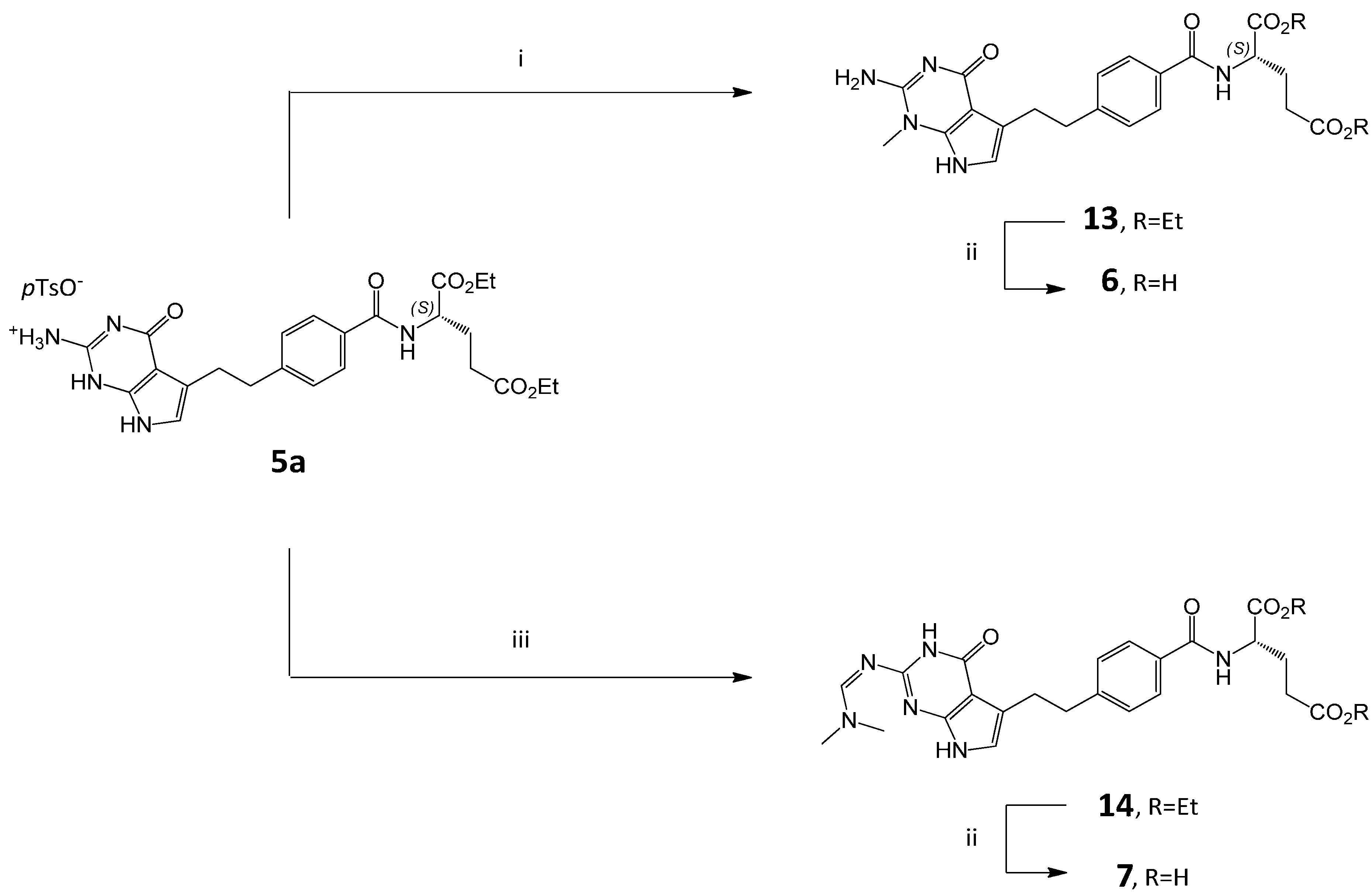
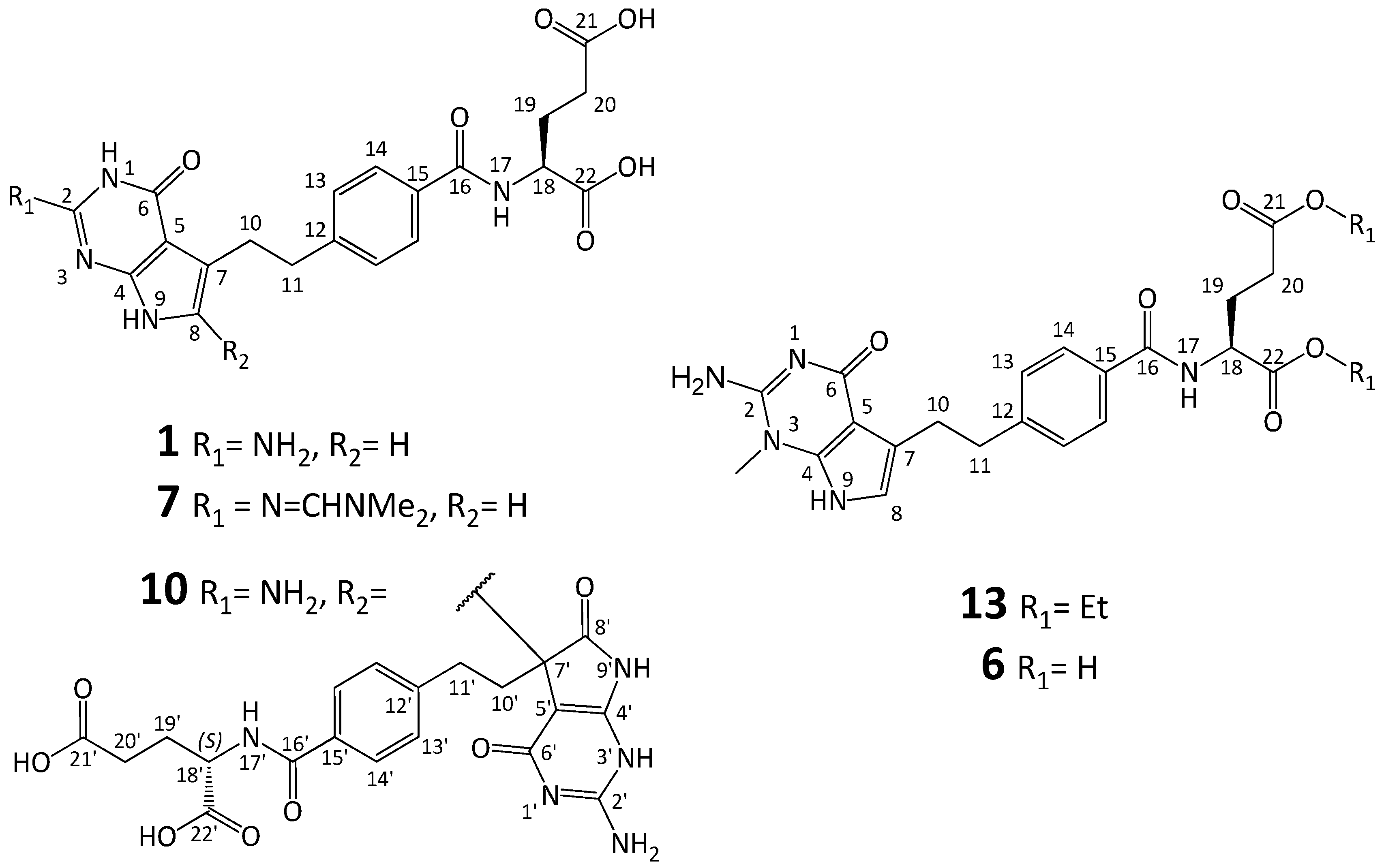
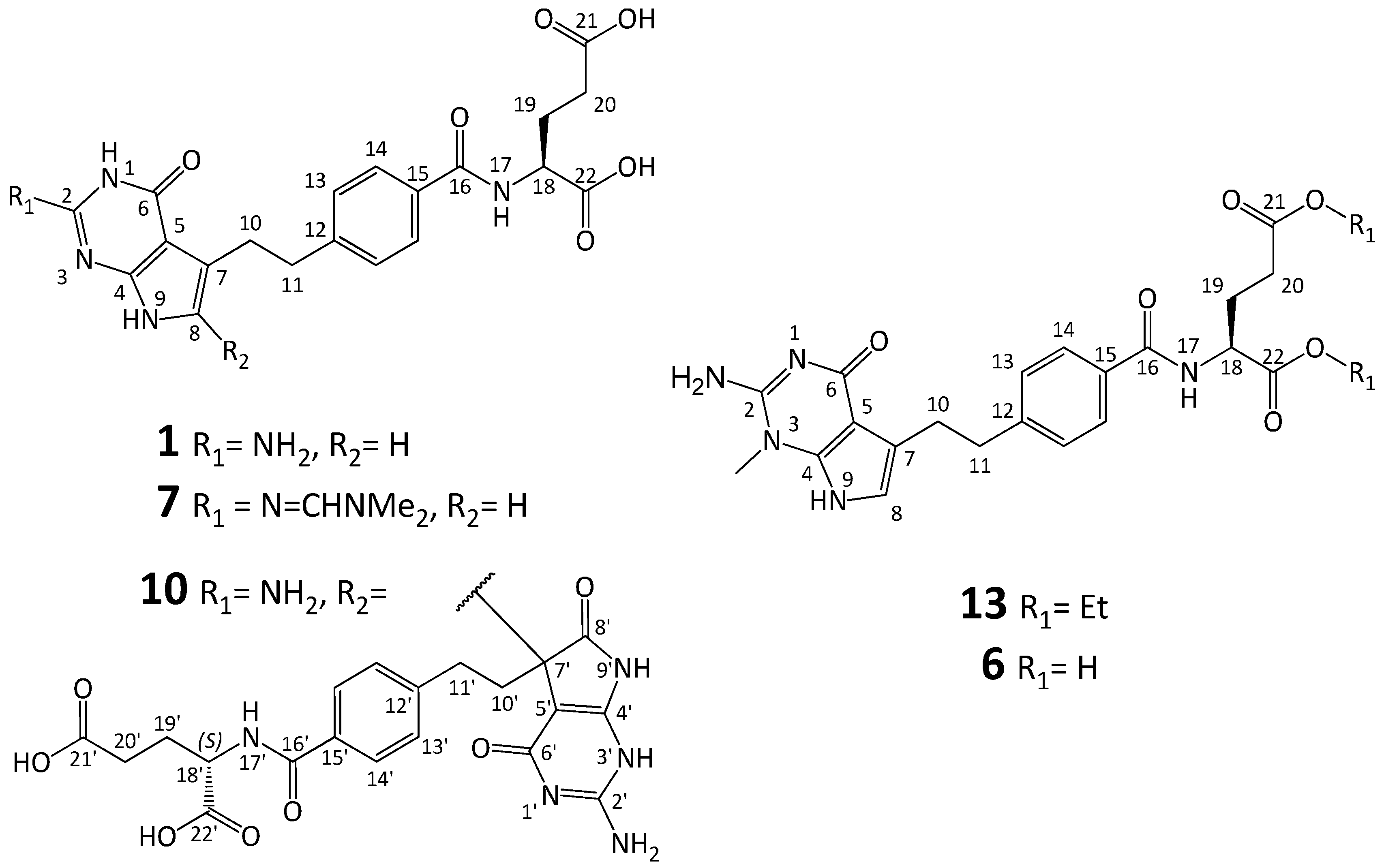
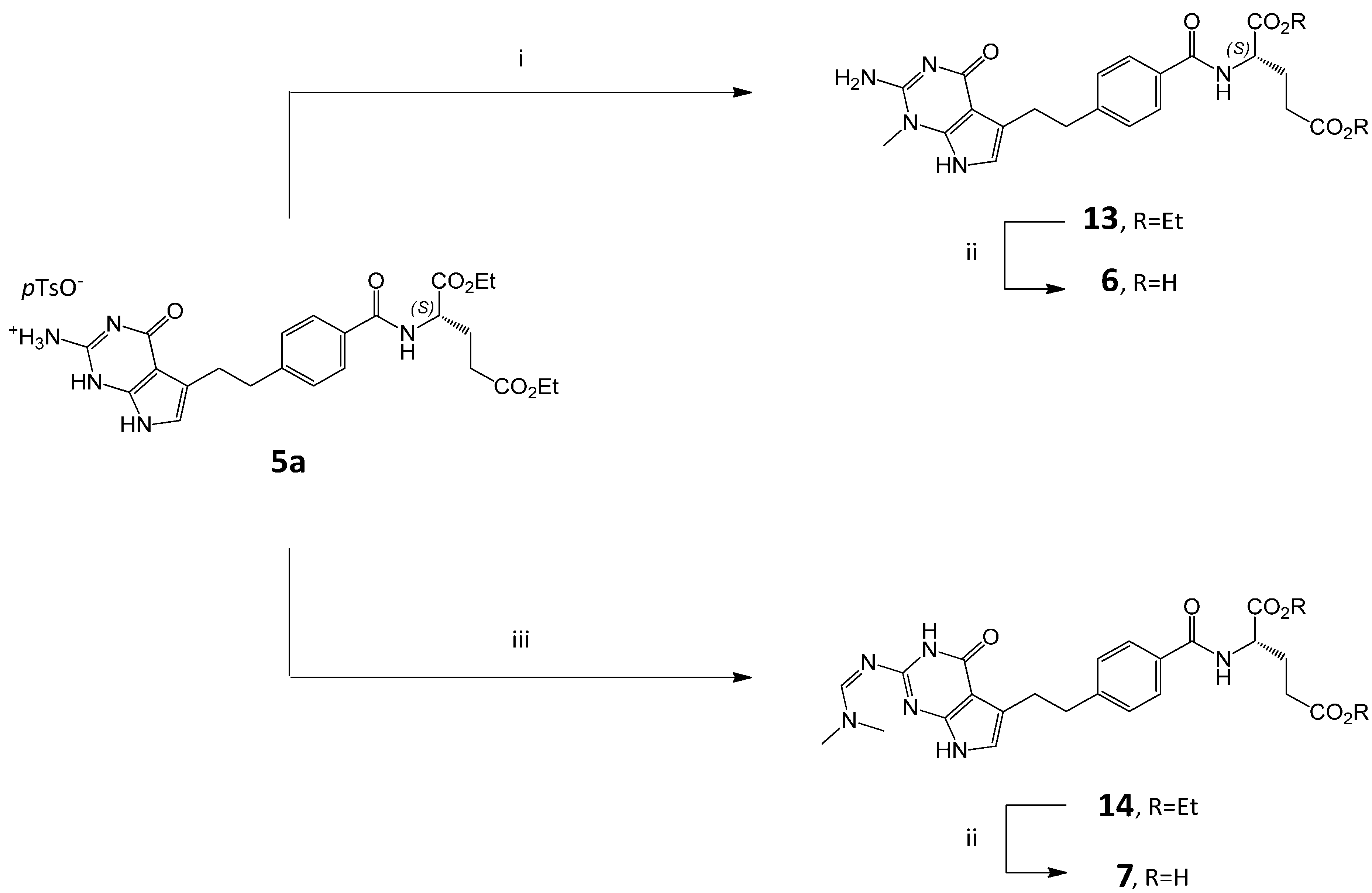
3.2. Synthesis of Impurity 6
3.2.1. (2S)-2-[[4-[2-(2-Amino-1-methyl-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioic Acid Diethyl Ester (13)
To the solution of 5a (6.0 g, 9.16 mmol) in DMF (40 mL) triethylamine was added (3.20 mL, 22.96 mmol), followed by methyl iodide (2.06 mL, 33.09 mmol) and the solution was left at room temperature for 72 h. Then CH2Cl2 (80 mL) and water (80 mL) were added. The layers were separated and the aqueous layer was extracted with CH2Cl2 (1 × 40 mL).The combined organic layers were dried over anhydrous MgSO4 and concentrated. The residue was dissolved in MeOH (10 mL) and iPr2O (50 mL) was added. The resulted precipitate was filtered, washed with iPr2O (2 × 10 mL), and dried to give 13 (4.0 g, 88%).
TLC: RF = 0.18 (CHCl3/MeOH/Et3N 8:2:1)
1H-NMR: δ: 8.65 (1H, d, J = 7.5 Hz, N17-H), 7.81 (2H, d, J = 8.0 Hz, H14), 7.32 (2H, d, J = 8.0 Hz, H13), 6.64 (1H, s, H8), 4.44 (1H, m, H18), 4.11 (2H, m, -CH2CH3 at C22), 4.05 (2H, q, J = 7.1 Hz, -CH2CH3 at C21), 3.56 (3H, s, CH3 group at N3), 3.00 (2H, m, H11), 2.95 (2H, m, H10), 2.44 (2H, m, H20), 2.12 and 2.02 (2H, 2 × m, both H19 protons), 1.19 and 1.17 (2 × 3H, 2 × q, J = 7.1 Hz, both CH3 groups of -CH2CH3 at C22 and C21);
13C-NMR: δ: 172.18 (CO, C21), 171.82 (CO, C22), 166.58 (CO, C16), 163.26 (probably C6), 152.11 (probably C2), 145.81 (C12), 139.31 (probably C4), 131.19 (C15), 128.17 (C13), 127.46 (C14), 119.69 (C7), 114.89 (C8), 100.31 (C5), 60.53 (-CH2CH3 at C22), 59.92 (-CH2CH3 at C21), 51.95 (C18), 35.79 (C11), 32.45 (CH3 group at N3), 30.17 (C20), 27.13 (C10), 25.69 (C19), 14.1 (2 × CH3 groups of -CH2CH3 at C22 and C21);
15N-NMR δ: −274.2 (N3), −266.8 (N17), −243.6 (N9), the 15N-NMR signals of NH2 at C2 and N1 not recorded in the 1H-15N g-HSQC/HMBC experiments.
HRMS: calcd for C25H32N5O6 m/z = 498.2353, found m/z = 498.2346.
3.2.2. (2S)-2-[[4-[2-(2-Amino-1-methyl-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioic Acid (6)
Compound 13 (3.50 g, 7.03 mmol) was treated with 1M NaOHaq (30 mL) and stirred at RT for 2 h. The reaction mixture was diluted with EtOH (30 mL) and water (30 mL) and adjusted to pH 3.0 with 1 M HCl. The resulting slurry was heated to 60–65 °C and then cooled to RT. The solid was filtered, washed with EtOH (2 × 10 mL) and dried in vacuo at 40 °C for 24 h. Crude 6 was purified by flash chromatography on a silica gel column using CH2Cl2/MeOH/H2O/NH3 as the mobile phase (40:40:5:2 v/v). The respective fractions were collected and concentrated. The residue was dissolved in water (50 mL) and the pH was adjusted to 2–3 with 1 M HCl. EtOH (220 mL) was added and stirred for 30 min. The suspension was filtered and the solid was washed with EtOH/H2O (20 mL) and dried at 40 °C to obtain 6 (2.51 g, 81%, HPLC purity 99.05%).
TLC: RF = 0.44, (CHCl3/MeOH/NH3, 2:2:1).
Mp. 270 °C;
= +10.03 (c = 1, DMSO);
1H-NMR (353 K) δ: 8.12 (1H, s, N17-H), 7.75 (2H, m, H14), 7.28 (2H, m, H13), 6.42 (1H, s, H8), 4.39 (1H, m, H18), 3.49 (3H, s, CH3 at N3), 2.99 (2H, m, H11), 2.94 (2H, m, H10), 2.34 (2H, m, H20), 2.08 and 2.00 (2H, m, H19);
13C-NMR (353 K) δ: 173.54 (CO, C21), 173.06 (CO, C22), 165.85 (CO, C16), 163.75 (C6), 152.04 (C2), 145.47 (C12), 139.03 (C4), 131.48 (C15), 127.72 (C13), 126.75 (C14), 119.43 (C7), 113.18 (C8), 100.94 (C5), 52.06 (C18), 35.63 (C11), 31.53 (N3-CH3), 30.59 (C20), 26.77 (C10), 26.41 (C19);
HRMS: calcd for C21H24N5O6 m/z = 442.1727, found m/z = 442.1724.
FT-IR: [cm−1] 3226–3127 (N-Hν, O-Hν); 2926 (C-Hν); 1682–1640 (C=Oν, C=Nν); 1612; 1504 (C=Cν); 1543 (N-Hδ, C=Nν); 1450–1402 (C-Hδ, C-Nν); 1236 (C-Oν); 697 (C-Hγ, N-Hγ).
3.3. Synthesis of Impurity 7
3.3.1. (2S)-2-[[4-[2-(2-(Dimethylamino)methyleneamino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioic Acid Diethyl Ester (14)
Compound 5a (6.5 g, 9.9 mmol) was suspended in N,N-dimethylformamide dimethyl acetal (DMF-DMA, 56 mL) and anhydrous DMF (18 mL) and stirred at RT for 3 days (TLC control). The reaction mixture was poured into water (100 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried over anhydrous Na2SO4 overnight, then filtered and concentrated to a thick liquid mass. Water (100 mL) was added to the mass and stirred (the use of a mechanical stirrer or a rotary evaporator is recommended) for 4 h at RT. The solid precipitate was filtered off and washed with water (25 mL), then dried. The solid was dissolved in MeOH (30 mL) in 60–65 °C and iPr2O (250 mL) was slowly added (over 45–60 min), then the mixture was cooled and stirred for 1 h at RT. The obtained precipitate was filtered and washed with iPr2O (2 × 15 mL) and dried under vacuum at 40 °C to afford 14 (4.37 g, 81.5%).
TLC: RF = 0.78 (CHCl3/MeOH, 8:2);
Mp. 193 °C;
1H-NMR δ: 10.82 (1H, s, N9-H), 10.77 (1H, s, N1-H), 8.63 (1H, d, J = 7.4 Hz), 8.49 (1H, s, -N=CH-N(CH3)2), 7.79 (2H, m, H14), 7.31 (2H, m, H13), 6.46 (1H, s, H8), 4.43 (1H, m, H18), 4.11 (2H, m, CH2 of -CH2CH3 at C22), 4.05 (2H, q, J = 7.1 Hz, CH2 of -CH2CH3 at C21), 3.12 (3H, s, one of the CH3 group of -N=CH-N(CH3)2), 3.01 (5H, m, one of the CH3 group of -N=CH-N(CH3)2) and both H11), 2.92 (2H, m, both H10), 2.44 (2H, m, both H20), 2.11 and 2.01 (2H, 2 × m, both H19), 1.19 (3H, s, CH3 of -CH2CH3 at C22) and 1.17 (3H, s, CH3 of -CH2CH3 at C21);
13C-NMR δ: 172.20 (CO, C21), 171.82 (CO, C22), 166.62 (CO, C16), 160.11 (probably C4), 156.91 (-N=CH-N(CH3)2), 155.52 (C2), 150.06 (probably C6), 146.28 (C12), 131.10 (C15), 128.18 (C13), 127.39 (C14), 117.73 (C7), 114.93 (C8), 101.64 (C5), 60.51 (CH2 of -O-CH2CH3 at C22), 59.90 (CH2 of -CH2CH3 at C21), 51.95 (C18), 40.44 (one of CH3 group of -N=CH-N(CH3)2), 36.17 (C11), 34.43 (one of CH3 group of -N=CH-N(CH3)2), 30.17 (C20), 27.89 (C10), 25.70 (C19), 14.05 (both CH3 groups of -CH2CH3 at C22 and C21);
15N-NMR δ: −281.3 (-N=CH-N(CH3)2), −266.9 (N17), −240.0 (N9), −175.8 (-N=CH-N(CH3)2. N1 and N3 not recorded in the 1H-15N g-HMBC experiment;
HRMS: calcd for C27H35N6O6 m/z = 539.2618, found m/z = 539.2610.
3.3.2. (2S)-2-[[4-[2-(2-(Dimethylamino)methyleneamino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioic Acid (7)
Compound 14 (0.77 g, 1.43 mmol) was treated with NaOHaq (0.25 g in 11 mL H2O), the mixture was cooled to 0–5 °C and stirred for 15 min. EtOH (11 mL) was added to the solution and stirred for 1 h at 0–5 °C. The pH was adjusted to 3 with 1N HCI, then EtOH was evaporated. The suspension was filtered and the solid was washed with EtOH:H2O (1:1, 1 × 5 mL) and dried at 40 °C to obtain 7 (0.41 g, 70.0%, HPLC purity 80.0%).
TLC: RF = 0.49 (CHCl3/MeOH, 1:2);
1H-NMR (600 MHz, DMSO): 12.4 (2H, broad s, probably protons of both COOH groups), 10.82 (1H, d, J = 1.8 Hz, N9-H), 10.78 (1H, s, N1-H), 8.51 (1H, d, J = 7.7 Hz, N17-H), 8.49 (1H, s, -N=CH-N(CH3)2), 7.80 (2H, m, H14), 7.31 (2H, m, H13), 6.47 (1H, d, J = 1.8 Hz, H8), 4.41 (1H, m, H18), 3.11 (3H, s, one of the CH3 groups of -N=CH-N(CH3)2), 3.01 (5H, m, one of the CH3 groups of -N=CH-N(CH3)2 and both protons at C11), 2.92 (2H, m, H10), 2.36 (2H, t, J = 7.5 Hz, C20), 2.10 (1H, m, one of the protons at C19), 1.96 (1H, m, one of the protons at C19);
13C-NMR (150 MHz, DMSO): 173.92 (CO, C21), 173.50 (CO, C22), 166.52 (CO, C16), 160.16 (probably C6), 156.94 (-N=CH-N(CH3)2), 155.55 (C2), 150.10 (probably C4), 146.17 (C12), 131.34 (C15), 128.20 (C13), 127.38 (C14), 117.76 (C7), 115.00 (C8), 101.67 (C5), 51.91 (C18), 40.47 (one of the CH3 groups of -N=CH-N(CH3)2), 36.20 (C11), 34.47 (one of the CH3 groups of -N=CH-N(CH3)2), 30.45 (C20), 27.94 (C10), 25.95 (C19);
15N-NMR: −281.2 (-N=CH-N(CH3)2), −265.9 (N17), −239.8 (N9), −222.3 (N1), −176.1 (-N=CH-N(CH3)2), N3 not recorded in 1H-15N g-HSQC/HMBC experiments;
HRMS: calcd for C23H27N6O6 m/z = 483.1992, found m/z = 483.1985.
FT-IR: ν [cm−1] 3320–3126(N-Hν, O-Hν); 2922(C-Hν); 1683 (C=Oν); 1635 (C=Nν); 1532 (N-Hδ, C=Nν); 1505 (C=Cν); 1420 (C-Hδ); 1354 (C-Nν); 1120 (C-Oν); 840 (C-Hγ).
Adjusting the reaction mixture’s pH to 8 enabled the isolation of trisodium salt
7a (details are given in the
Supplementary Part).
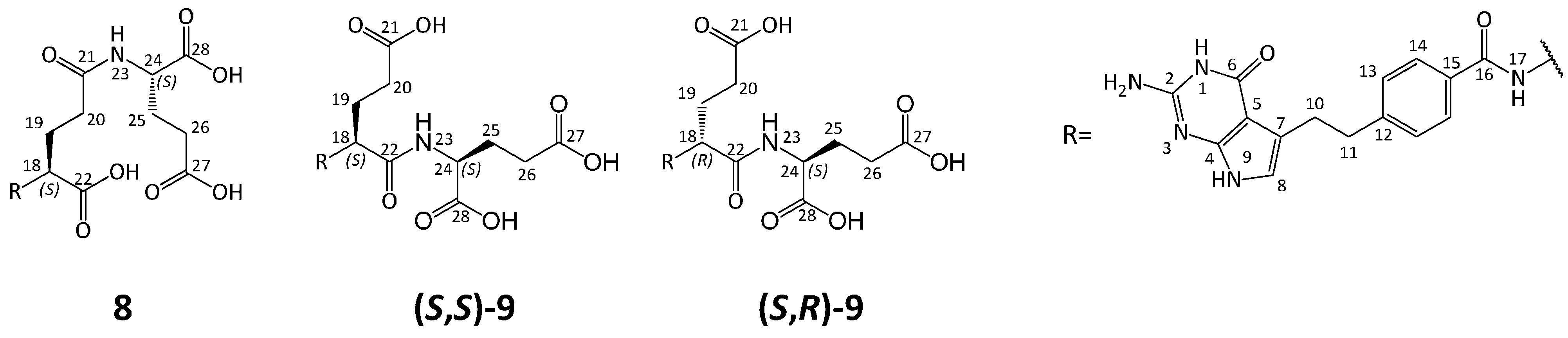
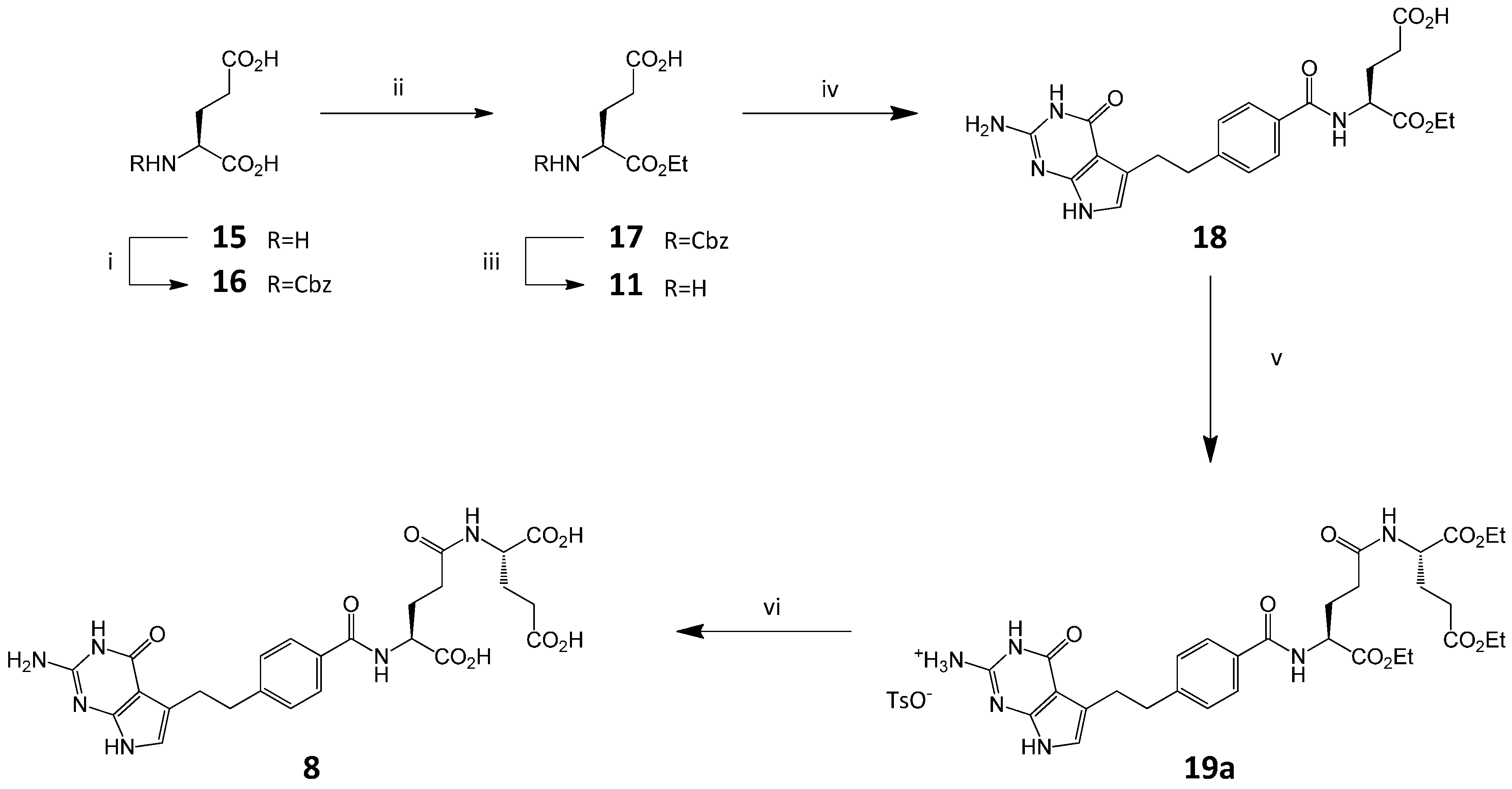
3.5. Synthesis of Impurity 8
3.5.1. N-Benzyloxycarbonyl-L-glutamic Acid (16)
N-Benzyloxycarbonyl-
L-glutamic acid was prepared according to literature [
21]. After recrystallization from ethyl acetate/hexanes (1:6), a white solid was obtained.
TLC RF = 0.48 (CHCl3/MeOH/NH3 2:2:1);
= −9.0 (c = 2, AcOH);
Mp. 118 °C;
1H-NMR (600 MHz, DMSO): 12.4 (2H, very broad s, both protons of -COOH); 7.57 (1H, d, J = 8.1 Hz, NH), 7.35 and 7.29 (5H, m, Ph), 5.03 (2H, s, CH2 of Cbz), 3.99 (1H, m, CH-NHCbz), 2.29 (2H, m, both H of CH2β), 1.96 and 1.75 (2H, 2m, both H of CH2α);
13C-NMR (150 MHz, DMSO): 173.77 (COγ), 173.62 (COα), 156.20 (CO of NHCbz), 137.00, 128.4, 127.8, 127.7 (Ph), 65.47 (CH2 of Cbz), 53.08 (CH-NHCbz), 30.12 (CH2β), 26.13 (CH2α);
HRMS calcd for C13H15NO6Na m/z = 304.0797, found m/z = 304.0799.
3.5.2. N-Benzyloxycarbonyl-L-glutamic Acid α-Ethyl Ester (17)
The dicyclohexylammonium (DCHA) salt of compound
16 was prepared according to literature [
25].
TLC RF = 0.75 (CHCl3/MeOH/NH3 2:2:1); DCHA salt
Mp. 160 °C (lit. [
25] 159–160 °C);
= −12.2 (c=0.5, MeOH) (lit. [
25] [α]23−25 D = −11.7 (c = 2, MeOH));
1H-NMR δ: 8.12 (1H, d, J = 6.6 Hz, NH), 7.44–7.25 (5H, m, all protons of Ph ring), 5.02 (2H, 2×d, J = 12.7 Hz, CH2 of Cbz), 4.09 (2H, CH2 of -O-CH2CH3), 4.01 (1H, m, CH-NH), 2.73, 2.15, 1.92–1.80, 1.75, 1.67, 1.56, 1.28–1.05;
13C-NMR δ: 174.69, 172.37, 156.05, 137.03, 128.32, 127.78, 127.66, 65.35, 60.32, 53.91, 51.94, 32.08, 31.17, 26.64, 25.41, 24.35, 14.06.
1 M H2SO4 aq (100 mL) was added to the suspension of DCHA salt of 16 (8.63 g, 17.58 mmol) in AcOEt (200 mL). After 10 min water (100 mL) was added to the reaction mixture, the organic layer was separated and the aqueous phase extracted with AcOEt (2 × 150 mL). The organic layers were collected, washed with water (2 × 50 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure to afford 17 (5.0 g, 92%).
= −20.7 (c = 0.5, MeOH);
1H-NMR δ: 7.76 (1H, d, J = 7.7 Hz, NH), 7.38–7.29 (5H, m, all protons of Ph ring), 5.03 (2H, 2×d, J = 12.5 Hz, CH2 of Cbz), 4.08 (2H, m, CH2 of -CH2CH3), 4.04 (1H, m, CH-NH), 2.28 (2H, m, -CH2-COOH), 1.93 (1H, m), 1.76 (1H, m), 1.17 (3H, t, J = 7.1Hz, CH3);
13C-NMR δ: 173.96, 172.12, 156.11, 136.94, 128.34, 127.83, 127.70, 65.46, 60.50, 53.27, 30.23, 26.09, 14.03;
HRMS calcd for C15H19NO6Na m/z = 332.1110, found m/z = 332.1114.
3.5.3. L-Glutamic Acid α-ethyl Ester (11)
The solution of 17 (5.0 g, 16.2 mmol) in EtOH (150 mL) was hydrogenated in the presence 10% Pd/C (0.4 g) for 2.5 h. After filtration through a Celite pad, the solution was evaporated and dropped into Et2O (50 mL). After cooling in the refrigerator, the white precipitate was filtered, washed with Et2O, and dried in vacuum at 30 °C to give 11 (2.6 g, 93%).
TLC: RF = 0.65 (CHCl3/MeOH/NH3 2:2:1);
= +27.0 (c = 2, 1 M HCl);
Mp. 90 °C;
1H-NMR δ: 5.00 (3H, very broad s, -NH2 and -COOH), 4.10 (2H, m, ‑CH2CH3), 3.37 (1H, m, -CHNH2), 2.29 (2H, m, both H of CH2β), 1.83 and 1.64 (2H, 2m, both H of CH2α), 1.20 (3H, t, J = 7.0 Hz, -CH2CH3);
13C-NMR δ: 174.80 (COα), 174.26 (COγ), 60.20 (-CH2CH3), 53.12 (-CHNH2), 30.84 (CH2β), 29.18 (CH2α), 14.07 (-CH2CH3);
15N-NMR δ: −351.5;
HRMS: calcd for C7H14NO4 m/z = 176.0923, found m/z = 176.0926.
3.5.4. (2S)-2-[[4-[2-(2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-pentanedioic Acid 1-Ethyl Ester (18)
NMM (1.99 mL, 18.11 mmol), followed by CDMT (1.94 g, 11.06 mmol) were added to the suspension of acid 2 (3 g, 10.06 mmol) in DMF (30 mL) and the resulting solution was stirred at RT for 2 h. α-ethyl L-glutamate 11 (1.85 g, 10.56 mmol) was added to this solution and the resulting mixture was stirred for 24 h. The reaction mixture was poured into water (30 mL) and extracted with CH2Cl2 (3 × 30 mL). The organic phases were collected, dried over anhydrous MgSO4 and concentrated to afford 18 (2.83 g, 61%).
TLC: RF = 0.67. (CHCl3/MeOH, 4:6);
1H-NMR δ: 11.1 (1H, broad s, probably N1-H), 10.57 (1H, s, N9-H), 10.38 (1H, s, N17-H), 7.80 (2H, m, H14), 7.23(2H, m, H13), 6.49 (2H, s, NH2 at C2), 6.27 (1H, s, H8), 4.20 (1H, m, H18), 4.07 (2H, m, -CH2CH3 at C22), 2.92 (2H, m, H11), 2.82 (2H, m, H10), 2.19 (2H, m, H20), 1.95 (2H, m, H19), 1.16 (3H, t, J = 7.1 Hz, -CH2CH3 at C22);
13C-NMR δ: 176.78 (CO, C21), 172.28 (CO, C22), 165.96 (C16), 159.70 (C6), 152.79 (C2), 151.52 (C4), 145.93 (C12), 131.16 (C15), 128.16 (C13), 127.30 (C14), 117.69 (C7), 113.09 (C8), 98.66 (C5), 60.09 (-O-CH2CH3), 54.12 (C18), 36.16 (C11), 33.97 (C20), 28.00 (C10), 26.37 (C19), 14.15 (-O-CH2CH3);
15N-NMR δ: −309.9 (NH2 at C2), −263.7 (N17), −241.8 (N9); N1 and N3 not recorded in 1H-15N g-HMBC experiment;
HRMS calcd for C22H25N5O6Na m/z = 478.1703, found m/z = 478.1694.
3.5.5. (2S)-2-[[(4S)-4-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-(ethoxycarbonyl)butanoyl]amino]pentanedioic Acid Diethyl Ester, p-TSA Salt (19a)
NMM (1.57 mL, 14.28 mmol) was added to the suspension of 18 (2.33 g, 5.12 mmol) in DMF (20 mL), followed by CDMT (1.08 g, 6.15 mmol), and the resulting solution was stirred at RT for 3 h. diethyl L-glutamate hydrochloride 4 (1.34 g, 5.61 mmol) was added to this solution and the resulting mixture was stirred for 24 h. Then water (30 mL) and CH2Cl2 (30 mL) were added to the reaction mixture which was stirred for 15 min. The organic layer was separated and the aqueous phase extracted with CH2Cl2 (2 × 25 mL). The organic layers were collected, washed with 1 M NaHCO3aq (1 × 35 mL), and concentrated under reduced pressure to afford oil.
EtOH (40 mL), followed by the solution of p-TSA·H2O in EtOH (2.46 g in 40 mL EtOH) were added to the oil and the resulting suspension was heated under reflux for 2 h. Then the mixture was cooled to RT, the crystals were filtered, washed with EtOH (2 × 15 mL) and dried in vacuo at 40 °C for 24 h to provide 19a (1.05 g, 25%).
TLC: RF = 0.46 (CHCl3/MeOH 8:2);
Mp. 232 °C;
1H-NMR δ: 11.39 (1H, s, probably N1-H), 11.26 (1H, s, N9-H), 8.67 (1H, d, J = 7.3 Hz, N-H17), 8.27 (1H, d, J = 7.6 Hz, N-H23), 7.80 (2H, m, H14), 7.53 (2H, d, J = 8.0 Hz, p-TSA), 7.30 (2H, m, H13), 7.15 (2H, d, J = 8.0 Hz, p-TSA), 6.51 (1H, s, H8), 4.39 (1H, m, H18), 4.24 (1H, m, H24), 4.11 (2H, q, J = 7.0 Hz, CH2 of -CH2CH3 at C22), 4.07 (2H, q, J = 7.0 Hz, CH2 of -CH2CH3 at C27), 4.04 (2H, q, J = 7.0 Hz, CH2 of -CH2CH3 at C26), 2.97 (2H, m, H11), 2.90 (2H, m, H10), 2.36 (2H, m, H26), 2.34–2.26 (5H, m, H20 and CH3 of p-TSA), 2.09 (1H, m, one of H19 protons), 1.96 (2H, m, one of the H19 protons and one of H25 protons), 1.81 (1H, m, one of the H25 protons);
13C-NMR δ: 172.10 (CO, C27), 172.00 (CO, C22), 171.74 (2×CO, C28 and C21), 166.47 (CO, C16), 157.45 (probably C6), 150.72 (probably C3), 145.70 (C12), 144.94 (CIV, p-TSA), 139.81 (probably C4), 138.12 (CIV, p-TSA), 131.32 (C15), 128.16 (C13), 128.16 (CH, p-TSA), 125.50 (CH, p-TSA), 118.92 (C7), 115.13 (C8), 99.00 (C5), 60.48 (-O-CH2CH3 at C27), 60.44 (-CH2CH3 at C22), 59.90 (-CH2CH3 at C26), 52.38 (C18), 51.22 (C24), 35.74 (C11), 31.38 (C20), 29.84 (C26), 27.25 (C10), 26.19 (C19), 26.03 (C25), 20.77 (CH3 group of p-TSA), 14.09, 14.05 and 14.00 (carbons of CH3 of -O-CH2CH3 groups);
15N-NMR δ: −266.8 (N23), −266.4 (N17), −242.1 (N9);
HRMS: calcd for C31H41N6O9 m/z = 641.2935, found m/z = 641.2930.
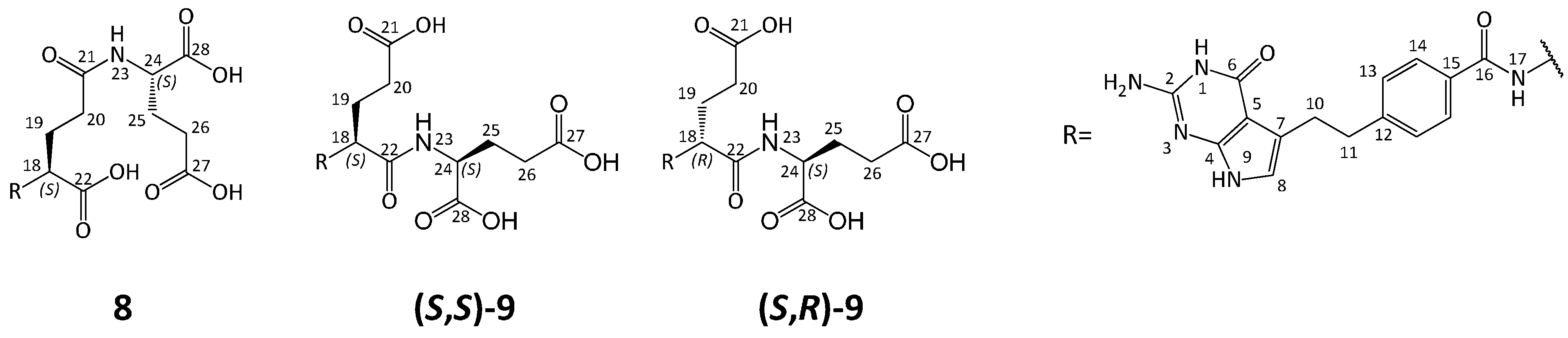
3.5.6. (2S)-2-[[(4S)-4-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-carboxybutanoyl]amino]pentanedioic Acid (8)
Compound 19a (1.86 g, 2.28 mmol) was treated with 1 M NaOHaq (15 mL), the mixture was stirred at RT. After 1 h the reaction mixture was adjusted to pH 3.0 with 1N HClaq, and the precipitate was formed. After 30 min the precipitate was filtered off, washed with water (2 × 10 mL) and dried to give crude 8. The crude material (1.60 g) was purified by column chromatography (SiO2, CHCl3-MeOH-H2O-25% NH3aq 40:40:5:2) to give fraction of 8, 668 mg (HPLC 98.6%). The obtained material (660 mg) was then dissolved in water (10 mL) and the solution was acidified to pH = 3 with 10% HClaq (ca. 2.5 mL), forming a precipitate. The mixture was stirred at RT for 30 min, then the precipitate was filtered off, washed with water (3 × 15 mL) and dried to give 8 (in triacid form, 0.48 g, 37.6% from 19a, HPLC 98.0%). A second fraction of 8 (390 mg, HPLC 91.5%) was obtained by chromatography and acidified as described above to give 8 in the triacid form (0.27 g, yield 21.4% from 19a, HPLC purity 97.6% ).
= −3.87 (c = 1, DMSO);
Mp. 155 °C;
1H-NMR δ: 12.40 (ca. 3H, broad s, 3×COOH), 10.61 (1H, s, N9-H), 10.18 (1H, s, N1-H), 8.57 (1H, d, J = 7.6 Hz, N17-H), 8.13 (1H, d, J = 7.8 Hz, N23-H), 7.80 (2H, m, H14), 7.29 (2H, m, H13), 6.31 (1H, s, H8), 6.03 (2H, s, NH2 at C2), 4.35 (1H, m, H18), 4.21 (1H, m, C24), 2.98 (2H, m, H11), 2.86 (2H, m, H10), 2.28 (2H, m, H20), 2.27 (2H, m, H26), 2.10 (1H, m, H19), 1.94 (2H, m, H19 and H25), 1.75 (H25);
13C-NMR δ: 173.77 (CO, C27), 173.54 (CO, C22), 173.39 (CO, C28), 171.75 (CO, C21), 166.43 (CO, C16), 159.30 (CO, C6), 152.22 (C2), 151.33 (probably C4), 146.12 (C12), 131.39 (C15), 128.15 (C13), 127.38 (C14), 117.64 (C7), 113.46 (C8), 98.75 (C5), 52.31 (C18), 51.19 (C24), 36.14 (C11), 31.76 (C20), 30.14 (C26), 28.02 (C10), 26.48 (C19), 26.39 (C25);
15N-NMR δ: −311.2 (C2-NH2), −265.6 (N17), −260.0 (N23), −241.4 (N9), −236.4 (N1), −208.0 (N3);
HRMS: calcd for C25H27N6O9 m/z = 555.1840, found m/z = 555.1851.
FT-IR: [cm−1] 3312 (N-Hν, O-Hν); 2926 (C-Hν); 1702 (C=Oν); 1647 (C=Oν, C=Nν); 1504 (C=Cν); 1541 (N-Hδ, C=Nν); 1448, 1407 (C-Hδ); 1330 (C-Nν); 1223 (C-Oν, C-Nν); 666 (C-Hγ, N-Hγ).
Adjusting the reaction mixture’s pH to 8 enabled the isolation of the trisodium salt
8a (details are given in the
Supplementary Part).
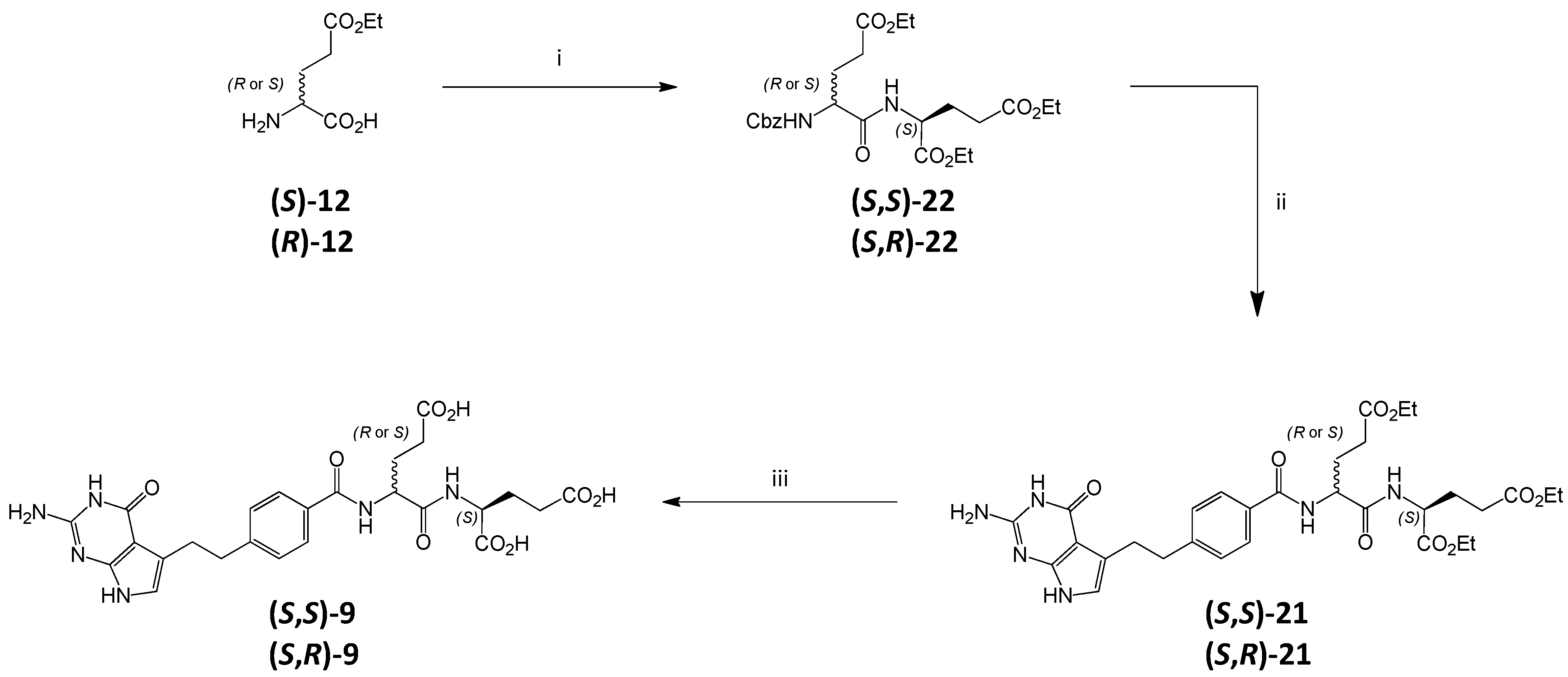
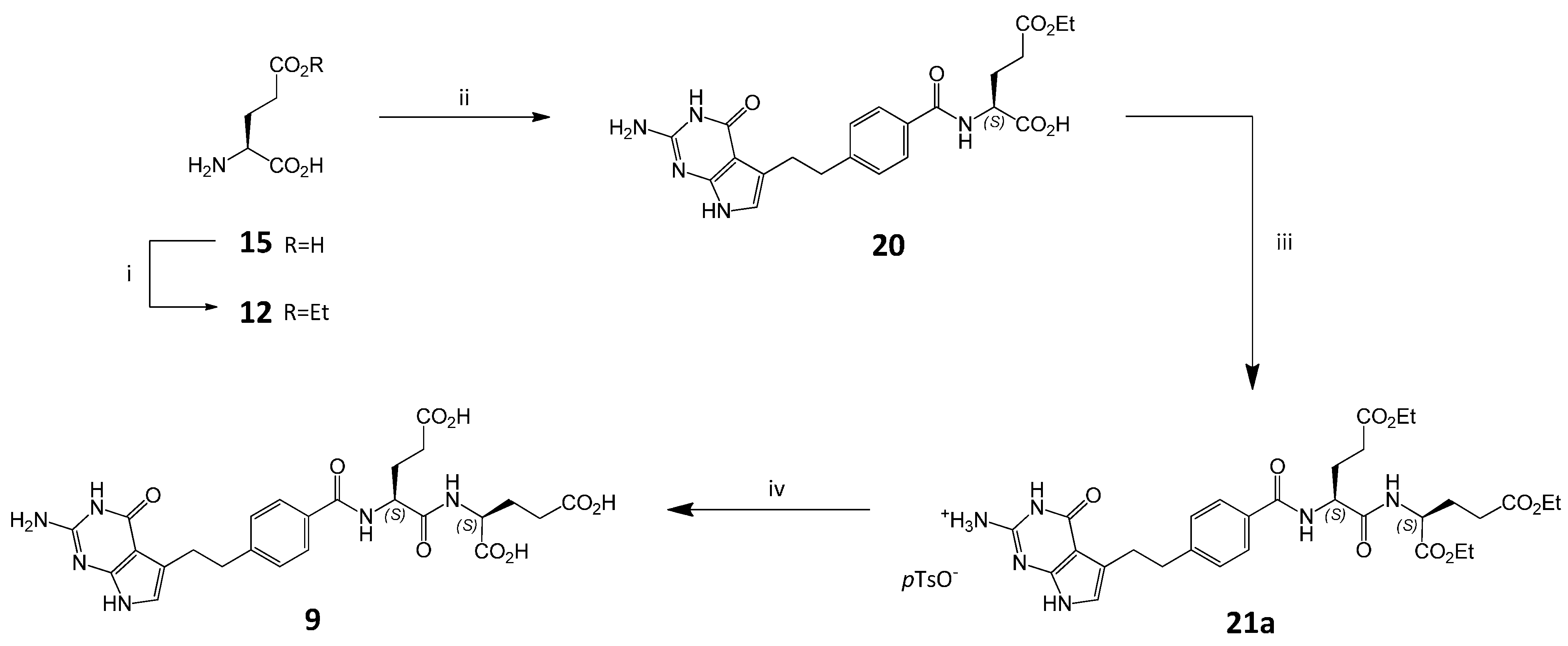
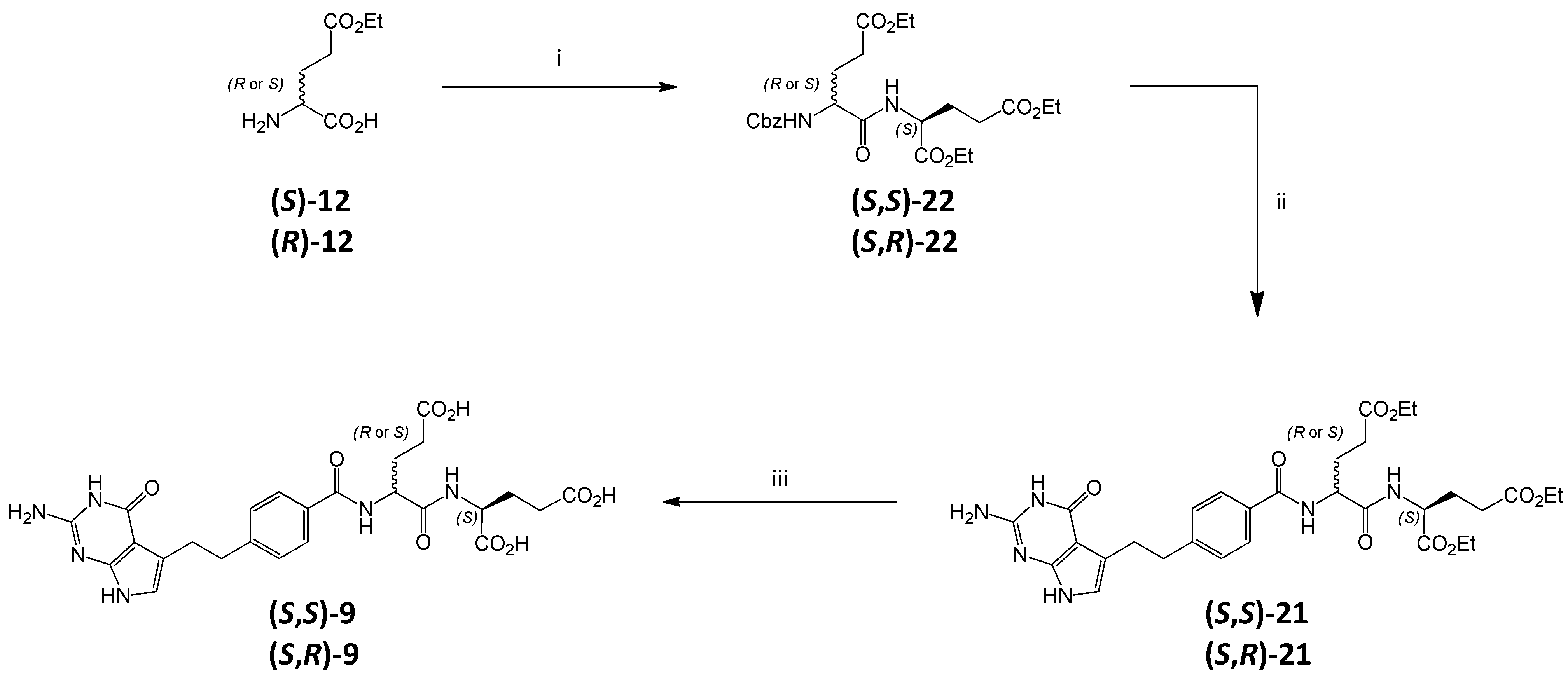
3.6. Synthesis of Impurity (S,S)-9 and Diastereoisomer (S,R)-9
3.6.1. (2S)-2-[(2S)-2-Carboxybenzylamino-4-(ethoxycarbonyl)butanoyl]aminopentanedioic Acid Diethyl Ester ((S,S)-22)
DIPEA (3.0 mL, 17.2 mmol) was added to the suspension of
N-Cbz protected
(S)-12 (1.4 g, 4.53 mmol prepared according to the literature procedure [
26]) in DMF (15 mL), followed by HATU (2.07 g, 5.43 mmol). The resulting solution was stirred at
RT for 1 h. Then diethyl
L-glutamate hydrochloride
4 (1.63 g, 6.79 mmol) was added to the solution and the resulting mixture was stirred for 24 h. The reaction mixture was diluted with water (70 mL) and extracted with AcOEt (3 × 25 mL). The combined organic layers were washed with 1 M HCl
aq (1 × 25 mL), 5% NaHCO
3 aq (1 × 25 mL), sat. brine (1 × 25 mL), dried over Na
2SO
4, and concentrated to give crude (
S,
S)
-22 as oil. The oil was crystallized from the AcOEt/hexanes mixture to obtain pure dipeptide (
S,
S)
-22 as a solid (53.2%).
TLC: RF = 0.52 (hexanes/AcOEt 1:1);
= −20.70 (c = 1.0 MeOH);
Mp. Phase trans.: peak endothermic. 1 = 84.3 °C; peak exothermic. = 89.2 °C; peak endothermic. 2 = 109.8 °C;
1H-NMR δ: 8.34 (1H, d, J = 7.8 Hz, NH), 7.46 (1H, d, J = 7.8 Hz, Cbz-NH), 7.38–7.28 (5H, Ph), 5.02 (2H, dd, J = 12.5 Hz, CH2 of Cbz group), 4.26 (1H, m, CHα), 4.10–4.02 (7H, m, all CH2 of ester -CH2CH3 groups and CHα), 2.40–2.32 (4H, m, all CHγ protons), 2.00+1.84 (2H, 2 × m, both CHβ protons), 1.91 + 1.78 (2H, 2 × m, both CHβ protons), 1.17 (9H, three overlapping triplets, all CH3 of ester -CH2CH3 groups);
13C-NMR δ: 172.3, 172.1, 171.4 (CO of ester groups), 171.6 (CO of amide group), 155.8 (CO of Cbz group), 137.0 (CIV of Ph), 128.3, 127.8 and 127.6 (Ph), 65.4 (CH2 of Cbz group), 60.6, 59.9 and 59.8 (CH2 carbons of ester -CH2CH3 groups), 53.6 (CHα), 51.2 (CHα) 30.0, 29.8 (both CHγ carbons), 27.2, 25.8 (both CHβ carbons), 14.0 and 13.9 (all CH3 carbons of ester -CH2CH3 groups).
HRMS calcd for [M + H]+ C24H35N2O9 m/z = 495.2343, found m/z = 495.2344.
3.6.2. (2S)-2-[(2R)-2-Carboxybenzylamino-4-(ethoxycarbonyl)butanoyl]aminopentanedioic Acid Diethyl Ester ((S,R)-22)
(
R)
-12 prepared according to the literature procedure [
26] was coupled with
4 analogically to the preparation of (
S,
S)
-22 to give triester (
S,
R)
-22.
TLC: RF = 0.44 (hexanes/AcOEt 3:2);
= −5.12 (c = 1.0 MeOH);
Mp. 90.3 °C;
1H-NMR δ: 8.34 (1H, d, J = 7.8 Hz, NH), 7.40 (1H, d, J = 7.8 Hz, Cbz-NH), 7.38–7.28 (5H, Ph), 5.02 (2H, m, CH2 of Cbz group), 4.21 (1H, m, CHα), 4.10–4.02 (7H, m, all CH2 of ester -CH2CH3 groups and CHα), 2.36–2.28 (4H, m, all CHγ protons), 2.00 + 1.84 (2H, 2 × m, both CHβ protons), 1.88 + 1.78 (2H, 2 × m, both CHβ protons), 1.17 (9H, three overlapping triplets, all CH3 of ester -CH2CH3 groups);
13C-NMR δ: 172.4, 172.3, 171.7 (CO of ester groups), 171.5 (CO of amide group), 156.0 (CO of Cbz group), 137.0 (CIV of Ph), 128.4, 127.9 and 127.7 (Ph), 65.6 (CH2 of Cbz group), 60.8, 60.1 and 60.0 (CH2 carbons of ester -CH2CH3 groups), 53.9 (CHα), 51.3 (CHα) 30.1, 29.8 (both CHγ carbons), 27.4, 26.0 (both CHβ carbons), 14.1 and 14.0 (all CH3 carbons of ester -CH2CH3 groups).
HRMS calcd for [M + Na]+ C24H34N2O9Na m/z = 517.2162, found m/z = 517.2154.
3.6.3. (2S)-2-[[(2S)-2-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-(ethoxycarbonyl)butanoyl]amino]pentanedioic Acid Diethyl Ester ((S,S)-21)
The solution of triester (S,S)-22 (800 mg, 1.62 mmol) in EtOH (20 mL) was hydrogenated in the presence of 10% Pd/C (0.4 g) for 2.5 h. After filtrating through a Celite pad, the solution was evaporated to obtain amino-triester as oil (574 mg).
Acid 2 (296 mg, 0.99 mmol) was dissolved in DMF (10 mL), then DIPEA (0.52 mL, 2.97 mmol) was added followed by HATU (498 mg, 1.29 mmol) and the resulting solution was stirred at RT for 30 min. Then the solution of the previously obtained amino-triester (500 mg, 1.39 mmol) in 10 mL DMF was added and the resulting mixture was stirred overnight.
The reaction mixture was diluted with water (25 mL) and extracted with AcOEt (3 × 25 mL). The combined organic layers were washed with 5% NaHCO3 aq (1 × 25 mL), dried over anhydrous Na2SO4 and concentrated to give crude (S,S)-21 as oil. The oil was crystallized from the AcOEt/MTBE mixture to obtain pure dipeptide (S,S)-21 as a solid (596 mg, 65% from (S,S)-22).
TLC: RF = 0.35 (CH2Cl2/MeOH 10:0.8);
= −1.20 (c = 1.0 MeOH);
Mp. 149.1 °C;
1H-NMR δ: 10.60 (1H, d, J = 2.0 Hz, H9); 10.15 (1H,s, H1); 8.37 (1H, d, J = 7.5 Hz, H23); 8.35 (1H, d, J = 7.8 Hz, H17); 7.79 and 7.28 (2×2H, AA′BB′, H13 and H14); 6.30 (1H, m, H8); 6.00 (2H, s, C2-NH2); 4.45 (1H, m, H18); 4.27 (1H, ddd, J = 5.3 Hz, J = 7.5Hz, J = 9.3 Hz, H24); 4.10–4.00 (6H, ov, m, 3 × -CH2CH3); 2.97 (2H, m, H11); 2.85 (2H, m, H10); 2.41 (2H, m, ov, H20); 2.39 (2H, m, ov, H26); 2.05 (1H, m, ov, H19); 2.01 (1H, m, ov, H25); 1.95 (1H, m, ov, H19); 1.86 (1H, m, H25); 1.18–1.14 (9H, 3 t, ov, J = 7.1 Hz, 3 × -CH2CH3);
13C-NMR δ: 172.4 (C21); 172.2 (C27); 171.6 (C22); 171.5 (C28); 166.4 (C16); 159.3 (C6); 152.2 (C2); 151.3 (C4); 146.1 (C12); 131.4 (C15); 128.1 (C13); 127.4 (C14); 117.6 (C7); 113.4 (C8); 98.8 (C5); 60.6 (CH2CH3); 59.9 (CH2CH3); 59.8 (CH2CH3); 52.5 (C18); 51.2 (C24); 36.1 (C11); 30.3 (C20); 29.8 (C26); 28.0 (C10); 26.9 (C19); 25.8 (C25); 14.04 (2C, -CH2CH3); 13.9 (-CH2CH3);
15N-NMR δ: −208.0 (N3); −236.3 (N1); −241.4 (N9); −263.7 (N23); −265.9 (N17); −311.2 (C2-NH2);
HRMS calcd for [M+H]+ C31H41N6O9 m/z = 641.2935, found m/z = 641.2930.
3.6.4. (2S)-2-[[(2R)-2-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-(ethoxycarbonyl)butanoyl]amino]pentanedioic Acid Diethyl Ester ((S,R)-21)
Triester (S,R)-21 was obtained in a similar manner as (S,S)-21 starting from triester (S,R)-22.
Recrystallization from TBME gave (S,R)-21 as a white solid (yield 88.3%). This material can be additionally purified by chromatography (SiO2, CH2Cl2-MeOH, 99:1 → 93:7, v/v).
TLC: RF = 0.70 (CH2Cl2/MeOH 10:1.4);
= −9.56 (c = 1.0 MeOH);
1H-NMR δ: 10.60 (1H, d, J = 2.0 Hz, H9); 10.14 (1H,s, H1); 8.35 (1H, d, J = 7.8 Hz, H23); 8.32 (1H, d, J = 7.9 Hz, H17); 7.79 and 7.28 (2 × 2H, AA′BB′, H13 and H14); 6.3 (1H, m, H8); 6 (2H, s, H2); 4.48 (1H, m, H18); 4.24 (1H, ddd, J = 5.3 Hz, J = 7.8Hz, J = 9.3 Hz, H24); 4.10–4.00 (6H, 3 × m, ov, 3 × -CH2CH3); 2.97 (2H, m, H11); 2.85 (2H, m, H10); 2.38 (2H, m, ov, H20); 2.34 (2H, m, ov, H26); 2.02 (1H, m, ov, H19); 2 (1H, m, ov, H25); 1.96 (1H, m, ov, H19); 1.84 (1H, m, H25); 1.18–1.14 (9H, 3 × t, ov, J = 7.1 Hz, 3 × -CH2CH3);
13C-NMR δ: 172.4 (C21); 172.2 (C27); 171.7 (C22); 171.5 (C28); 166.4 (C16); 159.4 (C6); 152.2 (C2); 151.4 (C4); 146.2 (C12); 131.4 (C15); 128.1 (C13); 127.5 (C14); 117.7 (C7); 113.5 (C8); 98.8 (C5); 60.6 (-CH2CH3); 59.9 (2C, -CH2CH3); 52.7 (C18); 51.2 (C24); 36.2 (C11); 30.4 (C20); 29.8 (C26); 28.0 (C10); 27.0 (C19); 25.9 (C25); 14.0 (2C, -CH2CH3); 13.9 (‑CH2CH3);
15N-NMR δ: −208.0 (N3), −236.3 (N1); −241.4 (N9); −264.8 (N23); −266.0 (N17); −311.1 (C2-NH2);
HRMS calcd for [M + Na]+ C31H40N6O9Na m/z = 663.2754, found m/z = 663.2734.
3.6.5. (2S)-2-[[(2S)-2-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-carboxybutanoyl]amino]pentanedioic Acid ((S,S)-9)
Triester (S,S)-21 (100 mg, 0.156 mmol) was treated with 1 M NaOHaq (6 mL), the mixture was stirred at RT. After 30 min the reaction was completed (TLC control) and the mixture was acidified with 1 M HClaq to pH ≈ 3. The formed precipitate was filtered off, washed with water and dried (vacuum drier, 38 °C, overnight) to obtain (S,S)-9 (58 mg, 66.8%).
TLC: RF = 0.37 (2×CHCl3-MeOH 1:2); RF = 0.50 (CHCl3-MeOH-H2O-25% NH3aq 40:40:10:2)
= +18.9 (c = 1, DMSO);
Mp. 171.5 °C;
1H-NMR δ: 10.60 (1H, s, H9); 10.17 (1H, s, H1); 8.34 (1H, d, J = 7.8 Hz, H17); 8.21 (1H, d, J = 7.8 Hz, H23); 7.78 (2H, BB′, H14); 7.28 (2H, AA′, H13); 6.31 (1H, m, H8); 6.01 (2H, s, C2-NH2); 4.46 (1H, m, H18); 4.22 (1H, m, H24); 2.97 (2H, m, H11); 2.85 (2H, m, H10); 2.34 (2H, m, H20); 2.30 (2H, m, H26); 2.03 (1H, ov, m, H19); 1.99 (1H, ov, m, H25); 1.92 (1H, m, H19); 1.81 (1H, m, H25);
13C-NMR δ: 174.3 (C21); 173.9 (C27); 173.3 (C28); 171.7 (C22); 166.6 (C16); 159.5 (C6); 152.3 (C2); 151.4 (C4); 146.2 (C12); 131.5 (C15); 128.2 (C13); 127.5 (C14); 117.5 (C7); 113.6 (C8); 98.8 (C5); 52.8 (C18); 51.3 (C24); 36.2 (C11); 30.5 (C20); 30.1 (C26); 28.1 (C10); 27.1 (C19); 27.1 (C19); 26.3 (C25); 26.3 (C25);
15N-NMR δ: −208 (N3); −236.3 (N1); −241.4 (N9); −263.2 (N23); −265.5 (N17); −311.1 (C2-NH2);
HRMS calcd for C25H29N6O9 m/z = 557.1996, found m/z = 557.1989.
FT-IR: [cm−1] 3315 (N-Hν, O-Hν); 2927 (C-Hν); 1702 (C=Oν); 1636 (C=Oν, C=Nν); 1534 (N-Hδ, C=Nν); 1504 (C=Cν); 1405 (C-Hδ); 1221 (C-Oν, C-Nν); 669 (C-Hγ, N-Hγ).
3.6.6. (2S)-2-[[(2R)-2-[[4-[2-Amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]-4-carboxybutanoyl]amino]pentanedioic Acid ((S,R)-9)
Triester (S,R)-21 was hydrolyzed in the same manner as (S,S)-21 ( yield of (S,R)-21, 47.8%).
TLC: RF = 0.37 (2 × CHCl3-MeOH 1:2); RF = 0.50 (CHCl3-MeOH-H2O-25% NH3aq 40:40:10:2)
= −23.46 (c = 0.8, DMSO);
Mp. 156.7 °C;
1H-NMR δ: 10.60 (s, C9); 10.15 (1H, s, C1); 8.30 (1H, d, J = 8.1 Hz, C17); 8.20 (1H, d, J = 7.9 Hz, C24); 7.79 (2H, BB′, C14); 7.28 (2H, AA′, C13); 6.32 (m, C8); 6.01 (2H, s, C2-NH2); 4.50 (1H, m, C18); 4.23 (1H, m, C25); 2.98 (2H, m, C11); 2.86 (2H, m, C10); 2.32 (2H, m, C21); 2.25 (2H, m, C28); 2.01 (1H, m, ov, C20); 1.99 (1H, m, ov, C27); 1.94 (1H, m, ov, C19); 1.79 (1H, m, C26);
13C-NMR δ: 174.1 (C22); 173.7 (C29); 173.1 (C30); 171.6 (C23); 166.4 (C16); 159.3 (C6); 152.2 (C2); 151.3 (C4); 146.1 (C12); 131.5 (C15); 128.1 (C13); 127.5 (C14); 117.7 (C7); 113.4 (C8); 98.7 (C5); 52.8 (C18); 51.1 (C25); 36.1 (C11); 30.5 (C21); 29.9 (C28); 28 (C10); 27.2 (C19); 27.2 (C20); 26.4 (C26); 26.4 (C27);
HRMS calcd for C25H29N6O9 m/z = 557.1996, found m/z = 557.1989.
3.7. Synthesis of (2S,2′S)-2,2′-[[2,2′-Diamino-4,4′,6-trioxo-1,4,4′,6,7,7′-hexahydro-1′H,5H-5,6′-bipyrrolo[2,3-d]pyrimidine-5,5′-diyl]bis(ethylenebenzene-4,1-diylcarbonylimino)]dipentanedioic Acid (Impurity 10)
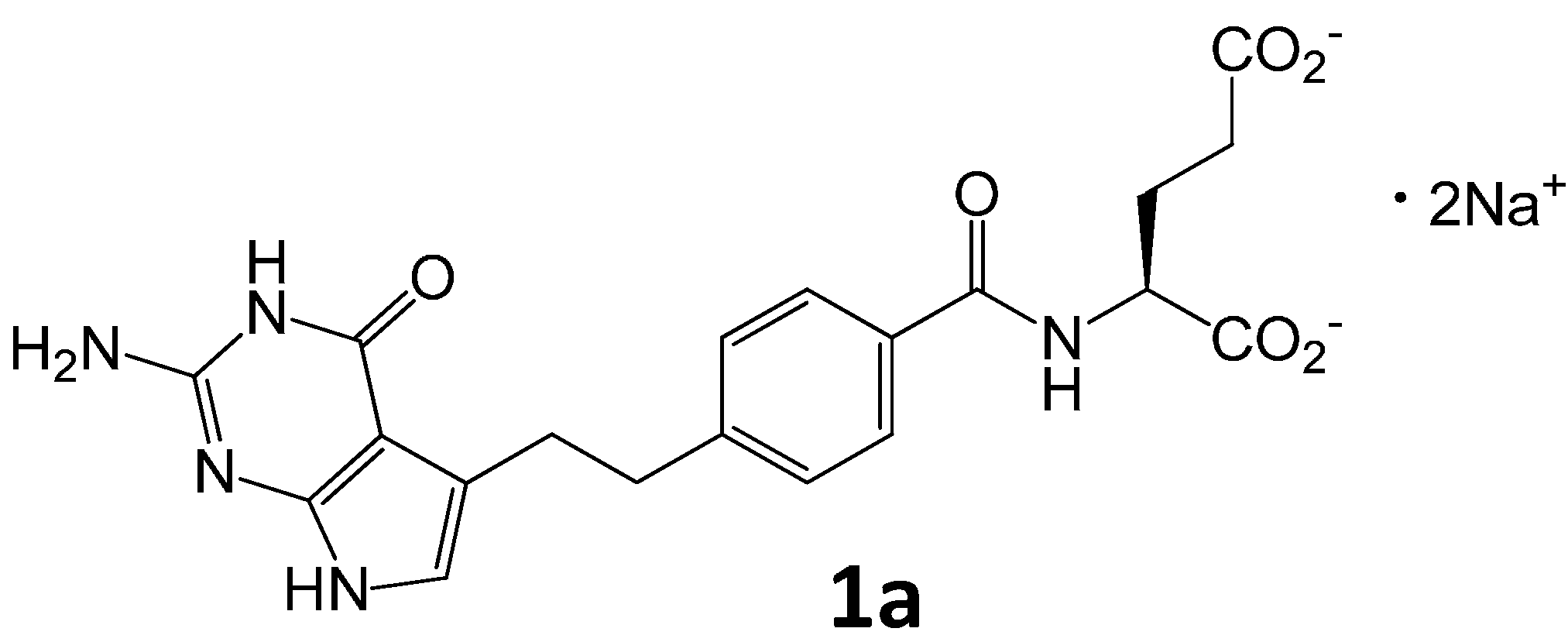
Pemetrexed disodium heptahydrate (1a, 2.0 g) was dissolved in 0.1 M NaOHaq (400 mL) and heated under reflux for 3 days (TLC control). Then the mixture was cooled and evaporated under reduced pressure to get crude diastereoisomeric mixture 10 as brown oil.
The obtained mixture was purified by chromatography (SiO2, EtOH-MeOH-AcOEt-4%NH3 aq, 40:30:10:12, v/v).The respective fractions were collected and concentrated. The residue was dissolved in water (10 mL) and the pH was adjusted to 2–3 with 1 M HClaq. The suspension was filtered, then the solid was washed with H2O (2 × 2 mL) and dried at 40 °C to obtain 10 (407 mg, 10%).
TLC: RF = 0.59 (EtOH/MeOH/AcOEt/4%NH3 aq 40:30:10:12 v/v)
HPLC purity 84.3%
Mp. 230 °C
HRMS: calcd for C40H39N10O13 m/z = 867.2698, found m/z = 867.2709
1H-NMR δ: 12.39 (3H, ov, total for 21, 21′, 22 and 22′ –CO2H), 10.87 (1H, s, N9′-H), 10.74 (1H, s, N9-H), 10.60 (1H, bs, probably N1′-H), 10.09 (1H, s, N1-H), 8.52 (2H, 2 × d, ov, N17-H and N17′-H), 7.80 (4H, m, H14 and H14′), 7.30 (2H, d, J = 8.0 Hz, H13′), 7.26 (2H, d, J = 8.0 Hz, H13), 6.90 (2H, bs, probably NH2 at C2′), 6.02 (2H, NH2 at C2), 4.41 (2H, m, H18 and H18′), 2.72–2.54 (6H, H11, H10 and H10′), 2.45 (2H, H11′), 2.40–2.32 (4H, H20 and H20′), 2.10 and 1.97 (4H, H19 and H19′);
13C-NMR δ: 179.59 (C8′), 173.93, 173.90 (CO, C21 and C21′), 173.51 and 173.48 (CO, C22, C22′), 166.59 and 166.41 (CO, C16 and C16′), 164.03 (probably C4′), 159.08 (C6), 157.76 (probably C6′), 157.63 (unknown, C4 or C2′), 152.10 (unknown, C4 or C2′), 150.29 (probably C2), 146.52 (C12), 145.00 (C12′), 131.49 and 131.39 (C15 and C15′), 128.08 (C13′), 127.97 (C13), 127.54 and 127.42 (C14 and C14′), 114.22 (C7), 99.51 (C5), 92.71 (C5′), 51.91 (C18 and C18′), 51.72 (C7′), 37.76 (C11), 34.18 (C10′), 30.44 (C20, C20′), 29.75 (C11′), 28.08 (C10), 25.98 (C19 and C19′);
15N-NMR δ: −311.0 (NH2 at C2), −265.9 (N17 and N17′), −238.9 (N9), −236.2 (N1), −233.7 (N9′), all remaining 15N-NMR signals not recorded in the 1H-15N g-HMBC experiment.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}