
Post-Synthetic Shaping of Porosity and Crystal Structure of Ln-Bipy-MOFs by Thermal Treatment

Abstract
:
1. Introduction
2. Results and Discussion
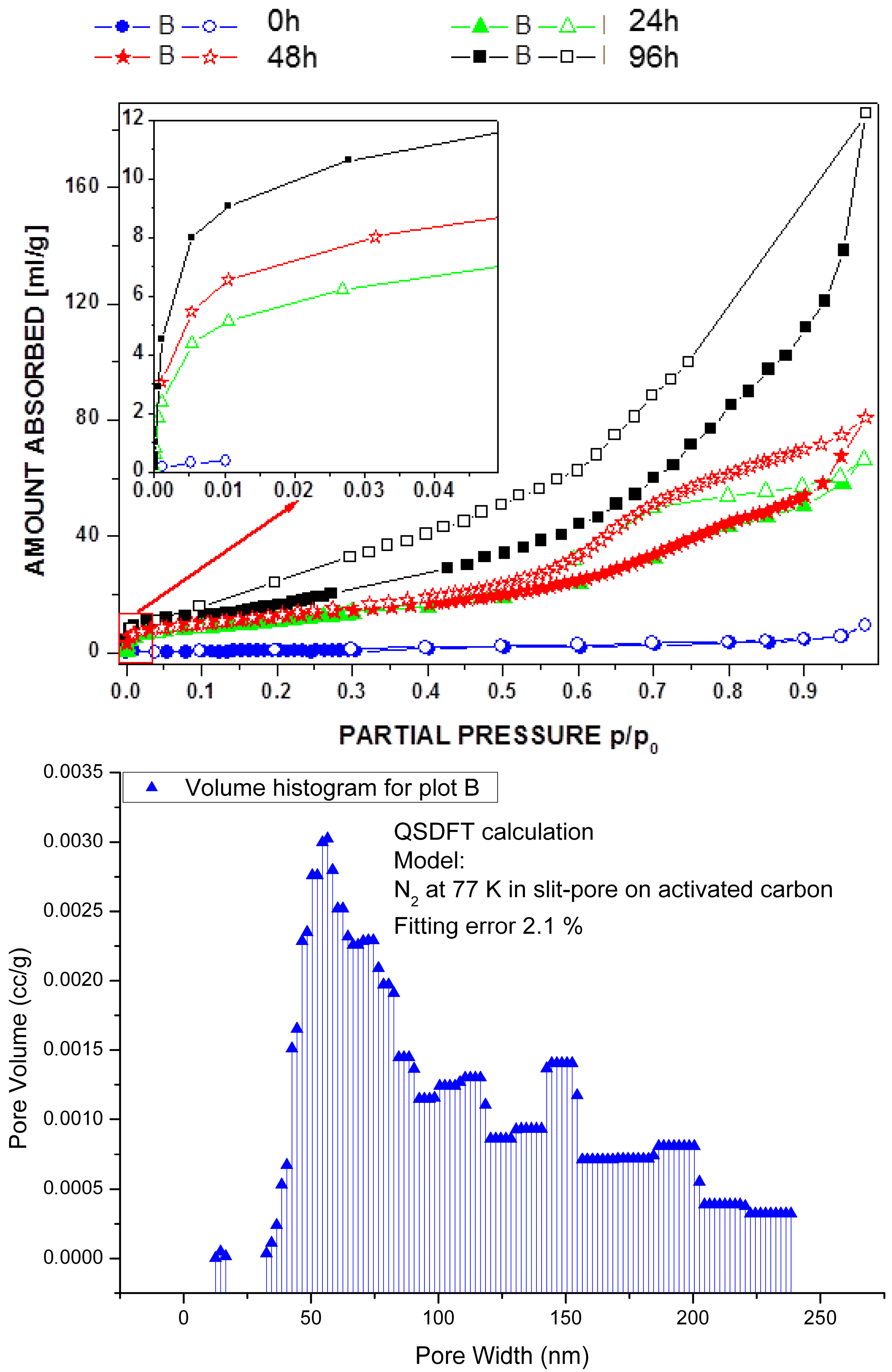
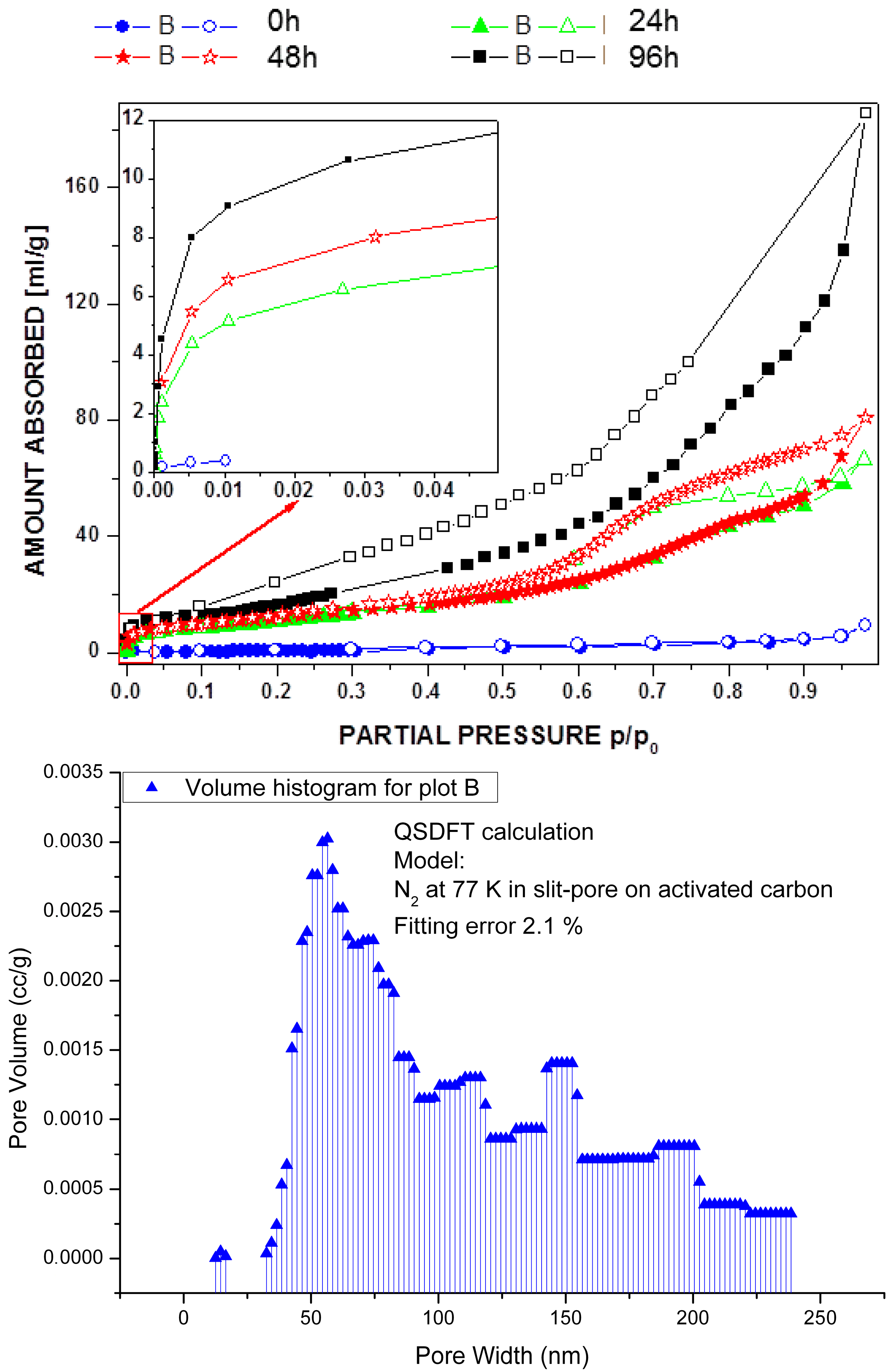
2.1. Post-Synthetic Surface Modification and Morphology Control

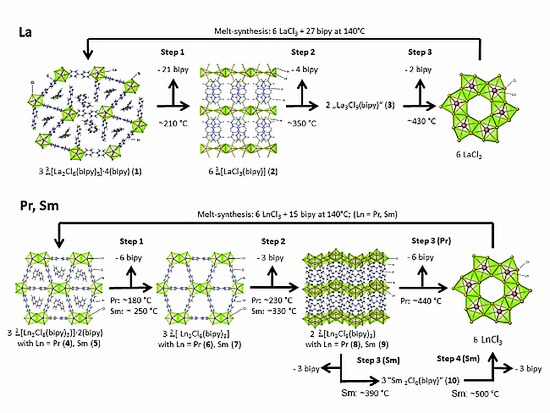
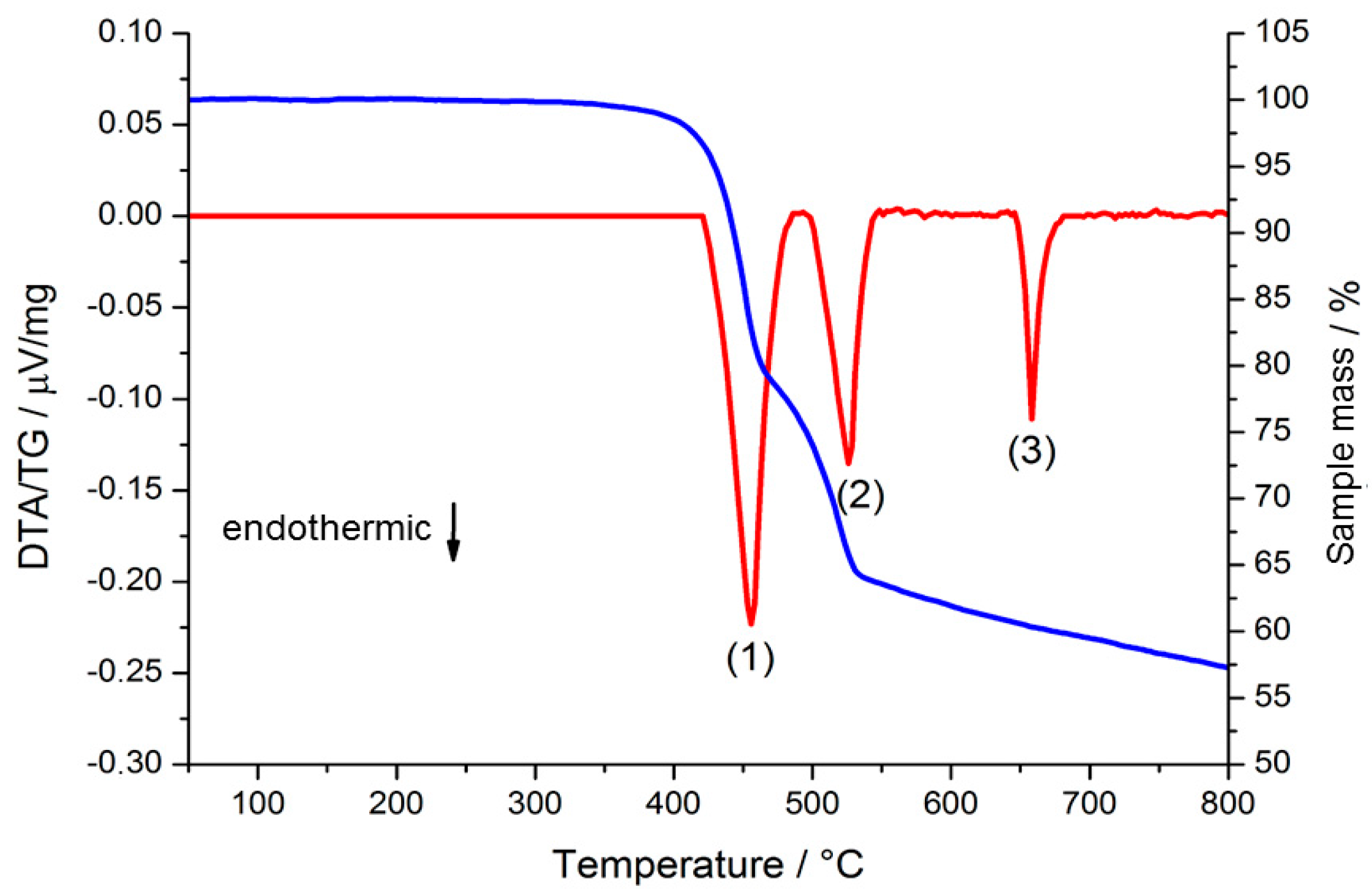
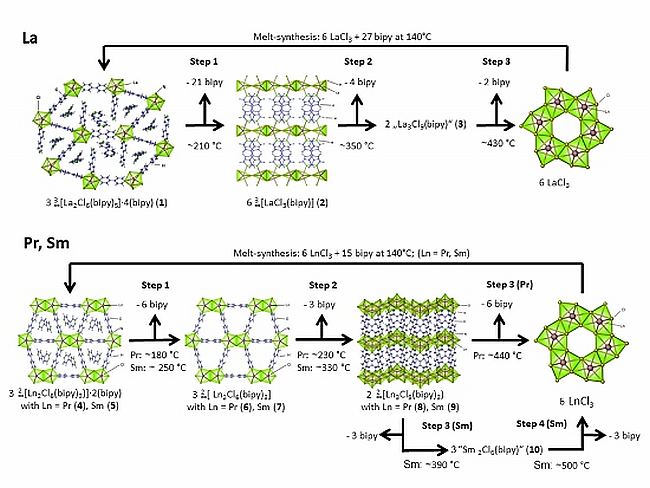
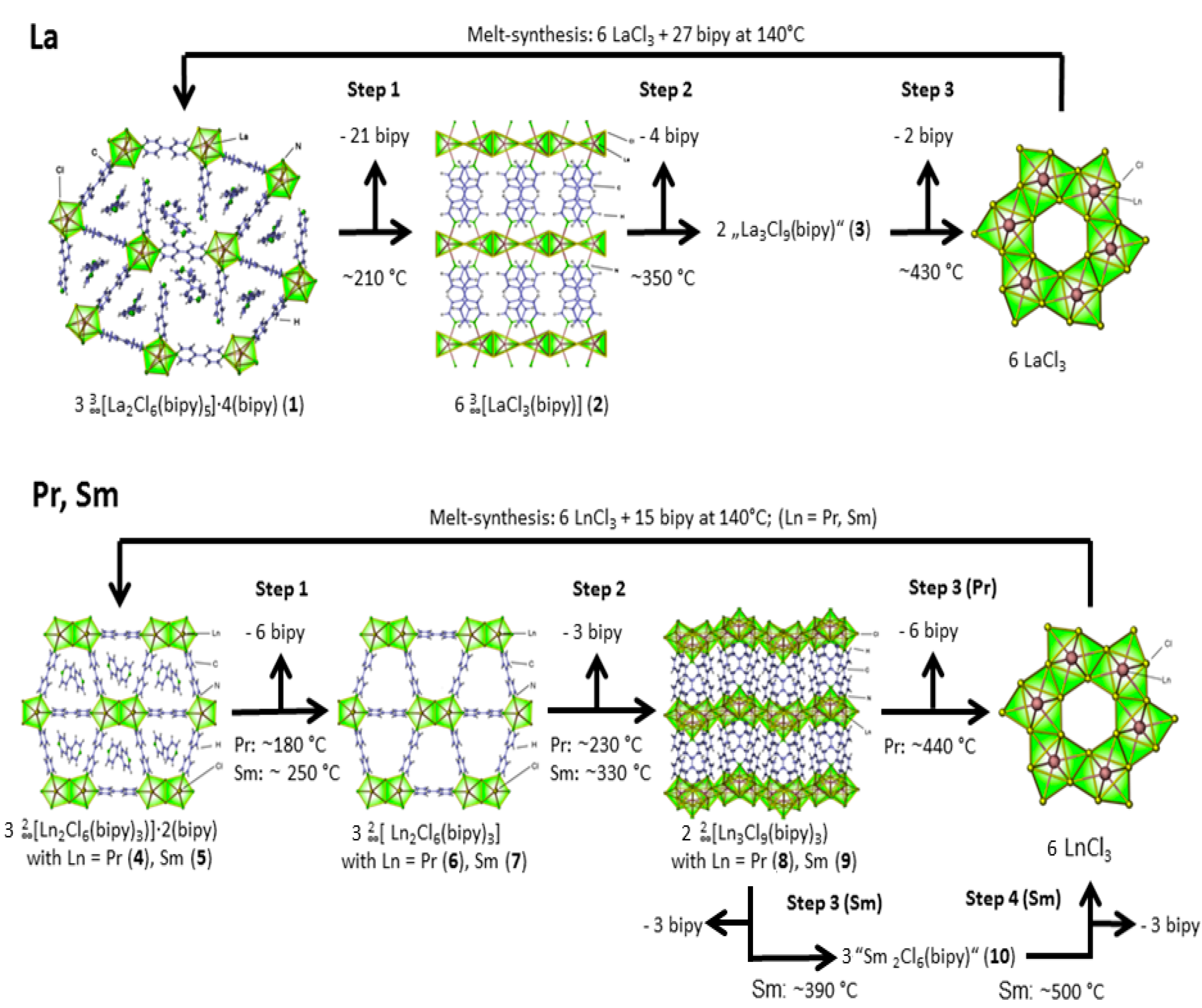
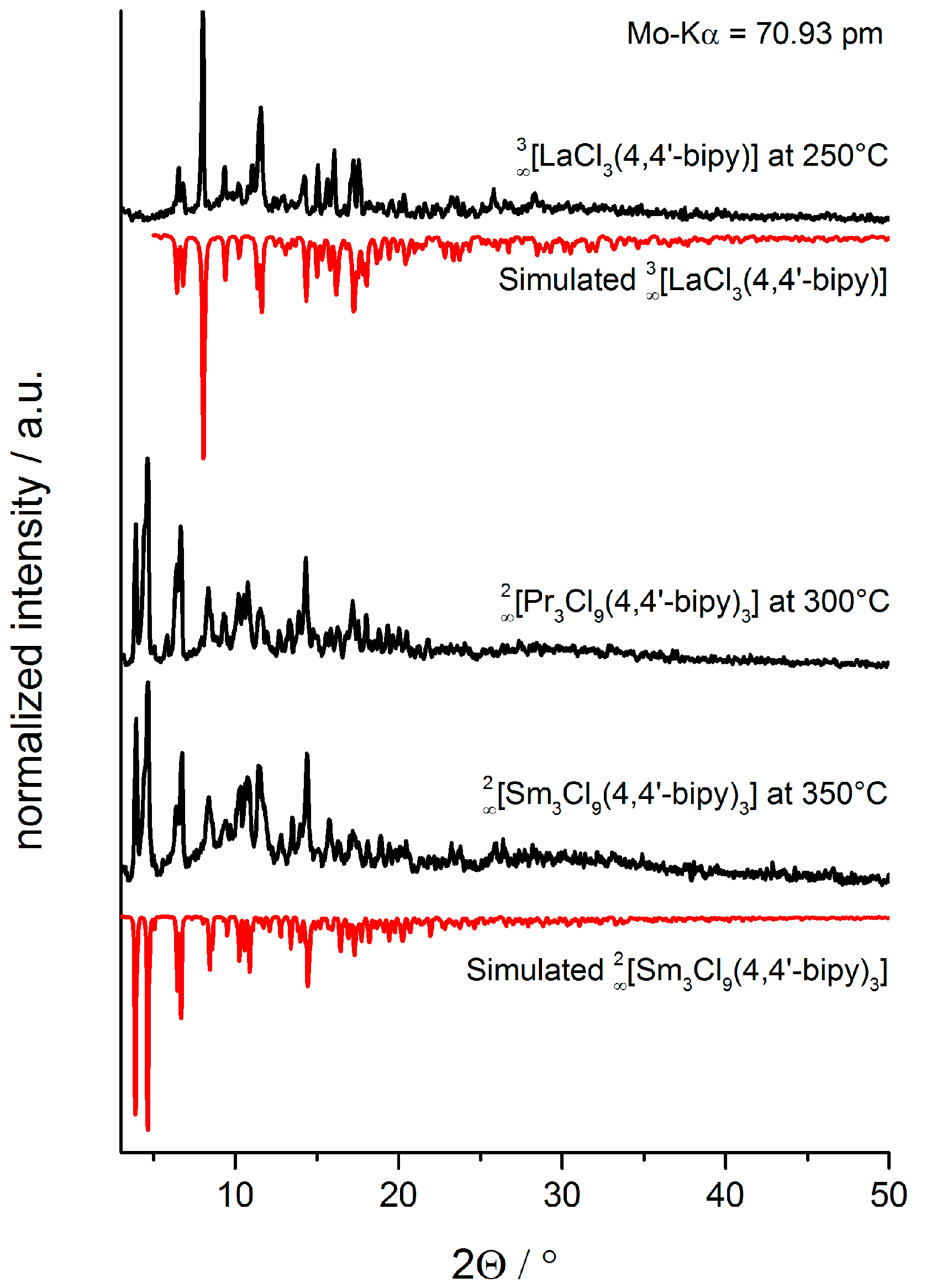
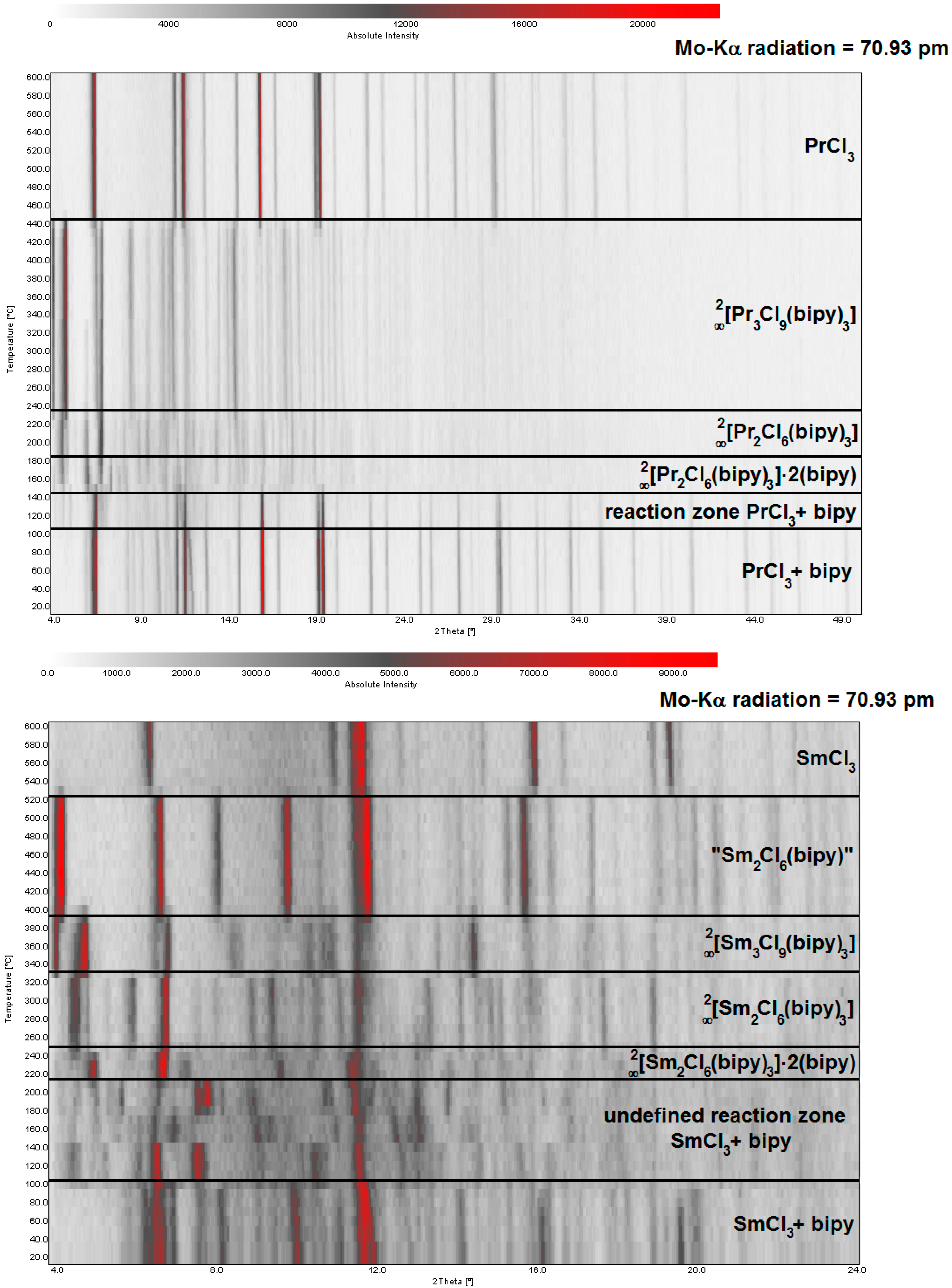
2.2. Thermal Conversion Processes
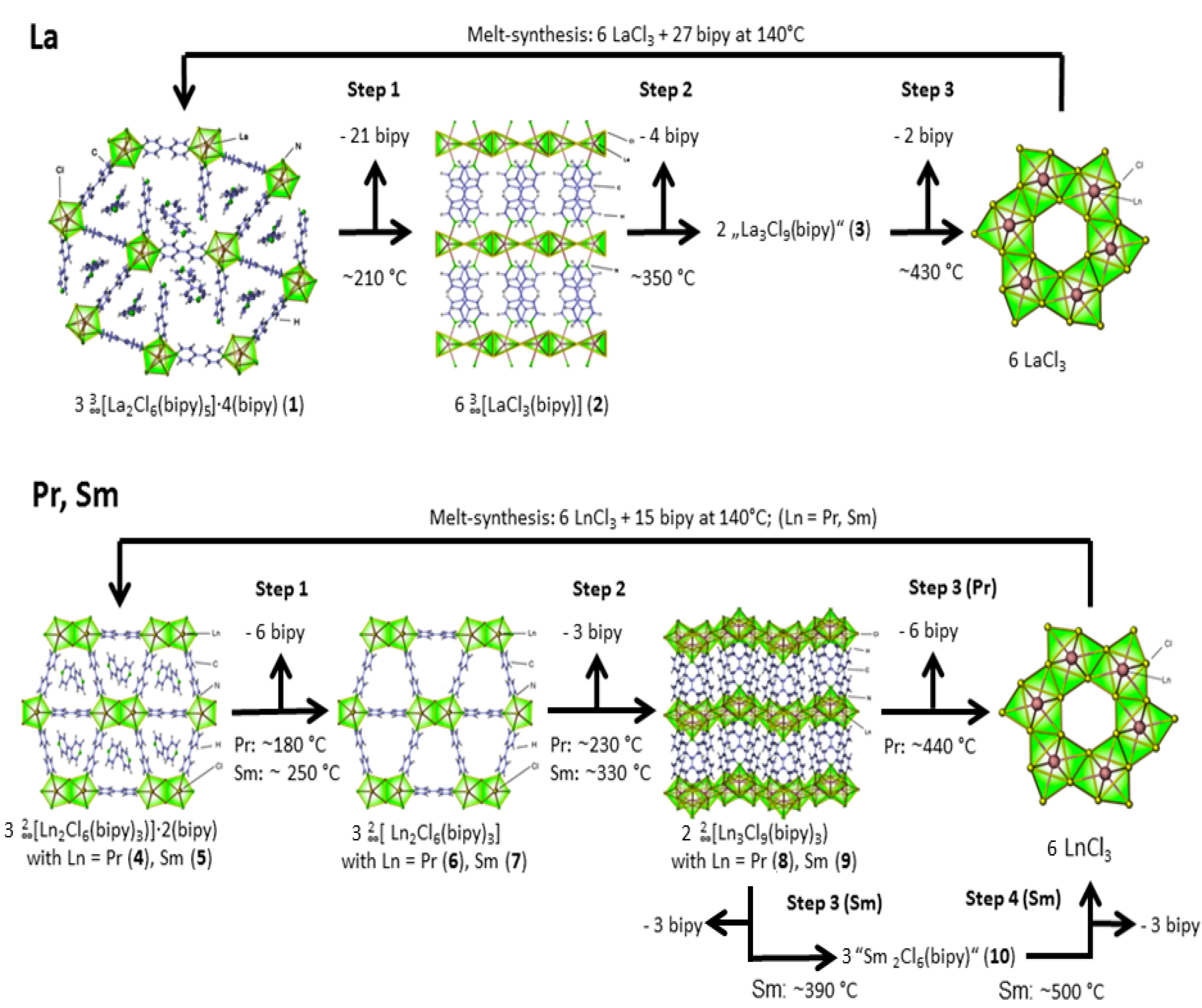
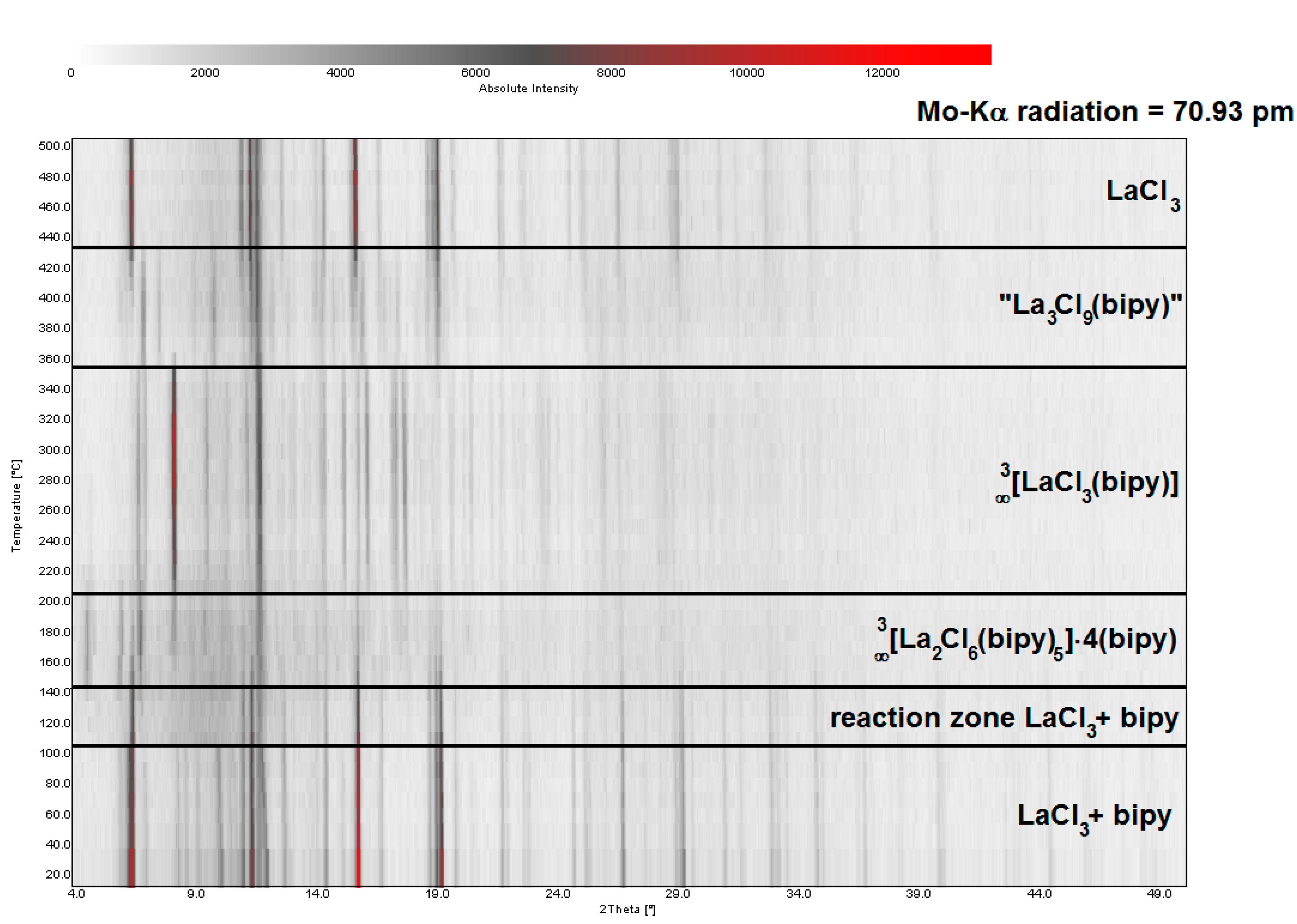
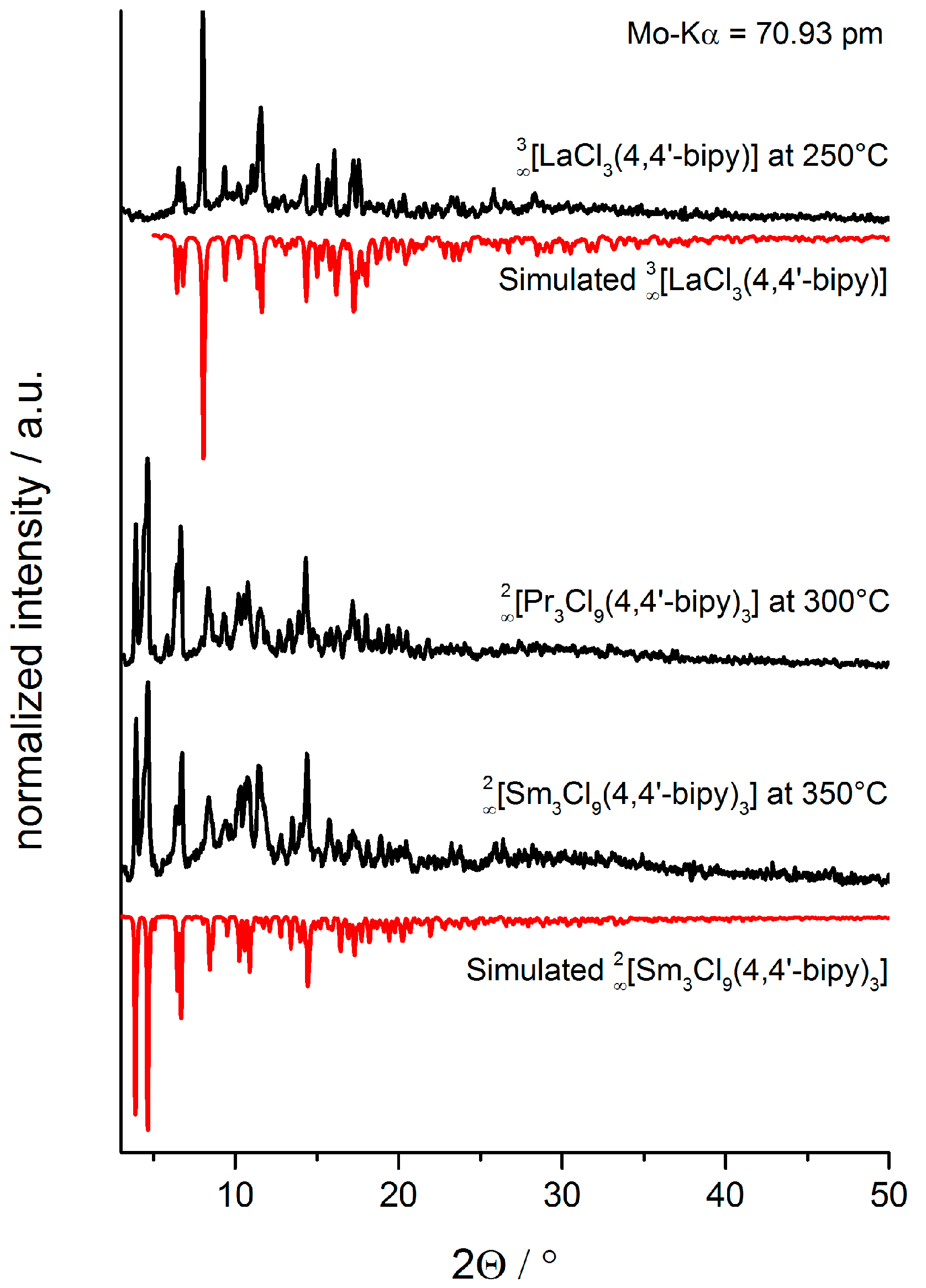

2.3. Synthesis of Single Crystalline High-Temperature Phases via a Solvothermal Approach
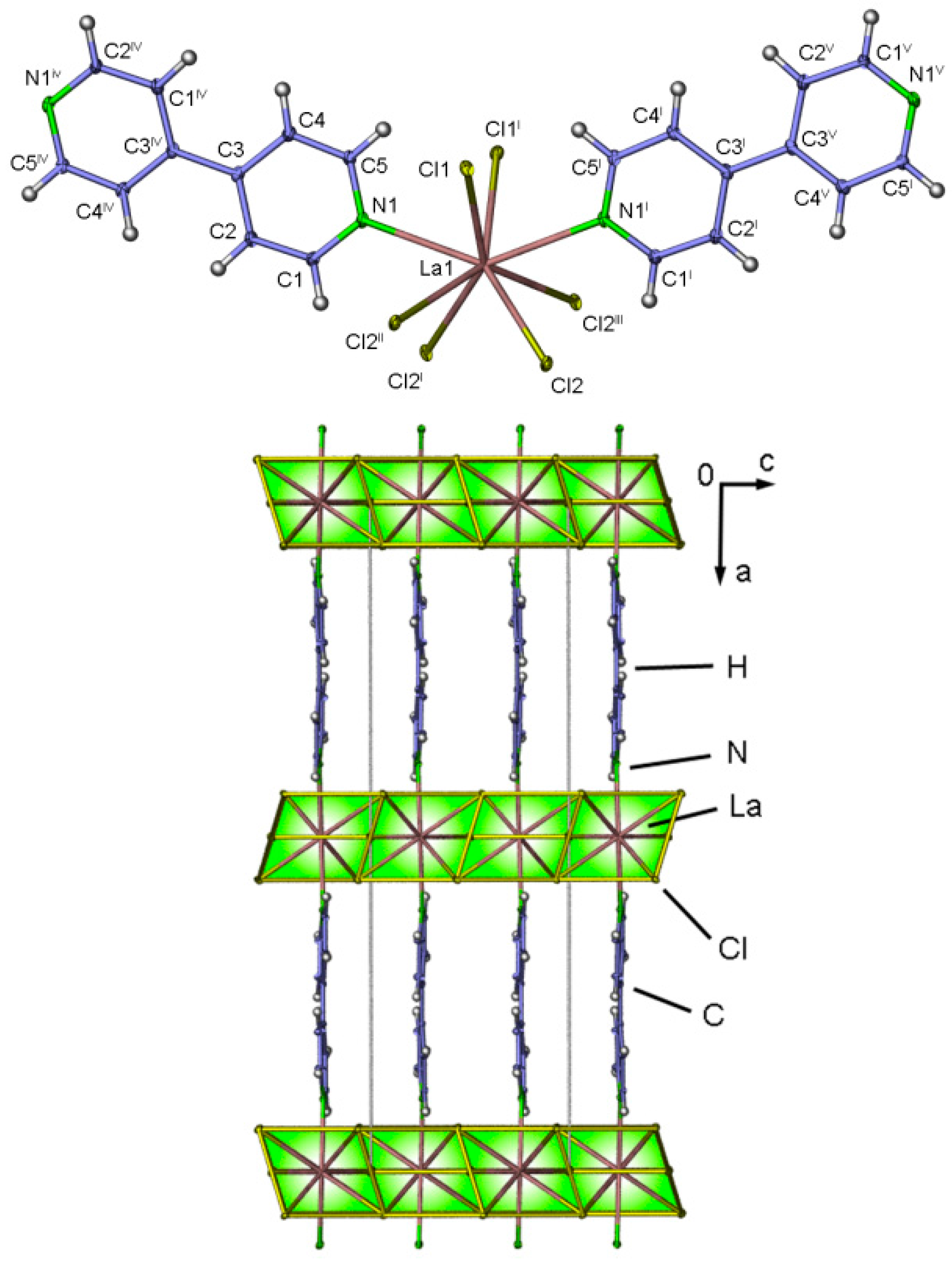
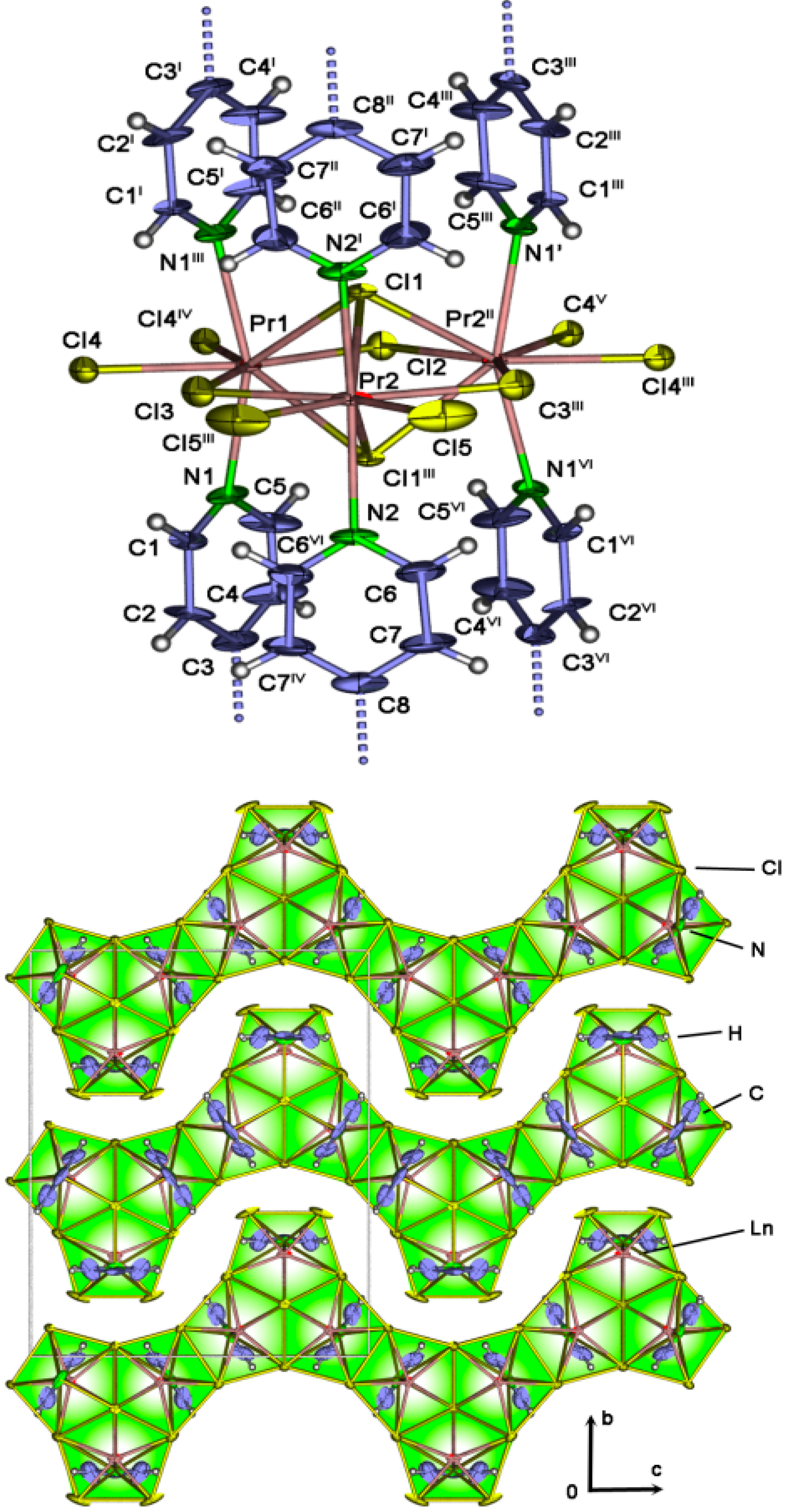
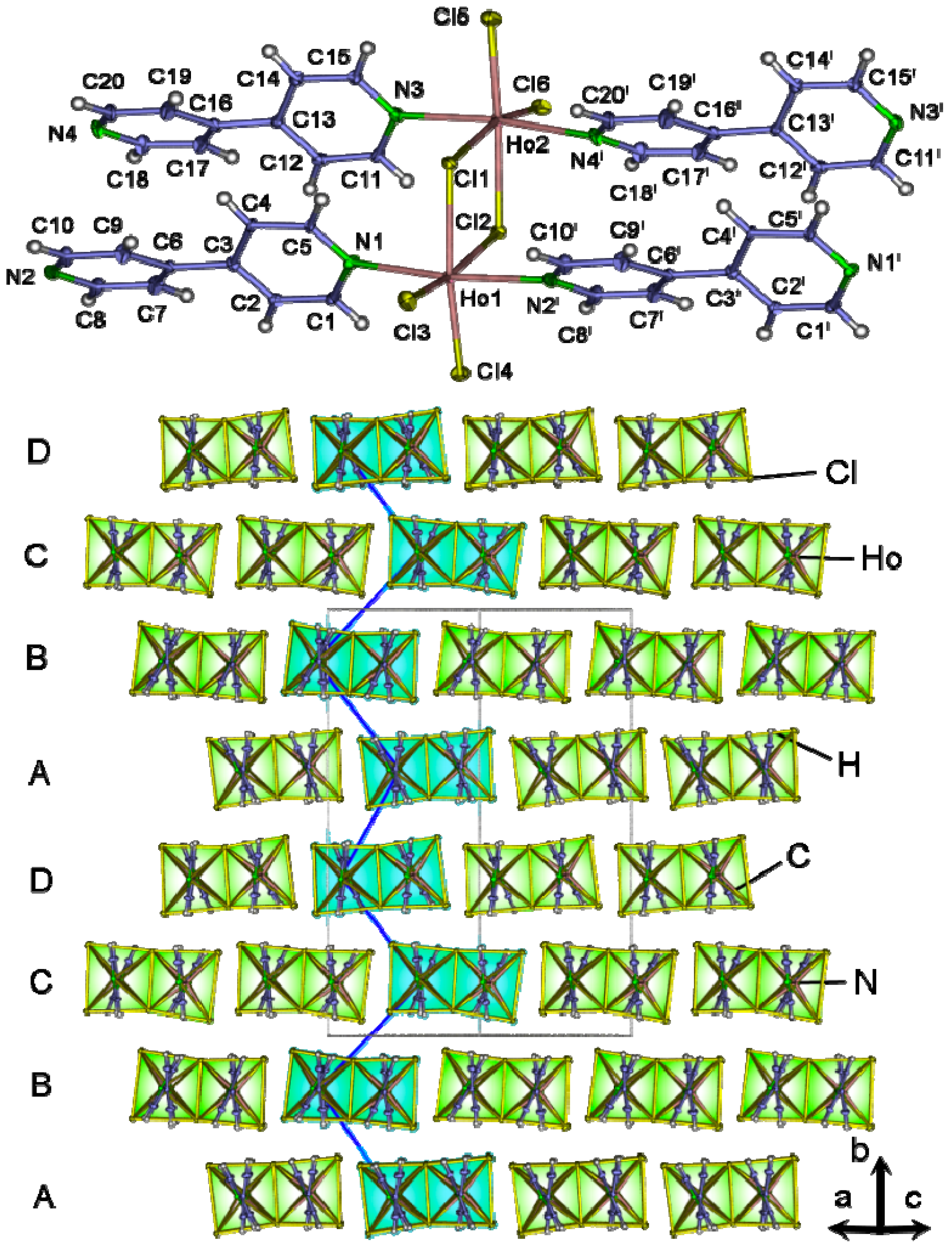
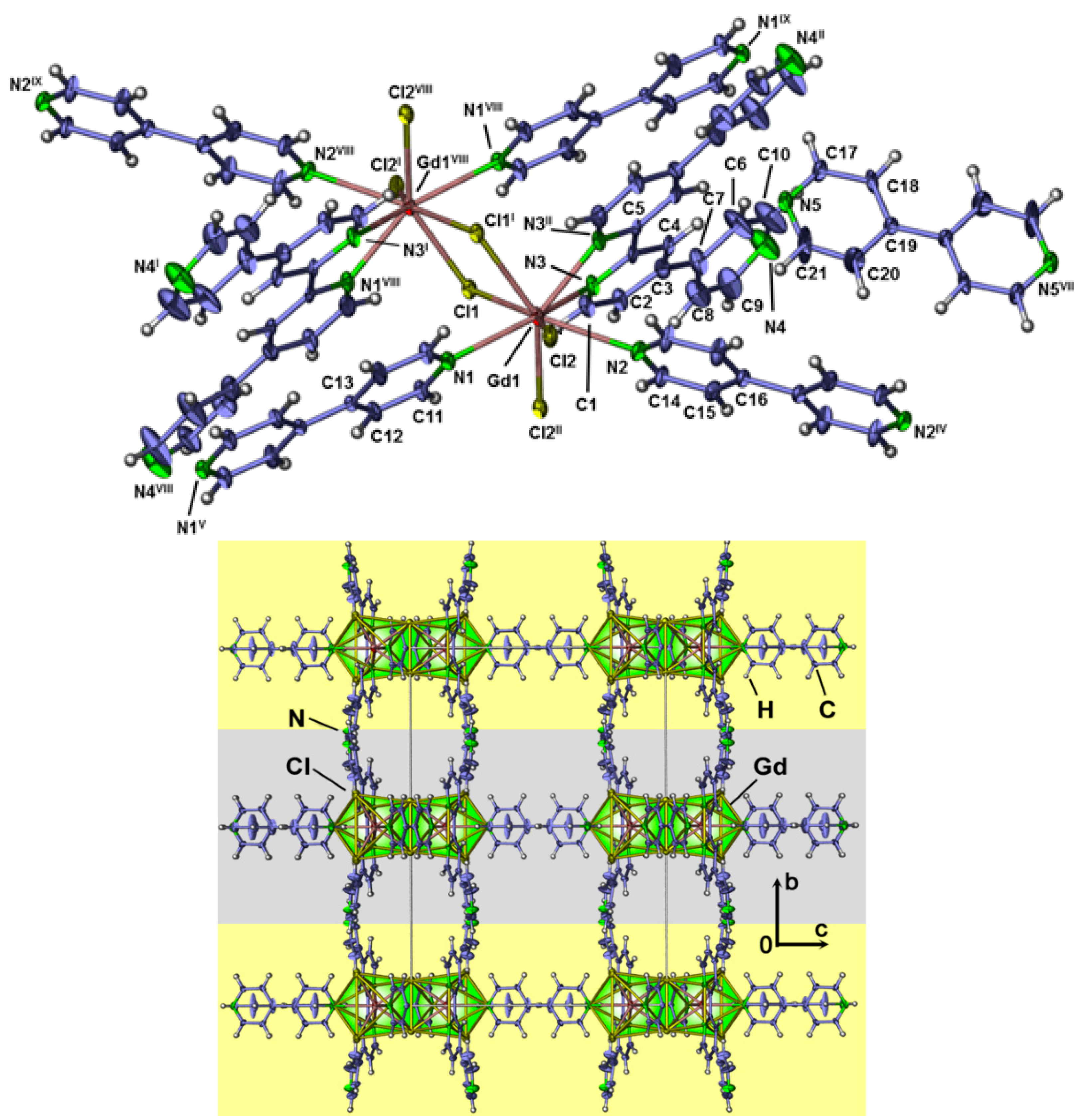
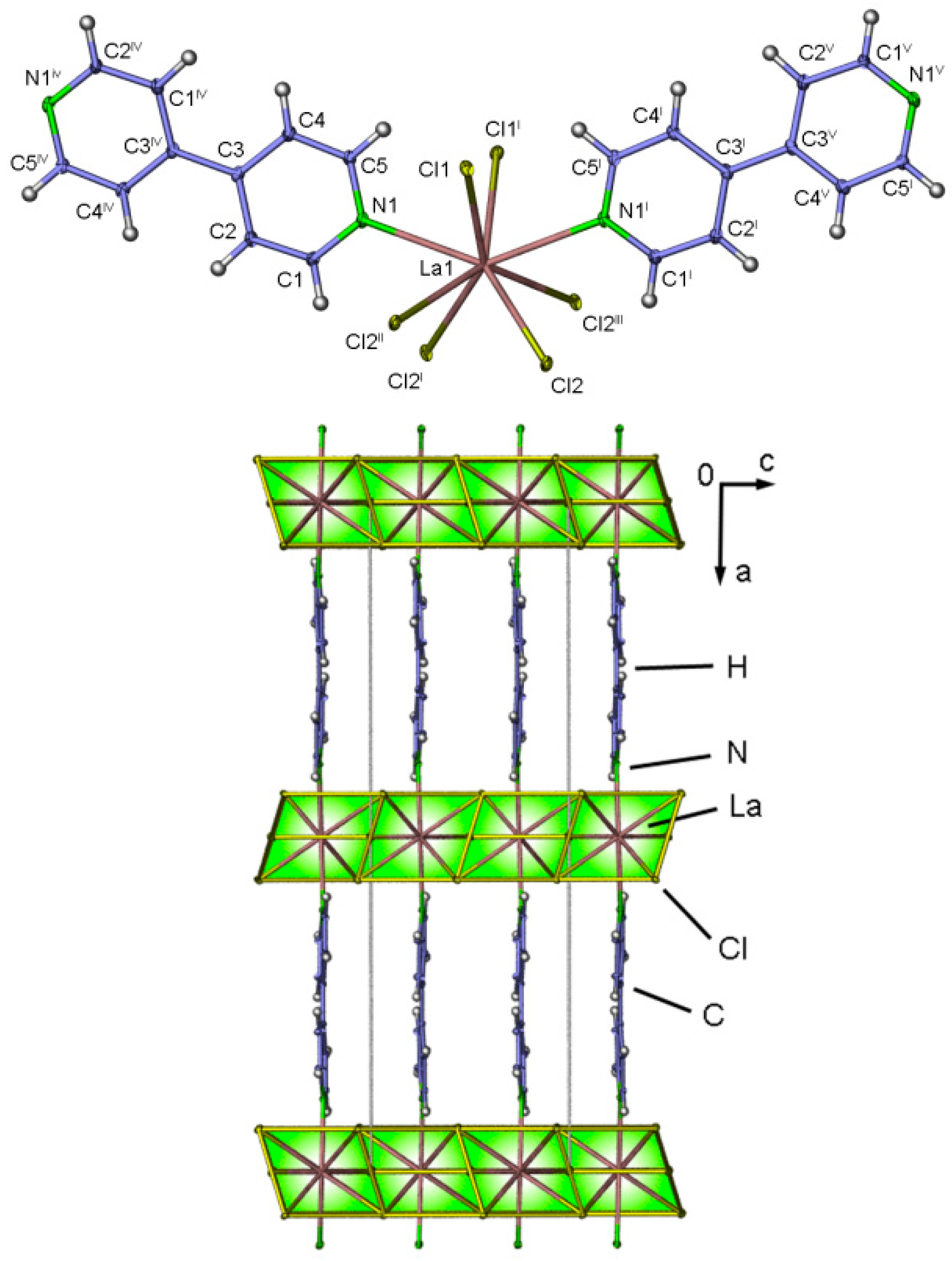
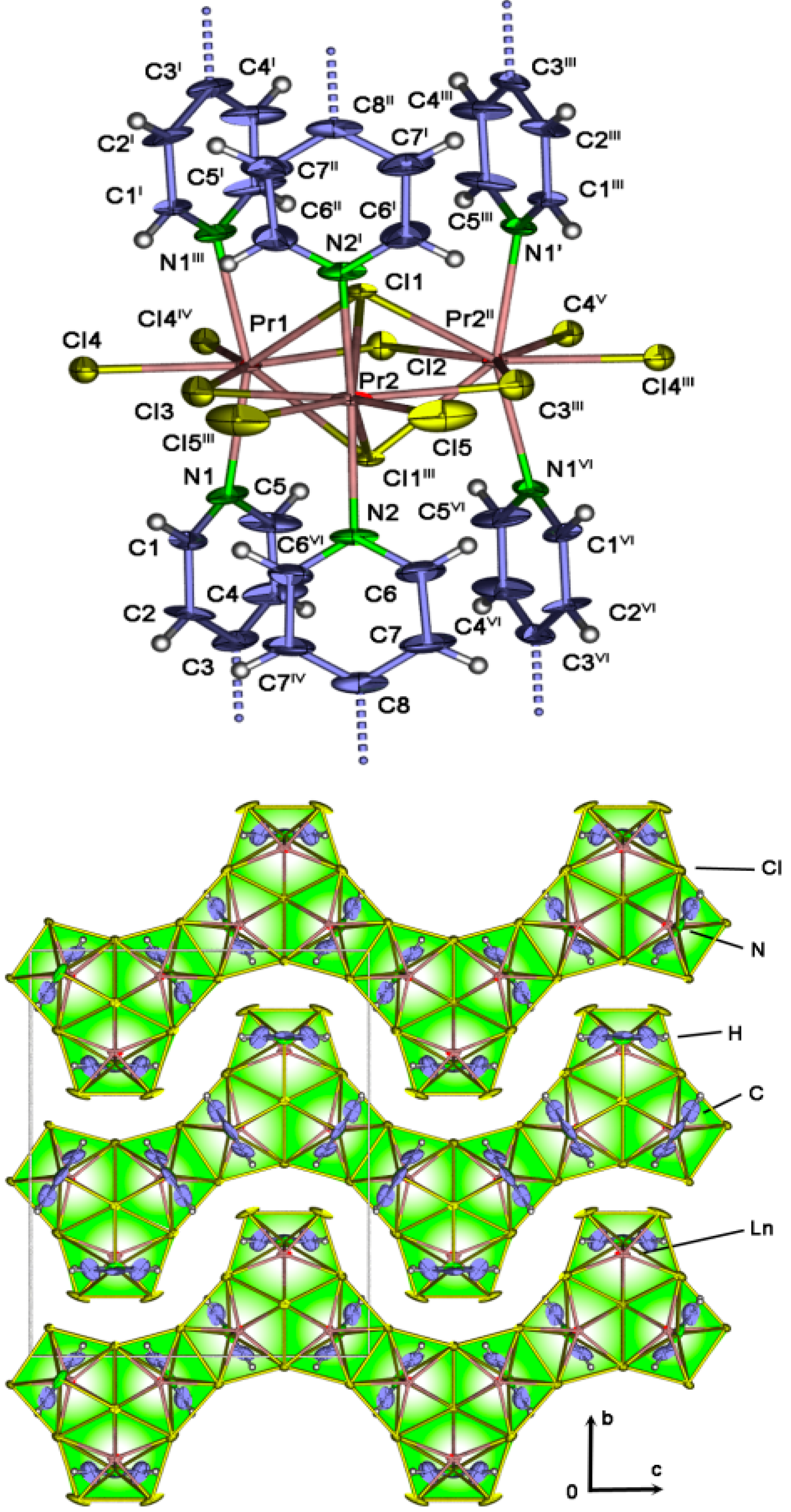
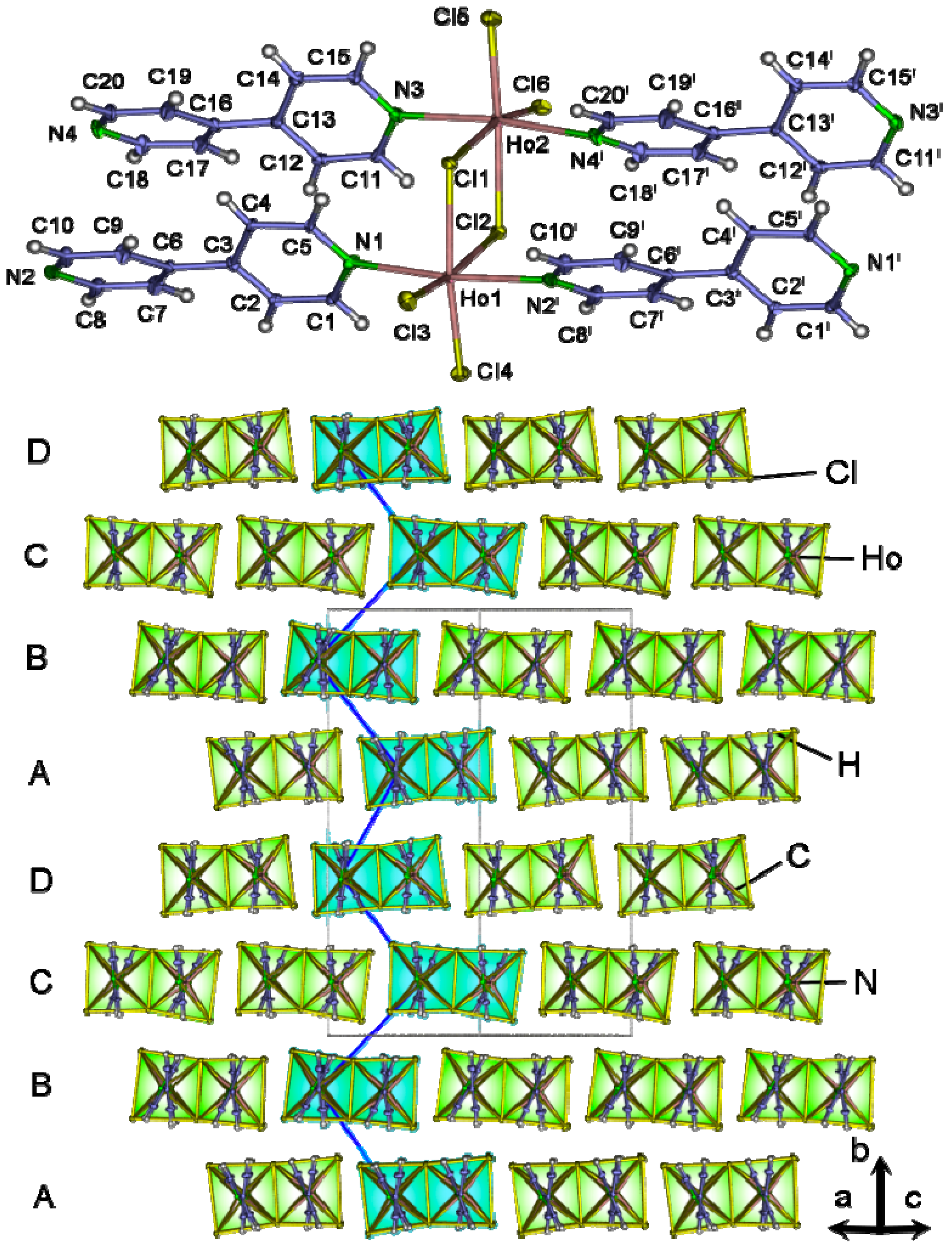
2.4. Crystal Structures of[LaCl3(bipy)] (2),[Ln3Cl9(bipy)3], Ln = Pr (8), Sm (9),[Ho2Cl6(bipy)2] (11), and[Gd2Cl6(qtpy)2(bipy)2]·bipy (12)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [LaCl3(bipy)] (2) | [Pr3Cl9(bipy)3] (8) | [Sm3Cl9(bipy)3] (9) | [Ho2Cl6(bipy)2] (11) | [Gd2Cl6(qtpy)2 (bipy)2]·bipy (12) | |
|---|---|---|---|---|---|
| Formula weight/g·mol−1 | 401.44 | 1210.36 | 1238.65 | 854.95 | 1608.40 |
| Crystal system | orthorhombic | orthorhombic | orthorhombic | monoclinic | monoclinic |
| Space group | Pcca | Cmcm | Cmcm | P21/c | C2/m |
| a/pm | 2387.4(5) | 1213.49(14) | 1206.99(5) | 988.4(2) | 1177.53(13) |
| b/pm | 744.9(2) | 2015.5(2) | 2015.93(9) | 2463.1(6) | 2351.7(2) |
| c/pm | 717.8(2) | 1606.2(2) | 1590.81(7) | 1137.2(3) | 1687.2 (2) |
| β/° | - | - | - | 112.142(7) | 107.710(3) |
| Volume/pm3 | 1276.6(4) | 3928.5(8)·106 | 3870.8(3)·106 | 2564.4(11)·106 | 4450.8(8)·106 |
| Z | 4 | ||||
| dc/g/cm3 | 2.089 | 2.0463 | 2.126 | 2.214 | 2.400 |
| Diffractometer | Bruker Apex II | SMART Bruker Apex | Bruker Apex II | Bruker Apex II | Bruker Apex II |
| Monochromator | Helios-mirror | Graphite | Helios-mirror | Helios-mirror | Helios-mirror |
| Radiation | 70.93 pm (Mo-Kα) | ||||
| Temperature/K | 100(3) | 168(5) | 100(3) | 100(3) | 100(3) |
| 2θ range | 3.42 ≤ 2θ ≤ 60.7° | 3.92 ≤ 2θ ≤ 60.08° | 3.94 ≤ 2θ ≤ 60.22° | 4.2 ≤ 2θ ≤ 61.82° | 2.54 ≤ 2θ ≤ 60.18° |
| μ/mm−1 | 3.946 | 4.305 | 5.144 | 6.768 | 1.697 |
| F000 | 761.5 | 2308.8 | 2340.0 | 1604.0 | 1584.0 |
| Reflections collected | 17347 | 22073 | 28523 | 39177 | 33891 |
| Independent reflections | 1834 | 3091 | 3023 | 7326 | 6484 |
| [R(int) = 0.0355] | [R(int) = 0.1876] | [R(int) = 0.0768] | [R(int) = 0.1042] | [R(int) = 0.2972] | |
| Data/restraints/parameters | 1834/0/74 | 3091/0/122 | 3023/0/123 | 7326/0/288 | 6484/0/225 |
| S | 1.143 | 1.001 | 1.077 | 1.018 | 1.083 |
| R1 for n reflections > [I ≥ 2σ (I)] [a] | 0.0255 | 0.0580 | 0.0356 | 0.0432 | 0.1241 [a] |
| R1[all data] [a] | 0.0372 | 0.0993 | 0.0541 | 0.0915 | 0.3063 [a] |
| wR2[all data] [b] | 0.0458 | 0.1341 | 0.0700 | 0.0789 | 0.3879 [b] |
| Largest diff. peak/hole/(e pm−3)·10−6 | 1.02/−1.33 | 2.78/−3.25 | 2.53/−1.08 | 3.69/−4.07 | 2.00/−2.05 |

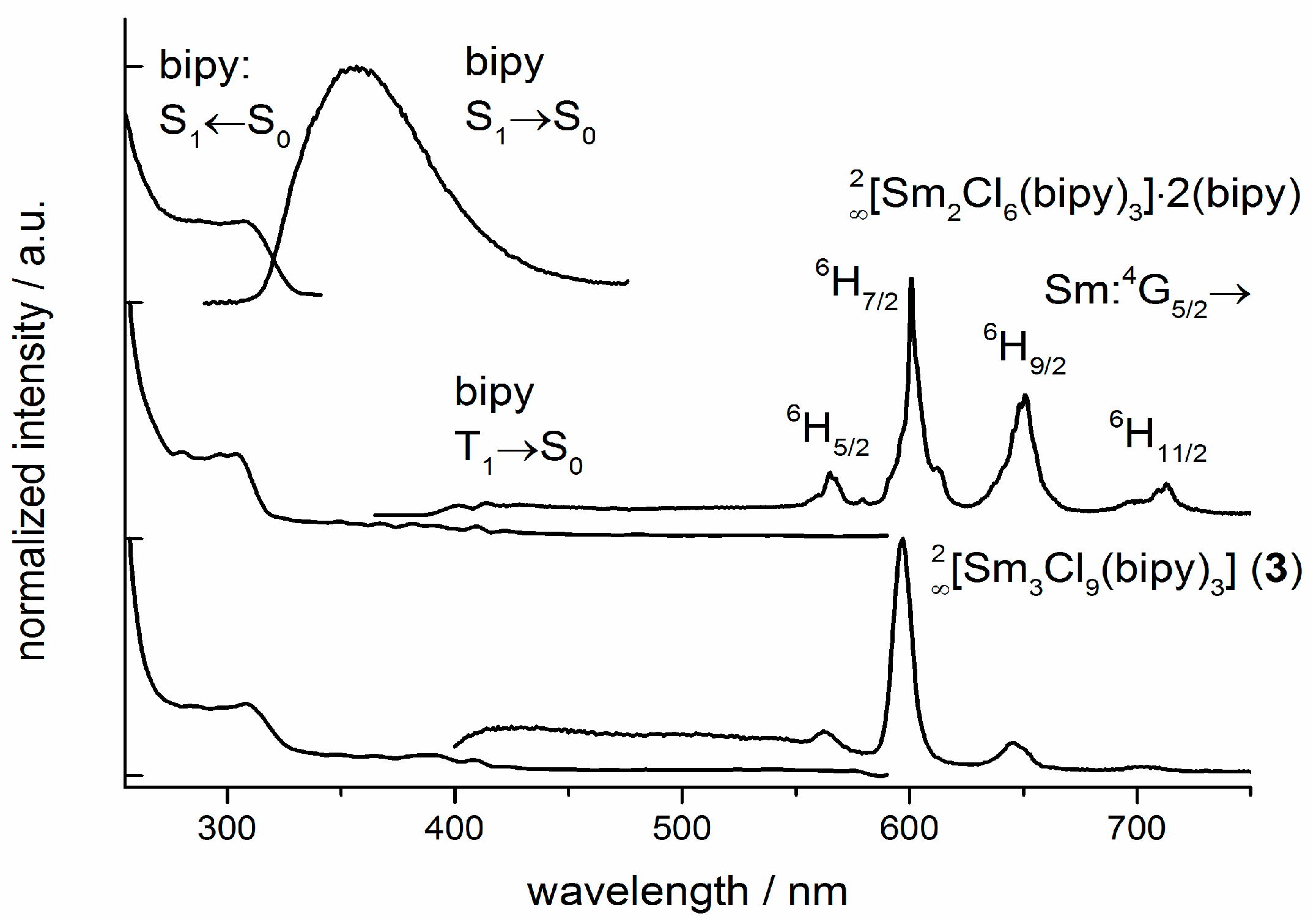
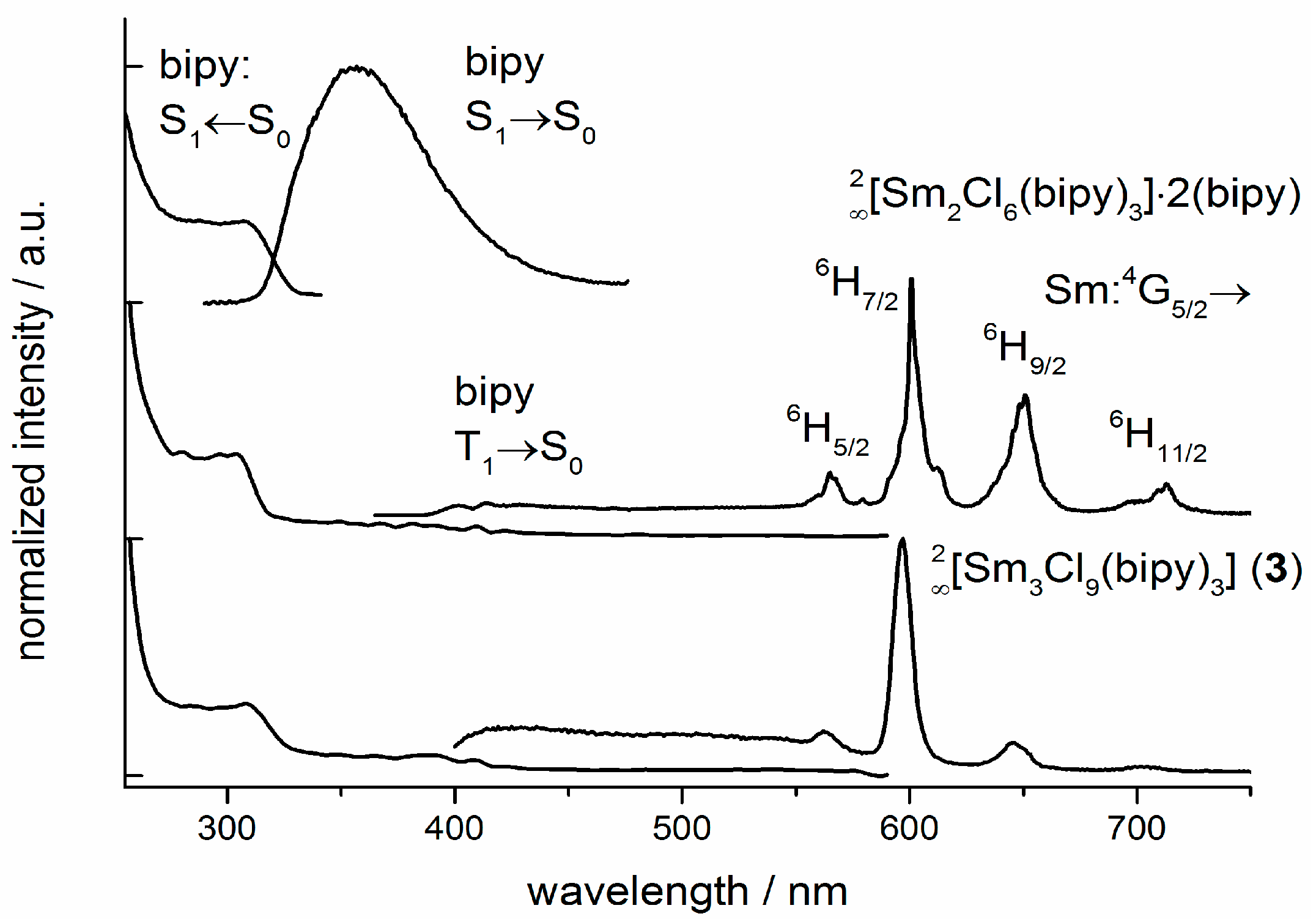
2.5. Photoluminescence and Vibrational Spectroscopy
3. Experimental Section
3.1. Synthesis
3.1.1. Synthesis of[La2Cl6(bipy)5]·4(bipy) (1),[Ln2Cl6(bipy)3]·2(bipy), Ln = Pr (4), Sm (5), and[Ln2Cl6(bipy)3], Ln = Pr (6), Sm (7), Eu (13), Eu/Tb (14)
3.1.2. Synthesis of[LaCl3(bipy)] (2)
3.1.3. Synthesis of[Pr3Cl9(bipy)3] (8)
3.1.4. Synthesis of[Sm3Cl9(bipy)3] (9)
3.1.5. Synthesis of[Ho2Cl6(bipy)2] (11)
3.1.6. Synthesis of[Gd2Cl6(qpy)2(bipy)2]·bipy (12)
3.2. Crystal Structure Determination
3.3. Powder X-ray Diffraction
3.4. Photoluminescence- and Vibrational Spectroscopy
3.5. Thermal and Elemental Analysis
3.6. Gas Adsorption Experiments
3.7. SEM and EDX Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kitagawa, S.; Kitaura, R.; Noro, S. Funktionale poröse Koordinationspolymere. Angew. Chem. 2004, 116, 2388–2430. [Google Scholar] [CrossRef]
- Janiak, C.; Vieth, J.K. MOFs, MILs and more: Concepts, properties and applications for porous coordination networks (PCNs). New J. Chem. 2010, 34, 2366–2388. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Metal-organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Janiak, C. Engineering coordination polymers towards applications. Dalton Trans. 2003, 14, 2781–2804. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Yaghi, O.M. Deconstructing the crystal structures of metal-organic frameworks and related materials into their underlying nets. Chem. Rev. 2012, 112, 675–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, S. Microporous lanthanide metal-organic frameworks. Rev. Inorg. Chem. 2012, 32, 81–100. [Google Scholar] [CrossRef]
- Kurmoo, M. Magnetic metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1353–1379. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.; Carlos, L.D.; Paz, F.A.A.; Ananias, D. Luminescent multifunctional lanthanides-based metal-organic frameworks. Chem. Soc. Rev. 2011, 40, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.; Müller-Buschbaum, K. Engineering metal-based luminescence in coordination polymers and metal-organic frameworks. Chem. Soc. Rev. 2013, 42, 9232–9242. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.U.; Trukhan, N.; Müller, U. Industrial applications of metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Lanthanide-based luminescent hybrid materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Duyne, R.P.V.; Hupp, J.T. Metal-organic framework materials as chemical sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Hupp, J.T. Rational design, synthesis, purification, and activation of metal-organic framework materials. Acc. Chem. Res. 2010, 43, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Li, H.; Yaghi, O.M. Highly porous and stable metal-organic frameworks: Structure design and sorption properties. J. Am. Chem. Soc. 2000, 122, 1391–1397. [Google Scholar] [CrossRef]
- Férey, G.; Serre, C. Large breathing effects in three-dimensional porous hybrid matter: Facts, analyses, rules and consequences. Chem. Soc. Rev. 2009, 38, 1380–1399. [Google Scholar] [CrossRef] [PubMed]
- Vittal, J.J. Supramolecular structural transformations involving coordination polymers in the solid state. Coord. Chem. Rev. 2007, 251, 1781–1795. [Google Scholar] [CrossRef]
- Kole, G.K.; Vittal, J.J. Solid-state reactivity and structural transformations involving coordination polymers. Chem. Soc. Rev. 2013, 42, 1755–1775. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.-C.; Schellenberg, I.; Pöttgen, R.; Müller-Buschbaum, K. MOFs by transformation of 1D-coordination polymers II: The homoleptic divalent rare-earth 3D-Benzotriazolate[Eu(Btz)2] initiating from[Eu(Btz)2(BtzH)2]. Z. Anorg. Allg. Chem. 2010, 636, 1720–1725. [Google Scholar] [CrossRef]
- Bhunia, M.K.; Hughes, J.T.; Fettinger, J.C.; Navrotsky, A. Thermochemistry of paddle-wheel MOFs: Cu-HKUST-1 and Zn-HKUST-1. Langmuir 2013, 29, 8140–8145. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lim, D.W.; Yang, W.S.; Oh, T.R.; Suh, M.P. A highly porous metal-organic framework: Structural transformations of a guest-free MOF depending on activation method and temperature. Chem. Eur. J. 2011, 17, 7251–7260. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Englert, U. Kristall-zu-Kristall-Umwandlung von einem Kettenpolymer zu einem zweidimensionalen Netz bei tiefen Temperaturen. Angew. Chem. 2005, 117, 2321–2323. [Google Scholar] [CrossRef]
- Hu, C.; Englert, U. Crystal-to-Crystal transformation from a chain polymer to a two-dimensional network at low temperatures. Angew. Chem. Int. Ed. 2005, 44, 2281–2285. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wang, Z.U.; Sun, L.B.; Chen, Y.P.; Zhou, H.C. Introduction of functionalized mesopores to metal-organic frameworks via metal-ligand-fragment coassembly. J. Am. Chem. Soc. 2012, 134, 20110–20116. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Jeon, H.J.; Jeung Ku Kang, J.K.; Yaghi, O.M. Heterogeneity within order in crystals of a porous metal-organic framework. J. Am. Chem. Soc. 2011, 133, 11920–11923. [Google Scholar] [CrossRef] [PubMed]
- Barin, G.; Krungleviciute, V.; Gutov, O.; Hupp, J.T.; Yildirim, T.; Farha, O.K. Defect creation by linker fragmentation in metal-organic frameworks and its effects on gas uptake properties. Inorg. Chem. 2014, 53, 6914–6919. [Google Scholar] [CrossRef] [PubMed]
- Karagiaridi, O.; Vermeulen, N.A.; Klet, R.C.; Wang, T.C.; Moghadam, P.Z.; Al-Juaid, S.S.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. Functionalized defects through solvent-assisted linker-exchange: Synthesis, Characterization, and Partial Postsynthesis Elaboration of a metal-organic framework containing free carboxylic acid moieties. Inorg. Chem. 2015, 54, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Vermoortele, F.; Bueken, B.; le Bars, G.; van de Voorde, B.; Vandichel, M.; Houthoofd, K.; Vimont, A.; Daturi, M.; Waroquier, M.; van Speybroeck, V.; et al. Synthesis Modulation as a tool to increase the catalytic activity of metal-organic frameworks: The unique case of UiO-66(Zr). J. Am. Chem. Soc. 2013, 135, 11465–11468. [Google Scholar] [CrossRef] [PubMed]
- Noei, H.; Amirjalayer, S.; Müller, M.; Zhang, X.; Schmid, R.; Muhler, M.; Fischer, R.A.; Wang, Y. Low-temperature CO-Oxidation over Cu-based metal-organic frameworks monitored by using FTIR spectroscopy. ChemCatChem 2012, 4, 755–759. [Google Scholar] [CrossRef]
- Kozachuk, O.; Luz, I.; Llabres i Xamena, F.X.; Noei, H.; Kauer, M.; Albada, H.B.; Bloch, E.D.; Marler, B.; Wang, Y.; Muhler, M.; et al. Multifunctional, defect-engineered metal-organic frameworks with ruthenium centres: sorption and catalytic properties. Angew. Chem. Int. Ed. 2014, 53, 7058–7062. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.; Kleist, W.; Baiker, A. Synthesis, structural properties, and catalytic behavior of Cu-BTC and mixed-linker Cu-BTC-PyDC in the oxidation of benzene derivatives. J. Catal. 2011, 281, 76–87. [Google Scholar] [CrossRef]
- Umemura, A.; Diring, S.; Furukawa, S.; Uehara, H.; Tsuruoka, T.; Kitagawa, S. Morphology design of porous coordination polymer crystals by coordination modulation. J. Am. Chem. Soc. 2011, 133, 15506–15513. [Google Scholar] [CrossRef] [PubMed]
- McGuire, C.V.; Forgan, R.S. The surface-chemistry of metal-organic frameworks. Chem. Commun. 2015, 51, 5199–5217. [Google Scholar] [CrossRef] [PubMed]
- Höller, C.J.; Mai, M.; Feldmann, C.; Müller-Buschbaum, K. The interaction of rare earth chlorides with 4,4ʹ-bipyridine for the reversible formation of template based luminescent Ln-N-MOFs. Dalton Trans. 2010, 39, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Matthes, P.R.; Höller, C.J.; Mai, M.; Heck, J.; Sedlmaier, S.J.; Schmiechen, S.; Feldmann, C.; Schnick, W.; Müller-Buschbaum, K. Luminescence tuning of MOFs via ligand to metal and metal to metal energy transfer by co-doping of[Gd2Cl6(bipy)3]∙2bipy with europium and terbium. J. Mater. Chem. 2012, 22, 10179–10187. [Google Scholar] [CrossRef] [Green Version]
- Höller, C.J.; Matthes, P.R.; Beckmann, J.; Müller-Buschbaum, K. MOF formation vs. reversible high ligand uptake in anhydrous halides: Two opposing aspects of[La2Cl6(4,4ʹ-bipy)5]∙4(4,4ʹ-bipy). Z. Anorg. Allg. Chem. 2010, 636, 395–399. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS-Academic; Coelho Software: Brisbane, Australia, 2007. [Google Scholar]
- Inc., A.S. Materials Studio v.5.5.0.0; Accelrys: San Diego, CA, USA, 2010. [Google Scholar]
- Masset, P. Thermochemical properties of lanthanides (Ln = La, Nd) and actinides (An = U, Np, Pu, Am) in the molten LiCl-KCl eutectic. J. Nuc. Mat. 2005, 344, 173–179. [Google Scholar] [CrossRef]
- Jantsch, G.; Gruibtsch, H.; Hoffmann, F.; Alber, H. Zur Kenntnis der Halogenide der Seltenen Erden. Über die Iodide der Ceriterdenelemente und die Neubestimmung der Schmelzpunkte der Chloride. Z. Anorg. Allg. Chem. 1930, 185, 49–53. [Google Scholar] [CrossRef]
- Jantsch, G.; Jawurek, H.; Skalla, N.; Gawalonski, H. Zur Kenntnis der Halogenide der Seltenen Erden. VI. Über die Halogenide der Terbin- und Erbinerdengruppe. Z. Anorg. Allg. Chem. 1932, 207, 353–367. [Google Scholar] [CrossRef]
- Matthes, P.R.; Nitsch, J.; Kuzmanoski, A.; Feldmann, C.; Marder, T.B.; Müller-Buschbaum, K. The series of rare earth complexes [Ln2Cl6(μ-4,4ʹ-bipy)(py)6], Ln = Y, Pr, Nd, Sm-Yb: A molecular model system for luminescence properties in MOFs based on LnCl3 and 4,4ʹ-bipyridine. Chem. Eur. J. 2013, 19, 17369–17378. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Baker, A.D. 2,2ʹ:4,4ʹʹ:4ʹ,4ʹʹʹ-quaterpyridyl: A building block for the preparation of novel redox agents. 1. Preparation and Quaternization. J. Org. Chem. 1990, 55, 1986–1993. [Google Scholar] [CrossRef]
- Burstall, F.H. Researches on the polypyridyls. J. Chem. Soc. 1938, 1662–1672. [Google Scholar] [CrossRef]
- Kende, A.S.; Lieberskind, L.S.; Braitsch, D.M. In situ generation of a solvated zerovalent nickel reagent. Tetrahedron Lett. 1975, 16, 3375–3378. [Google Scholar] [CrossRef]
- Höller, C.J.; Matthes, P.R.; Adlung, M.; Wickleder, C.; Müller-Buschbaum, K. Antenna- and metal-triggered luminescence in dense 1,3-benzodinitrile metal-organic frameworks[LnCl3(1,3-Ph(CN)2)], Ln = Eu, Tb. Eur. J. Inorg. Chem. 2012, 5479–5484. [Google Scholar] [CrossRef]
- Höller, C.J.; Müller-Buschbaum, K. The first dinitrile frameworks of the rare earth elements:[LnCl3(1,4-Ph(CN)2] and[Ln2Cl6(1,4-Ph(CN)2], Ln = Sm, Gd, Tb, Y; Access to novel metal-organic frameworks by solvent free synthesis in molten 1,4-benzodinitrile. Inorg. Chem. 2008, 47, 10141–10149. [Google Scholar] [CrossRef] [PubMed]
- Baisch, U.; Dell’Amico, D.B.; Calderazzo, F.; Conti, R.; Labella, L.; Marchetti, F.; Quadrelli, E.A. The mononuclear and dinuclear dimethoxyethane adducts of lanthanide trichlorides [LnCl3(DME)2]n, n = 1 or 2, fundamental starting materials in lanthanide chemistry: preparation and structures. Inorg. Chim. Acta 2004, 357, 1538–1543. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P. TOPOS 4.0; ToposPro: Samara, Russia, 2011. [Google Scholar]
- Liang, L.L.; Ren, S.B.; Zhang, J.; Li, Y.Z.; Du, H.B.; You, X.Z. Two unprecedented NLO-active coordination polymers constructed by a semi-rigid tetrahedral linker. Dalton Trans. 2010, 39, 7723–7726. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.M.; Singh, A.; Mehrotra, R.C.; Goel, S.C.; Chiang, M.Y.; Buhro, W.E. Synthesis, reactivity and X-ray crystallographic characterization of chloro(propan-2-ol)bis(tetraisopropoxoaluminato)praseodymium(III) dimer, [{Pr(Al(OPri)4]2(PriOH)(μ-Cl)}2]. J. Chem. Soc. Chem. Commun. 1992, 152–153. [Google Scholar] [CrossRef]
- Paraskevopoulou, P.; Makedonas, C.; Psaroudakis, N.; Mitsopoulou, C.A.; Floros, G.; Seressioti, A.; Ioannou, M.; Sanakis, Y.; Rath, N.; Garcia, C.J.G.; et al. Isolation, characterization, and computational studies of the novel [Mo3(μ3-Br)2(μ-Br)3Br6]2− cluster anion. Inorg. Chem. 2010, 49, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Fagin, A.A.; Bochkarev, M.N.; Kozimor, S.A.; Ziller, J.W.; Evans, W.J. Comparative reductive reactivity of SmI2 with TmI2 in the synthesis of lanthanide arene complexes. Z. Anorg. Allg. Chem. 2005, 631, 2848–2853. [Google Scholar] [CrossRef]
- Zhang, L.P.; Wan, Y.H.; Jin, L.P. Hydrothermal synthesis and crystal structures of three novel lanthanide coordination polymers with glutarate and 1,10-phenanthroline. J. Mol. Struct. 2003, 646, 169–178. [Google Scholar] [CrossRef]
- Evans, W.J.; Johnston, M.A.; Greci, M.A.; Ansari, M.A.; Brady, J.C.; Ziller, J.W. Synthesis of arene-soluble mixed-metal Zr/Ce, Zr/Y, and related {[Zr2(OiPr)9]LnX2}n. Inorg. Chem. 2000, 39, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Toma, M.; Sanchez, A.; Castellano, E.; Berdan, I.; Garcia-Tasende, M.S. The crystallographic study of the coordinative compounds of TiCl3 with nicotinamide and isonicotinamide. Rev. Chim. (Bucharest Rom.) 2003, 54, 476–479. [Google Scholar]
- Yamamotoa, Y.; Suzuki, H.; Tajima, N.; Tatsumi, K. Stepwise formation of quasi-octahedral macrocyclic complexes of rhodium (III) and iridium (III) bearing a pentamethylcyclopentadienyl group. Chem. Eur. J. 2002, 8, 372–379. [Google Scholar] [CrossRef]
- Wang, J.Q.; Ren, C.X.; Jin, G.X. Synthesis and structural characterization of macrocyclic half-sandwich rhodium(III) and iridium(III) complexes bearing bipyridyl derivatives and terephthalate. Organometallics 2006, 25, 74–81. [Google Scholar] [CrossRef]
- Song, X.M.; Hu, F.; Shi, H.T.; Chen, Q.; Zhang, Q.F. catena-Poly[[(triphenylphosphane-κP)silver(I)]-μ-4,4ʹ-bipyridine-κ2N:Nʹ-[(triphenylphosphane-κP)silver(I)]-di-μ-chlorido. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, m342. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.D.; Alcock, N.W.; Willey, G.R. A chloride-bridged chain polymer: Synthesis and X-ray structure of [GdCl(μ-Cl)2(H2O)2CH3CN]. Inorg. Chim. Acta 2003, 353, 276–279. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.; Zhang, H.; Li, C.; Sun, R.; Yang, Q. Study of structure and two-dimension correlation infrared spectroscopy on three rare earth/3-methylbenzoic acid complexes. J. Mol. Struct. 2009, 920, 441–449. [Google Scholar] [CrossRef]
- Figuerola, A.; Diaz, C.; El Fallah, M.S.; Ribas, J.; Maestro, M.; Mahía, J. Structure and magnetism of the first cyano-bridged hetero-one-dimensional GdIII-CrIII complexes. Chem. Commun. 2001, 1204–1205. [Google Scholar] [CrossRef]
- Ahmad, H.; Meijer, A.J.H.M.; Thomas, J.A. Tuning the excited state of photoactive building blocks for metal-templated self-assembly. Chem. Asian J. 2011, 6, 2339–2351. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, P.; Waywell, P.; Hanson, M.; Heath, S.L.; Meijer, A.J.H.M.; Teat, S.J.; Thomas, J.A. Self-assembled, kinetically locked, RuII-based metallomacrocycles: Physical, structural, and modeling studies. Chem. Eur. J. 2006, 12, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Das, M.; Chatterjee, S.; Chattopadhyay, S. Synthesis and characterization of two new nickel(II) complexes with azide: Formation of a two-dimensional coordination polymer with 63-hcb topology. Polyhedron 2014, 68, 205–211. [Google Scholar] [CrossRef]
- Wang, P.; Fan, R.Q.; Yang, Y.L.; Liu, X.R.; Xiao, P.; Li, X.Y.; Hasi, W.; Cao, W.W. 1-D helical chain, 2-D layered network and 3-D porous lanthanide-organic frameworks based on multiple coordination sites of benzimidazole-5,6-dicarboxylic acid: Synthesis, crystal structure, photoluminescence and thermal stability. Cryst. Eng. Commun. 2013, 15, 4489–4506. [Google Scholar] [CrossRef]
- Weissman, S.I. Intramolecular energy transfer: The fluorescence of complexes of europium. J. Chem. Phys. 1942, 10, 214–217. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.C.G. Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.C.G. Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 2010, 110, 2729–2755. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, S.V.; Bünzli, J.-C.G. Rare earths: Jewels for functional materials of the future. New J. Chem. 2011, 35, 1165–1176. [Google Scholar] [CrossRef]
- Topacli, A.; Akyüz, S. 4,4ʹ-Bipyridyl: Vibrational assignments and force field. Spectrochim. Acta 1994, 51, 633–641. [Google Scholar] [CrossRef]
- Czakis-Sulikowska, D.; Radwanska-Doczekalska, J. Synthesis and properties of 4,4ʹ-bipyridine complexes with some lanthanides. Rocz. Chem. 1975, 49, 197–203. [Google Scholar]
- Gill, N.S.; Nuttall, R.H.; Scaife, D.E.; A. Sharp, D.W. The infra-red spectra of pyridine complexes and pyridinium salts. J. Inorg. Nucl. Chem. 1961, 18, 79–87. [Google Scholar] [CrossRef]
- Corrsin, L.; Fax, B.J.; Lord, R.C. The vibrational spectra of pyridine and pyridine-d5. J. Chem. Phys. 1953, 21, 1170–1176. [Google Scholar] [CrossRef]
- Kline, C.H.; Turkevich, J. The vibrational spectrum of pyridine and the thermodynamic properties of pyridine vapors. J. Chem. Phys. 1944, 12, 300–309. [Google Scholar] [CrossRef]
- Meyer, G. The ammonium chloride route to anhydrous rare earth chlorides—The example of YCl3. Inorg. Synth. 1989, 25, 146–150. [Google Scholar]
- Bruker. SMART Apex Suite; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Babour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Stoe. In WIN-X-POW; Stoe & Cie GmbH: Darmstadt, Germany, 2007.
- Coelho, A. TOPAS-Academic, version 4.1; Coelho Software: Brisbane, Australia, 2007. [Google Scholar]
- Sample Availability: Samples of the compounds 1, 2, 4–9 and 13, 14 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthes, P.R.; Schönfeld, F.; Zottnick, S.H.; Müller-Buschbaum, K. Post-Synthetic Shaping of Porosity and Crystal Structure of Ln-Bipy-MOFs by Thermal Treatment. Molecules 2015, 20, 12125-12153. https://doi.org/10.3390/molecules200712125
Matthes PR, Schönfeld F, Zottnick SH, Müller-Buschbaum K. Post-Synthetic Shaping of Porosity and Crystal Structure of Ln-Bipy-MOFs by Thermal Treatment. Molecules. 2015; 20(7):12125-12153. https://doi.org/10.3390/molecules200712125
Chicago/Turabian StyleMatthes, Philipp R., Fabian Schönfeld, Sven H. Zottnick, and Klaus Müller-Buschbaum. 2015. "Post-Synthetic Shaping of Porosity and Crystal Structure of Ln-Bipy-MOFs by Thermal Treatment" Molecules 20, no. 7: 12125-12153. https://doi.org/10.3390/molecules200712125