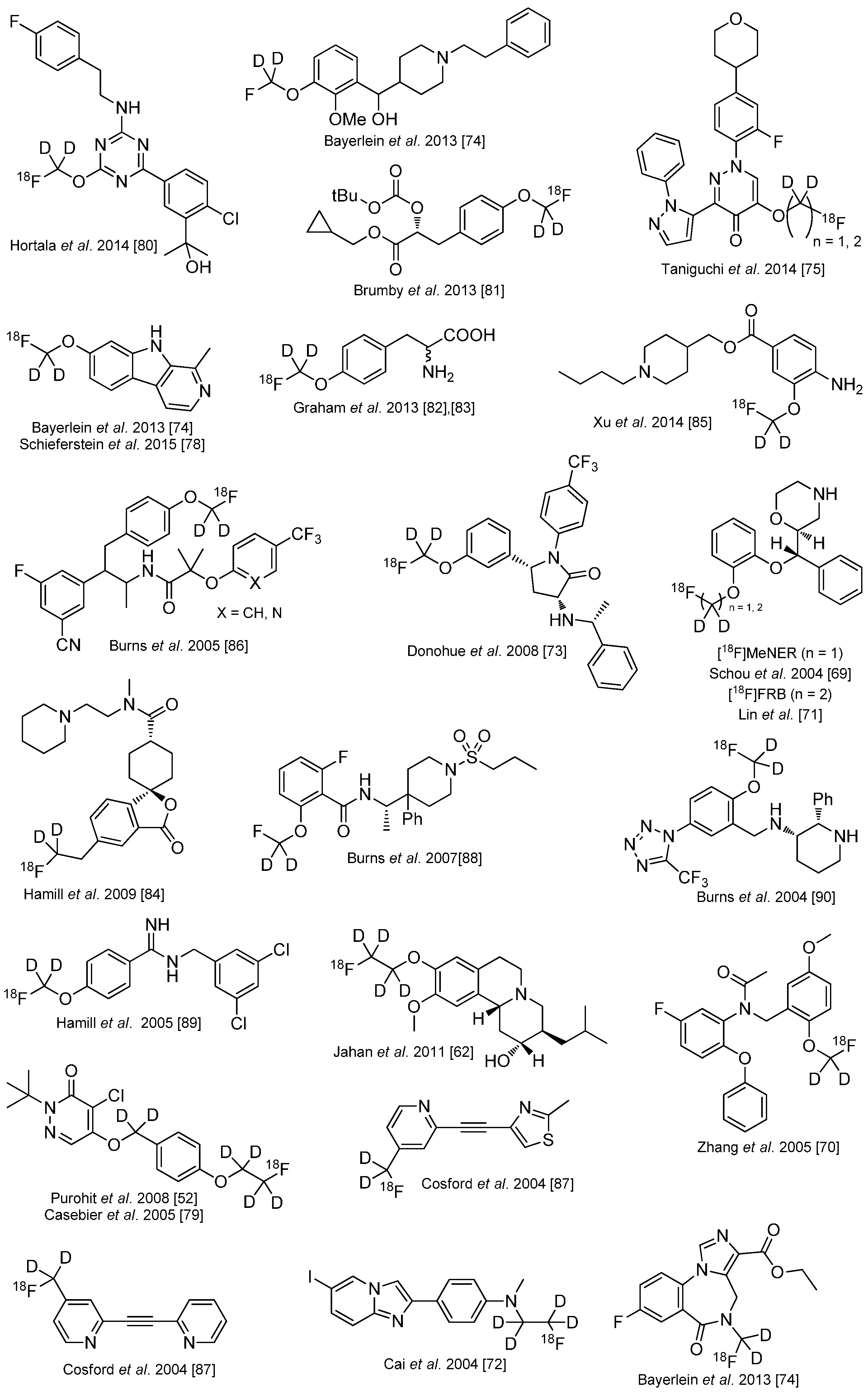
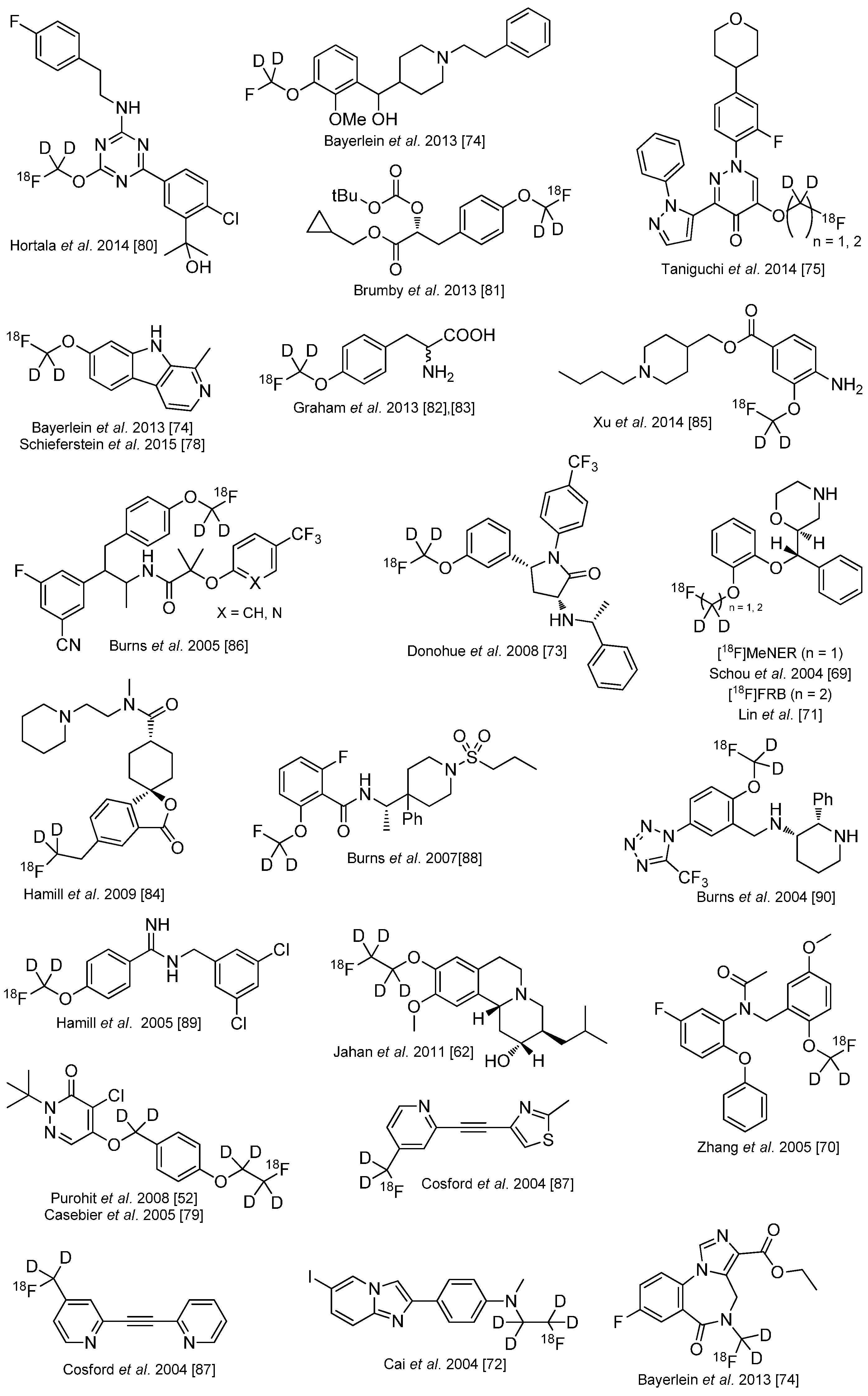
Several other 18F-tracers are known which are stabilized with deuterium. Selected examples are given in the following overview in Scheme 13 [83,84,85,86,87,88,89,90,91,92,93]. As stated before, the radiodefluorination process cannot fully be avoided, but it can be delayed considerably.
Scheme 13.
Overview over 18F-radiotracers stabilized with deuterium in direct neighborhood to 18F.
4. Deuteration on other Parts of the Molecule to Avoid Degradation
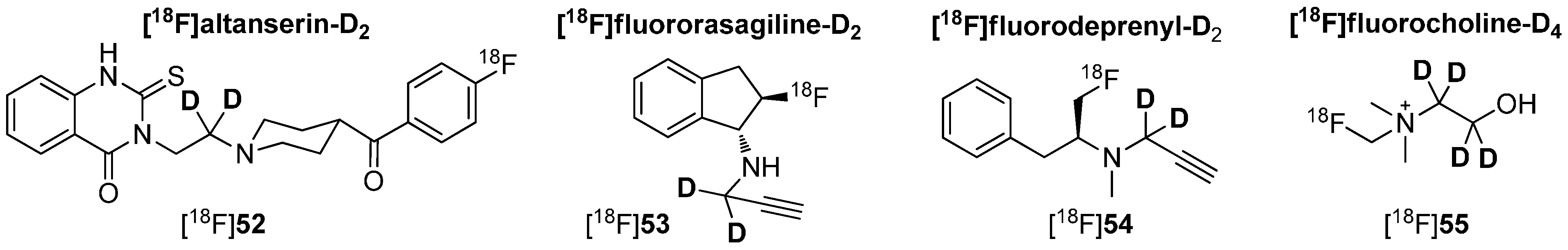
As already mentioned, hydrogen–deuterium exchange will not only be adopted in direct proximity to fluorine-18 to avoid radiodefluorination or other metabolic degradation. A similar effect could be achieved by the use of deuterium connected to endangered areas in the tracer molecule, which are prone to metabolic degradation. Such an additional stabilization was already shown for compound [18F]51 by Casebier and colleagues in Scheme 12 [82]. Furthermore, the application of deuterium leads occasionally to another metabolic pathways as it was figured out by Leyton, Smith and co-workers [94,95]. Some examples for 18F-radiotracers deuterated on other parts of the molecule are shown in Scheme 14.
Scheme 14.
Examples for radiotracers deuterated on other parts of the molecule.

[18F]Fluororasagiline-D2 ([18F]53) and [18F]fluorodeprenyl-D2 ([18F]54) (Scheme 14) are two examples of a successful enhancement of the metabolic stability using deuterium. Both tracers are known to be inhibitors of monoamnooxidase (MAO) and were applied for detection of psychiatric and neurological disorders such as depression, Alzheimer, and Parkinson diseases [96]. Although both lead structures (rasagiline and L-deprenyl) contain a propargyl group, there are significant differences in their metabolic products [97]. Inhibition experiments in vitro pointed out a high selectivity of MAO-B compared to MAO-A for both above mentioned radiotracers as well as for their non-deuterated derivatives. Moreover, it was found that the alkynyl chain of these molecules was cleaved under in vivo conditions. Thus, deuterium was selectively introduced into this residue to stabilize these molecules.
Comparing the deuterated and non-deuterated tracers in terms of their radiopharmacological behavior in vivo, [18F]fluorodeprenyl showed a fast and irreversible binding to the enzyme limited by blood flow rather than by the MAO-B enzyme concentration, whereas [18F]fluororasagiline expressed continuous increase of the radioactivity in the brain indicating a blood–brain barrier penetrating radiometabolite. In contrast, [18F]fluororasagiline-D2 ([18F]53) and [18F]fluorodeprenyl-D2 ([18F]54) exhibited fast clearance from the brain and less accumulation in cortical and sub-cortical regions. Furthermore, both deuterated analogues were more stable in monkey plasma compared to their non-deuterated analogues [98]; metabolic degradation was almost completely reduced. Thus, the deuterated tracers seemed to be more suitable for an application over the non-deuterated derivatives.
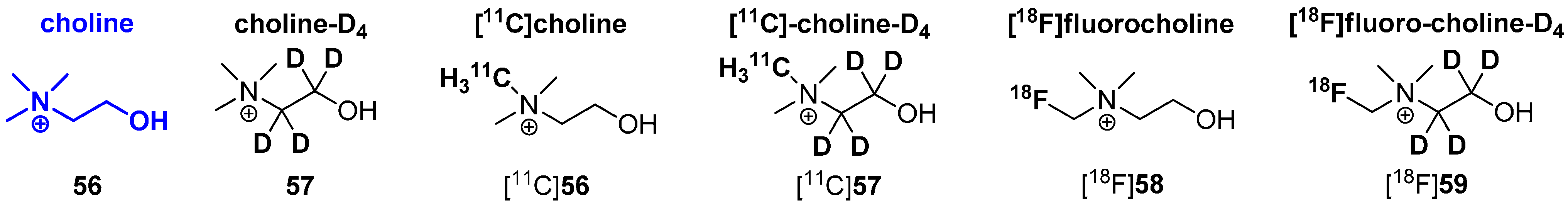
Another interesting example is the metabolic behavior of radiolabeled cholines. Known radiolabeled derivatives are exemplified in Scheme 15. In general, two main metabolic pathways are known for choline derivatives. The first pathway is based on the phosphorylation of choline via choline kinase (E.C. 2.7.1.32) to phosphocholine which is further transformed to phosphatidylcholine, a key component of the plasma membrane. This way is also known as The Kennedy pathway [99]. Once phosphorylated, phosphocholine is trapped within the cell, which is crucial for PET imaging with 11C and 18F radiotracers based on choline. The second main pathway of choline metabolism is based on the oxidation of choline to betaine. It was first described by Ikuta and co-workers in 1977 [100] and involves the conversion of choline to betain by choline oxidase (E.C. 1.1.3.17) via a four-electron oxidation using two sequential FAD-dependent reactions [101]. However, the second pathway is not preferred for PET imaging applications using radiolabeled choline derivatives. To overcome this obstacle, 11C and 18F labeled choline derivatives, which are deuterated at the ethylene moiety and not in the immediate neighborhood of the desired radionuclide were applied due to their altered pharmacological behavior.
Scheme 15.
Several choline derivatives with and without deuterium labeled with carbon-11 or fluorine-18.
Scheme 15.
Several choline derivatives with and without deuterium labeled with carbon-11 or fluorine-18.
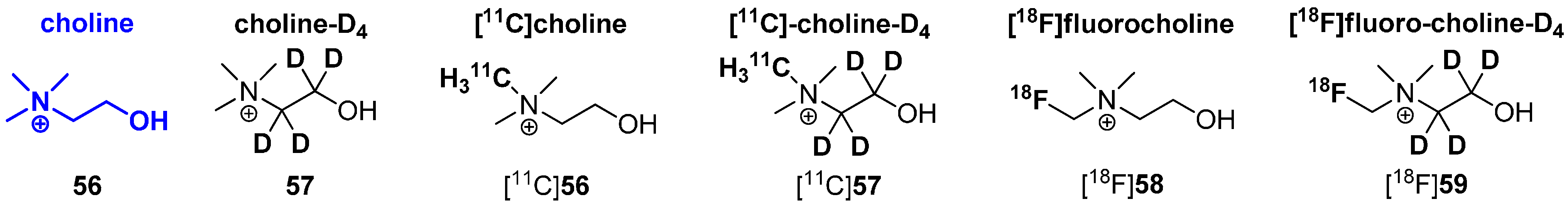
In 2003, Gadda investigated enzyme kinetics for choline oxidase with choline (56) and choline-D4 (57) as substrates to evaluate the impact of the kinetic isotope effect. It was shown that the oxidation of deuterated choline 57 was reduced to a minimum [101], which led to the successful development of choline-based radiotracers.
In 2009, Aboagye and colleagues compared the relative oxidation rates of the two isotopically radiolabeled choline species, [18F]fluorocholine ([18F]58) and [18F]fluorocholine-D4 ([18F]59) with respect to their metabolites [94]. Both betaine metabolites from [18F]58 and [18F]59 were obtained from mouse plasma after intravenous injection of both radiotracers. As a result, it was pointed out that [18F]59 was remarkably more stable to oxidation than [18F]58 with ~40% conversion of [18F]59 to the betaine at 15 min after intravenous injection into mice compared to ~80% conversion of [18F]58 to the respective betaine-metabolite.
In 2012, both 11C-labeled choline derivatives [11C]56 and [11C]57 as well as 18F-choline-D4 ([18F]59) were synthesized to compare their biodistribution and metabolic behavior. Additionally, the same group performed small-animal PET studies and kinetic analyses to evaluate the tracer uptake in human colon HCT116 xenograft-bearing mice [102]. It was found that the simple substitution of deuterium for hydrogen and the presence of 18F improves the stability and reduces degradation of the parent tracers. Furthermore, the availability is increased for phosphorylation and trapping within cells, which leads to a better signal-to-background contrast, thus improving tumor detection sensitivity of PET. In addition, deuterated 11C choline was demonstrated to have a higher stability compared to non-deuterated 11C-choline, but an increased rate of oxidation of betaine compared to 18F-D4-choline was observed. In 2014, the first promising human studies with healthy volunteers were accomplished [103].
4.1. Cycloalkyl Derivatives and Fluorine Connected to a Secondary Carbon Atom
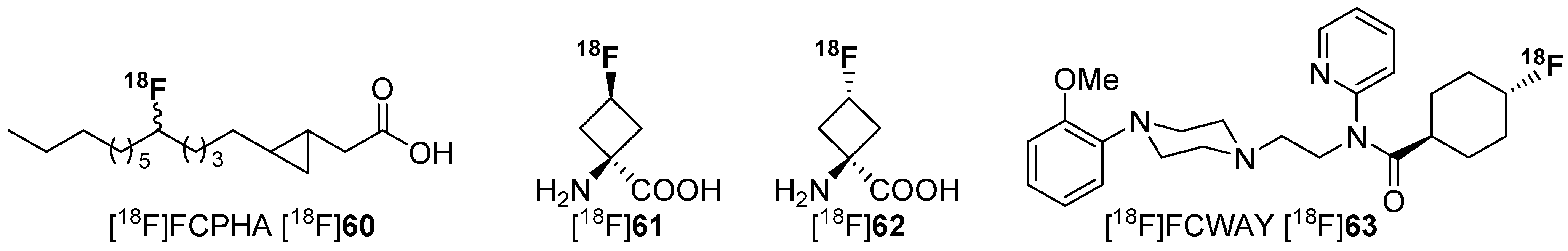
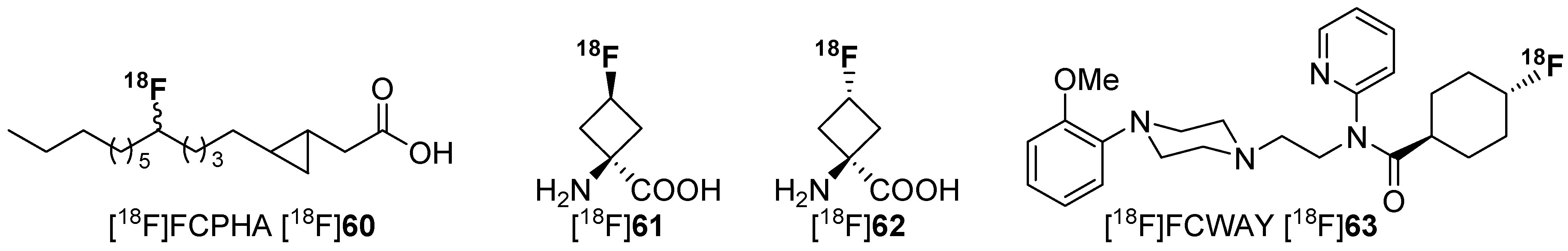
Several literature sources reported that the replacement of an alkyl chain by a cycloalkyl ring resulted in more metabolically stable compounds [104,105,106,107]. Examples are given in Scheme 16. Despite this increased stability, only a few reports exist on PET radiotracers containing cycloalkyl rings. One example describes a potential radiotracer for assessing myocardial fatty acid metabolism, [18F]FCPHA [18F]60, containing a cyclopropyl moiety which allows the tracer to be trapped in the cells [108]. Another example describes non-natural 18F-amino acids with fluorine-18 located at the cycloalkyl residue [109,110]. [18F]61 and [18F]62 show an increased metabolic stability compared to their non-cyclic counterparts. The placement of 18F is especially important for compound [18F]63, because the methoxy group itself and also the introduction of a fluoroalkoxy moiety instead of the methoxy group at the phenol part of the molecule lead to a fast cleavage [111].
Scheme 16.
Selected examples of fluorine-18 bound to secondary carbon for stabilization.
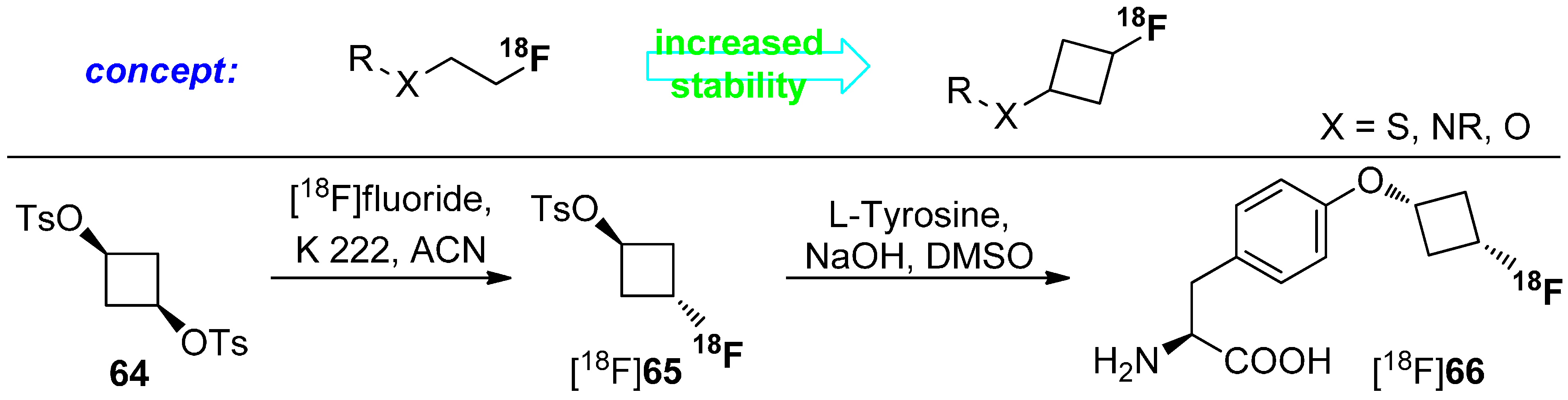
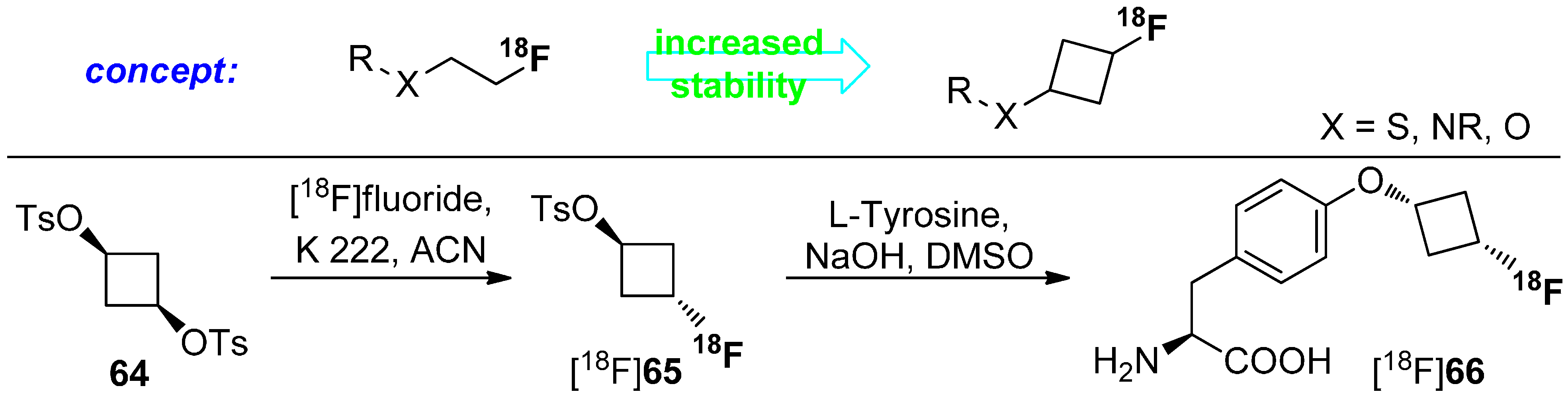
Both non-natural amino acids were used as brain tumor imaging agent and W. Yu et al. [112] found that the newly developed amino acid [18F]61 is comparable to [18F]62. However, the cyclic unnatural amino acids are not metabolized [113]. The major drawback of this approach is the stereoselective construction of the amino acid skeleton. Thus, Franck and colleagues reported a diverse approach using cyclic building blocks bearing the 18F-label. The research was focused on the metabolism of 18F-tracers with [18F]fluoroalkyl chains attached to hetereoatoms such as O, N, and S. Biotransformation (radiodefluorination) of these radiotracers was avoided by the utilization of cyclobutyl groups containing fluorine-18. Hence, cyclobutyl 1,3-ditosylate (64) was used as starting material. Radiofluorination was performed under standard conditions using [18F]F−, K 222 in anhydrous acetonitrile. After successful synthesis of the building block [18F]65, L-tyrosine was used and labeled. The concept and the full reaction path including radiolabeling are pointed out in Scheme 17.
Scheme 17.
Labeling concept to avoid radiodefluorination and radiolabeling of L-tyrosine with [18F]fluorocyclobutyl tosylate ([18F]65).
Scheme 17.
Labeling concept to avoid radiodefluorination and radiolabeling of L-tyrosine with [18F]fluorocyclobutyl tosylate ([18F]65).
The obtained [18F]fluorocyclobutyl derivative [18F]66 is comparable with the well-known amino acid O-(2-[18F]fluoroethyl)-l-tyrosine ([18F]FET) in the case of cell uptake and blocking and showed an excellent metabolic stability in phosphate buffer and in human and rat plasma for 120 min [114,115].
Further, the connection of fluorine to a secondary carbon could also help to prevent radiodefluorination in some cases. However, when using 18F-FCWAY [18F]63 (Scheme 16), the defluorination process is a major issue. To prevent degradation, the responsible enzyme (cytochrome P450 2E1 (CYP2E1) isozyme) is suppressed with miconazole nitrate prior to the injection of the radiotracer [116]. With this method it was possible to substantially avoid radiodefluorination and the combined uptake of [18F]fluoride in the skull.
4.2. SiFA-Techniology
The Si-F bond represents one of the strongest single bonds with a corresponding bond energy of 565 kJ/mol, which is 80 kJ/mol higher than the Si-C bond and suggest a high thermodynamically stability [117]. This fact led to the development of fluorine-18-radiotracers based on organosilanes, which should be unaffected against radiodefluoroination commonly associated with alkylfluorides. In 1985, Rosenthal and colleagues were the first who successfully radiolabeled [18F]fluorotrimethylsilane [118]. The reaction was performed using chlorotrimethylsilane as precursor with a yield of 65% and high radiochemical purity. However, subsequent in vivo investigation of [18F]fluorotrimethylsilane indicated a rapid hydrolysis followed by an enrichment of radioactivity in bones. For this reason, this concept was ineffective for the preparation of 18F-radiotracers [119,120].
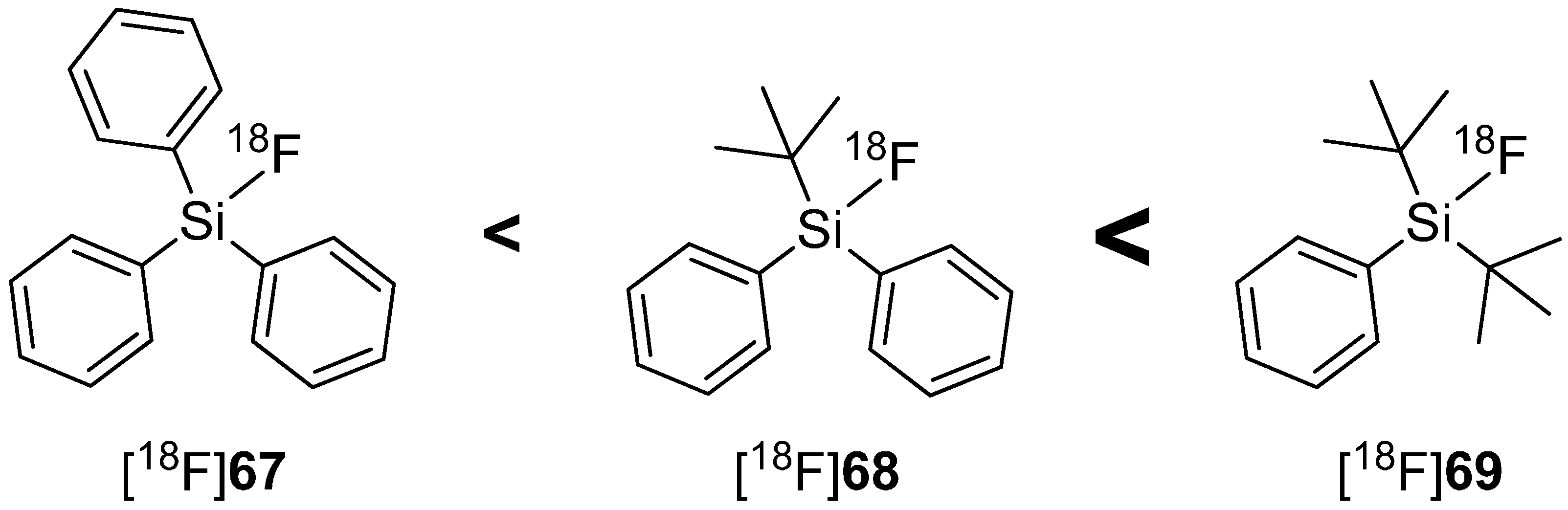
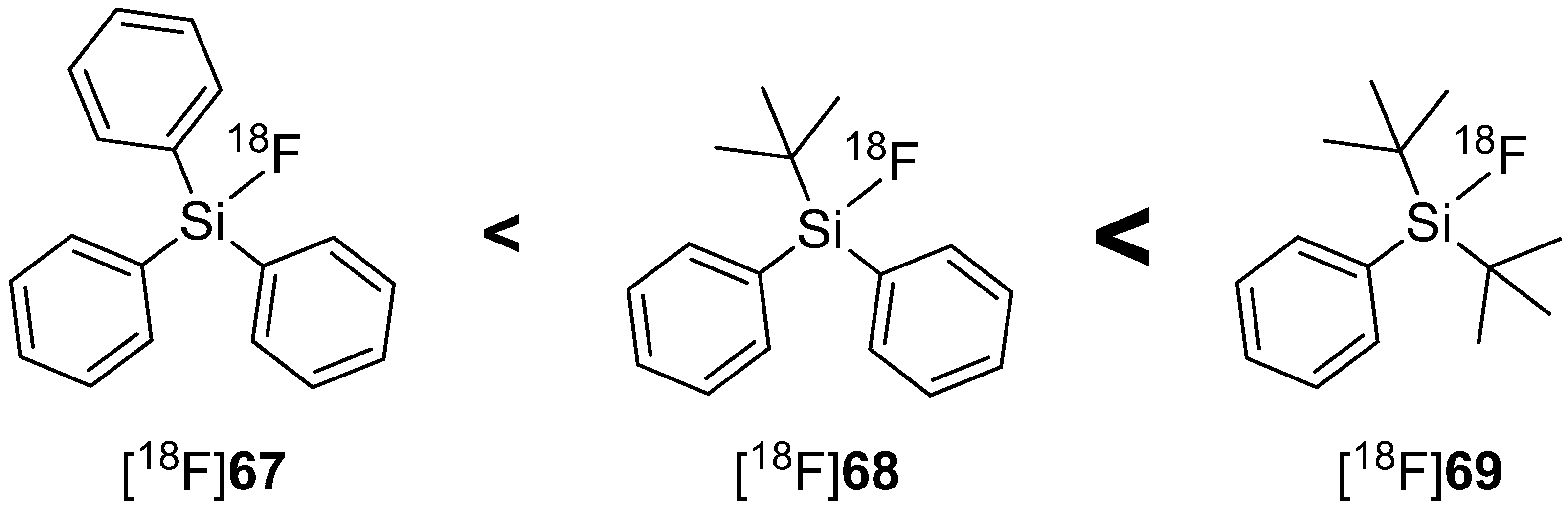
In 2000, Walsh and co-workers tried to induce the stabilization of Si-F-bond with bulky substituents such as phenyl or tert-butyl groups and confirmed the assumption of Rosenthal, who predicted the use of bulky substituents on silicon diminishes the hydrolysis of Si-F bond [121]. Furthermore, Choudhry and Blower investigated the behavior of different sized alkyl groups (Me, Ph, tert-Bu) and their combinations connected to fluorosilanes. The results showed that tert-butyldiphenyl[18F]fluorosilane ([18F]68) contained the highest stabilized Si-F-bond [122]. Contemporaneously, Schirrmacher and Jurkschat carried out comparable experiments and found di-tert-butylphenylfluorosilane ([18F]69) with the highest stability against hydrolysis of Si-F-bond and called this compound class SiFA (silicon-based fluoride-acceptor). The hydrolytic stability in dependence of the alkyl group is expressed in Scheme 18 [123].
Scheme 18.
In vitro hydrolytic stability of [18F]fluorosilanes in dependence of their organic groups in human serum.
Scheme 18.
In vitro hydrolytic stability of [18F]fluorosilanes in dependence of their organic groups in human serum.
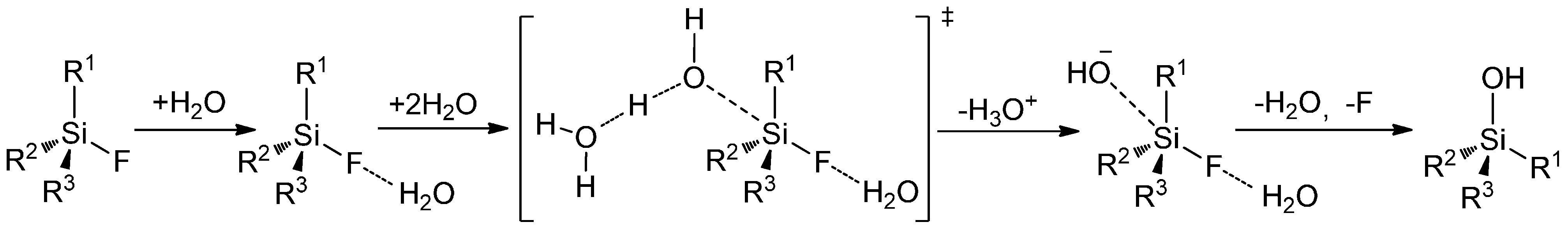
The high polarization of Si-F bond results in the kinetic instability of Si-F-bond [124] and allows an exchange under mild conditions. Due to the low energy of vacant d-orbitals tetravalent silicon as weak Lewis acid reacts with Lewis bases [125], which allows a nucleophilic attack by hydroxyl-groups in the case of aqueous conditions. Generally, nucleophilic displacement reactions on silicon proceed by the predicted SN2 mechanism in Scheme 19 [126,127].
Scheme 19.
Suggested SN2 mechanism of the hydrolysis reaction of organofluorosilanes.
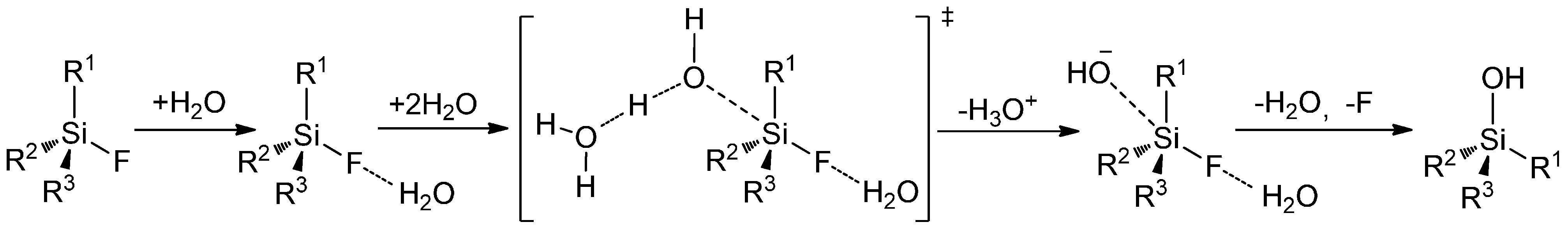
Contrary to carbon, a real pentagonal transition state including hypervalent silicon is formed and assists this substitution. The larger covalent radius of silicon compared to carbon contributes to this nucleophilic substitution [128], which led to the poor kinetic stability of Si-F-bond despite the high thermodynamic stability. Thus, a stabilization of Si-F bond to prevent a nucleophilic attack is only possible by raising the sterical bulkiness of the substituents. This fact explains the weak impact (plain structure) of phenyl moieties and they are also responsible for the augmented Lewis acid properties of silanes.
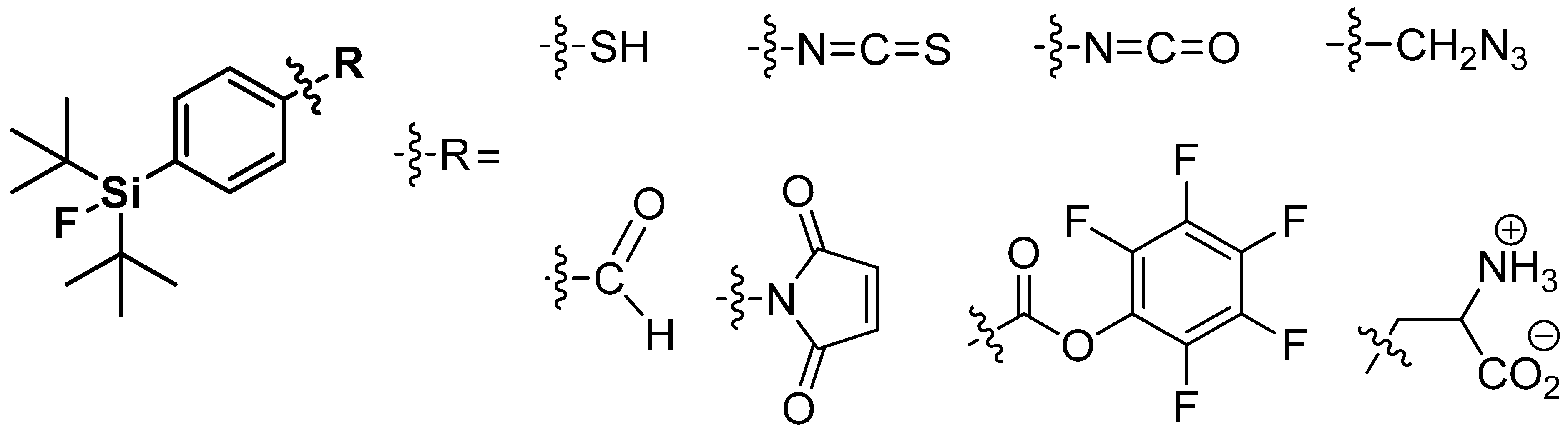
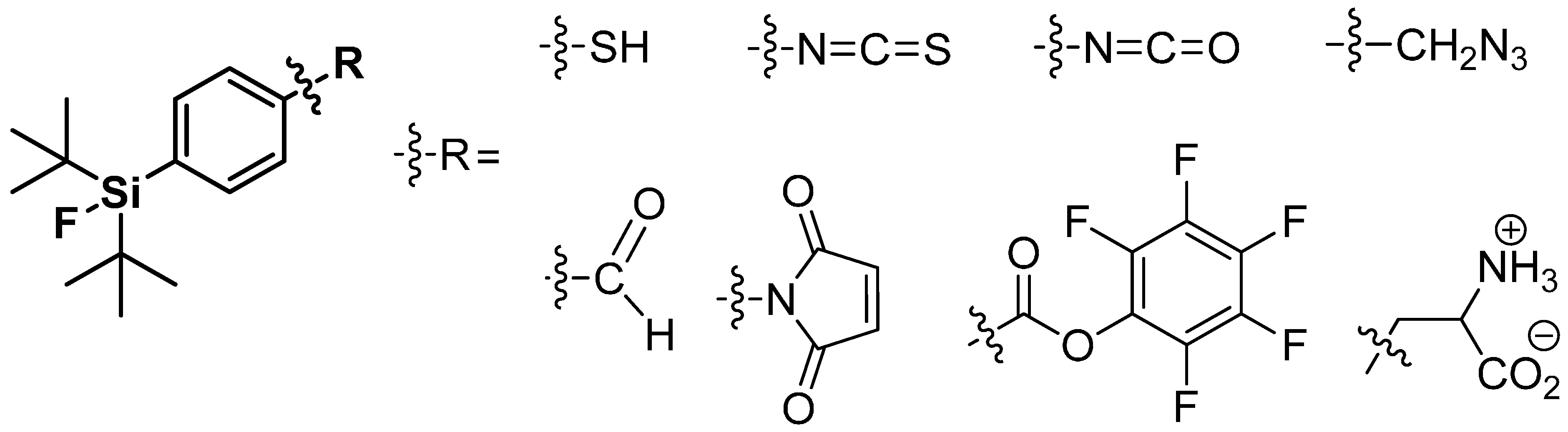
Only the use of tert-butyl groups located in direct neighborhood of Si-F-bond prevents hydrolysis due to their bulky three-dimensional structure. The third substituent on the silicon is utilized for further derivatization. Hence, the phenyl group seems to be the perfect choice for a functionalization with groups such as aldehydes, NCS-, or -SH in mainly para-position to the silyl residue. These resulting building blocks were often used for labeling of peptides and proteins [123,129,130,131]. An overview is given in Scheme 20. Furthermore, the use of alkyl-groups as third substituent with supplemental functionalization was proven, but exhibited a reduced hydrolytic stability compared to the phenyl tracers [126,132].
Scheme 20.
A summary of applied SiFA building blocks taken from the review by Bernard-Gauthier et al., 2014 [128].
Scheme 20.
A summary of applied SiFA building blocks taken from the review by Bernard-Gauthier et al., 2014 [128].
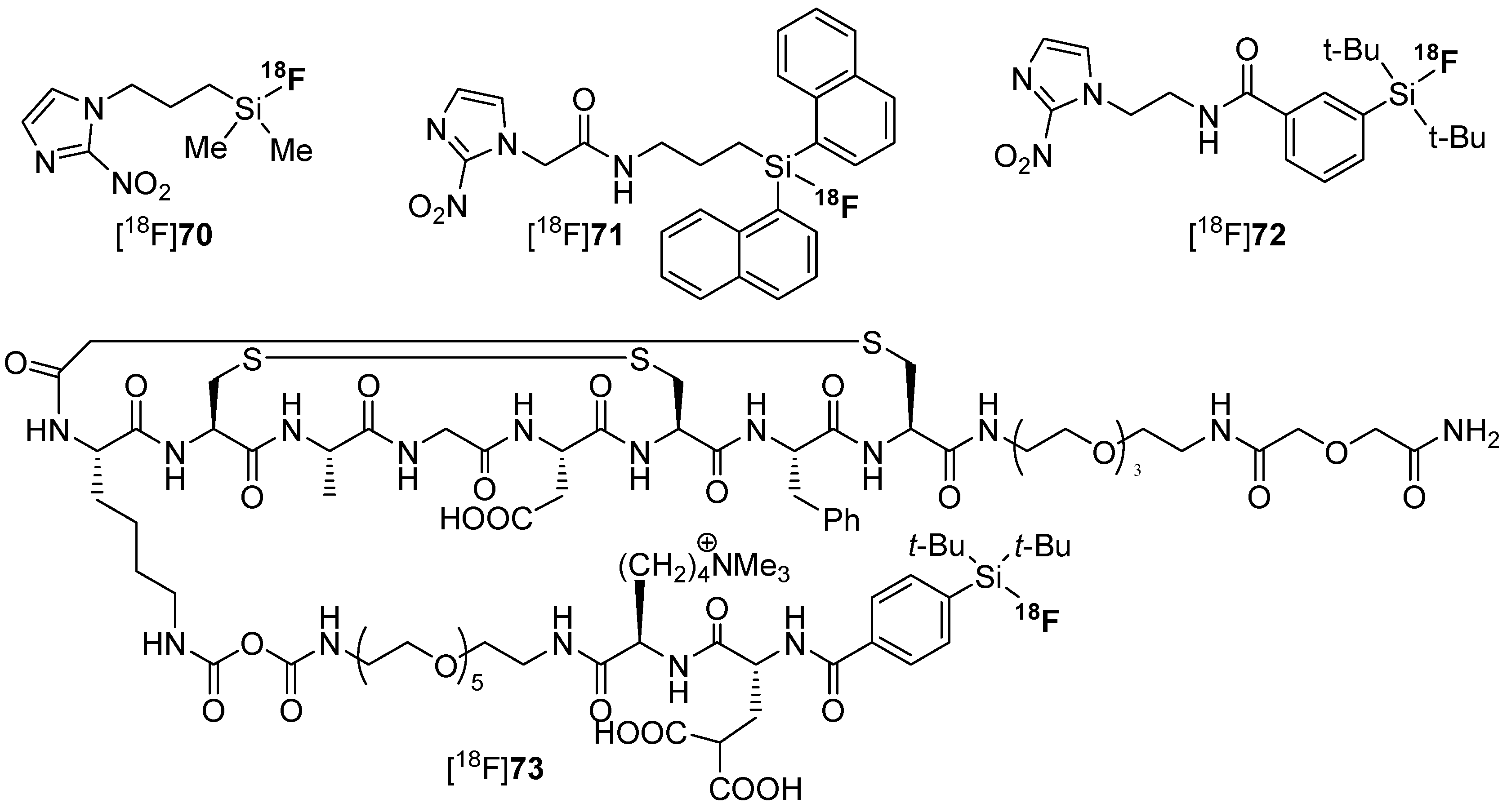
Next, the introduction of fluorine-18 was evaluated by the use of different leaving groups such as alkoxycarbonyl-groups [122,133], by halogen and isotope exchange [118,123] and by applying hydrosilanes [134]. Manifold examples for the use of the SiFA concept were octreotide, bombesin, RGD, PSMA, antibodies, simple molecules, carbohydrates, and biotin. An excellent overview is provided by an outstanding review, see [128]. In most of the cases, the respective building blocks were applied especially for the biomacromolecules, but also a direct introduction of fluorine-18 was accomplished. However, the direct introduction exhibited a rather low yield compared to the building block approaches. Examples of hypoxia tracers [18F]70–[18F]72 with rising metabolic stability and [18F]73 as SiFA-labeled peptide is found in Scheme 21.

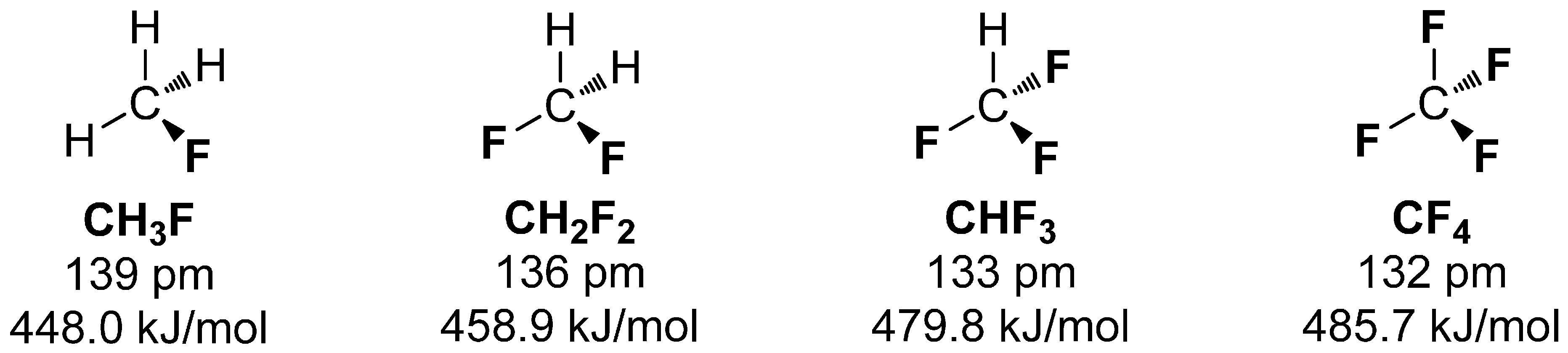{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5. Miscellaneous
5.1. Fluorosulfonamides
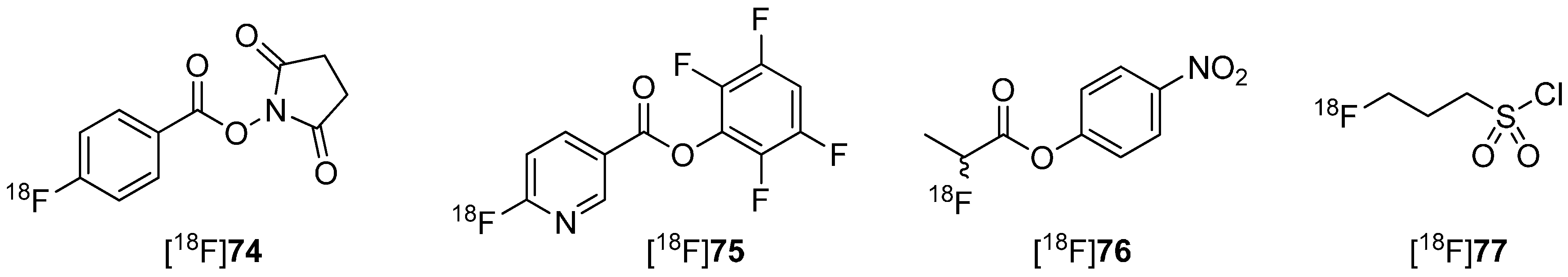
Metabolically stable building blocks also referred to as prosthetic groups were required especially for the radiolabeling of peptides or other biomacromolecules. Conventional building blocks such as [18F]SFB [18F]74 were used to radiolabel particularly with primary amine residues of peptides (N-terminus or lysine) under formation of amide (peptide) bonds. A selection of these 18F-building blocks is pointed out in Scheme 22.
Scheme 22.
Selected examples for 18F building blocks for radiolabeling of peptides.
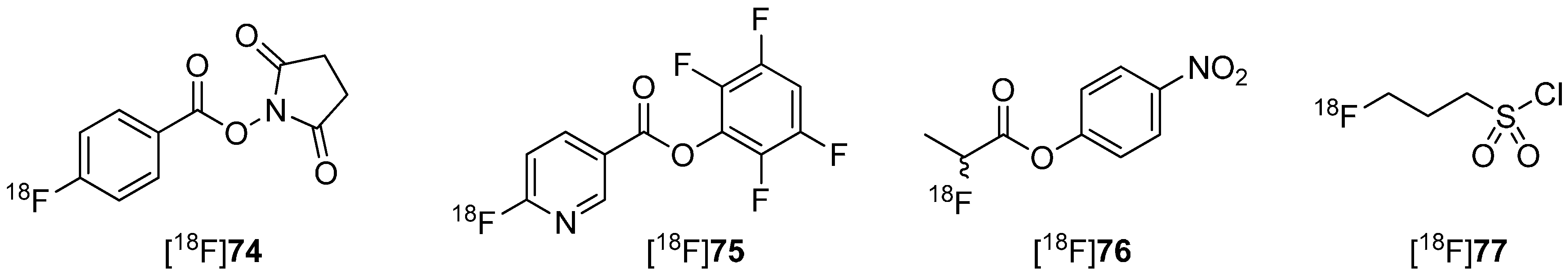
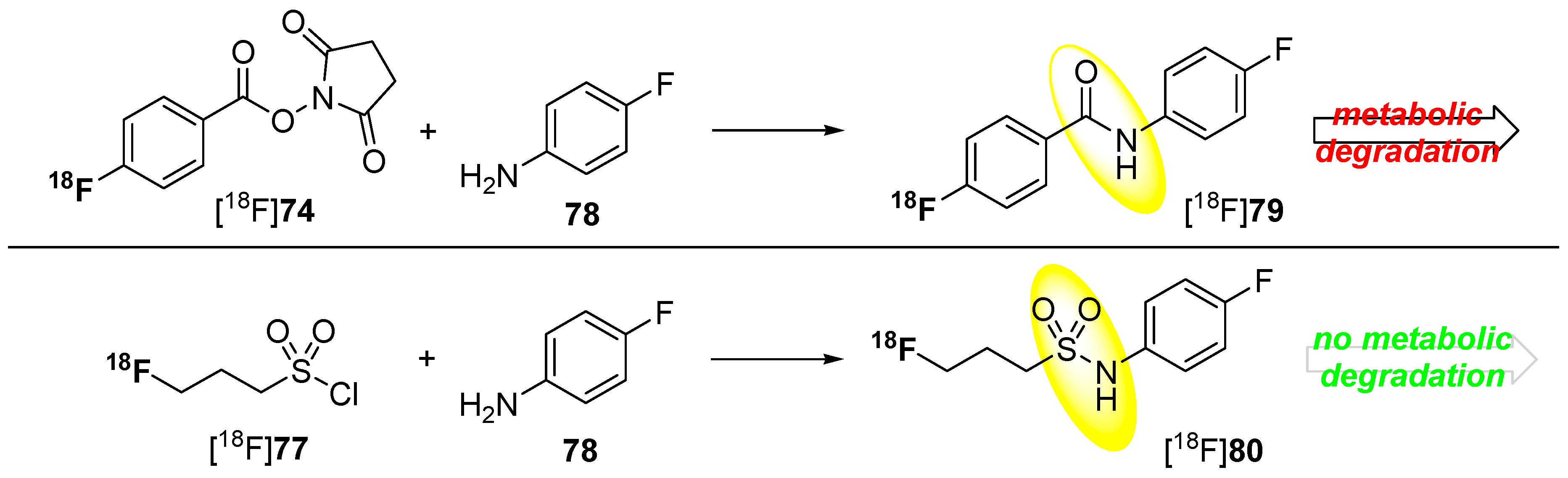
However, this kind of radiofluorinated aromatic fluoroacetamides turned out to be unstable in vivo and undergoes N-defluoroacylation [136]. It was reported that this degradation may be caused by the involvement of carboxylesterase (E.C. 3.1.1.1) or other hydrolases [137,138]. As an alternative to these acyl-based prosthetic groups, the 3-[18F]fluoropropanesulfonyl chloride ([18F]77) was introduced by Li et al. [139] and by Löser and co-workers [140]. They substantiate the metabolic integrity of fluorinated sulfonamide: N-(4-fluorophenyl)-3-fluoropropane-1-sulfonamide (80) compared to the aromatic acyl derivative N-(4-fluorophenyl)-fluoroacetamide (79) in a spectrophotometric enzyme assay using pig liver esterase. Both compounds are shown in Scheme 23.
Scheme 23.
Comparison in radiofluorination and metabolic stability for [18F]SFB [18F]74 and [18F]fluoropropylsulfonyl chloride [18F]77.
Scheme 23.
Comparison in radiofluorination and metabolic stability for [18F]SFB [18F]74 and [18F]fluoropropylsulfonyl chloride [18F]77.
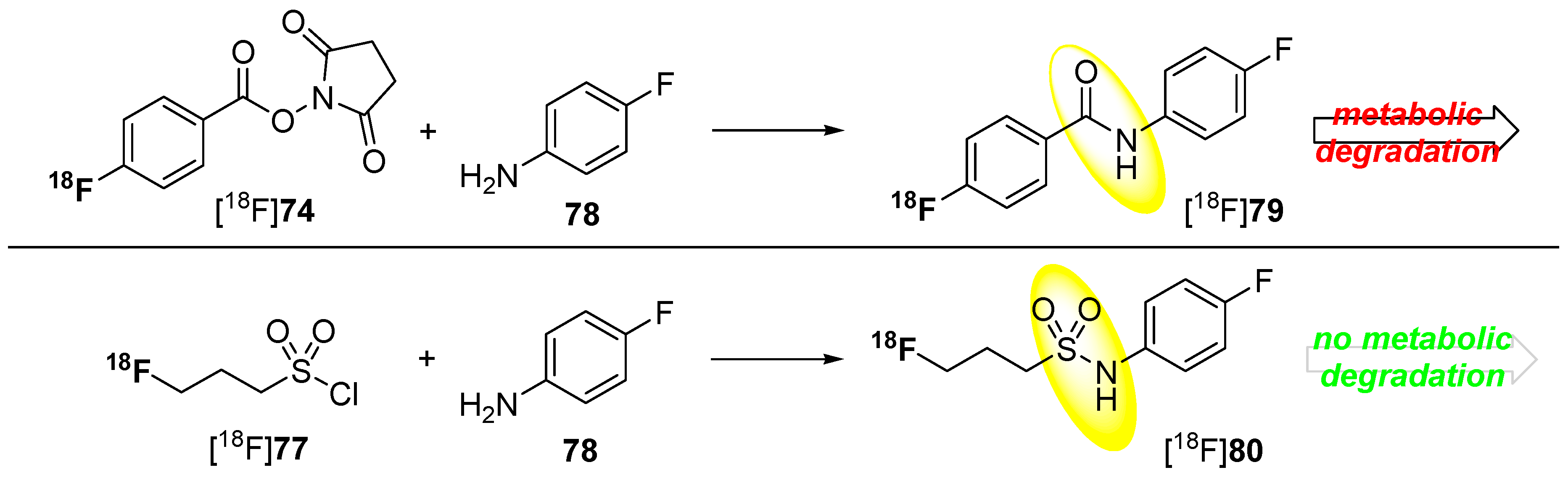
After 120 min (approx. one half-life of 18F), only 20% of the starting 19F-compound 79 was intact whereas, at the same time point, over 95% of the sulfonamide 80 was still detectable. Furthermore, pseudo-first order kinetics for the degradation of the acylamide could be determined.
5.2. Click-Chemistry
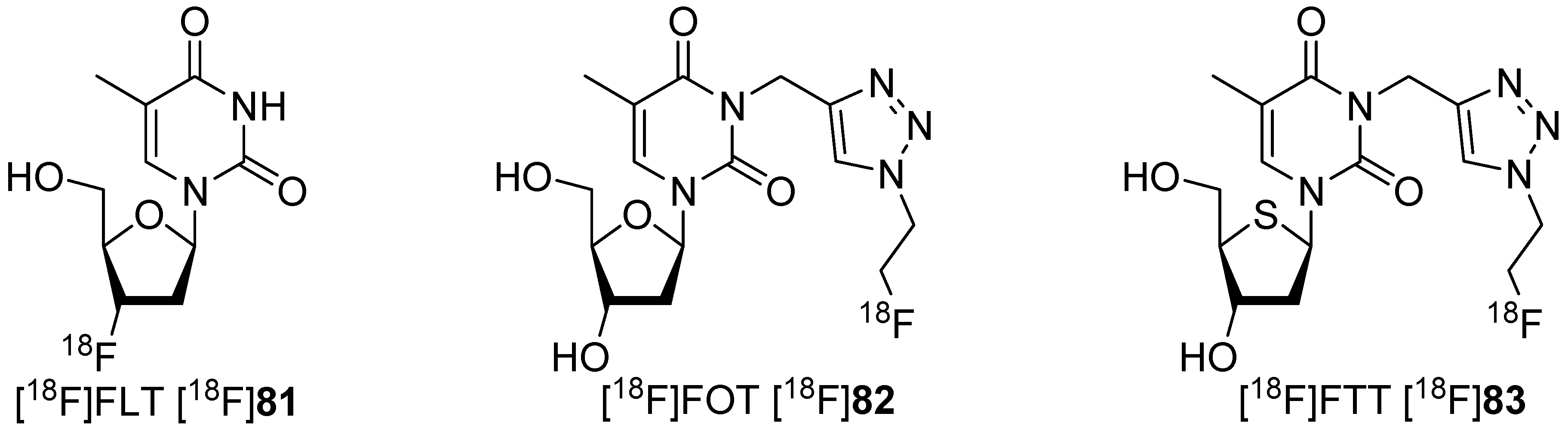
A further approach to avoid degradation was using triazoles [141], which were obtained by click chemistry [142,143]. Two different research groups investigated [18F]fluoroalkyl groups bound at position N-3 of the triazole moiety of thymidine derivatives like [18F]82 with conflicting results and uncertainty over the metabolic stability of the radiotracers in vivo. The prepared 18F-tracers are shown in Scheme 24. Smith and colleagues postulated that 1,4-disubstituted triazoles have a higher metabolic stability in vivo due to the greater steric bulk of the triazole. The metabolic stability is increased relative to simple fluoroalkyl substituents to thymidine-phosphorylase-mediated cleavage [144,145].
Scheme 24.
Presentation of 18F-labeled thymidine derivatives in the case of their labeling position.
Scheme 24.
Presentation of 18F-labeled thymidine derivatives in the case of their labeling position.
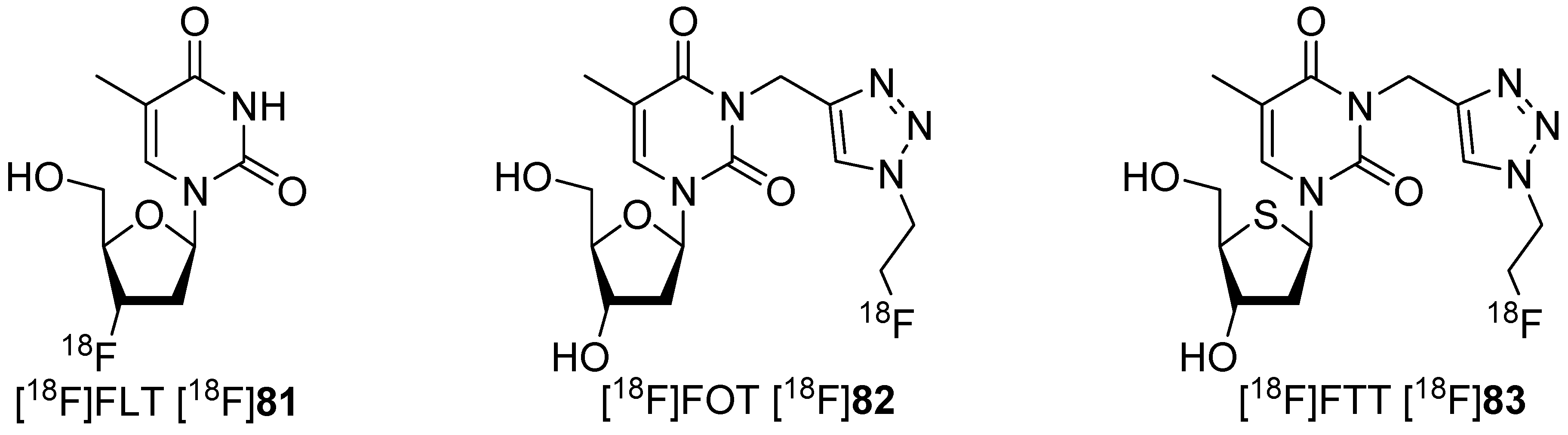
Initial studies of the ability of these nucleosides to undergo phosphorylation demonstrated that [18F]FLT [18F]81 was phosphorylated to approx. 7%–8% after 60 min incubation, whereas no phosphorylation was observed with [18F]FOT [18F]82 over the same time period. Comparison with [18F]FLT [18F]81 showed that [18F]FOT [18F]82 was poorly phosphorylated at the 5-position of the deoxyribose residue. The poor thymidine kinase 1 (TK1) substrate tolerance due to substitution at nitrogen N-3 was given as a possible reason for this finding.
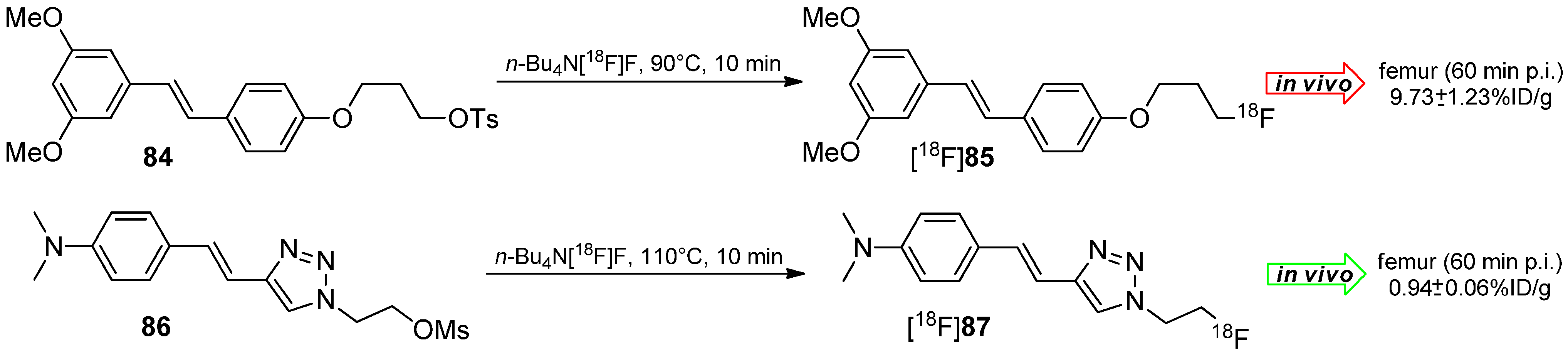
The working group of Choe developed 18F-Labeled styryltriazole and resveratrol derivatives such as [18F]85 and [18F]87 for β-amyloid plaque imaging [146]. Compounds 84 and 86 were labeled under standard labeling conditions (n-Bu4N[18F]F, acetonitrile, 90 °C or 110 °C, 10 min) and yielded both tracers in 20%–30% RCY for [18F]85 and 56% RCY for [18F]87 with a AS ≈ 38 GBq/µmol and a RCP > 99% (Scheme 25).
Scheme 25.
Radiolabeling and selected in vivo results of reservatrol [18F]85 and styryltriazole [18F]87.
Scheme 25.
Radiolabeling and selected in vivo results of reservatrol [18F]85 and styryltriazole [18F]87.
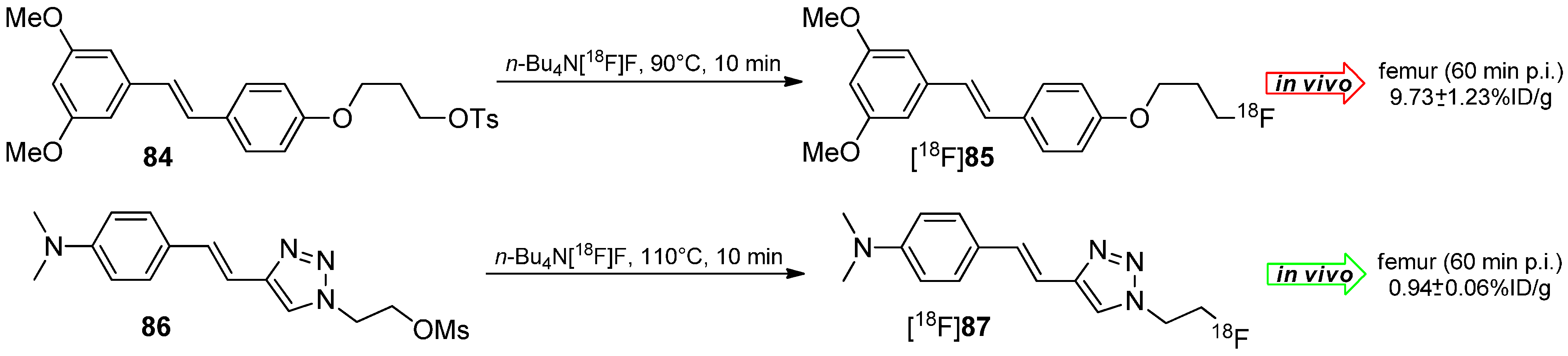
In vivo studies of both tracers showed a remarkable metabolic degradation of reservatrol derivative [18F]85 under elimination of [18F]fluoride which was accumulated in the femur (16.15% ± 3.10% ID/g after 120 min). Conversely, the styryltriazole compound [18F]87 showed almost no cleavage of [18F]fluoride (1.54% ± 0.02% ID/g after 120 min).
5.3. CF3-Derivatives
As stated in the introduction (Section 1.1), the use of CF3 groups could increase the metabolic stability of pharmacologically relevant compounds and radiotracers [147] due to the increased bond strength of the C-F bond in this group compared to single fluorine connected to carbon and due to the higher steric shielding of the carbon center. Furthermore, the trifluoromethyl group is present in a large number of agrochemicals, biologically active drugs and anesthetics, which led to attempts to introduce fluorine-18 to yield [18F]CF3 group containing radiotracers; see an excellent review by Lien and Riss [148].
Normally, the introduction of [18F]fluoride was accomplished via 18F/19F isotopic exchange [149,150,151], Lewis acid mediated reactions [152,153], halogen for 18F exchange [154,155,156] or H18F addition [157] and electrophilic reactions with [18/19F]F2 [158,159], but most of these reactions suffer from low specific activities due to the carrier added reactions and/or rough conditions.
Scheme 26.
Radiolabeling of [18F]CF3 containing Celecoxib derivative [18F]89.

An example is presented regarding the synthesis of 4-[5-(4-methylphenyl)-3-([18F]trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide ([18F]Celecoxib) ([18F]89) which is known to be a selective COX-2 inhibitor [160]. The labeling procedure was accomplished exchanging bromide with [18F]F− using [18F]TBAF in DMSO at 135 °C (Scheme 26). [18F]Celecoxib was achieved in 10% ± 2% RCY (end of synthesis) with >99% chemical and radiochemical purity and a specific activity, which was 4.40 ± 1.48 GBq/μmol (end of bombardment). In vitro stability experiments showed only a small amount of [18F]fluoride coming from radiodefluorination in 10% ethanol-saline (6.5% after 4 h). However, in vivo experiments of [18F]89 with Wistar rats showed a higher skeleton uptake compared to brain or heart; regions where COX-2 is known to be present due to the radiodefluorination process. In contrast, no uptake in skull and skeleton was observed in baboon indicating only a low degree of defluorination of [18F]89 in vivo. In addition, metabolite analyses show that [18F]89 undergoes fast metabolism. Polar metabolites were found in baboon plasma and 17.0% of unmetabolized tracer was determined at 60 min after injection; no evidence was obtained for free [18F]fluoride.
5.4. 18F-Fluoroborates
An impressive stability was found for the B-F bond (645 kJ/mol) in BF3 [117]. Thus, the introduction of fluorine-18 directly connected to boron represents a further promising alternative to avoid radiodefluorination. The non-binding electrons of fluorine atoms in BF3 form π-bonds with boron, which represent partial double bonds with an average bond length of 130 pm. Based on this fact, the still electron demanding boron center is less hydrolytic unstable. The previously sp2-hybridized boron center is changed to sp3 by accepting an electron pair of an additional fluoride in the former pz-orbital to form a tetrafluoroborate anion (BF4−). Thereupon, the bonds in BF4− are single bonds with also a high hydrolytic stability [117,161].
This basic principle is used for the creation of fluourine-18-containing boron derivatives. Exchange of F− in these species is rare due to the aforementioned high bond strength of the B-F bond. The fluorine atoms in BF4− are substitutable but the exchange should be advisedly chosen. In general, the exchange of fluorine by other halogens leads to weaker bonds [162]. Calculations for triarylfluoroborates predicted a weakening of remaining B-F bond [163]. Therefore, comparable functionalizations will be necessary to apply this concept for the development of radiotracers.
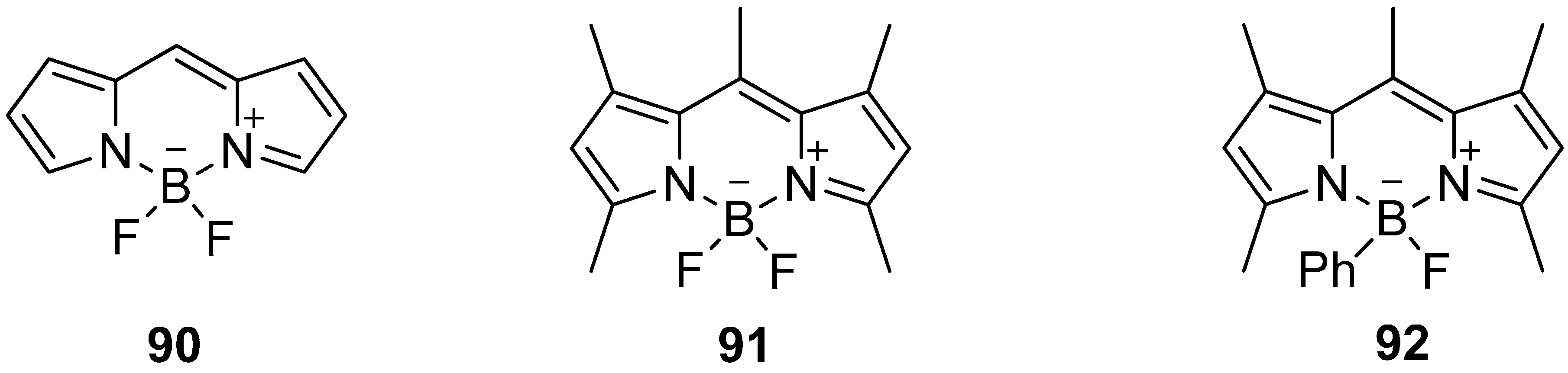
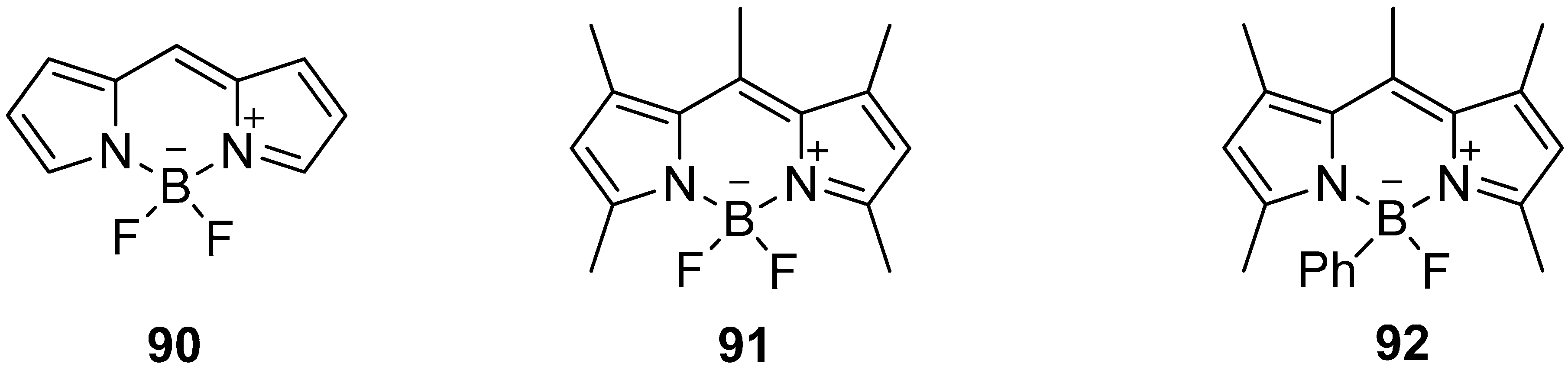
The application of bodipy derivatives represents a promising approach. Several methylated compounds such as 91 and 92 were described for the first time by Treibs and Kreuzer in 1968 in addition with the excellent fluorescence properties of these dyes [164]. However, the synthesis of the core structure 90 succeeded first in 2009 [165,166,167], see Scheme 27.
Scheme 27.
Core structure 90 of selected bodipy derivatives 91 and 92.
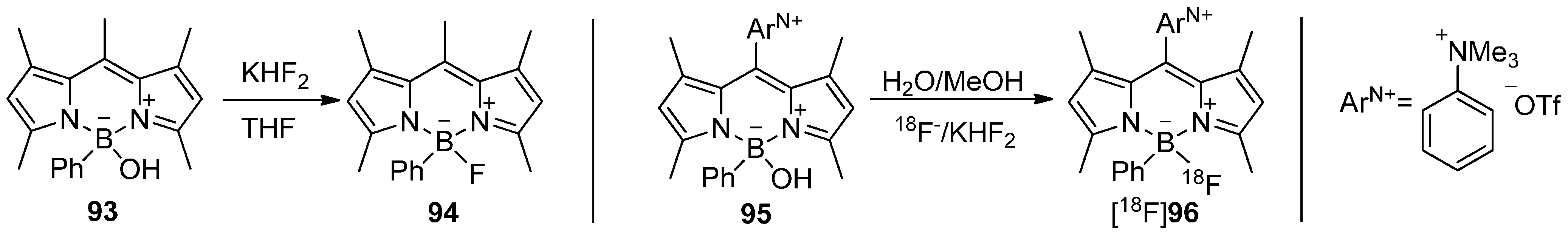
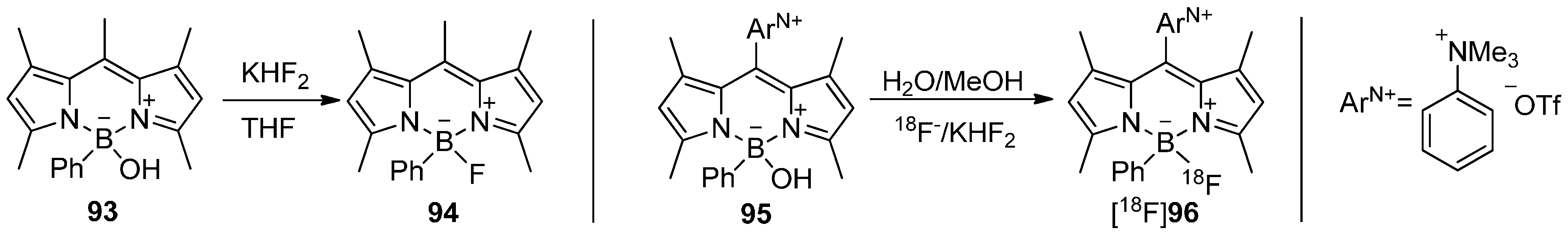
In 2011, bodipy dyes moved into focus for radiolabeling with fluorine-18. For that purpose, a radiolabeling building block based on a modified bodipy was created by Li and co-workers [168]. The radiosynthesis of the BPh[18F]F core containing derivative [18F]96 was simply realized by using KHF2/[18F]F− (directly from target water without drying) in water/methanol starting from BPhOH-precursor 95 with a AS = 0.9 GBq/µmol. The solubility of the precursor 95 and the desired bodipy derivative [18F]96 was achieved due to the ammonium triflate moiety ArN+. The results are summarized in Scheme 28.
Scheme 28.
Conversion of OH compound 93 to 94 and radiolabeling of 95 to yield [18F]96.
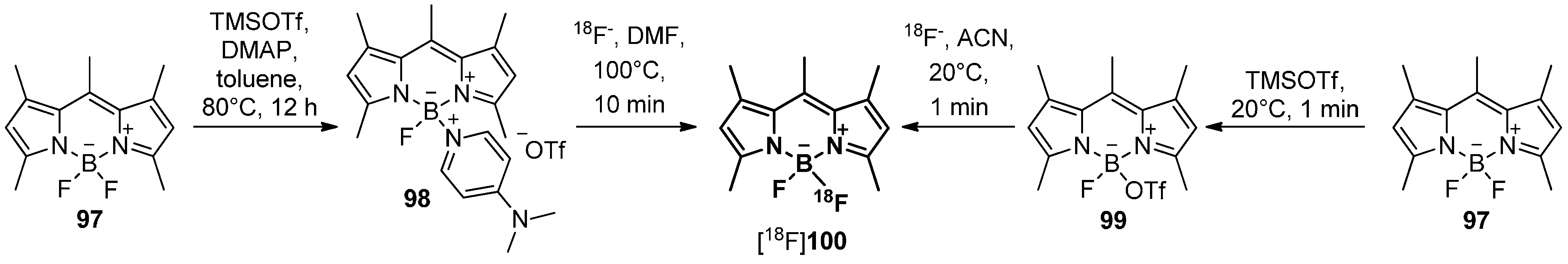
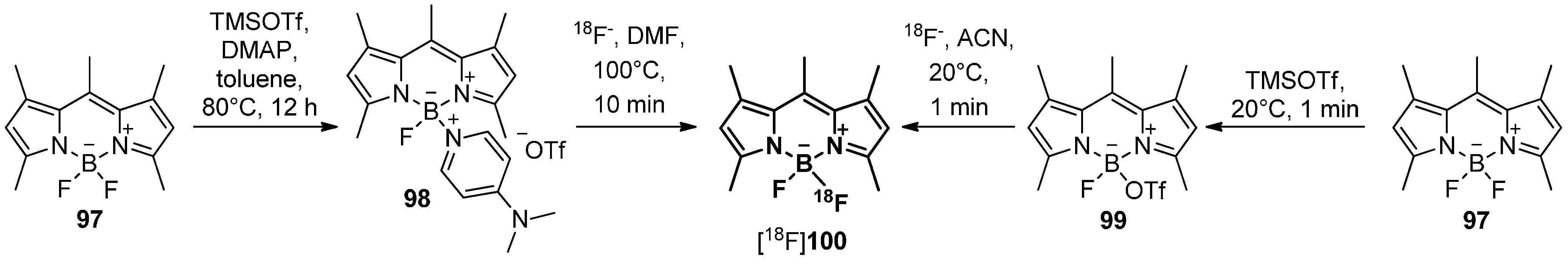
In 2012, an alternative radiolabeling building block based on the B[18F]F2 core containing bodipy derivative was prepared [169]. Thus, the respective precursors were either synthesized via an exchange of one of the fluorides of 97 by a DMAP leaving group using TMS triflate/DMAP to yield 98 or directly by changing one fluoride to triflate with TMS triflate to yield 99. The desired DMAP/18F exchange of 98 to [18F]100 succeed but the triflate precursor 99 was proven to be more effective due to the higher radiochemical yields of [18F]100, milder reaction conditions and a short reaction time during the labeling reaction (Scheme 29). A labeling building block based on [18F]100 was further used for the successful labeling of Trastuzumab with fluorine-18.
Scheme 29.
Preparation of the building block [18F]100 from compound 97 via either precursor 98 or 99 under mild labeling conditions.
Scheme 29.
Preparation of the building block [18F]100 from compound 97 via either precursor 98 or 99 under mild labeling conditions.
Both groups demonstrated the high metabolic stability of the desired bodipy derivatives in vitro as well as in vivo. No radiodefluorination in terms of an accumulation of activity in the skeleton due to free [18F]fluoride was observed. Based on these results, this concept has great potential to create stable radiotracers having a B-[18F]F bond.
6. Conclusions
Radiodefluorination is one of the most important metabolic degradation processes for 18F-radiotracers due to the release of [18F]fluoride in vivo, which is then accumulated in the skull and bones. This undesired accumulation leads to PET images that are false-positive in terms of skeleton imaging or comprise a bad signal to background ratio.
Several efforts have been made in the past to avoid this defluorination or to considerably reduce it. The insertion of deuterium to stabilize the C-F bond seems to be the most successful approach. Thus, a building block strategy was developed using small deuterated molecules with 18F-label. In many cases, radiodefluorination could be reduced in an appreciable manner.
Other approaches can show reduced radiodefluorination in a remarkable manner as well. The introduction of deuterium in other positions relative to 18F (or 11C) is also promising. In this case, the stabilization is used to reduce cleavage of other parts of the molecule. Furthermore, the introduction of C[18F]F3 groups leads to a reduced degradation since the bond strength of the C-F bond is increased in the CF3 group. Finally, the insertion of a [18F]fluorocyclobutyl moiety is favored over open fluoroalkyl chains due to the increased steric demand and, therefore, reduced metabolism. The same effect can be reached by the utilization of special functional groups to avoid cleavage on this position.
Binding [18F]fluorine to heteroatoms like silicon or boron offers also the possibility to obtain radiotracers, which show reduced radiodefluorination. Though, in the case of silicon based 18F-radiotracers, the additional protection of the Si-[18F]F center with bulky substituents is mandatory. The use of [18F]bodipy derivatives offers the chance to use the same molecule for PET as for optical imaging.
Acknowledgments
The authors are grateful to Matthew D. Gott for proofreading the manuscript.
Author Contributions
Both authors (Manuela Kuchar and Constantin Mamat) contributed equally to this work in the case of writing this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Valk, P.E.; Baily, D.L.; Townsend, D.W.; Maisey, M.N. Positron Emission Tomography: Basic Science and Clinical Practice; Springer: London, UK, 2003. [Google Scholar]
- Fowler, J.S.; Ding, Y.-S. Radiotracer Chemistry. In Principles and Practice of PET and PET/CT, 2nd ed.; Wahl, R.L., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Ross, T.L.; Wester, H.J. 18F: Labeling Chemistry and Labeled Compounds. In Handbook of Nuclear Chemistry, 2nd ed.; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 2023–2025. [Google Scholar]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthese von 11C-, 18F-, 15O- und 13N-Radiotracern für die Positronenemissionstomographie. Angew. Chem. 2008, 120, 9136–9172. [Google Scholar] [CrossRef]
- Pretze, M.; Große-Gehling, P.; Mamat, C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules 2011, 16, 1129–1165. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Harper, D.B. Fluorine-containing natural products. J. Fluor. Chem. 1999, 100, 127–133. [Google Scholar] [CrossRef]
- Reddy, V.P. Organofluorine Compounds in Biology and Medicine, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1–23. [Google Scholar]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, F. 18F-Labeled Peptides: The Future Is Bright. Molecules 2014, 19, 20536–20556. [Google Scholar] [CrossRef] [PubMed]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and SPECT imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 48, 441–451. [Google Scholar] [CrossRef]
- Sharpe, A.G. The physical properties of the carbon-fluorine bond. In Ciba Foundation Symposium 2—Carbon-Fluorine Compounds: Chemistry, Biochemistry and Biological Activities; Elliot, K., Birch, J., Eds.; Associated Scientific Publishers: Amsterdam, The Netherlands, 1972; pp. 33–54. [Google Scholar]
- Peters, D. Problem of the Lengths and Strengths of Carbon-Fluorine Bonds. J. Chem. Phys. 1963, 38, 561–563. [Google Scholar] [CrossRef]
- Bent, H.A. An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Lemal, D.M. Perspective on Fluorocarbon Chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tozer, D.J. The conformation and internal rotational barrier of benzyl fluoride. Chem. Phys. Lett. 1999, 308, 160–164. [Google Scholar] [CrossRef]
- Kochi, J.K.; Hammond, G.S. Benzyl Tosylates. II. The Application of the Hammett Equation to the Rates of their Solvolysis. J. Am. Chem. Soc. 1953, 75, 3445–3451. [Google Scholar] [CrossRef]
- Wüst, F.; Müller, M.; Bergmann, R. Synthesis of 4-([18F]fluoromethyl)-2-chlorophenylisothiocyanate: A novel bifunctional 18F-labelling agent. Radiochim. Acta 2004, 92, 349–353. [Google Scholar] [CrossRef]
- Zavitsas, A.A.; Rogers, D.W.; Matsunag, N. Remote Substituent Effects on Allylic and Benzylic Bond Dissociation Energies. Effects on Stabilization of Parent Molecules and Radicals. J. Org. Chem. 2007, 72, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer: Berlin, Germany, 2000; p. 126. [Google Scholar]
- Frank, H.; Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths determined by X-ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar]
- Wiberg, K.B.; Rablen, P.R. Substituent Effects. 7. Phenyl Derivatives. When Is a Fluorine a π-Donor? J. Org. Chem. 1998, 63, 3722–3730. [Google Scholar] [CrossRef]
- Carroll, T.X.; Thomas, T.D.; Bergersen, H.; Børve, K.J.; Sæthre, L.J. Fluorine as a π Donor. Carbon 1s Photoelectron Spectroscopy and Proton Affinities of Fluorobenzenes. J. Org. Chem. 2006, 71, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Liotta, C.L.; Harris, H.P. Chemistry of naked anions. I. Reactions of the 18-crown-6 complex of potassium fluoride with organic substrates in aprotic organic solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Tressaud, A.; Haufe, G. Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008; pp. 35–42. [Google Scholar]
- Ermert, J. 18F-Labelled Intermediates for Radiosynthesis by Modular Build-Up Reactions: Newer Developments. BioMed. Res. Int. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Fischer, C.R.; Holland, J.P.; Becaud, J.; Schubiger, P.A.; Schibli, R.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Dinkelborg, L.M.; et al. 18F-Radiolabeling of Aromatic Compounds Using Triarylsulfonium Salts. Eur. J. Org. Chem. 2012, 2012, 889–892. [Google Scholar] [CrossRef]
- Sander, K.; Gendron, T.; Yiannaki, E.; Cybulska, K.; Kalber, T.L.; Lythgoe, M.F.; Årstad, E. Sulfonium Salts as Leaving Groups for Aromatic Labelling of Drug-like Small Molecules with Fluorine-18. Sci. Rep. 2015, 5, 9941. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.R.; Lee, E.; Boursalian, G.B.; Ritter, T. Mechanism of electrophilic fluorination with Pd(IV): Fluoride capture and subsequent oxidative fluoride transfer. Chem. Sci. 2014, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [18F]fluorination: New solutions to old problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.L.; Wester, H.J. 18F: Labeling Chemistry and Labeled Compounds. In Handbook of Nuclear Chemistry, 2nd ed.; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 2026–2032. [Google Scholar]
- Vallabhajosula, S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT; Springer: Dordrecht, The Netherlands, 2009; pp. 142–144. [Google Scholar]
- Tressaud, A.; Haufe, G. Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008; p. 7. [Google Scholar]
- Scott, P.J.H.; Hockley, B.G.; Kung, H.F.; Manchanda, R.; Zhang, W.; Kilbourn, M.R. Studies into radiolytic decomposition of fluorine-18 labeled radiopharmaceuticals for positron emission tomography. Appl. Radiat. Isot. 2009, 67, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Langguth, P.; Seydel, J. Überarbeitetes Glossar zu Begriffen der Pharmazeutik. Angew. Chem. 2011, 123, 3635–3651. [Google Scholar] [CrossRef]
- Middleton, R.K. Drug Interactions. In Textbook of Therapeutics: Drug and Disease Management, 8th ed.; Helms, R.A., Herfindal, E.T., Quan, D.J., Eds.; Lipincott Williams & Wilkins: Philadelphia, PA, USA, 2006; p. 50. [Google Scholar]
- Kharasch, E.D.; Thummel, K.E. Identification of cytochrome P450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. Anesthesiology 1993, 79, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Anders, M.W.; Jones, J.P. Metabolism of 1,2-dichloro-1-fluoroethane and 1-fluoro-1,2,2-trichloroethane: Electronic factors govern the regioselectivity of cytochrome P450-dependent oxidation. Chem. Res. Toxicol. 1996, 9, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the a1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F]cpfpx) in rodents and humans. Drug Metabol. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood–brain barrier and surviving metabolism. Trends Pharm. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R. Effects of fluorine substitution on drug metabolism: Pharmacological and toxicological implications. Drug Metab. Rev. 1994, 26, 605–643. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.J.; Katzenellenbogen, J.A.; Mathias, C.J.; Brodack, J.W.; Carlson, K.E.; Chi, D.Y.; Dence, C.S.; Kilbourn, M.R.; Perlmutter, J.S.; Raichle, M.E.; et al. N-(3-[18F]fluoropropyl)-spiperone: The preferred 18F labeled spiperone analog for positron emission tomographic studies of the dopamine receptor. Nucl. Med. Biol. 1988, 15, 83–97. [Google Scholar] [CrossRef]
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET Imaging of the Dopamine Transporter with 18F-FECNT: A Polar Radiometabolite Confounds Brain Radioligand Measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Ettlinger, D.E.; Wadsak, W.; Mien, L.-K.; Machek, M.; Wabnegger, L.; Rendl, G.; Karanikas, G.; Viernstein, H.; Kletter, K.; Dudczak, R.; et al. [18F]FETO: Metabolic considerations. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Agdeppa, E.D.; Kepe, V.; Liu, J.; Flores-Torres, S.; Satyamurthy, N.; Petric, A.; Cole, G.M.; Small, G.W.; Huang, S.-C.; Barrio, J.R. Binding Characteristics of Radiofluorinated 6-Dialkylamino-2-Naphthylethylidene Derivatives as Positron Emission Tomography Imaging Probes for β-Amyloid Plaques in Alzheimer’s Disease. J. Neurosci. 2001, 21, RC189. [Google Scholar] [PubMed]
- Mitterhauser, M.; Wadsak, W.; Wabnegger, L.; Mien, L.K.; Tögel, S.; Langer, O.; Sieghart, W.; Viernstein, H.; Kletter, K.; Dudczak, R. Biological evaluation of 2ʹ-[18F]fluoroflumazenil ([18F]FFMZ), a potential GABA receptor ligand for PET. Nucl. Med. Biol. 2004, 31, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Gottumukkala, V.; Heinrich, T.K.; Baker, A.; Dunning, P.; Fahey, F.H.; Treves, S.T.; Packard, A.B. Biodistribution and Stability Studies of [18F]Fluoroethylrhodamine B, a Potential PET Myocardial Perfusion Agent. Nucl. Med. Biol. 2010, 37, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Pourghiasian, M.; Hundal, N.; Lau, J.; Bénard, F.; Dedhar, S.; Lin, K.-S. 2-[18F]Fluoroethanol and 3-[18F]fluoropropanol: Facile preparation, biodistribution in mice, and their application as nucleophiles in the synthesis of [18F]fluoroalkyl aryl ester and ether PET tracers. Nucl. Med. Biol. 2013, 40, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Tewson, T.J.; Welch, M.J. Preparation and preliminary biodistribution of “no carrier added” fluorine F-18 fluoroethanol. J. Nucl. Med. 1980, 21, 559–564. [Google Scholar] [PubMed]
- Lee, K.C.; Lee, S.-Y.; Choe, Y.S.; Chi, D.Y. Metabolic Stability of [18F]Fluoroalkylbiphenyls. Bull. Korean Chem. Soc. 2004, 25, 1225–1230. [Google Scholar]
- Milicevic Sephton, S.; Dennler, P.; Leutwiler, D.S.; Mu, L.; Wanger-Baumann, C.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis, radiolabelling and in vitro and in vivo evaluation of a novel fluorinated ABP688 derivative for the PET imaging of metabotropic glutamate receptor subtype 5. Am. J. Med. Mol. Imaging 2012, 2, 14–18. [Google Scholar]
- French, A.N.; Napolitano, E.; van Brocklin, H.F.; Brodack, J.W.; Hanson, R.N.; Welch, M.J.; Katzenellenbogen, J.A. The β-heteroatom effect in metabolic defluorination: The interaction of resonance and inductive effects may be a fundamental determinant in the metabolic liability of fluorine-substituted compounds. J. Label. Compd. Radiopharm. 1991, 30, 431–433. [Google Scholar]
- Purohit, A.; Radeke, H.; Azure, M.; Hanson, K.; Benetti, R.; Su, F.; Yalamanshili, P.; Yu, M.; Hayes, M.; Guaraldi, M.; et al. Synthesis and biological evaluation of pyridazinone analogues as potential cardiac positron emission tomography tracers. J. Med. Chem. 2008, 51, 2954–2970. [Google Scholar] [CrossRef] [PubMed]
- Dollé, F. Fluorine-18-labelled fluoropyridines: Advances in radiopharmaceutical design. Curr. Pharm. Des. 2005, 11, 3221–3235. [Google Scholar] [CrossRef] [PubMed]
- Gant, T.G. Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Limbach, H.-H. Isotope Effects in Chemistry and Biology; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Roston, D.; Islam, Z.; Kohen, A. Isotope Effects as Probes for Enzyme Catalyzed Hydrogen-Transfer Reactions. Molecules 2013, 18, 5543–5567. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, V.; Mikkola, K.K.; Ishizu, T.; Arponen, E.; Kauhanen, S.; Nagren, K.; Solin, O.; Nuutila, P.; Haaparanta, M. Assessment of islet specificity of dihydrotetrabenazine radiotracer binding in rat pancreas and human pancreas. J. Nucl. Med. 2010, 51, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.R.; Souza, F.; Witkowski, P.; Maffei, A.; Raffo, A.; Herron, A.; Kilbourn, M.; Jurewicz, A.; Herold, K.; Liu, E.; et al. Visualizing pancreatic-cell mass with [11C]DTBZ. Nucl. Med. Biol. 2006, 33, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Goland, R.; Freeby, M.; Parsey, R.; Saisho, Y.; Kumar, D.; Simpson, N.; Hirsch, J.; Prince, M.; Maffei, A.; Mann, J.J.; et al. 11C-Dihydrotetrabenazine PET of the pancreas in subjects with long-standing type 1 diabetes and in healthy controls. J. Nucl. Med. 2009, 50, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Jahan, M.; Johnström, P.; Korsgren, O.; Sundin, A.; Halldin, C.; Johansson, L. In vivo and in vitro characterization of [18F]-FE-(+)-DTBZ as a tracer for beta-cell mass. Nucl. Med. Biol. 2010, 37, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Weng, Y.H.; Wey, S.P.; Hsiao, I.T.; Lu, C.S.; Skovronsky, D.; Chang, H.P.; Kung, M.P.; Yen, T.C. Whole-body biodistribution and radiation dosimetry of 18F-FP-(+)-DTBZ (18F-AV-133): A novel vesicular monoamine transporter 2 imaging agent. J. Nucl. Med. 2010, 51, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Jahan, M.; Eriksson, O.; Johnström, P.; Korsgren, O.; Sundin, A.; Johansson, L.; Halldin, C. Decreased defluorination using the novel betacell imaging agent [18F]FE-DTBZ-d4 in pigs examined by PET. EJNMMI Res. 2011, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-S.; Fowler, J.S.; Wolf, A.P. Rapid, regiospecific syntheses of deuterium substituted 6-[18F]fluorodopamine (α,α-D2; β,β-D2 and α,α,β,β-D4) for mechanistic studies with positron emission tomography. J. Labelled Compd. Radiopharm. 1993, 33, 645–654. [Google Scholar] [CrossRef]
- DeWolf, W.E.J., Jr.; Carr, S.A.; Varrichio, A.; Goodhart, P.J.; Mentzer, M.A.; Roberts, G.D.; Southan, C.; Dolle, R.E.; Kurse, L.I. Inactivation of dopamine β-hydroxylase by p-cresol: Isolation and characterization of covalently modified active site peptides. Biochemistry 1988, 27, 9093–9101. [Google Scholar] [CrossRef] [PubMed]
- Coleman, A.A.; Hindsgaul, O.; Palcic, M.M. Stereochemistry of copper amine oxidase reactions. J. Biol. Chem. 1989, 264, 19500–19505. [Google Scholar] [PubMed]
- Yu, P.H. Three types of stereospecificity and the kinetic deuterium isotope effect in the oxidative deamination of dopamine as catalyzed by different amine oxidases. Biochem. Cell Biol. 1988, 66, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.H.; Bailey, B.A.; Durden, D.A.; Boulton, A.A. Stereospecific deuterium substitution at the alpha-carbon position of dopamine and its effect on oxidative deamination catalyzed by MAO-A and MAO-B from different tissues. Biochem. Pharmacol. 1986, 35, 1027–1036. [Google Scholar] [CrossRef]
- Ding, Y.-S.; Fowler, J.S.; Gatley, S.J.; Logan, J.; Volkow, N.D.; Shea, C. Mechanistic Positron Emission Tomography Studies of 6-[18F]Fluorodopamine in Living Baboon Heart: Selective Imaging and Control of Radiotracer Metabolism Using the Deuterium Isotope Effect. J. Neurochem. 1995, 65, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Halldin, C.; Sóvágó, J.; Pike, V.W.; Hall, H.; Gulyás, B.; Mozley, P.D.; Dobson, D.; Shchukin, E.; Innis, R.B.; et al. PET evaluation of novel radiofluorinated reboxetine analogs as norepinephrine transporter probes in the monkey brain. Synapse 2004, 53, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-R.; Maeda, J.; Ito, T.; Okauchi, T.; Ogawa, M.; Noguchi, J.; Suhara, T.; Halldin, C.; Suzuki, K. Synthesis and evaluation of N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethoxy-d2–5-methoxybenzyl)acetamide: A deuterium substituted radioligand for peripheral benzodiazepine receptor. Bioorg. Med. Chem. 2005, 13, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-S.; Ding, Y.-S.; Kim, S.-W.; Kil, K.-E. Synthesis, enantiomeric resolution, F-18 labeling and biodistribution of reboxetine analogs: Promising radioligands for imaging the norepinephrine transporter with positron emission tomography. Nucl. Med. Biol. 2005, 32, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Chin, F.T.; Pike, V.W.; Toyama, H.; Liow, J.-S.; Zoghbi, S.S.; Modell, K.; Briard, E.; Shetty, H.U.; Sinclair, K.; et al. Synthesis and Evaluation of Two 18F-Labeled 6-Iodo-2-(4-N,N-dimethylamino)phenylimidazo[1,2-a]pyridine Derivatives as Prospective Radioligands for β-Amyloid in Alzheimer’s Disease. J. Med. Chem. 2004, 47, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Donohue, S.R.; Krushinski, J.H.; Pike, V.W.; Chernet, E.; Phebus, L.; Chesterfield, A.K.; Felder, C.C.; Halldin, C.; Schaus, J.M. Synthesis, Ex Vivo Evaluation, and Radiolabeling of Potent 1,5-Diphenylpyrrolidin-2-one Cannabinoid Subtype-1 Receptor Ligands as Candidates for in Vivo Imaging. J. Med. Chem. 2008, 51, 5833–5842. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, F.; Piel, M.; Hoehnemann, S.; Roesch, F. Automated synthesis and purification of [18F]fluoro-[di-deutero]methyl tosylate. J. Label. Compd. Radiopharm. 2013, 56, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miura, S.; Hasui, T.; Halldin, C.; Stepanov, V.; Takano, A. Radiolabeled Compounds and Their Use as Radiotracers for Quantitative Imaging of Phosphodiesterase (PDE10A) in Mammals. WO 2013027845 A1, 28 February 2013. [Google Scholar]
- Schieferstein, H.; Piel, M.; Beyerlein, F.; Lueddens, H.; Bausbacher, N.; Buchholz, H.-G.; Ross, T.L.; Roesch, F. Selective binding to monoamine oxidase A: In vitro and in vivo evaluation of 18F-labeled β-carboline derivatives. Bioorg. Med. Chem. 2015, 23, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Rami-Mark, C.; Zhang, M.-R.; Mitterhauser, M.; Lanzenberger, R.; Hacker, M.; Wadsak, W. [18F]FMeNER-D2: Reliable fully-automated synthesis for visualization of the norepinephrine transporter. Nucl. Med. Biol. 2013, 40, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Halldin, C.; Sóvágó, J.; Pike, V.W.; Gulyás, B.; Mozley, P.D.; Johnson, D.P.; Hall, H.; Innis, R.B.; Farde, L. Specific in vivo binding to the norepinephrine transporter demonstrated with the PET radioligand, (S,S)-[11C]MeNER. Nucl. Med. Biol. 2003, 30, 707–714. [Google Scholar] [CrossRef]
- Casebier, D.S.; Robinson, S.P.; Purohit, A.; Radeke, H.S.; Azure, M.T.; Dischino, D.D. Contrast Agents for Myocardial Perfusion Imaging. WO 2005079391 A2, 1 September 2005. [Google Scholar]
- Hortala, L.; Arnaud, J.; Roux, P.; Oustric, D.; Boulu, L.; Oury-Donat, F.; Avenet, P.; Rooney, T.; Alagille, D.; Barret, O.; et al. Synthesis and preliminary evaluation of a new fluorine-18 labelled triazine derivative for PET imaging of cannabinoid CB2 receptor. Bioorg. Med. Chem. Lett. 2014, 24, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Brumby, T.; Graham, K.; Krueger, M. Direct Synthesis of [18F]fluoromethoxy Compounds for PET Imaging and New Precursors for Direct Radiosynthesis of Protected Derivatives of O-([18F]fluoromethyl)tyrosine. WO 2013001088 A1, 3 January 2013. [Google Scholar]
- Graham, K.; Ede, S. Simplified Radiosynthesis of O-[18F]fluoromethyl Tyrosine Derivatives. WO 2013026940 A1, 28 February 2013. [Google Scholar]
- Graham, K.; Zitzmann-Kolbe, S.; Brumby, T. Preparation of Fluorodeuteriomethyl Tyrosine Derivatives. WO 2012025464 A1, 1 March 2012. [Google Scholar]
- Hamill, T.G.; Sato, N.; Jitsuoka, M.; Tokita, S.; Sanabria, S.; Eng, W.; Ryan, C.; Krause, S.; Takenaga, N.; Patel, S.; et al. Inverse agonist histamine H3 receptor PET tracers labeled with carbon-11 or fluorine-18. Synapse 2009, 63, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hong, J.; Morse, C.L.; Pike, V.W. Synthesis, Structure-Affinity Relationships, and Radiolabeling of Selective High-Affinity 5-HT4 Receptor Ligands as Prospective Imaging Probes for Positron Emission Tomography. J. Med. Chem. 2010, 53, 7035–7047. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.H.; Chen, A.M.; Gibson, R.E.; Goulet, M.T.; Hagmann, W.K.; Hamill, T.G.; Jewell, J.P.; Lin, L.S.; Liu, P.; Peresypkin, A.V. Heterocyclic Radiolabeled Cannabinoid-1 Receptor Modulators. WO 2005009479 A1, 3 February 2005. [Google Scholar]
- Cosford, N.D.P.; Govek, S.P.; Hamill, T.G.; Kamenecka, T.; Roppe, J.R.; Seiders, T.J. Alkyne Derivatives as Tracers for Metabotropic Glutamate Receptor Binding. WO 2004038374 A2, 6 May 2004. [Google Scholar]
- Burns, H.D.; Hamill, T.G.; Lindsley, C.W. Radiolabeled Glycine Transporter Inhibitors. WO 2007041025 A2, 12 April 2007. [Google Scholar]
- Hamill, T.G.; McCauley, J.A.; Burns, H.D. The Synthesis of A Benzamidine-containing NR2B-selective NMDA Receptor Ligand Labelled with Tritium or Fluorine-18. J. Label. Compd. Radiopharm. 2005, 48, 1–10. [Google Scholar] [CrossRef]
- Burns, H.D.; sEng, W.-S.; Gibson, R.E.; Hamill, T.G. Radiolabeled Neurokinin-1 Receptor Antagonists. WO 2004029024 A2, 8 April 2004. [Google Scholar]
- Leyton, J.; Smith, G.; Zhao, Y.; Perumal, M.; Nguyen, Q.-D.; Robins, E.; Årstad, E.; Aboagye, E.O. [18F]Fluoromethyl-[1,2-2H4]-Choline: A Novel Radiotracer for Imaging Choline Metabolism in Tumors by Positron Emission Tomography. Cancer Res. 2009, 69, 7721–7728. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Zhao, Y.; Leyton, J.; Shan, B.; de Nguyen, Q.; Perumal, M. Radiosynthesis and pre-clinical evaluation of [18F]fluoro-[1,2-2H4]choline. Nucl. Med. Biol. 2011, 38, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Lehmann, L.; Kettschau, G.; Toth, M.; Heinrich, T.; Thiele, A.; Varrone, A.; Halldin, C. Development of a novel fluorine-18 labeled deuterated fluororasagiline ([18F]fluororasagiline-D2) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg. Med. Chem. 2013, 21, 6634–6641. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raya, S.; Tabakman, R.; Blaugrund, E.; Trembovler, V.; Lazarovici, P. Neuroprotective and neurotoxic effects of monoamine oxidase-B inhibitors and derived metabolites under ischemia in PC12 cells. Eur. J. Pharmacol. 2002, 434, 109–116. [Google Scholar] [CrossRef]
- Nag, S. Development of Novel Fluorine-18 Labeled PET Radioligands for Monoamine Oxidase B (MAO-B). Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2013. [Google Scholar]
- Ramirez de Molina, A.; Gallego-Ortega, D.; Sarmentero-Estrada, J.; Lagares, D.; Bandrés, E.; Gomez del Pulgar, T.; García-Foncillas, J.; Lacal, J.C. Choline kinase as a link connecting phospholipid metabolism and cell cycle regulation: Implications in cancer therapy. Int. J. Biochem. Cell Biol. 2008, 40, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, S.; Imamura, S.; Misaki, H.; Horiuti, Y. Purification and Characterization of Choline oxidase from Arthrobacter globiformis. J. Biochem. 1977, 82, 1741–1749. [Google Scholar] [PubMed]
- Gadda, G. pH and deuterium kinetic isotope effects studies on the oxidation of choline to betaine-aldehyde catalyzed by choline oxidase. Biochim. Biophys. Acta 2003, 1650, 4–9. [Google Scholar] [CrossRef]
- Witney, T.H.; Alam, I.S.; Turton, D.R.; Smith, G.; Carroll, L.; Brickute, D.; Twyman, F.J.; Nguyen, Q.-D.; Tomasi, G.; Awais, R.O.; et al. Evaluation of Deuterated 18F- and 11C-Labeled Choline Analogs for Cancer Detection by Positron Emission Tomography. Clin. Cancer Res. 2012, 18, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Challapalli, A.; Sharma, R.; Hallett, W.A.; Kozlowski, K.; Carroll, L.; Brickute, D.; Twyman, F.; Al-Nahhas, A.; Aboagye, E.O. Biodistribution and Radiation Dosimetry of Deuterium-Substituted 18F-Fluoromethyl-[1, 2–2H4]Choline in Healthy Volunteers. J. Nucl. Med. 2014, 55, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.; Barta, N.; Bikker, J.; Borosky, S.; Campbell, B.; Crawford, T.; Denny, L.; Evans, L.; Gray, D.; Lee, P. Synthesis and pharmacological evaluation of aminopyrimidine series of 5-HT1A partial agonists. Bioorg. Med. Chem. Lett. 2009, 19, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Manoury, P.M.; Binet, J.; Rousseau, J.; Lefevre-Borg, F.; Cavero, I. Synthesis of a series of compounds related to betaxolol, a new β1-adrenoceptor antagonist with a pharmacological and pharmacokinetic profile optimized for the treatment of chronic cardiovascular diseases. J. Med. Chem. 1987, 30, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, B.; Rohde, J.; Wang, J.; Fung, S.; Monzon, K.; Chiou, W.; Pan, L.; Deng, X.; Stolarik, D.A.; Frevert, E.U. Adamantane 11-β-HSD-1 inhibitors: Application of an isocyanide multicomponent reaction. Bioorg. Med. Chem. Lett. 2006, 16, 5958–5962. [Google Scholar] [CrossRef] [PubMed]
- Wrobleski, M.; Reichard, G.; Paliwal, S.; Shah, S.; Tsui, H.; Duffy, R.; Lachowicz, J.; Morgan, C.; Varty, G.; Shih, N. Cyclobutane derivatives as potent NK1 selective antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3859–3863. [Google Scholar] [CrossRef] [PubMed]
- Shoup, T.; Elmaleh, D.; Bonab, A.; Fischman, A. Evaluation of trans-9–18F-fluoro-3,4-methyleneheptadecanoic acid as a PET tracer for myocardial fatty acid imaging. J. Nucl. Med. 2005, 46, 297–304. [Google Scholar] [PubMed]
- Martel, F.; Berlinguet, L. Impairment of tumor growth by unnatural amino acids. Can. J. Biochem. Physiol. 1959, 37, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Connors, T.; Elson, L.; Haddow, A.; Ross, W. The pharmacology and tumour growth inhibitory activity of 1-aminocyclopentane-1-carboxylic acid and related compounds. Biochem. Pharmacol. 1960, 5, 108–129. [Google Scholar] [CrossRef]
- Wooten, D.W.; Moraino, J.D.; Hillmer, A.T.; Engle, J.W.; DeJesus, O.J.; Murali, D.; Barnhart, T.E.; Nickles, R.J.; Davidson, R.J.; Schneider, M.L.; et al. In Vivo Kinetics of [F-18]MEFWAY: A comparison with [C-11]WAY100635 and [F-18]MPPF in the nonhuman primate. Synapse 2011, 65, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Williams, L.; Camp, V.; Malveaux, E.; Olson, J.; Goodman, M. Stereoselective synthesis and biological evaluation of syn-1-amino-3-[18F]fluorocyclobutyl-1-carboxylic acid as a potential positron emission tomography brain tumor imaging agent. Bioorg. Med. Chem. 2009, 17, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, T.; Oku, T.; Uehara, H.; Desai, R.; Beattie, B.; Tjuvajev, J.; Blasberg, R. Facilitated amino acid transport is upregulated in brain tumors. J. Celeb. Blood Flow. Metab. 1998, 18, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Franck, D.; Kniess, T.; Steinbach, J.; Zitzmann-Kolbe, S.; Friebe, M.; Dinkelborg, L.M.; Graham, K. Investigations into the synthesis, radiofluorination and conjugation of a new [18F]fluorocyclobutyl prosthetic group and its in vitro stability using a tyrosine model system. Bioorg. Med. Chem. 2013, 21, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Franck, D. Radiofluorinated Cyclobutyl Group for Increased Metabolic Stability Using Tyrosine Derivatives as Model System. Ph.D. Thesis, TU Dresden, Dresden, Germany, 2012. [Google Scholar]
- Tipre, D.N.; Zoghbi, S.S.; Liow, J.S.; Green, M.V.; Seidel, J.; Ichise, M.; Innis, R.B.; Pike, V.W. PET Imaging of Brain 5-HT1A Receptors in Rat in Vivo with 18F-FCWAY and Improvement by Successful Inhibition of Radioligand Defluorination with Miconazole. J. Nucl. Med. 2006, 47, 345–353. [Google Scholar] [PubMed]
- Holleman, A.; Wiberg, N.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 102nd ed.; De Gruyter Verlag: Berlin, Germany, 2008; pp. 1097–1103. [Google Scholar]
- Rosenthal, M.S.; Bosch, A.L.; Nickles, R.J.; Gatley, S.J. Synthesis and some characteristics of no-carrier added [18F]fluorotrimethylsilane. Int. J. Appl. Radiat. Isot. 1985, 36, 318–319. [Google Scholar] [CrossRef]
- Gatley, S.J. Rapid production and trapping of [F-18]fluorotrimethylsilane, and its use in nucleophilic F-18 labeling without an aqueous evaporation step. Appl. Radiat. Isot. 1989, 40, 541–544. [Google Scholar] [CrossRef]
- Mulholland, G.K. Recovery and purification of no-carrier-added [18F]fluoride with bistrimethylsilylsulfate (BTMSS). Int. J. Radiat. Appl. Instr. 1991, 42, 1003–1008. [Google Scholar] [CrossRef]
- Walsh, J.C.; Fleming, L.M.; Satyamurthy, N.; Barrio, J.R.; Phelps, M.E.; Gambhir, S.S.; Toyokuni, T. Application of silicon-fluoride chemistry for the development of amine-reactive F-18-labeling agents for biomolecules. J. Nucl. Med. 2000, 41, 249. [Google Scholar]
- Choudhry, U.; Martin, K.E.; Biagini, S.; Blower, P.J. Alkoxysilane groups for instant labeling of biomolecules with 18F. Nucl. Med. Commun. 2006, 27, 293. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; et al. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, R.; Kostikov, A.; Wängler, C.; Jurkschat, K.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, B. Silicon Fluoride Acceptors (SIFAs) for Peptide and Protein Labeling with 18F. In Radiochemical Syntheses, 1st ed.; Scott, P.J.H., Ed.; J. Wiley & Sons: Hoboken, NJ, USA, 2015; Volume 2, pp. 149–162. [Google Scholar]
- Fleischer, H. Molecular “Floppyness” and the Lewis Acidity of Silanes: A Density Functional Theory Study. Eur. J. Inorg. Chem. 2001, 393–404. [Google Scholar] [CrossRef]
- Höhne, A.; Yu, L.; Mu, L.; Reiher, M.; Voigtmann, U.; Klar, U.; Graham, K.; Schubiger, P.A.; Ametamey, S.M. Organofluorosilanes as model compounds for 18F-labeled silicon-based PET tracers and their hydrolytic stability: Experimental data and theoretical calculations (PET = Positron Emission Tomography). Chemistry 2009, 15, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Bickelhaupt, F.M. Nucleophilic Substitution at Silicon (SN2@Si) via a Central Reaction Barrier. J. Org. Chem. 2007, 72, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Wängler, C.; Schirrmacher, E.; Kostikov, A.; Jurkschat, K.; Wängler, B.; Schirrmacher, R. 18F-Labeled Silicon-Based Fluoride Acceptors: Potential Opportunities for Novel Positron Emitting Radiopharmaceuticals. BioMed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Neto, P.; Wängler, B.; Iovkova, L.; Boening, G.; Reader, A.; Jurkschat, K.; Schirrmacher, E. [18F]SiFA-isothiocyanate: A new highly effective radioactive labeling agent for lysine-containing proteins. ChemBioChem 2009, 10, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Wängler, C.; Niedermoser, S.; Chin, J.; Orchowski, K.; Schirrmacher, E.; Jurkschat, K.; Kostikov, A.P.; Iovkova-Berends, L.; Schirrmacher, R.; Wängler, B. One-step 18F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat. Prot. 2012, 7, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.; Michler, C.; Leidner, S.; Rensch, C.; Wängler, C.; Schirrmacher, R.; Bartenstein, P.; Wängler, B. Synthesis and in Vitro and in Vivo Evaluation of SiFA-Tagged Bombesin and RGD Peptides as Tumor Imaging Probes for Positron Emission Tomography. Bioconjugate Chem. 2014, 25, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Balentova, E.; Collet, C.; Lamandé-Langle, S.; Chrétien, F.; Thonon, D.; Aerts, J.; Lemaire, C.; Luxen, A.; Chapleuret, Y. Synthesis and hydrolytic stability of novel 3-[18F]fluoroethoxybis(1-methylethyl)silyl]propanamine-based prosthetic groups. J. Fluor. Chem. 2011, 132, 250–257. [Google Scholar] [CrossRef]
- Ting, R.; Adam, M.J.; Ruth, T.J.; Perrin, D.M. Arylfluoroborates and alkylfluorosilicates as potential PET imaging agents: High-yielding aqueous biomolecular18F-labeling. J. Am. Chem. Soc. 2005, 127, 13094–13095. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Höhne, A.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Cyr, J.E.; Dinkelborg, L.; Stellfeld, T.; Srinivasan, A.; Voigtmann, U.; et al. Silicon-Based Building Blocks for One-Step 18F-Radiolabeling of Peptides for PET Imaging. Angew. Chem. Int. Ed. 2008, 47, 4922–4925. [Google Scholar] [CrossRef] [PubMed]
- Bohn, P.; Deyine, A.; Azzouz, R.; Bailly, L.; Fiol-Petit, C.; Bischoff, L.; Fruit, C.; Marsais, F.; Vera, P. Design of siliconbased misonidazole analogues and 18F-radiolabelling. Nucl. Med. Biol. 2009, 36, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Briard, E.; Zoghbi, S.S.; Siméon, F.G.; Imaizumi, M.; Gourley, J.P.; Shetty, H.U.; Lu, S.; Fujita, M.; Innis, R.B.; Pike, V.W. Single-Step High-Yield Radiosynthesis and Evaluation of a Sensitive 18F-Labeled Ligand for Imaging Brain Peripheral Benzodiazepine Receptors with PET. J. Med. Chem. 2009, 52, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Mayer, J.M. Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2003. [Google Scholar]
- Fukami, T.; Yokoi, T. The Emerging Role of Human Esterases. Drug Metabol. Pharmacokin. 2012, 27, 466–477. [Google Scholar] [CrossRef]
- Li, Z.; Lang, L.; Ma, Y.; Kiesewetter, D.O. [18F]Fluoropropylsulfonyl chloride: A new reagent for radiolabeling primary and secondary amines for PET imaging. J. Label. Compd. Radiopharm. 2008, 51, 23–27. [Google Scholar] [CrossRef]
- Löser, R.; Fischer, S.; Hiller, A.; Köckerling, M.; Funke, U.; Maisonial, A.; Brust, P.; Steinbach, J. Use of 3-[18F]fluoropropanesulfonyl chloride as a prosthetic agent for the radiolabelling of amines: Investigation of precursor molecules, labelling conditions and enzymatic stability of the corresponding sulfonamides. Beilstein J. Org. Chem. 2013, 9, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Sala, R.; Carroll, L.; Behan, K.; Glaser, M.; Robins, E.; Nguyen, Q.-D.; Aboagye, E.O. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl. Med. Biol. 2012, 39, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent Trends in Bioorthogonal Click-Radiolabeling Reactions Using Fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, J.; Hayashi, A.; Gogami, A.; Hamada, M.; Hamashima, Y.; Katoh, T.; Node, M.; Fujibayashi, Y. Alkyl- fluorinated thymidine derivatives for imaging cell proliferation I. The in vitro evaluation of some alkyl-fluorinated thymidine derivatives. Nucl. Med. Biol. 2006, 33, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.; Soghomonyan, S.; Yeh, H.H.; Flores, L.G.; Shavrin, A.; Volgin, A.Y.; Gelovani, J.G.; Alauddin, M.M. Synthesis and preliminary PET imaging of N3-[18F]fluoroethyl thymidine and N3-[18F]fluoropropyl thymidine. Nucl. Med. Biol. 2008, 35, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I. (Ed.) Wiley-Blackwell: Sussex, UK, 2009.
- Lee, I.; Choe, Y.S.; Choi, J.Y.; Lee, K.-H.; Kim, B.-T. Synthesis and Evaluation of 18F-Labeled Styryltriazole and Resveratrol Derivatives for β-Amyloid Plaque Imaging. J. Med. Chem. 2012, 55, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Lien, V.T.; Riss, P.J. Radiosynthesis of [18F]Trifluoroalkyl Groups: Scope and Limitations. BioMed. Res. Int. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Ido, T.; Irie, T.; Kasida, Y. Isotope exchange with 18F on superconjugate system. J. Label. Compd. Radiopharm. 1979, 16, 153–154. [Google Scholar]
- Satter, M.R.; Martin, C.C.; Oakes, T.R.; Christian, B.; Nickles, R.J. Synthesis of the fluorine-18 labeled inhalation Anesthetics. Appl. Radiat. Isot. 1994, 45, 1093–1100. [Google Scholar] [CrossRef]
- Suehiro, M.; Yang, G.; Torchon, G.; Ackerstaff, E.; Humm, J.; Koutcher, J.; Ouerfelliet, O. Radiosynthesis of the tumor hypoxia marker [18F]TFMISO via O-[18F]trifluoroethylation reveals a striking difference between trifluoroethyl tosylate and iodide in regiochemical reactivity toward oxygen nucleophiles. Bioorg. Med. Chem. 2011, 19, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Speranza, M.; Shiue, C.-Y.; Wolf, A.P. H18F + Sb2O3: A new selective radiofluorinating agent. Chem. Commun. 1986, 12, 924–925. [Google Scholar] [CrossRef]
- Angelini, G.; Speranza, M.; Wolf, A.P.; Shiue, C.-Y. Synthesis of N-(α,α,α-tri[18F]fluoro-m-tolyl)piperazine. A potent serotonin agonist. J. Label. Compd. Radiopharm. 1990, 28, 1441–1448. [Google Scholar] [CrossRef]
- Kilbourn, M.R.; Pavia, M.R.; Gregor, V.E. Synthesis of fluorine-18 labeled GABA uptake inhibitors. Appl. Radiat. Isot. 1990, 41, 823–828. [Google Scholar] [CrossRef]
- Das, M.K.; Mukherjee, J. Radiosynthesis of [F-18]fluoxetine as a potential radiotracer for serotonin reuptake sites. Appl. Radiat. Isot. 1993, 44, 835–842. [Google Scholar] [CrossRef]
- Johnstrom, P.; Stone-Elander, S. The 18F-labelled alkylating agent 2,2,2-trifluoroethyl triflate: Synthesis and specific activity. J. Label. Compd. Radiopharm. 1995, 36, 537–547. [Google Scholar] [CrossRef]
- Riss, P.J.; Aigbirhio, F.I. A simple, rapid procedure for nucleophilic radiosynthesis of aliphatic [18F]trifluoromethyl groups. Chem. Commun. 2011, 47, 11873–11875. [Google Scholar] [CrossRef] [PubMed]
- Dolbier, W.R., Jr.; Li, A.-R.; Koch, C.J.; Shiue, C.-Y.; Kachur, A.V. [18F]-EF5, a marker for PET detection of hypoxia: Synthesis of precursor and a new fluorination procedure. Appl. Radiat. Isot. 2001, 54, 73–80. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Alauddin, M.M.; Hu, J.; Conti, P.S.; Olah, G.A. Expedient synthesis of [18F]-labeled α-trifluoromethyl ketones. J. Label. Compd. Radiopharm. 2003, 46, 1087–1092. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of [18F]-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorg. Med. Chem. 2007, 15, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Kaufmann, D.E. Organometallics: Boron Compounds. In Science of Synthesis, Houben-Weyl Methods of Molecular Transformations; Kaufmann, D.E., Matteson, D.S., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2004; Volume 6, pp. 247–256. [Google Scholar]
- Hartman, J.S.; Miller, J.M. Adducts of Mixed Trihalides of Boron. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Academic Press: New York, NY, USA, 1978; Volume 21, pp. 147–177. [Google Scholar]
- Wade, C.R.; Broomsgrove, E.J.; Aldridge, S.; Gabbaï, F.P. Fluoride Ion Complexation and Sensing Using Organoboron Compounds. Chem. Rev. 2010, 110, 3958–3984. [Google Scholar] [CrossRef] [PubMed]
- Treibs, A.; Kreuzer, F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Schmitt, A.; Hinkeldey, B.; Wild, M.; Jung, G. Synthesis of the Core Compound of the BODIPY Dye Class: 4,4-Difluoro-4-bora-(3a,4a)-diaza-s-indacene. J. Fluoresc. 2009, 19, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Tram, K.; Yan, H.; Jenkins, H.A.; Vassiliev, S.; Bruce, D. The synthesis and crystal structure of unsubstituted 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY). Dyes Pigments 2009, 82, 392–395. [Google Scholar] [CrossRef]
- Arroyo, I.J.; Hu, R.; Merino, G.; Tang, B.Z.; Pena-Cabrera, E. The smallest and one of the brightest. Efficient preparation and optical description of the parent borondipyrromethene system. J. Org. Chem. 2009, 74, 5719–5722. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, T.-P.; Liu, S.; Huang, C.-W.; Hudnall, T.W.; Gabbaï, F.P.; Conti, P.S. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem. Commun. 2011, 47, 9324–9326. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.A.; Keliher, E.J.; Wan, D.; Hilderbrand, S.A.; Weissleder, R.; Mazitschek, R. Synthesis of [18F]BODIPY: Bifunctional Reporter for Hybrid Optical/Positron Emission Tomography Imaging. Angew. Chem. 2012, 124, 4681–4684. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuchar, M.; Mamat, C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules 2015, 20, 16186-16220. https://doi.org/10.3390/molecules200916186
AMA Style
Kuchar M, Mamat C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules. 2015; 20(9):16186-16220. https://doi.org/10.3390/molecules200916186
Chicago/Turabian StyleKuchar, Manuela, and Constantin Mamat. 2015. "Methods to Increase the Metabolic Stability of 18F-Radiotracers" Molecules 20, no. 9: 16186-16220. https://doi.org/10.3390/molecules200916186