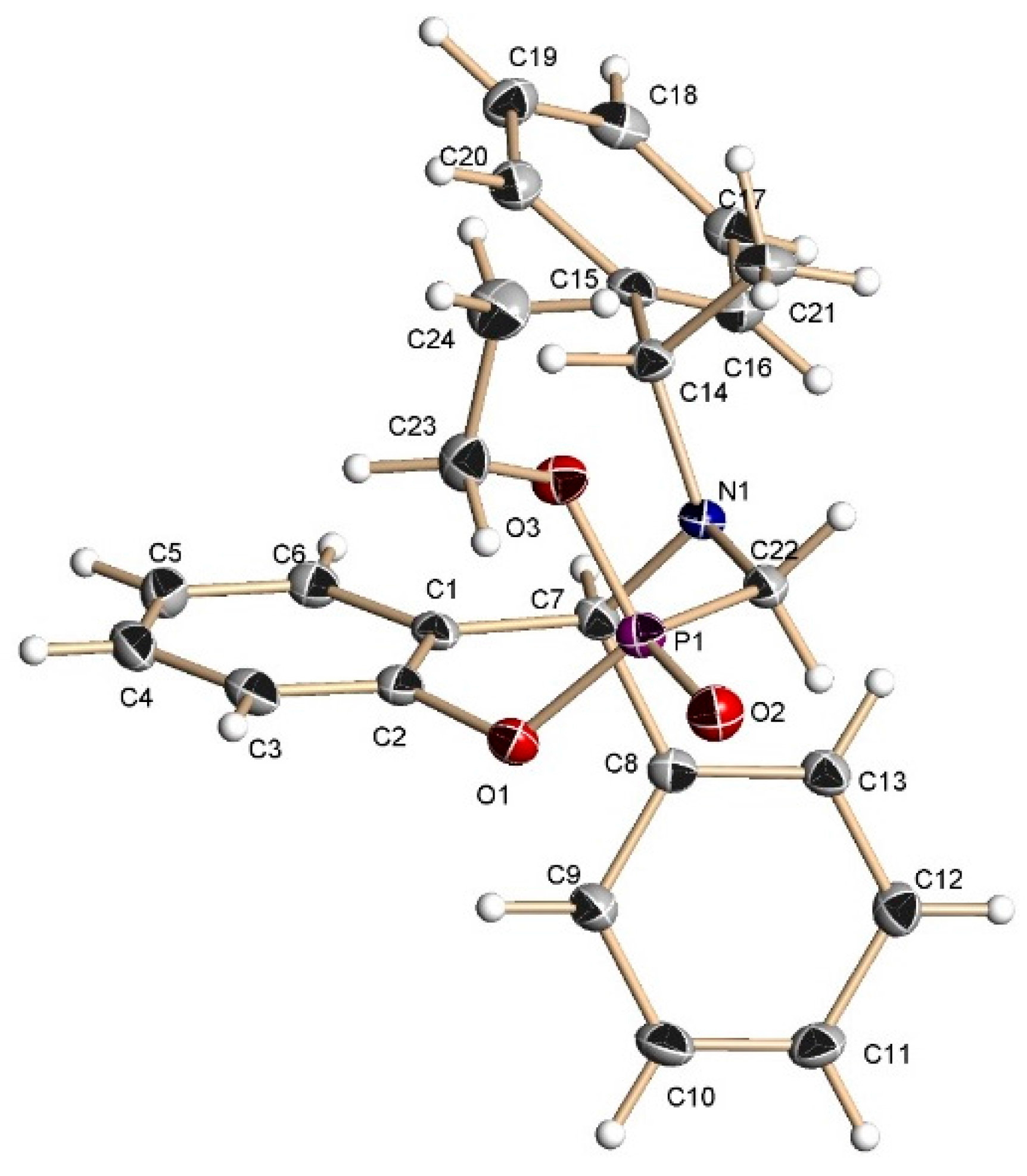
3.2. Preparation of Aminophenols
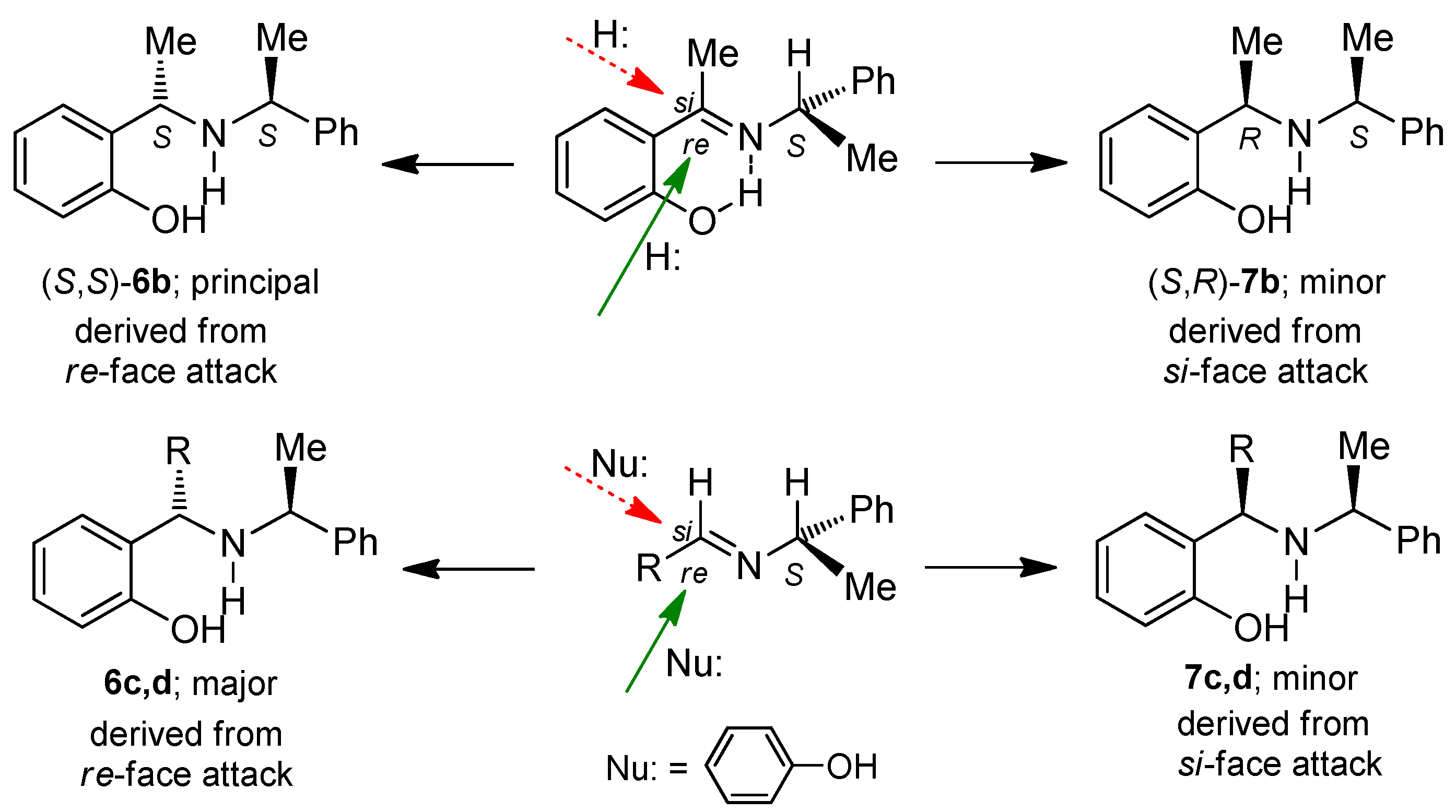
3.2.1. Preparation of 2-{(1S)-1-{[(1S)-1-Phenyethyl]amino}ethyl}phenol (6b)
A mixture of 2-hydroxyacetophenone 1.0 g, 1.37 mL (7.3 mmol), (
S)-α-methylbenzylamine 0.89 g, 0.93 mL (7.3 mmol) and toluene (25 mL), was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure; the crude product was dissolved in methanol (21 mL) and treated with cerium trichloride heptahydrate 1.36 g (3.7 mmol). The solution was cooled at −78 °C, and sodium borohydride 0.55 g (1.5 mmol) was added. The reaction mixture was allowed the room temperature and stirred for 16 h. The solvent was removed under vacuum, the crude product was dissolved in dichloromethane (175 mL), treated with a saturated solution of ammonium chloride (35 mL), and extracted with dichloromethane (3 × 30 mL). The organic layers were dried over anhydrous Na
2SO
4, filtered and evaporated under reduced pressure, obtaining (1.72 g, 97%) as a mixture of two diastereoisomers (90:10 d.r.). The mixture was dissolved in ethyl ether (50 mL), washed with 1.5 M hydrochloric acid (5 mL), 1.0 M sodium hydroxide (7 mL), and extracted with ethyl acetate (3 × 30 mL). The organic phase was dried over anhydrous Na
2SO
4, filtered and evaporated under reduced pressure, to give the (
S,
S)-diastereoisomer (
6b) [
21] (1.55 g, 88%) as a colorless oil. [α]
D = −70.9° (
c = 0.0127, CHCl
3).
1H-NMR (CDCl
3, 200 MHz): δ 1.34 (d,
J = 7.0 Hz, 3H), 1.40 (d,
J = 6.7 Hz, 3H), 3.64 (q,
J = 6.7 Hz, 1H), 3.70 (q,
J = 6.7 Hz, 1H), 6.75–7.38 (m, 9H).
13C-NMR (CDCl
3, 50 MHz): δ 23.0, 23.6, 55.5, 56.3, 116.9, 119.3, 126.4, 127.6, 128.4, 128.8, 143.5, 157.6.
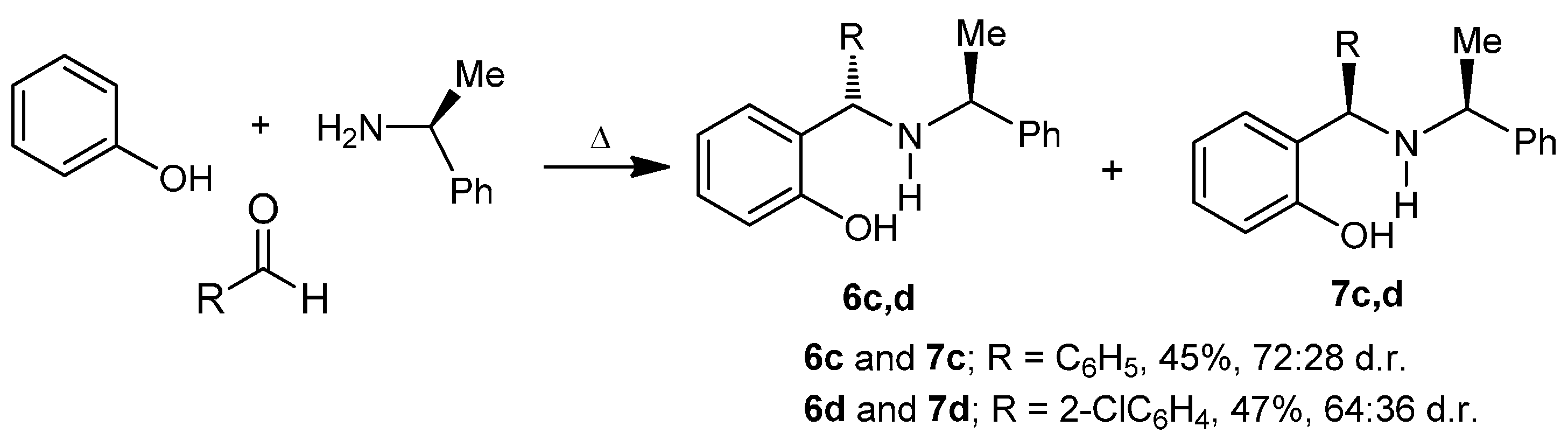
3.2.2. Preparation of Aminophenols (6c) and (7c)
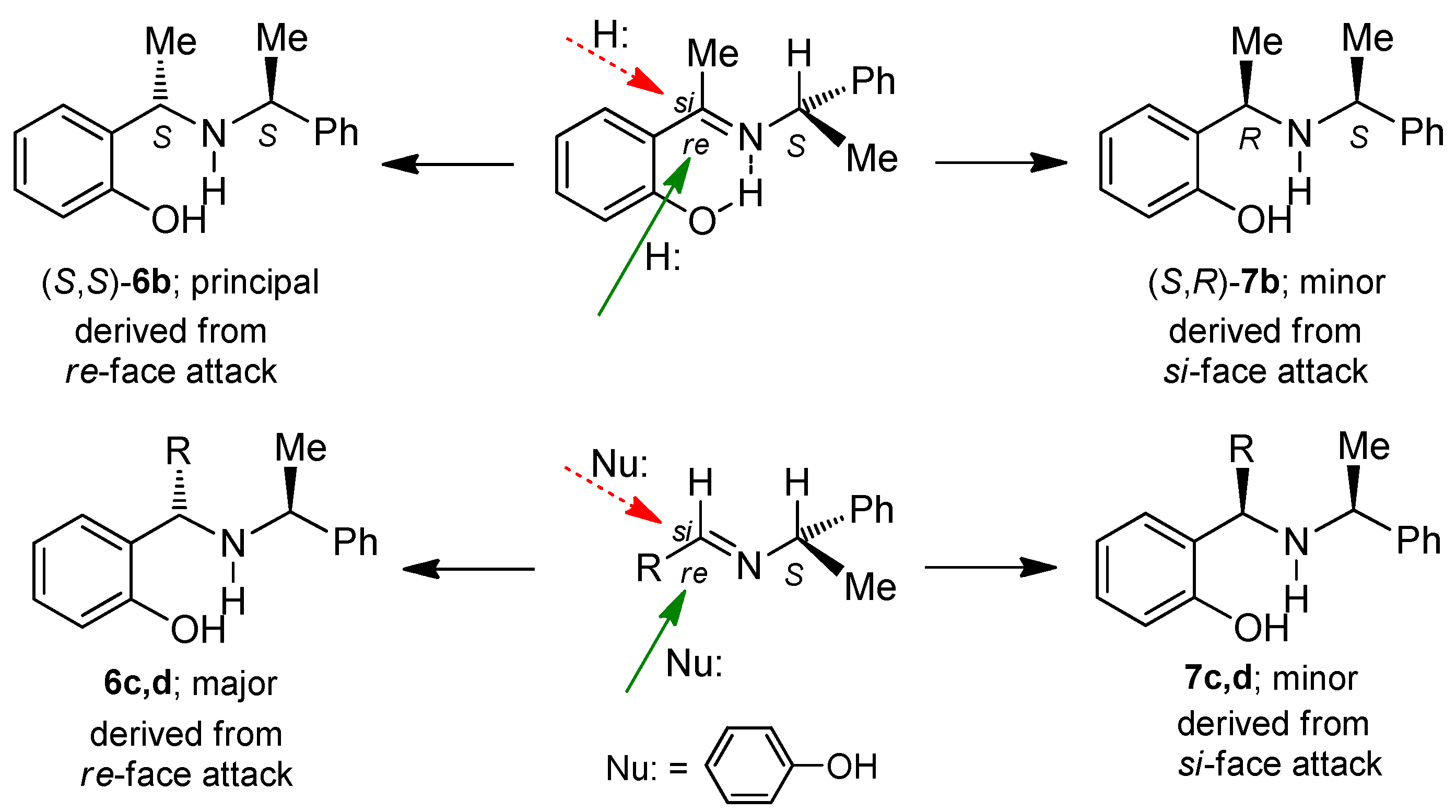
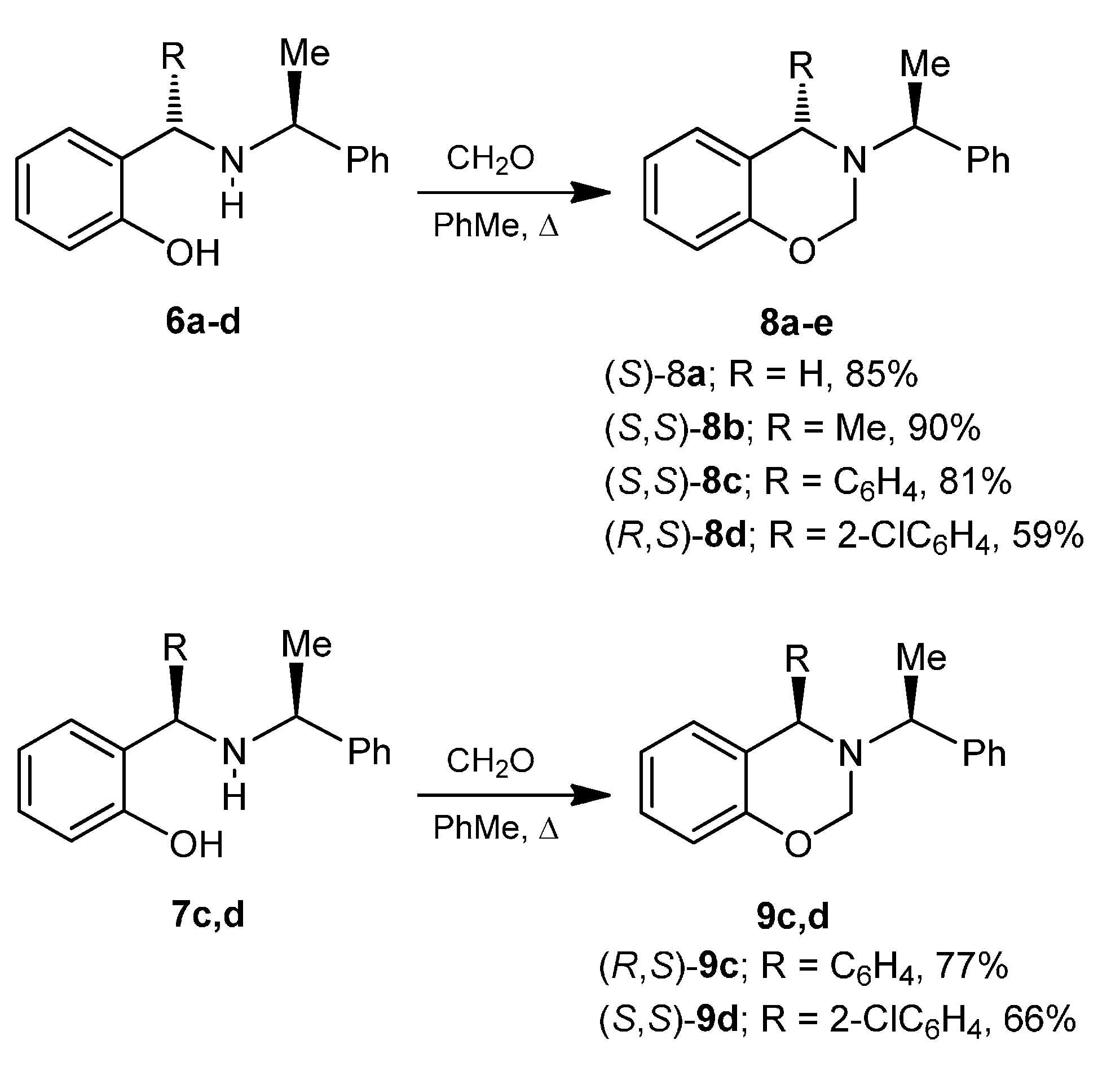
A mixture of benzaldehyde 3.0 g, 2.8 mL (28.3 mmol), phenol 3.2 g (33.9 mmol) and (S)-α-methylbenzylamine 3.4 g, 3.6 mL (28.3 mmol), was heated at 60–70 °C for 48 h. The reaction mixture was percolated on a column chromatography, eluting with hexane:EtOAc (98:2), obtaining (6c) and (7c) (3.85 g, 45%) as a diastereoisomeric mixture 72:28 d.r., which was separated by column chromatography eluting with hexane:EtOAc (99:1), to give both aminophenols (6c) as viscous oil (2.8 g, 32%) and (7c) as viscous oil (1.1 g, 13%).
2-[(S)-Phenyl-{[(1S)-1-phenylethyl]amino}methyl]phenol (6c). [α]D = +132.8° (c = 0.0179, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.47 (d, J = 6.8 Hz, 3H), 3.85 (q, J = 6.8 Hz, 1H), 4.70 (s, 1H), 6.77–7.45 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 23.0, 56.3, 64.9, 117.3, 119.7, 126.8, 127.7, 128.1, 129.1, 129.6, 142.1, 142.4, 157.8.
2-[(R)-Phenyl-{[(1S)-1-phenylethyl]amino}methyl]phenol (7c). [α]D = −102.1° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.42 (d, J = 6.6 Hz, 3H), 3.71 (q, J = 6.6 Hz, 1H), 4.83 (s, 1H), 6.46–7.41 (m, 14 H). 13C-NMR (CDCl3, 50 MHz): δ 23.2, 55.2, 63.2, 117.0, 119.1, 125.9, 127.3, 128.0, 128.1, 128.3, 129.0, 129.1, 129.2, 140.2, 142.3, 157.8.
3.2.3. Preparation of Aminophenols (6d) and (7d)
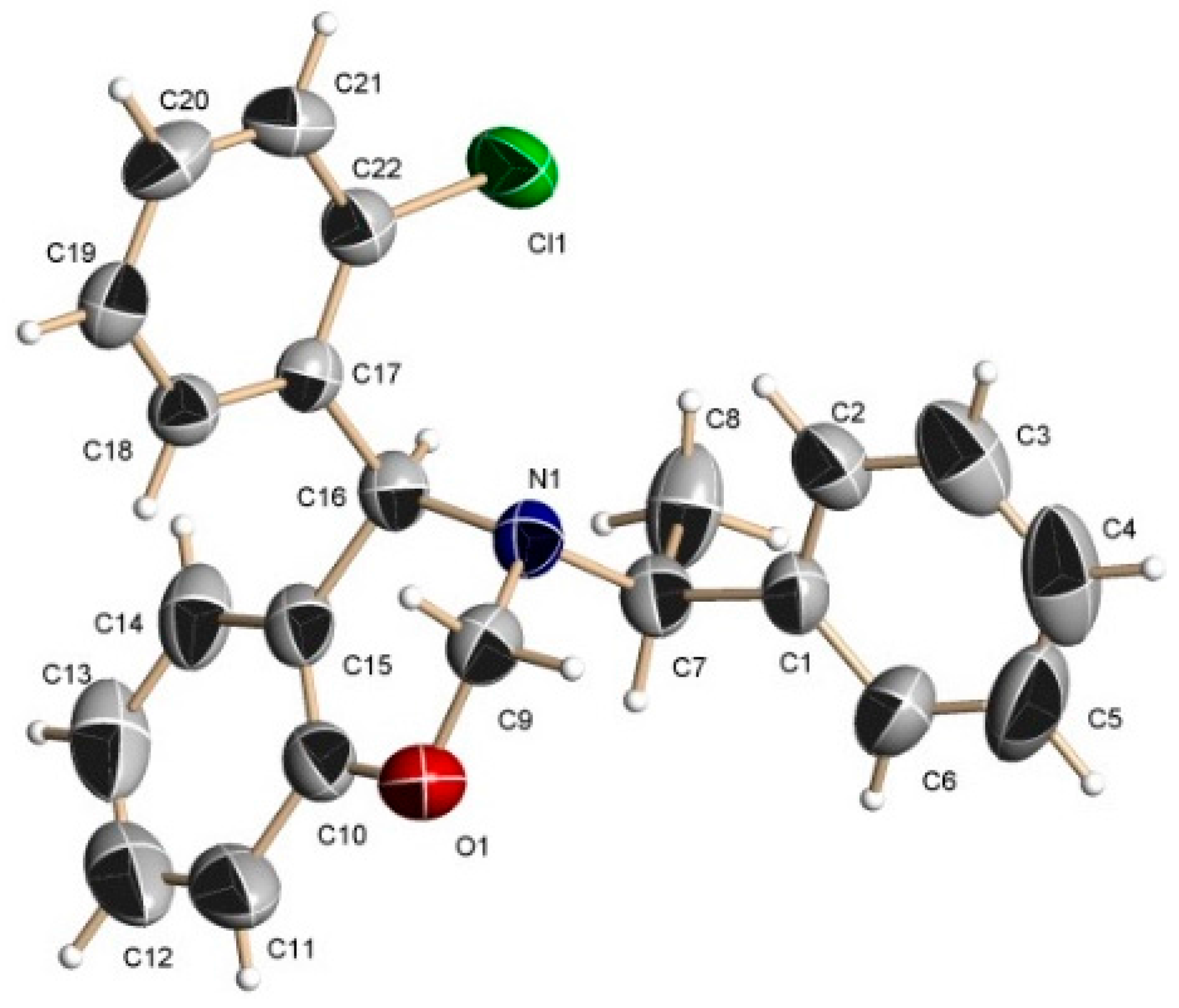
A mixture of phenol 3.0 g (31.9 mmol), o-chlorobenzaldehyde 4.48 g (31.9 mmol) and (S)-α-methylbenzylamine 3.86 g, 4.05 mL (31.9 mmol), was heated at 60–70 °C for 24 h. After this time, the reaction mixture was purified by column chromatography eluting with hexane:EtOAc (98:2), to give (3.4 g, 47%) as a diastereoisomeric mixture 64:36 d.r., which was separated by column chromatography using a mixture of hexane:EtOAc (99:1) as eluent, obtaining the aminophenols (6d) as viscous oil (2.2 g, 30%) and (7d) as viscous oil (1.2 g, 17%).
2-[(R)-2-Chlorophenyl-{[(1S)-1-phenylethyl]amino}methyl]phenol (6d). [α]D = +125.95° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.49 (d, J = 6.8 Hz, 3H), 3.85 (q, J = 6.7 Hz, 1H), 5.23 (s, 1H), 6.73–7.41 (m, 14H). 13C-NMR (CDCl3, 100 MHz): δ 22.2, 56.6, 61.3, 117.4, 119.8, 123.5, 127.2, 127.7, 128.1, 128.8, 129.3, 129.4, 130.0, 130.3, 131.1, 133.4, 137.0, 138.5, 142.5, 158.6. HRMS (CI+): m/z calculated for C21H20ClNO [M + H] 337.1233; found for [M + H]+, m/z 338.1325.
2-[(S)-2-Chlorophenyl-{[(1S)-1-phenylethyl]amino}methyl]phenol (7d). [α]D = −63.81° (c = 0.0118, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.46 (d, J = 6.6 Hz, 3H), 3.75 (q, J = 6.6 Hz, 1H), 5.34 (s, 1H), 6.48–7.46 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 22.8, 55.8, 61.0, 117.1, 119.3, 124.1, 127.3, 127.7, 128.0, 128.6, 129.0, 129.2, 129.4, 130.6, 130.8, 134.1, 137.1, 142.4, 158.0. HRMS (CI+): m/z calculated for C21H20ClNO [M + H] 337.1233; found for [M + H]+, m/z 338.1325.
3.3. Preparation of 1,3-Benzoxazines
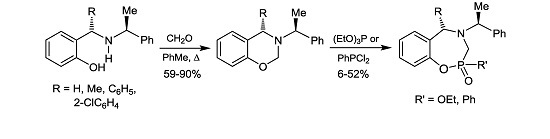
3.3.1. (4S)-4-Methyl-3-[(1ʹS)-1-phenylethyl]-3,4-dihydro-2H-1,3-benzoxazine (8b)
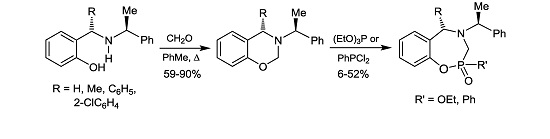
A mixture of (6b) 1.5 g (5.9 mmol), formaldehyde 0.23 g, 0.6 mL (7.7 mmol) and dichloromethane (25 mL), was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography on neutral alumina, using hexane as eluent, obtaining the compound (8b) (1.4 g, 90%) as viscous oil. [α]D = +39.18° (c = 0.012, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.36 (d, J = 7.0 Hz, 3H), 1.44 (d, J = 6.6 Hz, 3H), 3.60 (q, J = 6.6 Hz, 1H), 3.88 (q, J = 6.6 Hz, 1H), 5.00 (AB system, J = 11.0 Hz, 1H), 5.15 (AB system, J = 11.0 Hz, 1H), 6.78–7.32 (m, 9H). 13C-NMR (CDCl3, 50 MHz): δ 22.6, 24.5, 52.4, 59.3, 74.5, 116.7, 120.6, 125.7, 127.3, 127.5, 128.7, 128.8, 145.9, 154.6. HRMS (CI+): m/z calculated for C17H19NO [M + H] 253.1467; found for [M + H]+, m/z 254.1474.
3.3.2. (4S)-4-Phenyl-3-[(1ʹS)-1-phenylethyl]-3,4-dihydro-2H-1,3-benzoxazine (8c)
A mixture of (6c) 0.5 g (1.6 mmol), formaldehyde 60 mg, 0.16 mL (2.1 mmol) and dichloromethane (15 mL), was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure and the crude product was purified by recrystallization from cold methanol, to give the compound (8c) (420 mg, 81%) as a white solid, mp = 98–100 °C. [α]D = +37.3° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.51 (d, J = 6.6 Hz, 3H), 3.96 (q, J = 6.6 Hz, 1H), 4.70 (s, 1H), 4.80 (AB system, J = 11.0 Hz, 1H), 5.05 (ABX system, J = 10.8, 2.0 Hz, 1H), 6.77–7.45 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 21.6, 59.0, 59.4, 74.5, 116.6, 120.3, 120.4, 127.2, 127.6, 127.9, 128.1, 128.3, 128.5, 128.8, 129.0, 130.4, 143.8, 145.3, 154.6. HRMS (CI+): m/z calculated for C22H21NO [M + H] 315.1623; found for [M + H]+, m/z 316.1694.
3.3.3. (4R)-4-(2-Chlorophenyl)-3-[(1ʹS)-1-phenylethyl]-3,4-dihydro-2H-1,3-benzoxazine (8d)
A mixture of (6d) 1.7 g (4.9 mmol), formaldehyde 180 mg, 0.5 mL, (6.3 mmol) and dichloromethane (25 mL), was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of hexane:EtOAc (99:1) as eluent, obtaining the compound (8d) (1.2 g, 59%) as a colorless highly viscous liquid. [α]D = +66.65° (c = 0.0108, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.53 (d, J = 6.6 Hz, 3H), 4.34 (q, J = 6.7 Hz, 1H), 4.71 (AB system, J = 11.0 Hz, 1H), 4.78 (AB system, J = 11.0 Hz, 1H), 5.30 (s, 1H), 6.77–7.40 (m, 14 H). 13C-NMR (CDCl3, 50 MHz): δ 18.4, 58.2, 60.3, 74.4, 116.8, 120.6, 122.0, 126.3, 127.5, 127.8, 128.2, 128.4, 128.6, 128.8, 128.9, 129.2, 130.0, 132.0, 134.6, 141.3, 142.6, 155.4. HRMS (CI+): m/z calculated for C22H20ClNO [M + H] 349.1233; found for [M + H]+, m/z 350.1321.
3.3.4. (4R)-4-Phenyl-3-[(1ʹS)-1-phenylethyl]-3,4-dihydro-2H-1,3-benzoxazine (9c)
A mixture of (7c) 0.5 g (1.6 mmol), formaldehyde 60 mg, 0.16 mL, (2.1 mmol) in dichloromethane (15 mL), was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure and the crude product was purified by recrystallization from cold methanol obtaining (9c) (0.42 g, 77%) as a white solid, mp = 79–81 °C. [α]D = −68.3° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.57 (d, J = 6.0 Hz, 3H), 4.11 (q, J = 6.4 Hz, 1H), 4.37 (ABX system, J = 10.4, 2.4 Hz, 1H), 4.57 (AB system, J = 10.8 Hz, 1H), 5.24 (s, 1H), 6.88–7.43 (m, 14H). 13C-NMR (CDCl3, 100 MHz): δ 21.5, 57.1, 57.9, 76.1, 116.7, 119.9, 120.4, 127.0, 127.3, 127.7, 128.0, 128.3, 128.4, 128.9, 129.7, 143.7, 144.0, 154.8.
3.3.5. (4S)-4-(2-Chlorophenyl)-3-[(1ʹS)-1-phenylethyl]-3,4-dihydro-2H-1,3-benzoxazine (9d)
A mixture of (7d) 0.63 g (1.8 mmol), formaldehyde 70 mg, 0.19 mL, (2.3 mmol) and dichloromethane (25 mL) was heated for 1 h under azeotropic removal of water. The solvent was evaporated under reduced pressure and crude was purified by column chromatography using hexane:EtOAc (99:1) as eluent, obtaining the compound (9d) (340 mg, 66%) as a white solid, mp = 100–104 °C. [α]D = −118.26° (c = 0.0108, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.63 (d, J = 6.6 Hz, 3H), 4.10 (q, J = 6.7 Hz, 1H), 4.34 (ABX system, J = 10.6, 1.4 Hz, 1H), 4.64 (AB system, J = 10.8 Hz) 5.66 (s, 1H), 6.80–7.47 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 21.3, 56.1, 58.7, 75.6, 116.6, 120.0, 120.5, 126.3, 127.4, 127.7, 128.5, 128.8, 129.2, 130.3, 132.5, 134.6, 140.9, 144.5, 154.9. HRMS (CI+): m/z calculated for C22H20ClNO [M + H] 349.1233; found for [M + H]+, m/z 350.1349.
3.5. General Procedure for the Preparation of 1,4,2-Oxazaphosphepines (12), (13) and (14)
Under anhydrous conditions, the corresponding benzoxazine in dry dichloromethane was treated with boron trifluoride etherate and triethyl phosphite. The reaction mixture was stirred at room temperature for 72 h. The solvent was evaporated under reduced pressure, and the crude was dissolved in ethyl acetate, and treated with a saturated solution of ammonium chloride and stirred for 15 min. The organic phase was extracted with ethyl acetate, and the organic extracts were dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was purified by column chromatography.
3.5.1. (S)-2,2,2-Triethoxy-4-(1-phenylethyl)-2,3,4,5-tetrahydro-1,4,2λ5-benzoxazaphosphepine (12a)
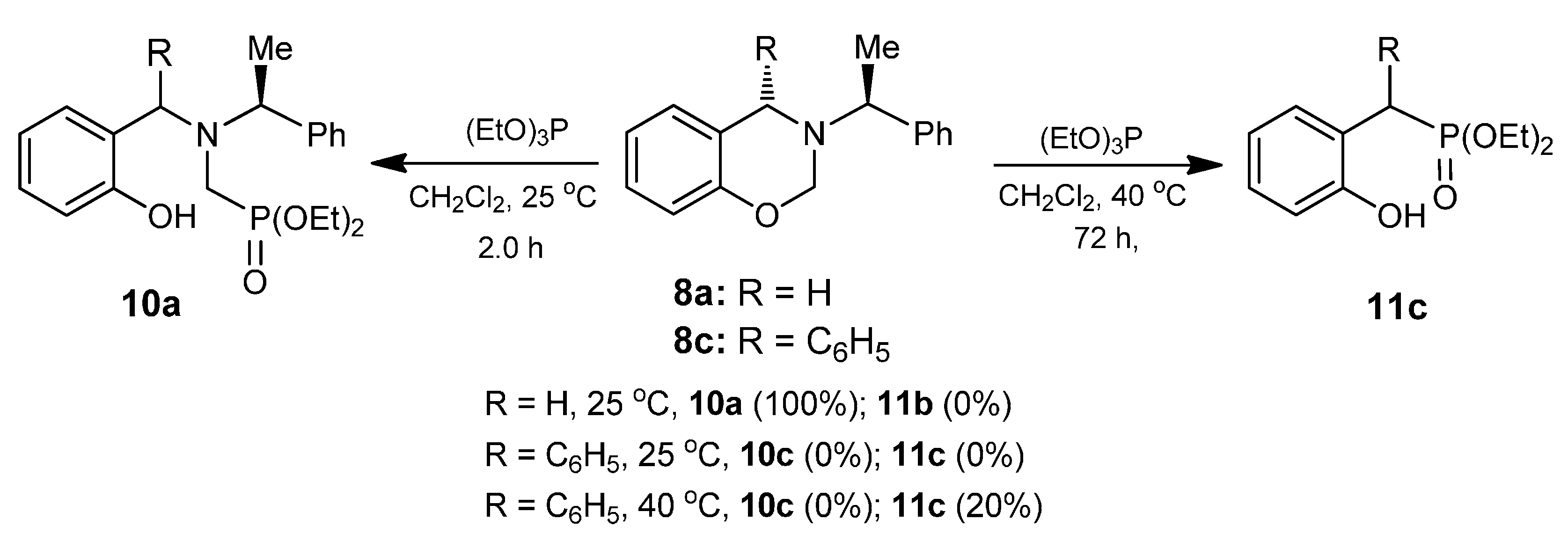
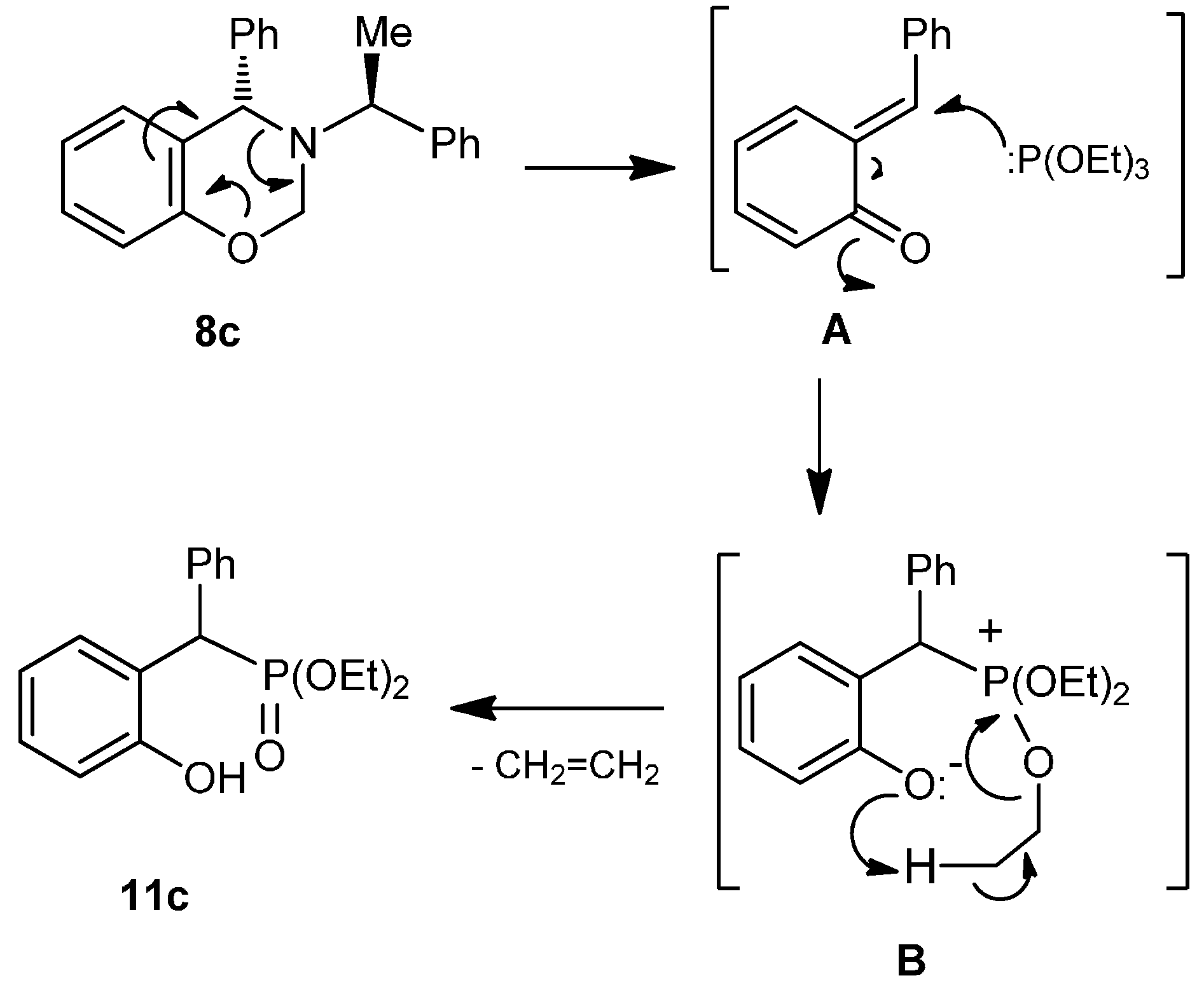
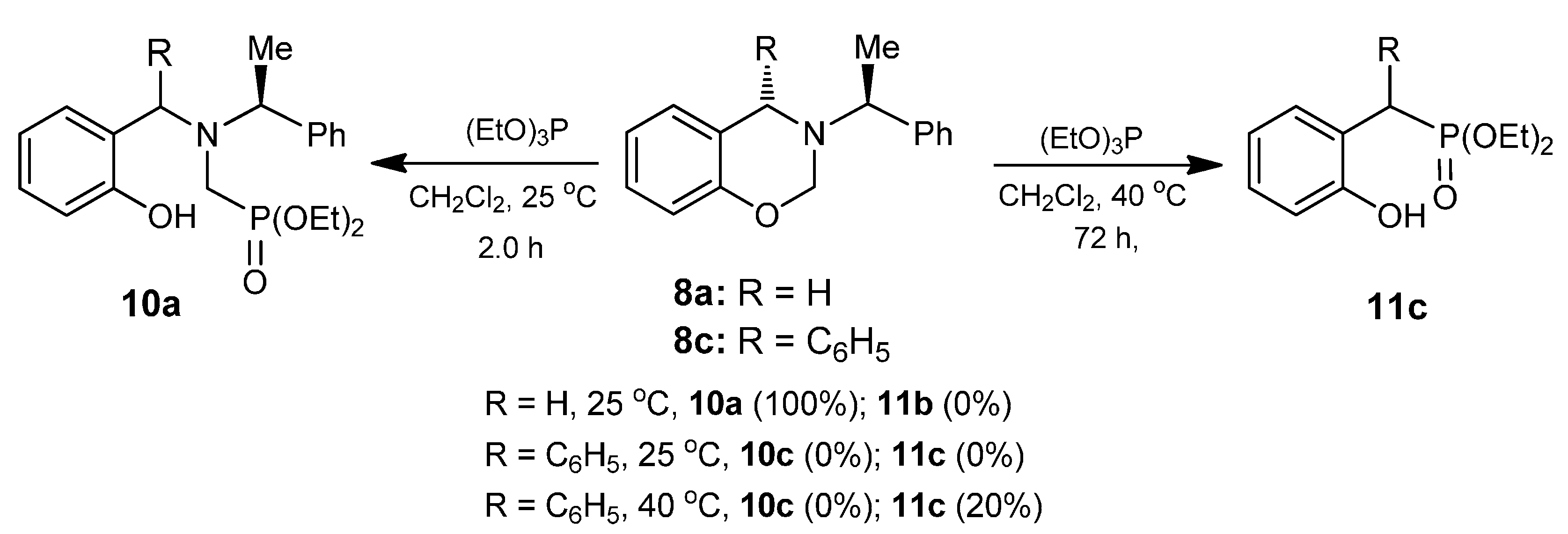
A mixture of benzoxazine (8a) 0.75 g (3.1 mmol) boron trifluoride etherate 80 mg, 0.08 mL (0.6 mmol) and triethyl phosphite 0.52 g, 0.53 mL, (3.1 mmol) in dry dichloromethane (10 mL), was reacted at room temperature for 72 h. The solvent was eliminated and the crude product was purified by column chromatography using hexane: i-PrOH (98:2) as eluent, obtaining the compound (12a) (155 mg, 15%), as colorless. The compound (10a) was also obtained (377 mg, 32%). [α]D = −28.4° (c = 0.011, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.24 (t, J = 6.8 Hz, 3H), 1.25 (t, J = 6.8 Hz, 3H), 1.39 (t, J = 6.8 Hz, 3H), 1.41 (d, J = 7.2 Hz, 3H), 2.80 (ABX system, JH/P = 15.4, 12.6 Hz, 1H), 2.97 (ABX system, JH/P = 15.6, 8.4 Hz, 1H), 3.71 (AB system, J = 14.8 Hz, 1H), 3.91 (AB system, J = 14.8 Hz, 1H), 3.96 (q, J = 7.2 Hz, 2H), 4.00 (q, J =6.8 Hz, 2H), 4.01 (q, J = 7.0 Hz, 2H), 4.19 (q, J = 6.8 Hz, 1H) 6.80–7.56 (m, 9H). 13C-NMR (CDCl3, 100 MHz): δ 14.1, 15.1, 16.60, 16.66, 45.5 (d, JC/P = 162.5 Hz), 48.6 (d, JC/P = 7.3 Hz), 58.3 (d, JC/P = 10.2 Hz), 61.7 (d, JC/P = 7.3 Hz), 61.8 (d, J = 7.3 Hz), 63.7, 111.3, 120.5, 126.9, 127.8, 128.1, 128.4, 130.7, 142.6, 157.2. 31P-NMR (CDCl3, 80.95 MHz): δ 10.24. HRMS (CI+): m/z calculated for C22H32NO4P [M + H] 405.2069; found for [M + H]+, m/z 406.2128.
3.5.2. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (13b) and (14b)
A mixture of benzoxazine (8b) 1.0 g (3.9 mmol), boron trifluoride etherate 110 mg, 0.09 mL, (0.8 mmol) and triethyl phosphite 0.65 g, 0.67 mL, (3.9 mmol) in dry dichloromethane (20 mL), was reacted at room temperature for 72 h. The solvent was eliminated and the crude product was purified by column chromatography using hexane:EtOAc (80:20) as eluent, obtaining the compounds (13b) (96 mg, 7% and (14b) (204 mg, 15%), both as colorless oil.
(2R,S)-2-Ethoxy-(5S)-5-methyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (13b). [α]D = −2.2° (c = 0.013, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.41 (d, J = 6.8 Hz, 3H), 1.43 (d, J = 6.8 Hz, 3H), 1.43 (t, J = 6.8 Hz, 3H), 3.67 (ABX system, JH/P = 16.4, 5.8 Hz, 1H), 3.74 (ABX system, JH/P = 16.4, 6.0 Hz, 1H), 3.79 (dq, J = 7.2, 5.3 Hz, 2H), 4.31 (q, J = 7.2 Hz, 1H), 4.33 (q, J = 7.2 Hz, 1H), 6.56–7.35 (m, 9H). 13C-NMR (CDCl3, 100 MHz): δ 16.4, 16.5, 18.7, 22.7, 41.3 (d, JC/P = 125.9 Hz), 59.0, 60.1, 62.1 (d, JC/P = 8.8 Hz), 122.5, 122.6, 124.9, 126.9, 127.1, 128.4, 129.3, 131.3, 134.1, 145.6, 147.9. 31P-NMR (CDCl3, 80.95 MHz): δ 15.52. HRMS (CI+): m/z calculated for C19H24NO3P [M + H] 345.1494; found for [M + H]+, m/z 346.1557.
(2R,S)-2-Ethoxy-(5S)-5-methyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide, (14b). [α]D = +5.40° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.23 (d, J = 7.0 Hz, 3H), 1.32 (d, J = 6.8 Hz, 3H), 1.44 (t, J = 7.2 Hz, 3H), 3.60 (dq, J = 6.8, 6.8 Hz, 1H), 3.75 (q, 7.2 Hz, 1H), 3.82 (ABX system, JH/P = 16.8, 1.6 Hz, 1H), 3.85 (ABX system, JH/P = 16.8, 3.6 Hz, 1H), 4.05–4.15 (m, 2H), 4.29–4.39 (m, 2H), 6.57–7.35 (m, 9H). 13C-NMR (CDCl3, 100 MHz): δ 16.3, 16.4, 18.4, 22.7, 41.3 (d, JC/P = 123.0 Hz), 58.6, 60.0, 61.5 (d, JC/P = 8.8 Hz), 121.9, 122.0, 124.7, 127.0, 127.1, 128.5, 129.0, 131.5, 134.1, 145.5, 148.4. 31P-NMR (CDCl3, 80.95 MHz): δ 18.75. HRMS (CI+): m/z calculated for C19H24NO3P [M + H] 345.1494; found for [M + H]+, m/z 346.1553.
3.5.3. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (13c) and (14c)
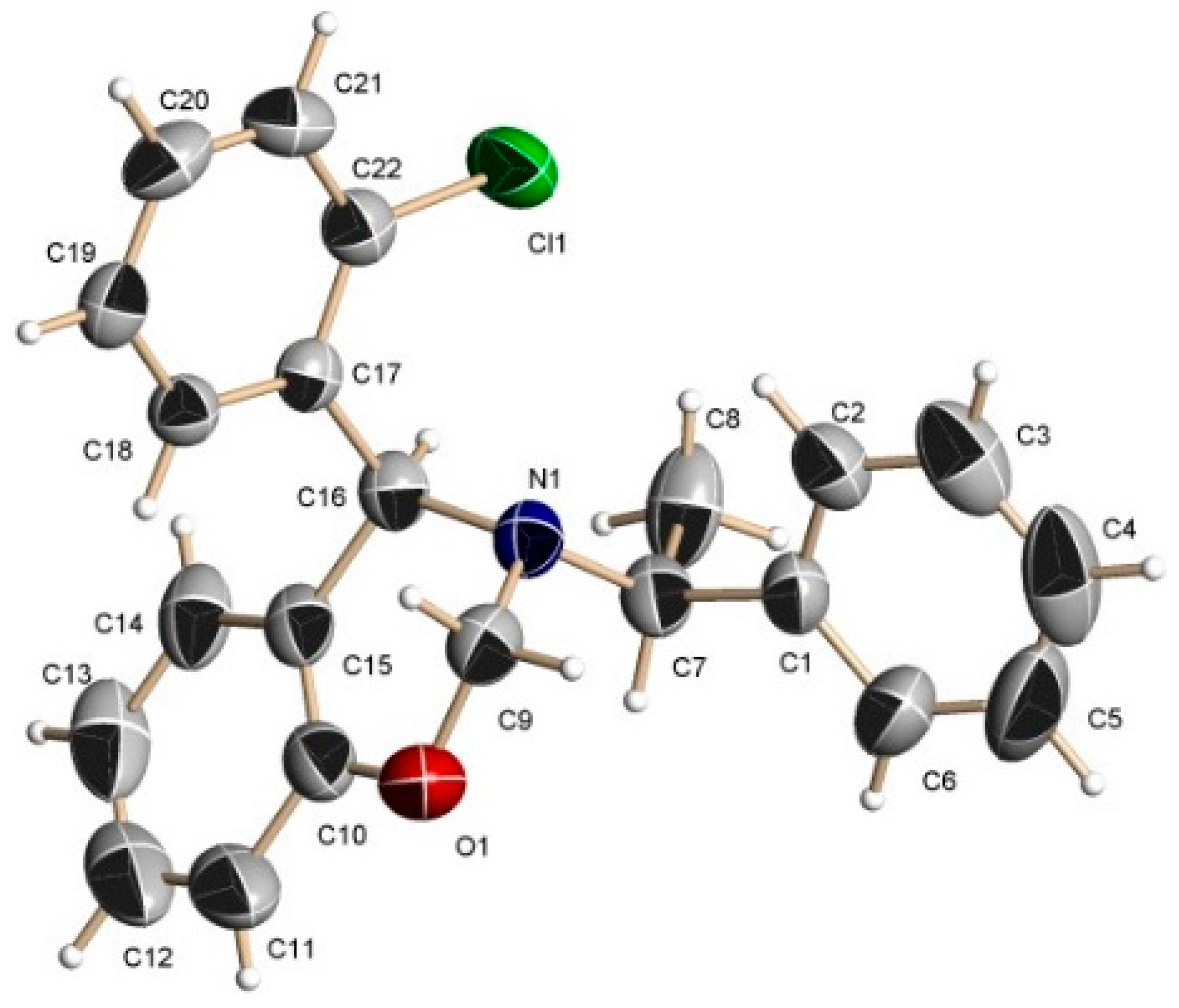
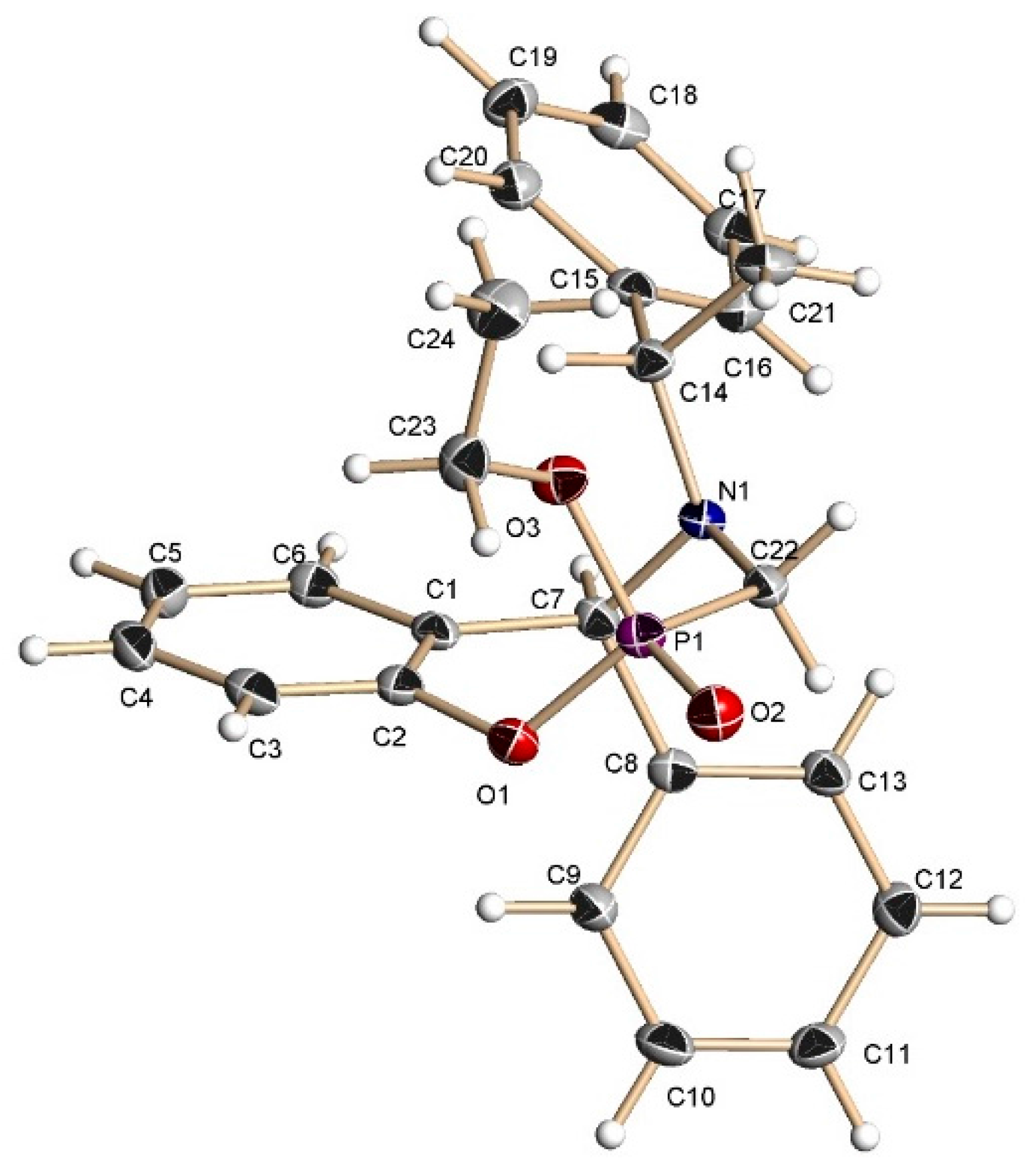
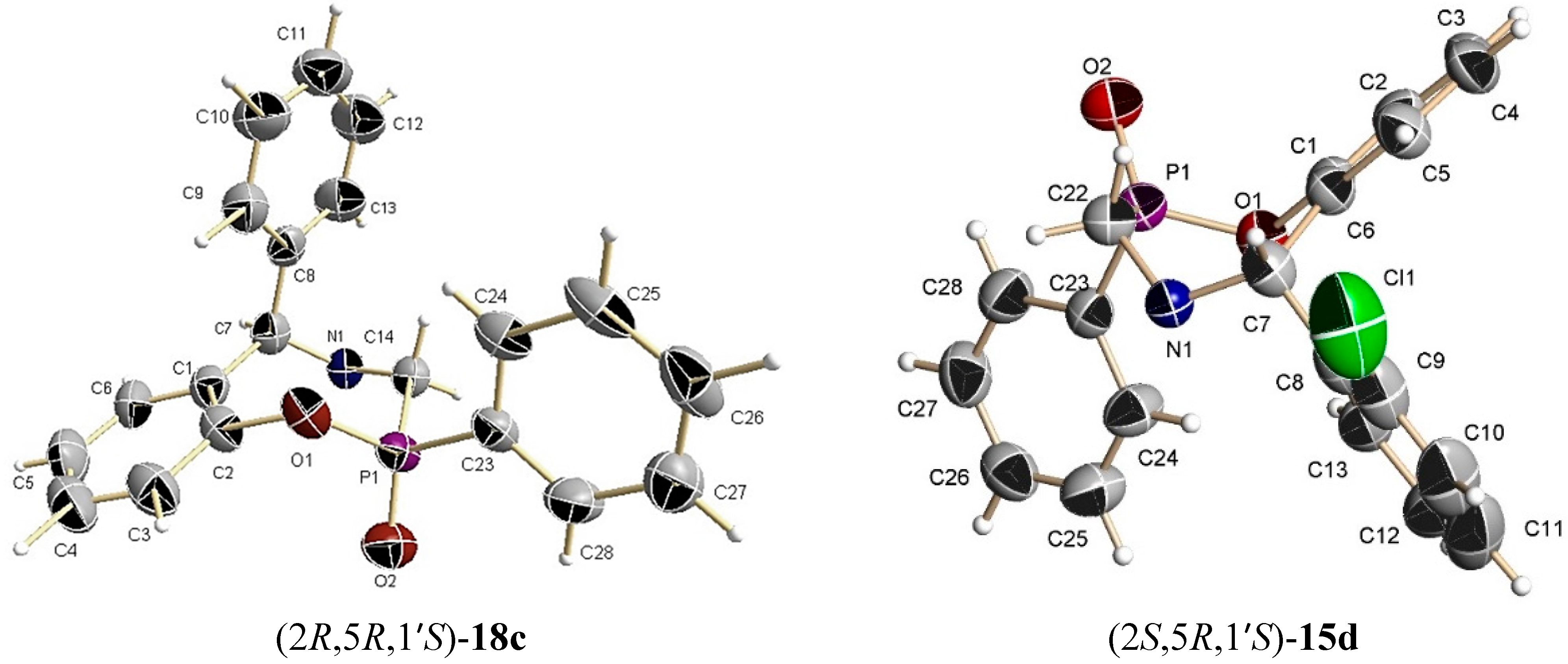
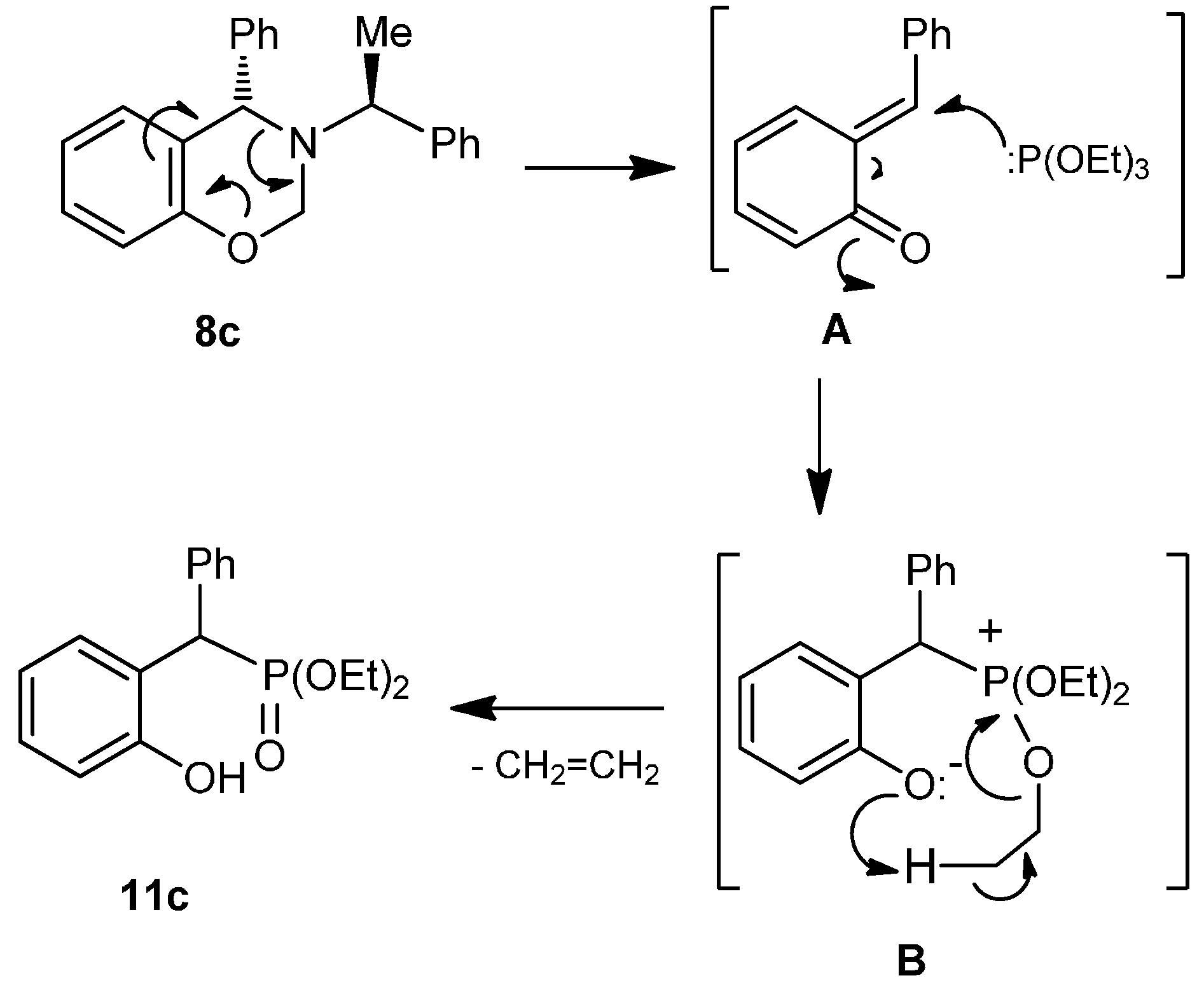
A mixture of benzoxazine (8c) 0.68 g (2.2 mmol), boron trifluoride etherate 60 mg, 0.05 mL, (0.4 mmol) and triethyl phosphite 0.36 g, 0.37 mL, (2.2 mmol) in dry dichloromethane (5 mL) was reacted at room temperature for 72 h. The solvent was eliminated and the crude product was purified by column chromatography using hexane:EtOAc (80:20) as eluent, obtaining the less polar compound (13c) (100 mg, 11%) as yellow oil, and the more polar compound (14c) (138 mg, 16%) as a white solid mp = 164–170 ºC. The compound (14c) was recrystallized from dichloromethane–hexane to give a crystal for X-ray studies.
(2S)-2-Ethoxy-(5S)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (13c). [α]D = +74.90° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.28 (t, J = 7.0 Hz, 3H), 1.51 (d, J = 6.6 Hz, 3H), 3.32 (ABX system, JH/P = 16.2, 8.4 Hz, 1H), 3.57 (ABX system, JH/P = 16.2, 3.8 Hz, 1H), 3.99–4.23 (m, 3H), 4.96 (s, 1H), 6.67–7.71 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 16.5 (d, JC/P = 5.85 Hz), 21.9, 40.2 (d, JC/P = 128.15 Hz), 59.9, 62.4 (d, JC/P = 7.3 Hz), 67.8, 123.1, 123.2, 125.5, 127.4, 127.5, 128.1, 128.5, 128.8, 130.3, 130.8, 133.2, 139.7, 145.5. 31P-NMR (CDCl3, 80.95 MHz): δ 10.97. HRMS (CI+): m/z calculated for C24H26NO3P [M + H] 407.1650; found for [M + H]+, m/z 408.1710.
(2R)-2-Ethoxy-(5S)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (14c). [α]D = +79.62° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.25 (t, J = 7.0 Hz, 3H), 1.42 (d, J = 6.8 Hz, 3H), 3.54 (ABX system, JH/P = 16.8, 6.8 Hz, 1H), 3.70 (ABX system, JH/P = 16.0, 5.2 Hz, 1H), 3.80 (q, J = 6.8 Hz, 1H), 4.03–4.45 (m, 2H), 4.92 (s, 1H), 6.66–7.39 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 16.5 (d, JC/P = 5.95 Hz), 22.6, 41.7 (d, JC/P = 123.3 Hz), 59.9, 61.6 (d, JC/P = 7.95 Hz), 67.2, 122.3, 122.4, 125.3, 127.3, 127.5, 127.6, 128.0, 128.7, 128.9, 130.0, 130.6, 130.7, 138.7, 145.4, 149.1 (d, JC/P = 7.2 Hz). 31P-NMR (CDCl3, 80.95 MHz): δ 13.52. HRMS (CI+): m/z calculated for C24H26NO3P [M + H] 407.1650; found for [M + H]+, m/z 408.1710.
3.5.4. Synthesis of (2R,S)-2-Ethoxy-(5R)-5-(2-chlrophenyl)-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (13d)
A mixture of benzoxazine (8d) 0.56 g (1.6 mmol), boron trifluoride etherate 40 mg, 0.04 mL (0.3 mmol) and triethyl phosphite 0.26 g, 0.27 mL, (1.6 mmol) in dry dichloromethane (10 mL) was reacted at room temperature for 72 h. The solvent was eliminated and the crude product was purified by column chromatography using hexane:EtOAc (80:20) as eluent, obtaining the compound (13d) (47 mg, 6%). [α]D = +163.41° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.28 (t, J = 7.0 Hz, 3H), 1.48 (d, J = 6.8 Hz, 3H), 3.11 (AB system, J = 16.4 Hz, 1H), 3.20 (AB system, J = 15.6 Hz, 1H), 3.89 (dq, J = 6.6, 3.2 Hz, 1H), 4.22 (m, 2H), 5.66 (s, 1H), 7.07–7.44 (m, 13H). 13C-NMR (CDCl3, 100 MHz): δ 13.8, 16.5, 16.6, 41.3 (d, JC/P = 153.75 Hz), 59.3 (d, JC/P = 11.7 Hz), 62.0, 65.8, 105.2, 123.0, 123.1, 125.9, 127.7, 127.8, 128.2, 128.3, 128.9, 129.6, 129.7, 131.3, 131.8, 132.1, 140.0, 141.8. 31P-NMR (CDCl3, 80.95 MHz): δ 21.21. HRMS (CI+): m/z calculated for C24H25ClNO3P [M + H] 441.1261; found for [M + H]+, m/z 442.1361.
3.6. General Procedure for the Preparation of 1,4,2-Oxazaphosphepines (15), (16), (17) and (18)
Under anhydrous conditions, the corresponding benzoxazine dissolved in dry dichloromethane was treated with dichlorophenylphosphine followed by the slow addition of triethylamine, and the reaction mixture was stirred at room temperature for 72 h. After this time, the solvent was evaporated under reduced pressure, and the residue was treated with a minimum amount of water and extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, evaporated under reduced pressure, and the crude product was purified by column chromatography.
3.6.1. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (16a)
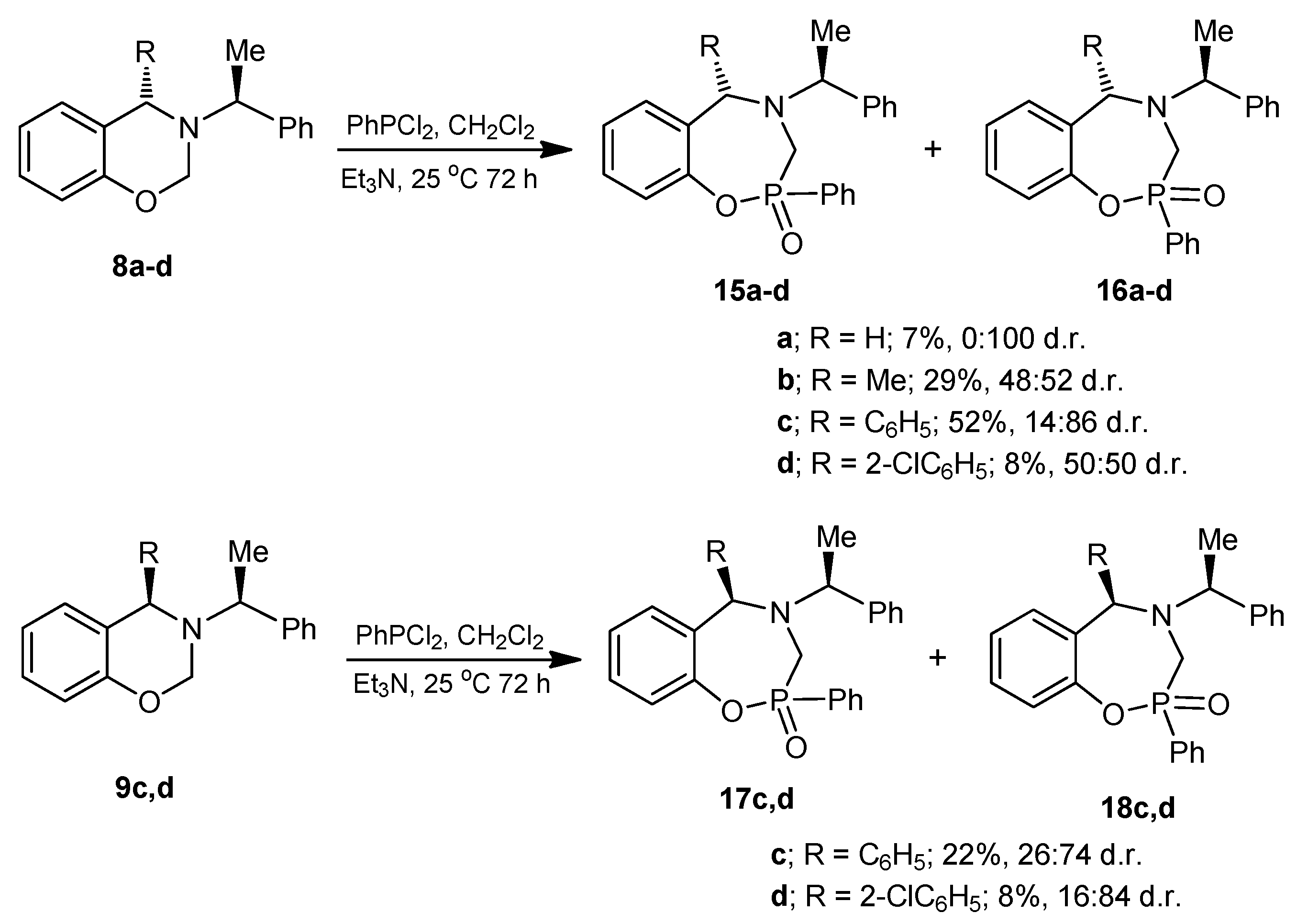
The benzoxazine (8a) 1.0 g (4.4 mmol) was reacted at room temperature with dichlorophenylphosphine 0.78 g, 0.6 mL, (4.4 mmol) and triethylamine 0.89 g, 1.22 mL, (8.8 mmol) in dichloromethane (25 mL). The solvent was evaporated under reduced pressure and the crude was purified by column chromatography using a mixture of hexane:EtOAc (8:2), obtaining the compound (16a) (110 mg, 7%) as a white solid, mp = 204–206 °C.
(2S)-2-Phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (16a). [α]D = +22.48° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.46 (d, J = 6.4 Hz, 3H), 3.51 (AB system, J = 15.2 Hz, 1H), 3.75 (ABX system, J = 15.2, 3.2 Hz, 1H), 3.81 (AB system, J = 14.4 Hz, 1H), 3.99 (dq, J = 7.0, 4.0 Hz, 1H), 4.09 (ABX system, J = 14.8, 1.6 Hz, 1H), 6.75–8.02 (m, 14H). 13C-NMR (CDCl3, 100 MHz): δ 21.7, 53.4 (d, JC/P = 89.5 Hz), 55.3, 61.3, 122.4 (d, JC/P = 3.0 Hz), 125.1, 127.4, 127.6, 128.6, 128.7, 128.8, 129.7, 130.7, 131.4, 131.5, 131.7, 133.0, 144.3, 150.1 (d, JC/P = 6.1 Hz). 31P-NMR (CDCl3, 80.95 MHz): δ 34.87. HRMS (CI+): m/z calculated for C22H22NO2P [M + H] 363.1388; found for [M + H]+, m/z 364.1454.
3.6.2. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (15b) and (16b)
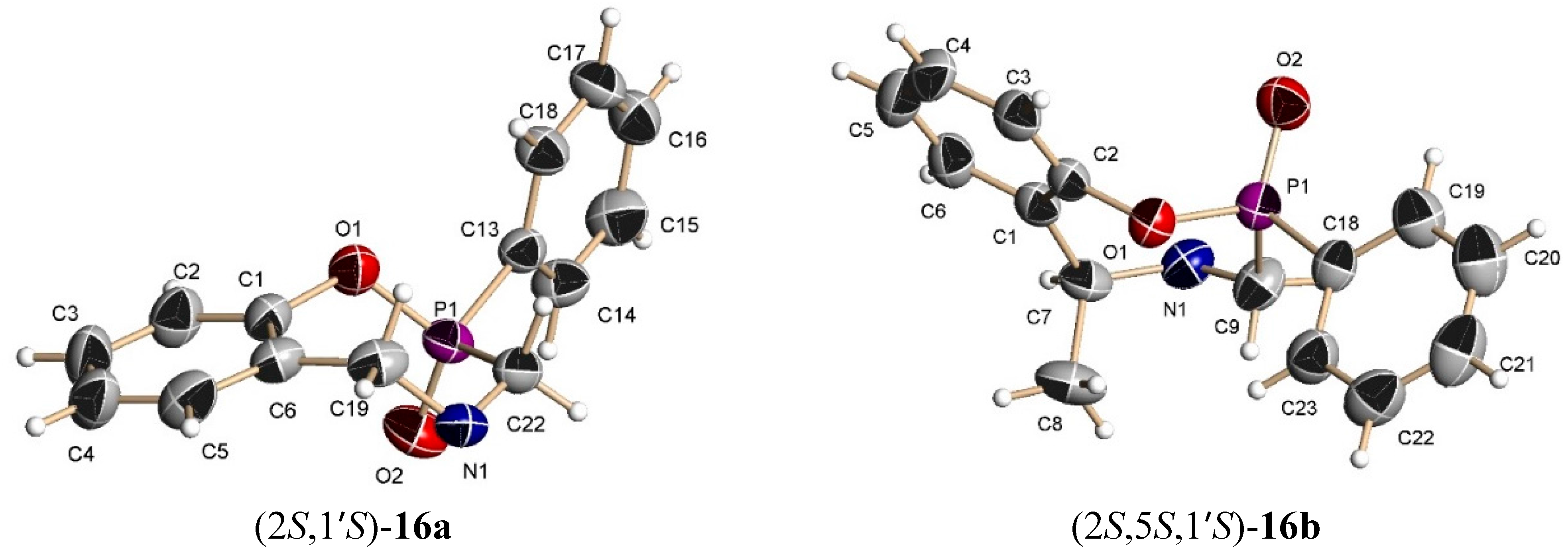
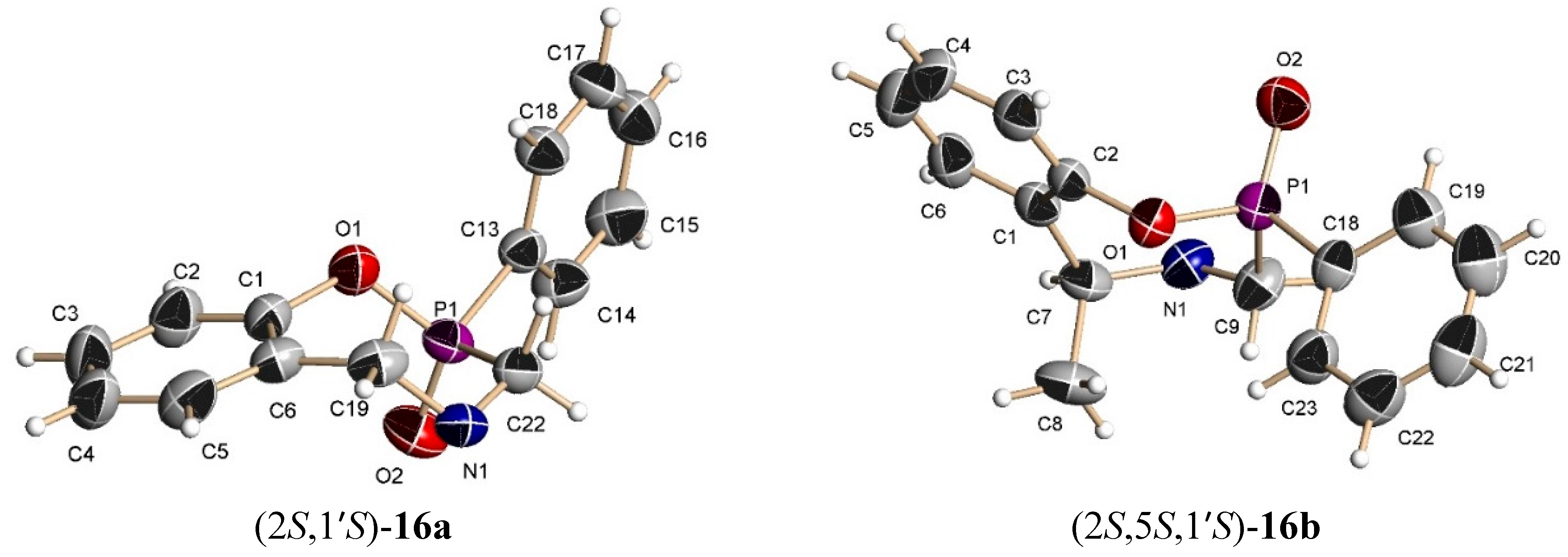
The benzoxazine (8b) 0.85 g (3.4 mmol) was reacted at room temperature with dichlorophenylphosphine 0.6 g, 0.46 mL, (3.4 mmol) and triethylamine 0.68 g, 0.94 mL, (6.8 mmol) in dichloromethane (25 mL). The solvent was evaporated under reduced pressure and the crude was analyzed by 31P-NMR, observing the two diastereoisomers with a 48:52 ratio, which was purified by column chromatography using a mixture of hexane:EtOAc (8:2), obtaining the compound (16b) (190 mg, 15%) as a white solid mp = 168–172 °C. The compound (16b) was recrystallized from dichloromethane-hexane to give a crystal for X-ray studies. The diastereoisomer (15b) was obtained as unstable colorless oil (14 %) and only the 1H-NMR spectrum was obtained.
(2R)-2-Phenyl-(5S)-5-methyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (15b). [α]D = +36.17° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.48 (d, J = 6.8 Hz, 3H), 1.51 (d, J = 7.6 Hz, 3H), 3.58 (ABX system, J = 15.4, 10.6 Hz, 1H), 3.67 (ABX system, J = 15.8, 7.8 Hz, 1H), 4.22 (q, J = 6.8 Hz, 1H), 4.36 (q, J = 7.2 Hz, 1H), 6.90–7.84 (m, 14H).
(2S)-2-Phenyl-(5S)-5-methyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (16b). [α]D = +71.55° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.48 (d, J = 6.6 Hz, 3H), 1.58 (d, J = 7.0 Hz, 3H), 3.56 (AB system, J = 16 Hz, 1H), 3.67 (AB system, J = 16.4 Hz, 1H), 3.91 (q, J = 6.9 Hz, 1H), 4.04 (q, J = 6.5 Hz, 1H), 6.8–7.94 (m, 14H). 13C-NMR (CDCl3, 50 MHz): δ 20.5, 20.6, 45.1 (d, JC/P = 87.9 Hz), 60.2, 60.6, 123.6, 125.3, 127.0, 127.3, 128.4, 128.5, 128.8, 129.4, 131.1, 131.5 (d, JC/P = 9.1 Hz), 132.8, 135.2, 144.9, 148.7 (d, JC/P = 7.6 Hz). 31P-NMR (CDCl3, 161.8 MHz): δ 37.23. HRMS (CI+): m/z calculated for C23H24NO2P [M + H] 377.1545; found for [M + H]+, m/z 378.1612.
3.6.3. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (15c) and (16c)
The benzoxazine (8c) 0.30 g (0.9 mmol) was reacted at room temperature with dichlorophenylphosphine 0.17 g, 0.13 mL, (0.9 mmol) and triethylamine 0.19 g, 0.26 mL, (1.9 mmol) in dichloromethane (5 mL). The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of hexane:EtOAc (80:20) as eleuent, obtaining the compounds (15c) as a colorless high viscosity oil (7%) and (16c) as a white solid (45%) mp = 166–172 °C.
(2R,S)-2-Phenyl-(5S)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (15c). [α]D = +88.4° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.29 (d, J = 6.8 Hz, 3H), 3.35 (ABX system, J = 16.4, 14.6 Hz, 1H), 3.49 (ABX system, J = 16.4, 6.4 Hz, 1H), 4.09 (dq, J = 6.8, 6.4 Hz, 1H), 5.29 (s, 1H), 6.70–7.59 (m, 19H). 13C-NMR (CDCl3, 50 MHz): δ 15.9, 29.8, 45.3 (d, JC/P = 95.2), 60.0 (d, J = 7.3 Hz), 69.1, 123.6, 123.7, 125.8, 127.6, 127.7, 127.9, 128.5, 128.6, 128.7, 128.8, 129.4, 131.4, 131.5, 132.0, 132.8, 132.9, 142.4 (d, JC/P = 99.5 Hz). 31P-NMR (CDCl3, 161.8 MHz): δ 36.74. HRMS (CI+): m/z calculated for C23H24NO2P [M + H] 439.1701; found for [M + H]+, m/z 440.1774.
(2R,S)-2-Phenyl-(5S)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (16c). (0.19 g, 45%) [α]D = +146.78° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.56 (d, J = 6.4 Hz, 3H), 3.40 (AB system, J = 16.4 Hz, 1H), 3.58 (AB system, J = 16.4 Hz, 1H), 4.14 (q, J = 6.0 Hz, 1H), 5.05 (s, 1H), 7.12–7.57 (m, 19H). 13C-NMR (CDCl3, 50 MHz): δ 21.3, 20.6, 45.9 (d, JC/P = 84.9 Hz), 60.3, 68.5, 123.9, 125.6, 127.4, 127.5, 127.6, 128.2, 128.6, 128.7, 130.2, 131.3, 131.4, 132.7, 132.9, 149.22. 31P-NMR (CDCl3, 161.8 MHz): δ 32.57. HRMS (CI+): m/z calculated for C23H24NO2P [M + H] 439.1701; found for [M + H]+, m/z 440.1783.
3.6.4. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (15d) and (16d)
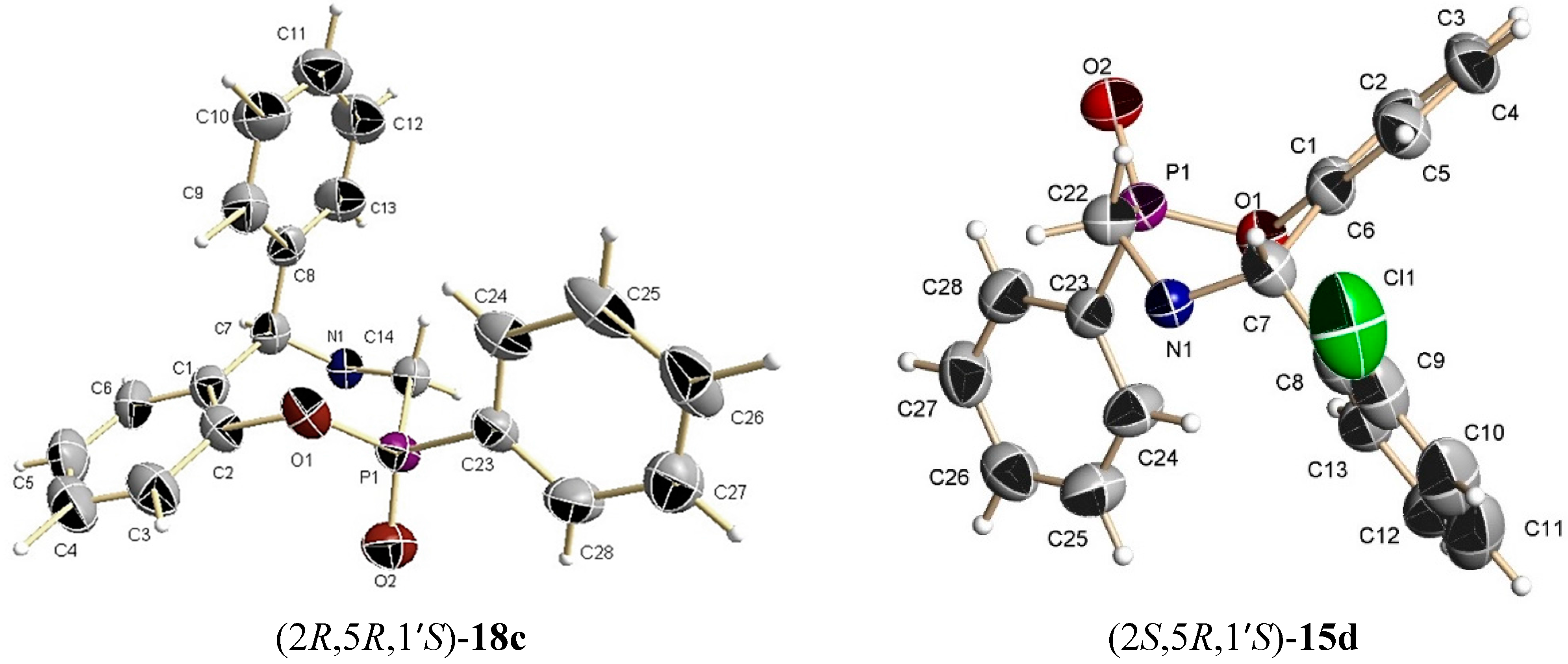
The benzoxazine (8d) 1.0 g (2.9 mmol) was reated at room temperature with dichlorophenylphosphine 0.5 g, 0.39 mL, (2.9 mmol) and triethylamine 0.57 g, 0.80 mL, (5.7 mmol) in dichloromethane (15 mL). The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of hexane:EtOAc (80:20) as eluent, obtaining the compounds (15d) (50 mg, 4%) as an orange solid, mp = 65–68 °C and (16d) (50 mg, 4%) as a white solid mp = 220–224 °C with a diastereoisomeric ratio 50:50.
(2R)-2-Phenyl-(5R)-5-(2-chlorophenyl)-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (15d). [α]D = +116.74° (c = 0.0036, CHCl3). 1H-NMR (CDCl3, 200 MHz): δ 1.44 (d, J = 7.0 Hz, 3H), 3.15 (ABX system, J = 15.2, 15.2 Hz, 1H), 3.43 (ABX system, J = 16.1, 9.9 Hz, 1H), 4.02 (q, J = 6.6 Hz, 1H), 5.90 (s, 1H), 6.70–8.54 (m, 18H). 13C-NMR (CDCl3, 50 MHz): δ 12.6, 43.7 (d, JC/P = 104.9 Hz), 59.5, 65.5, 124.2, 126.0, 127.7, 128.0, 128.3, 128.5, 128.9, 129.3, 131.7, 131.9, 132.7, 141.2, 148.3. 31P-NMR (CDCl3, 81 MHz): δ 38.60. HRMS (CI+): m/z calculated for C28H25ClNO2P [M + H] 473.1311; found for [M + H]+, m/z 474.1437.
(2S)-2-Phenyl-(5R)-5-(2-chlorophenyl)-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (16d). [α]D = +223.83° (c = 0.0072, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.54 (d, J = 6.8 Hz, 3H), 3.37 (ABX system, J = 16.0, 6.4 Hz, 1H), 3.42 (ABX system, J = 16.0, 8.4 Hz, 1H), 3.93 (bs, 1H), 5.85 (s, 1H), 6.94–8.11 (m, 18H). 13C-NMR (CDCl3, 50 MHz): δ 11.4, 44.0 (d, JC/P = 107.9 Hz), 58.8, 66.9, 124.7, 126.3, 127.3, 127.7, 127.9, 128.1, 128.3, 128.5, 129.0, 129.8, 130.0, 130.7, 131.2, 131.4, 132.1, 132.4, 133.3, 140.9, 147.2. 31P-NMR (CDCl3, 81 MHz): δ 42.36. HRMS (CI+): m/z calculated for C28H25ClNO2P [M + H] 473.1311; found for [M + H]+, m/z 474.1390.
3.6.5. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (17c) and (18c)
The benzoxazine (9c) 0.25 g (0.8 mmol) was reacted at room temperature with dichlorophenylphosphine 0.14 g, 0.10 mL, (0.8 mmol) and triethylamine 0.16 g, 0.22 mL, (1.6 mmol) in dichloromethane (5 mL). The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of hexane:EtOAc (80:20) as eluent, obtaining the compounds (17c) (18 mg, 5%) as a white solid mp = 60–65 °C, and (18c) which is unstable in solution, (60 mg, 17%) as a white solid mp = 193–195 °C.
(2S)-2-Phenyl-(5R)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (17c). 1H-NMR (CDCl3, 200 MHz): δ 1.30 (d, J = 6.6 Hz, 3H), 3.36 (ABX system, J = 16.0, 12.2 Hz, 1H), 3.52 (ABX system, J = 16.1, 5.1 Hz, 1H), 4.11 (dq, J = 7.0, 2.2 Hz, 1H), 5.30 (s, 1H), 6.91–7.63 (m, 19H). 13C-NMR (CDCl3, 50 MHz): δ 15.9, 45.3 (d, JC/P = 95.5 Hz), 59.9, 69.1, 123.5, 125.7, 127.6, 127.8, 128.4, 128.6, 128.6, 129.3, 131.3, 131.5, 132.0, 132.7, 141.8, 142.7. 31P-NMR (CDCl3, 80.95 MHz): δ 36.7. HRMS (CI+): m/z calculated for C23H24NO2P [M] 439.1701; found for [M + H]+, m/z 439.1772.
(2R)-2-Phenyl-(5R)-5-phenyl-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (18c). [α]D = −95.35° (c = 0.010, CHCl3). 1H-NMR (CDCl3, 400 MHz): δ 1.20 (d, J = 6.6 Hz, 3H), 3.04 (ABX system, J = 16.1, 4.7 Hz, 1H), 3.48 (ABX system, J = 16.1, 3.7 Hz, 1H), 4.16 (q, J = 7.0 Hz, 1H), 5.35 (s, 1H), 7.14–7.86 (m, 19H). 13C-NMR (CDCl3, 50 MHz): δ 21.3, 46.1 (d, JC/P = 98.7 Hz), 60.3, 69.7, 124.3, 126.1, 127.7, 127.8, 128.2, 128.5, 128.6, 128.9, 130.2, 131.9, 132.0, 132.1, 133.0, 142.3, 148.0. 31P-NMR (CDCl3, 161.8 MHz): δ 37.84. HRMS (CI+): m/z calculated for C23H24NO2P [M] 439.1701; found for [M + H]+, m/z 439.1639.
3.6.6. Synthesis of 1,4,2-Oxazaphosphepine 2-oxide (17d) and (18d)
The benzoxazine (9d) 300 mg (0.9 mmol) was reacted at room temperature with dichlorophenylphosphine 150 mg, 0.11 mL, (0.85 mmol) and triethylamine 170 mg, 0.24 mL, (1.7 mmol) in dichloromethane (10 mL). The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography using a mixture of hexane:EtOAc (80:20) as eluent, obtaining (430 mg, 9%) of diastereoisomeric mixture as high viscosity oil, the two compounds were identified by 1H- and 31P-NMR with a 16:84 diastereoisomeric ratio, which the separation was not possible.
(2R,S)-2-Phenyl-(5S)-5-(2-chlorophenyl)-4-[(1ʹS)-1-phenylethyl]-2,3,4,5-tetrahydro-1,4,2-benzoxazaphosphepine 2-oxide (17d) and (18d). The asterisk denotes the minor diastereoisomer.1H-NMR (CDCl3, 200 MHz): δ 1.06* (d, J = 7.0 Hz, 3H), 1.49 (d, J = 7.0 Hz, 3H), 2.99* (ABX system, J = 16.2, 4.2 Hz, 1H), 3.25 (ABX system, J = 16.3, 5.8 Hz, 1H), 3.42* (ABX system, J = 16.4, 5.4 Hz, 1H), 3.56 (ABX system, J = 16.5, 4.4 Hz, 1H), 3.96* (q, J = 6.9 Hz, 1H), 3.97 (q, J = 6.9 Hz, 1H), 5.76* (s, 1H), 5.85 (s, 1H), 6.72–8.45 (m, 36H). 31P-NMR (CDCl3, 81 MHz): δ 40.35*, 41.51. HRMS (FAB+): m/z calculated for C28H25ClNO2P [M + H] 473.1311; found for [M + H]+, m/z 474.1389.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}