
Functional Thermoplastic Materials from Derivatives of Cellulose and Related Structural Polysaccharides

Abstract
:1. Introduction
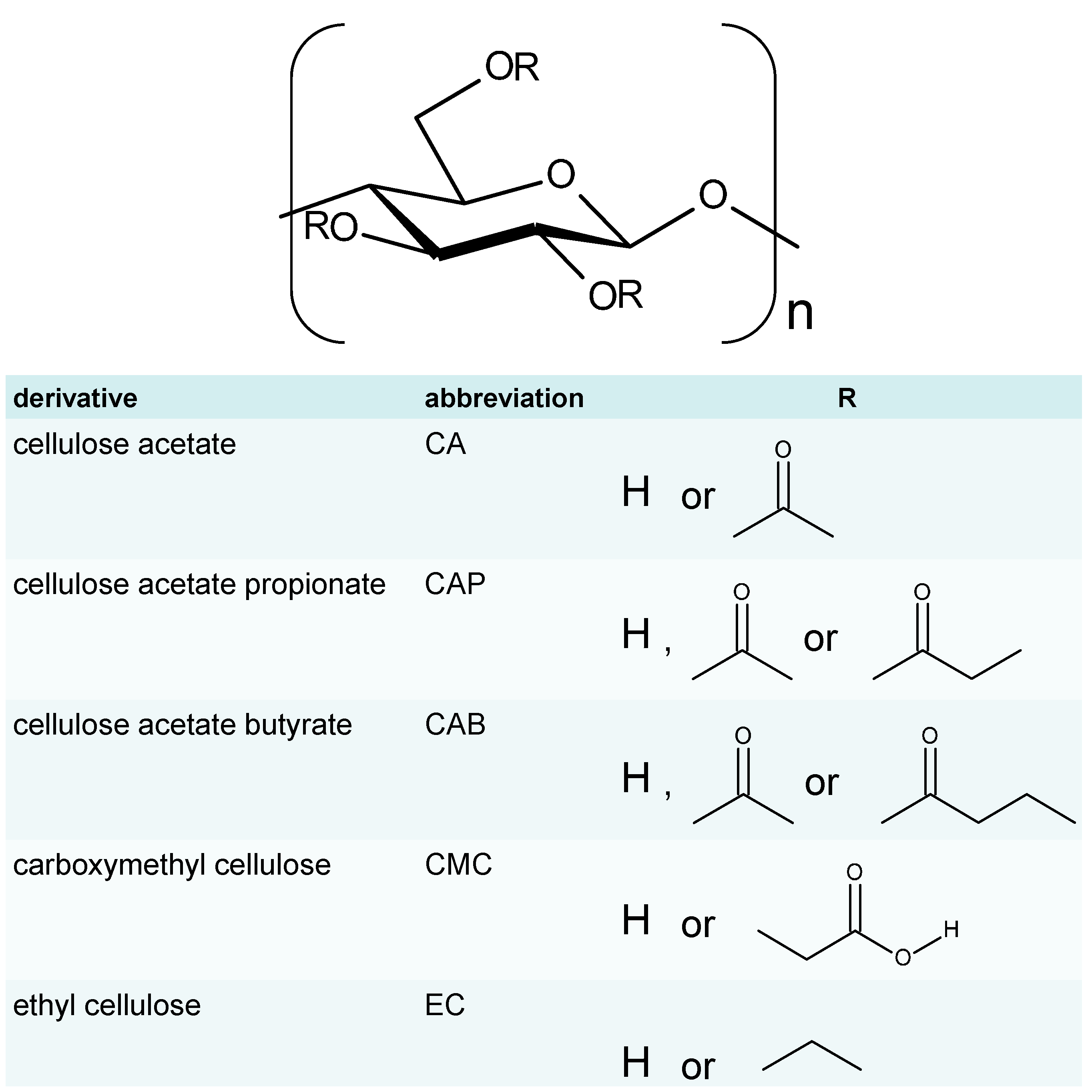
2. Single Substituent Derivatives
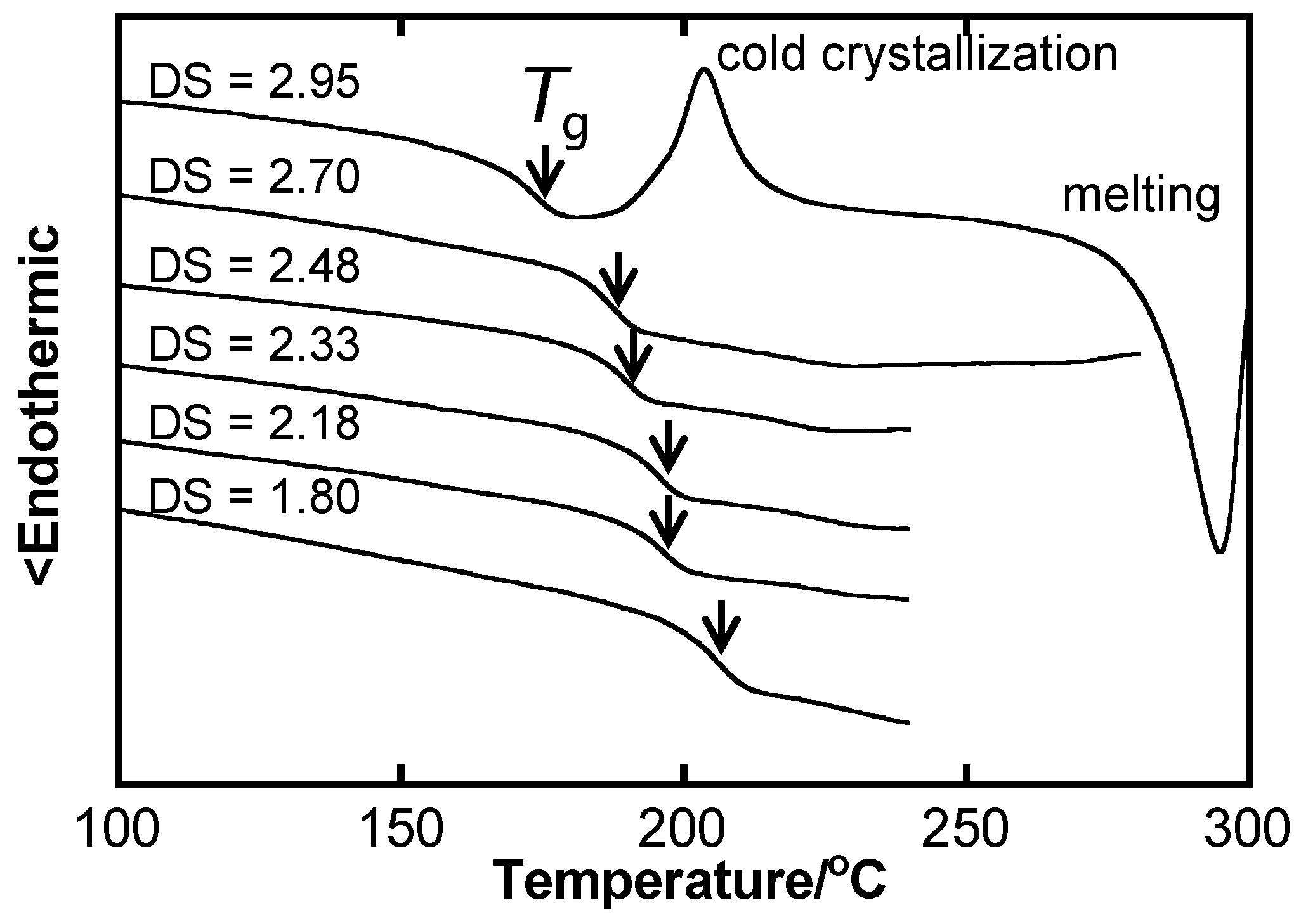
2.1. Fundamental Aspects of Thermal Properties and Phase Behavior
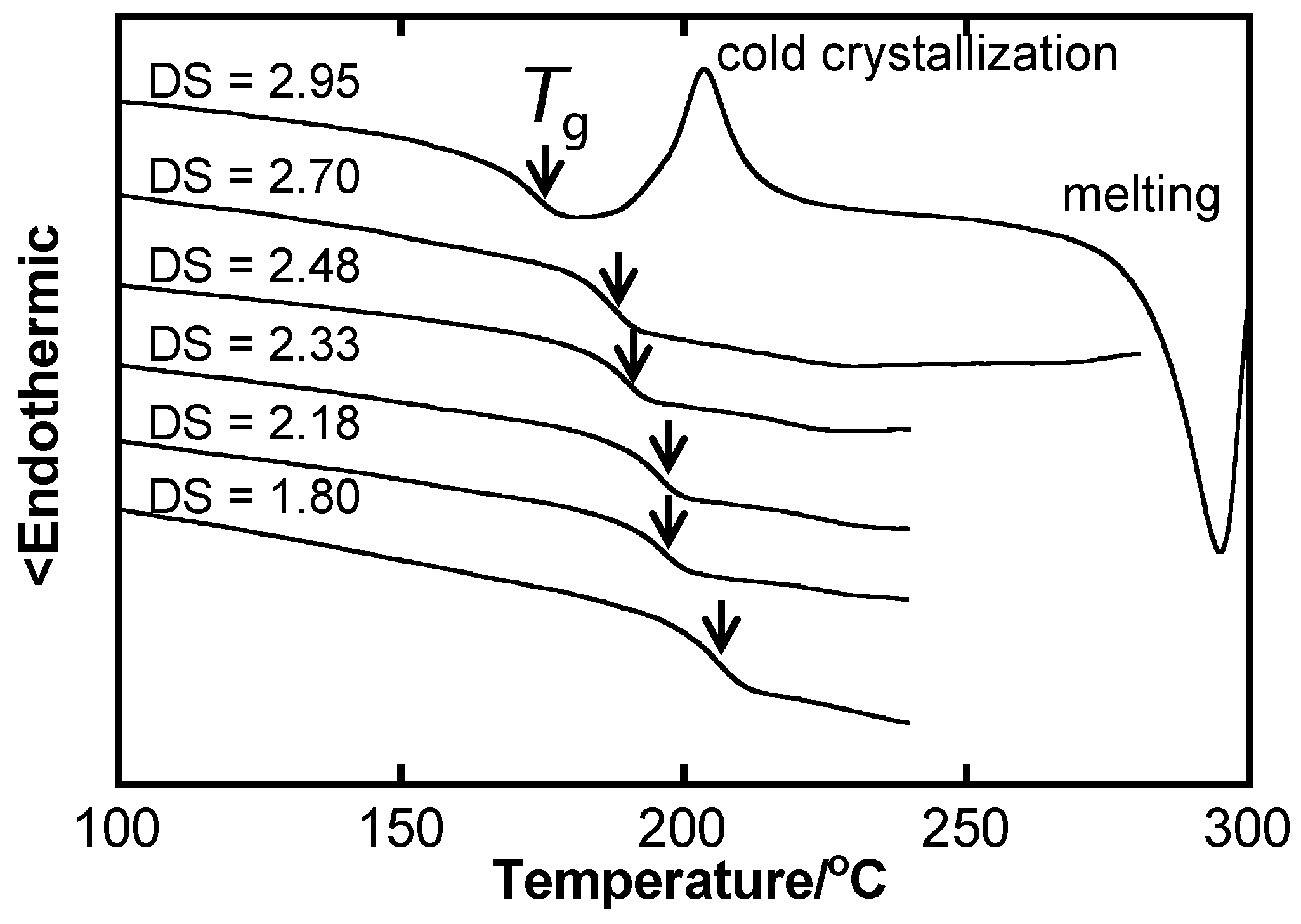
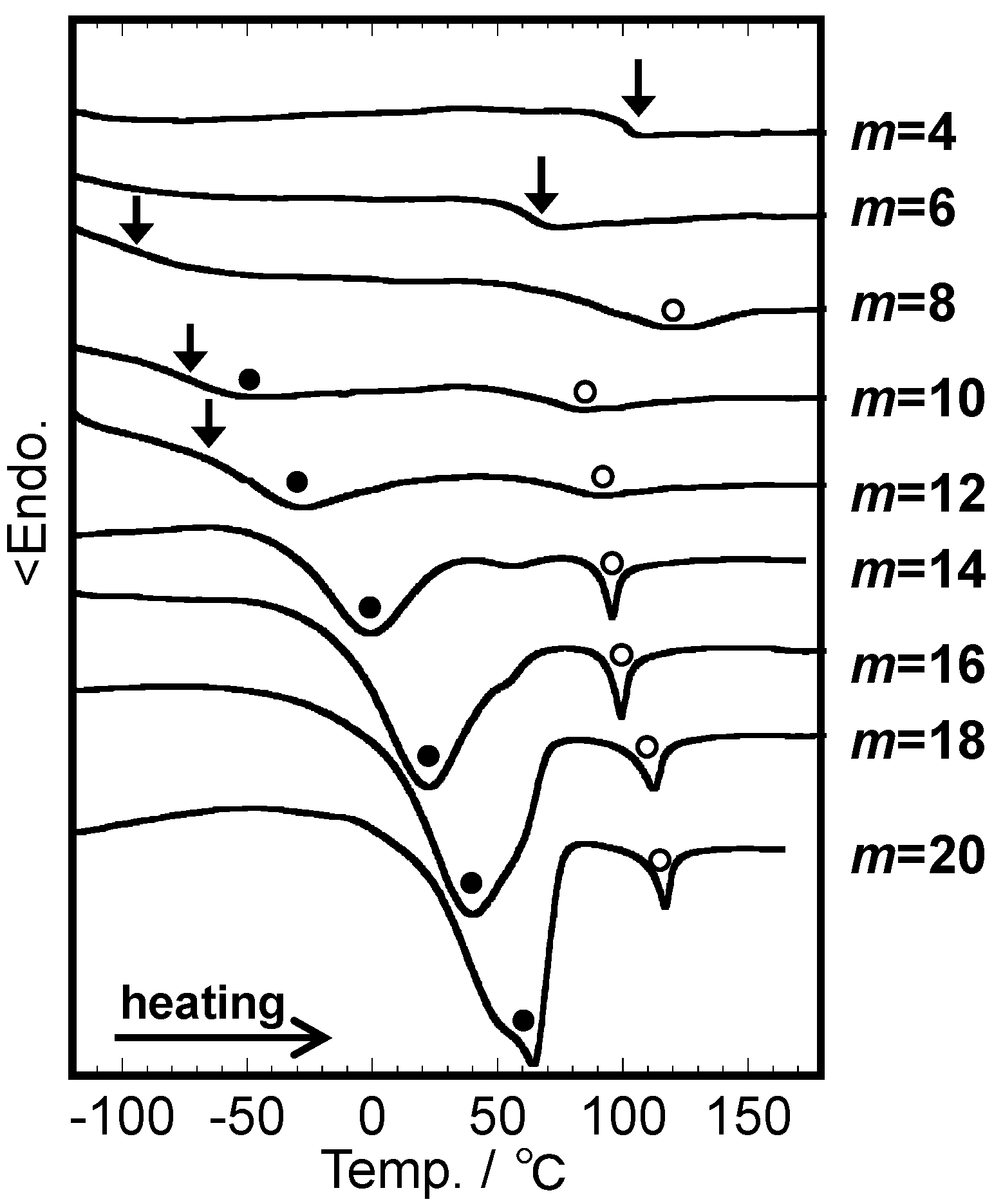

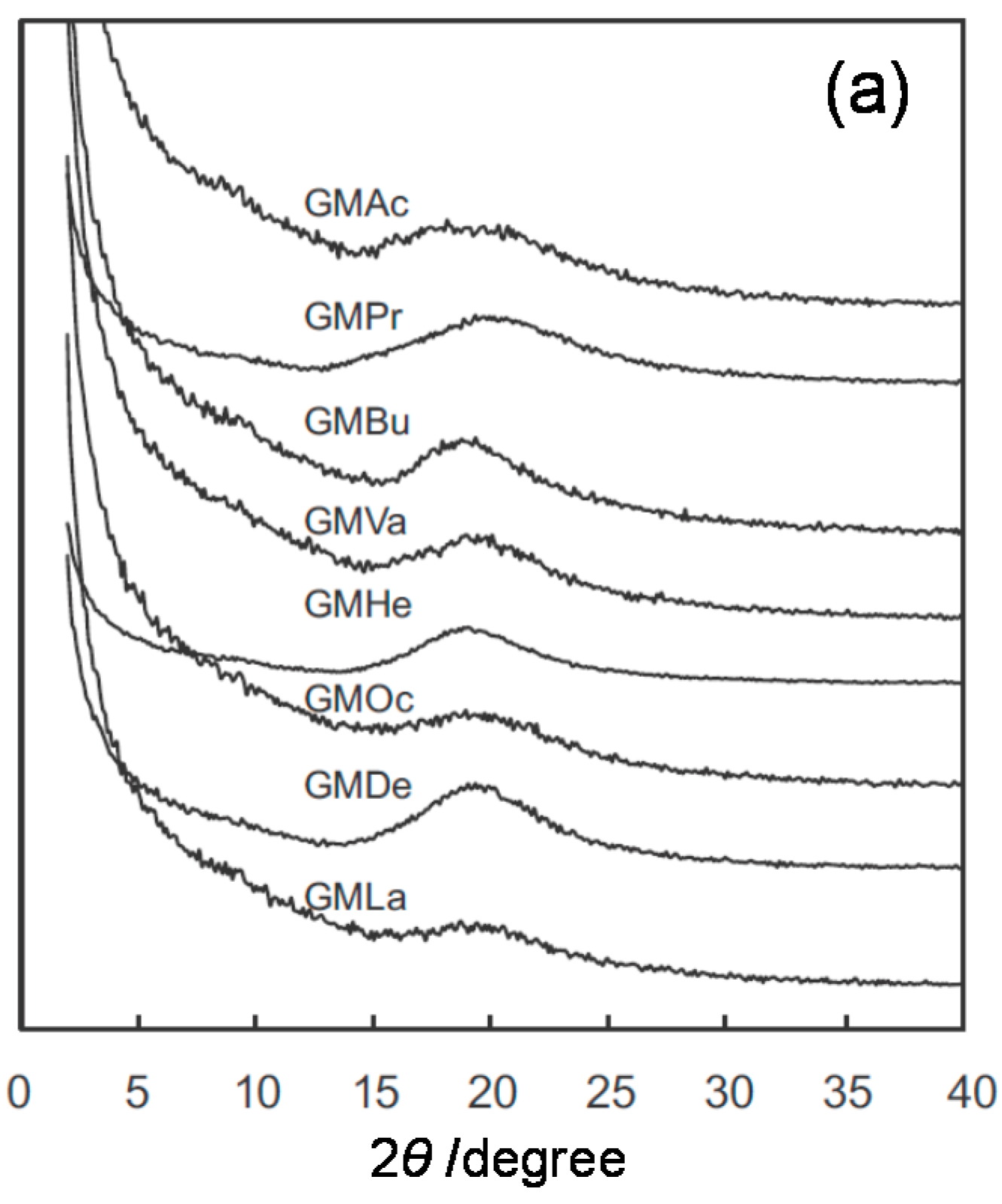
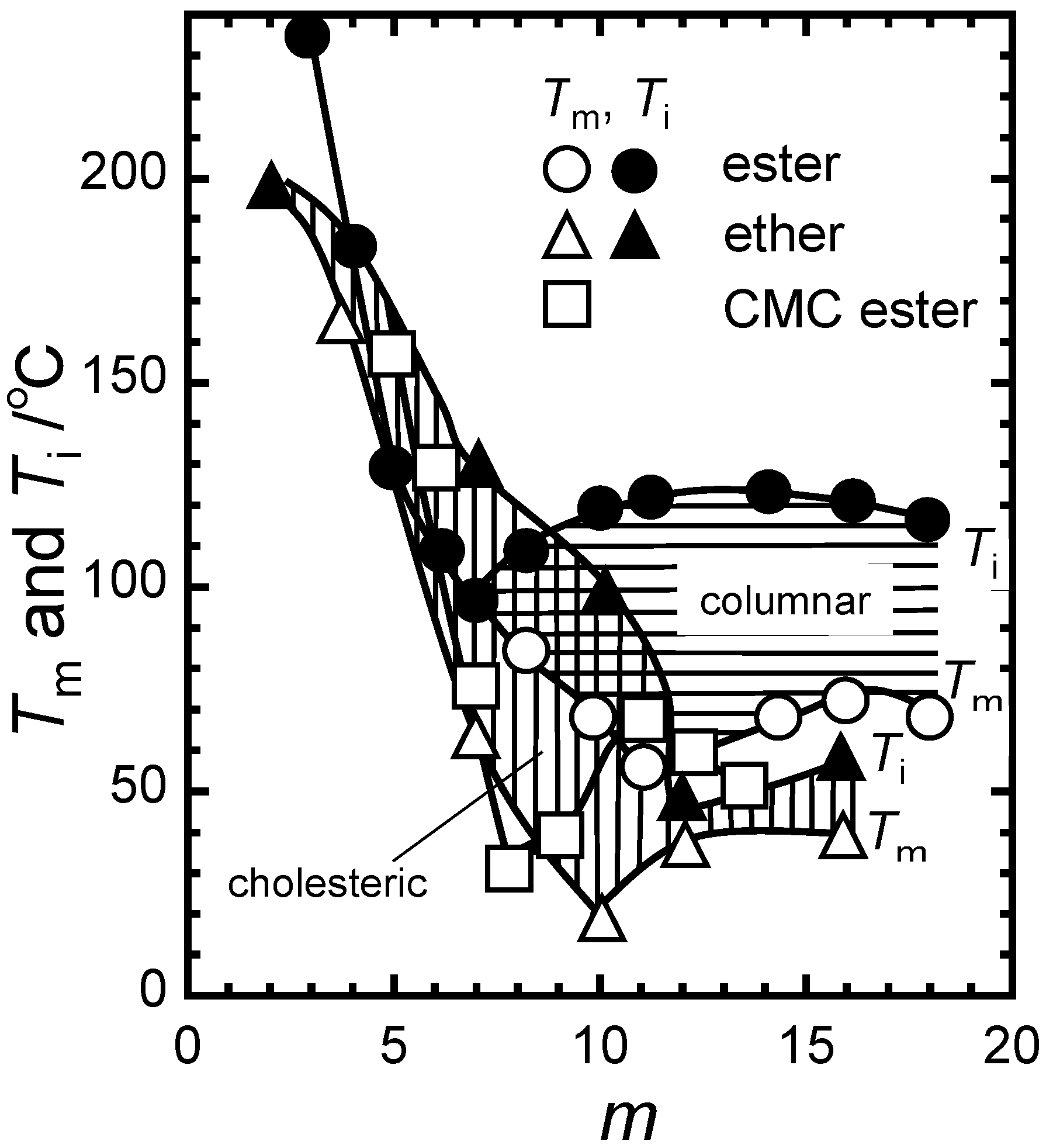
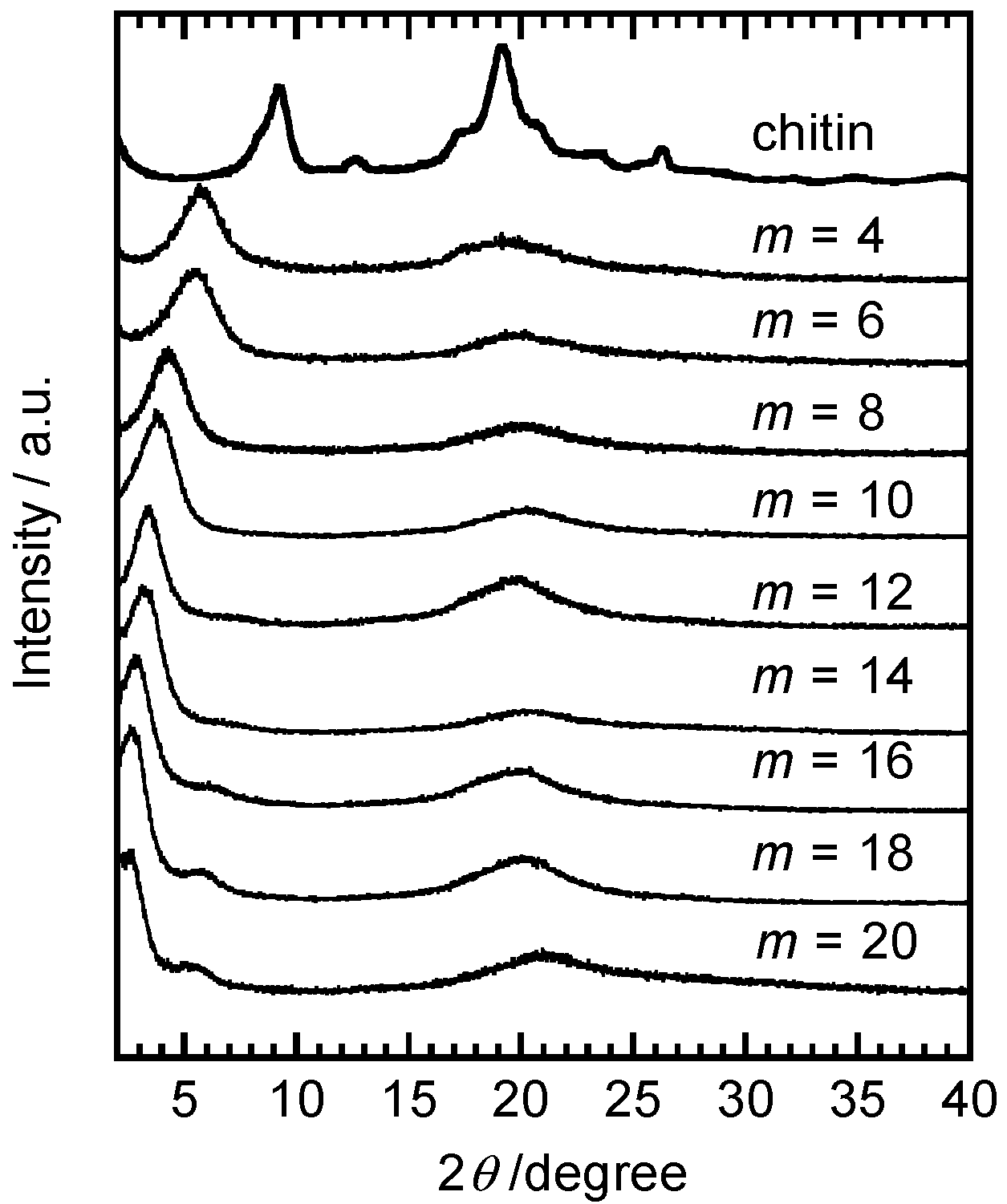
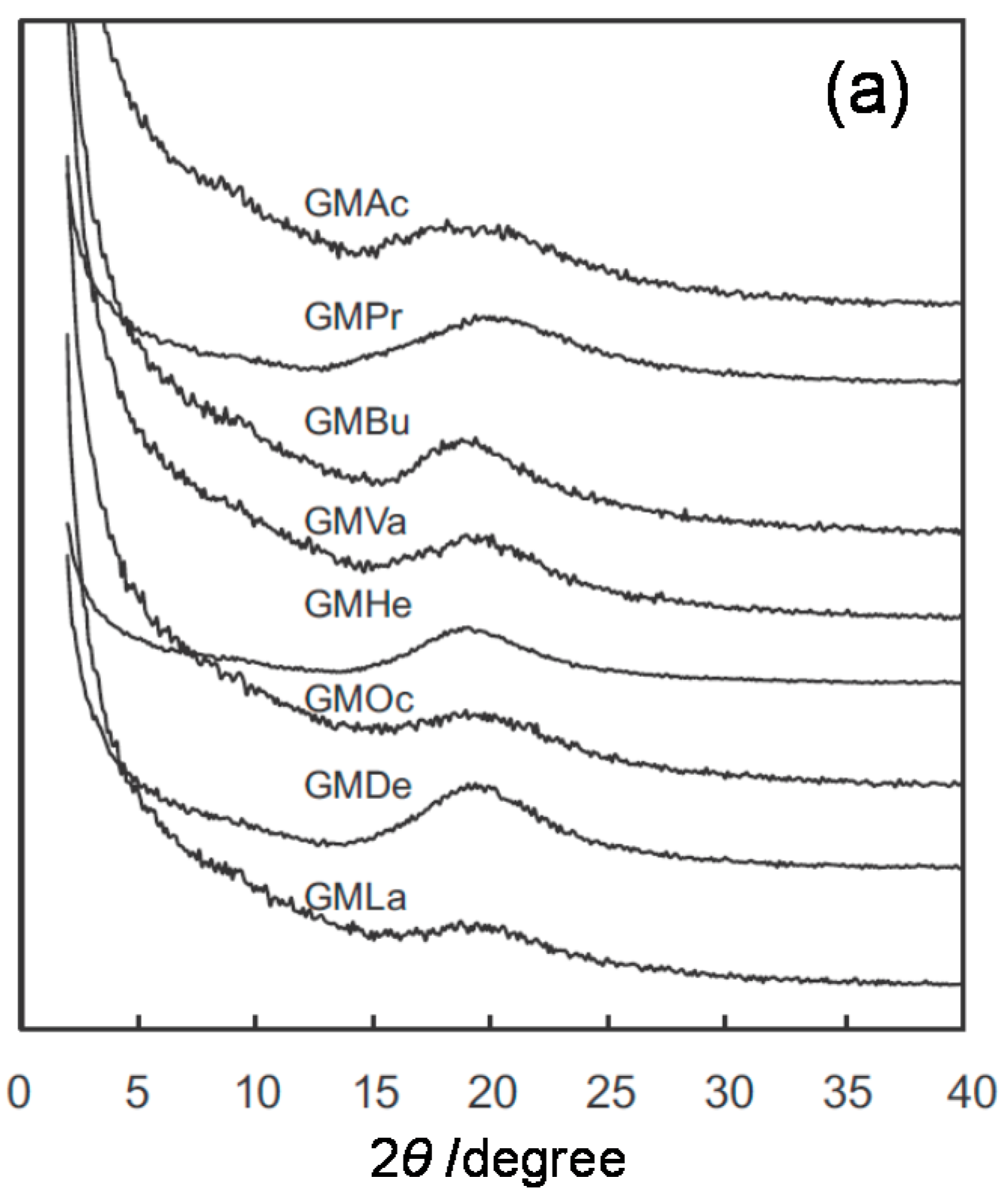
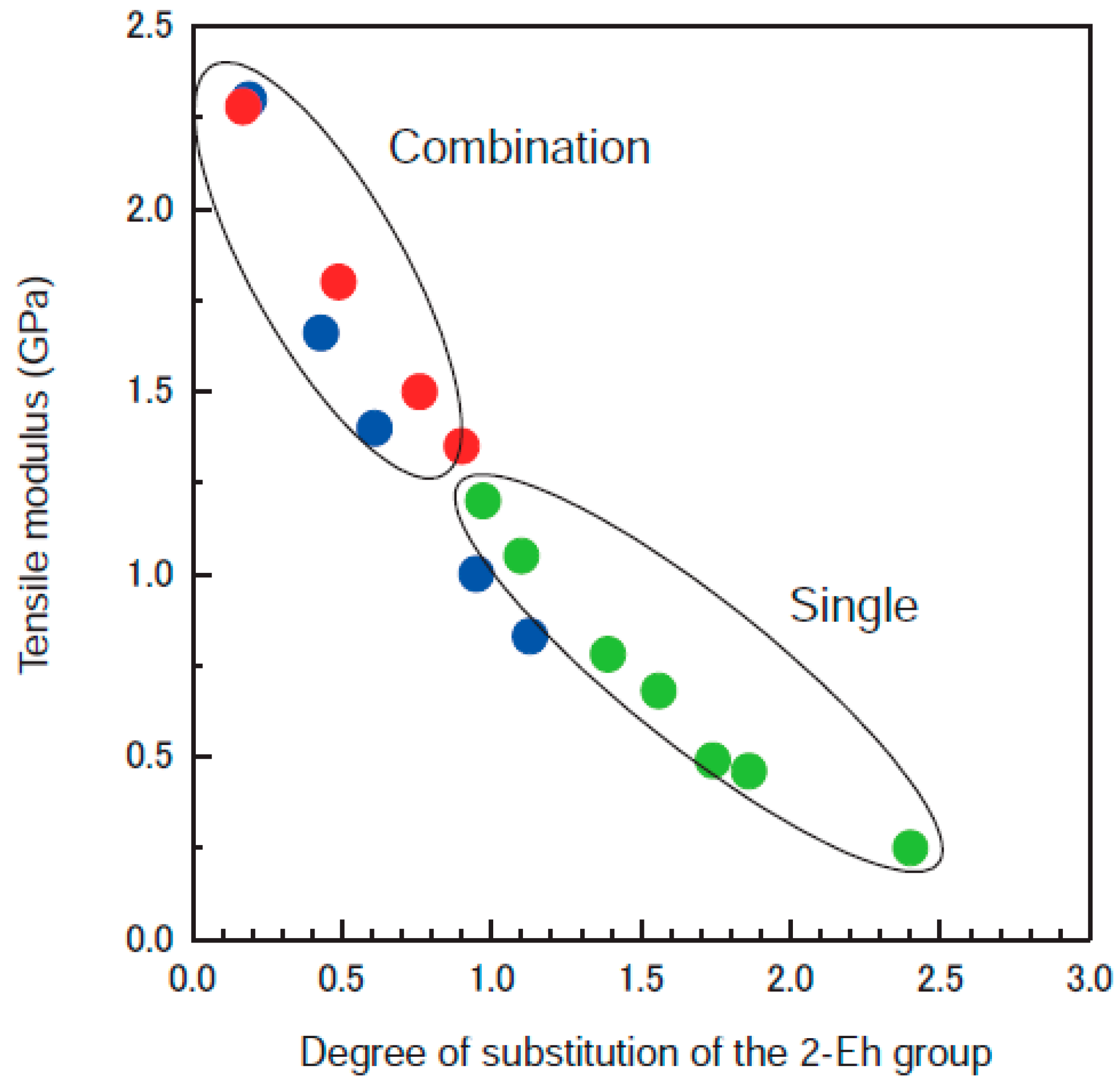
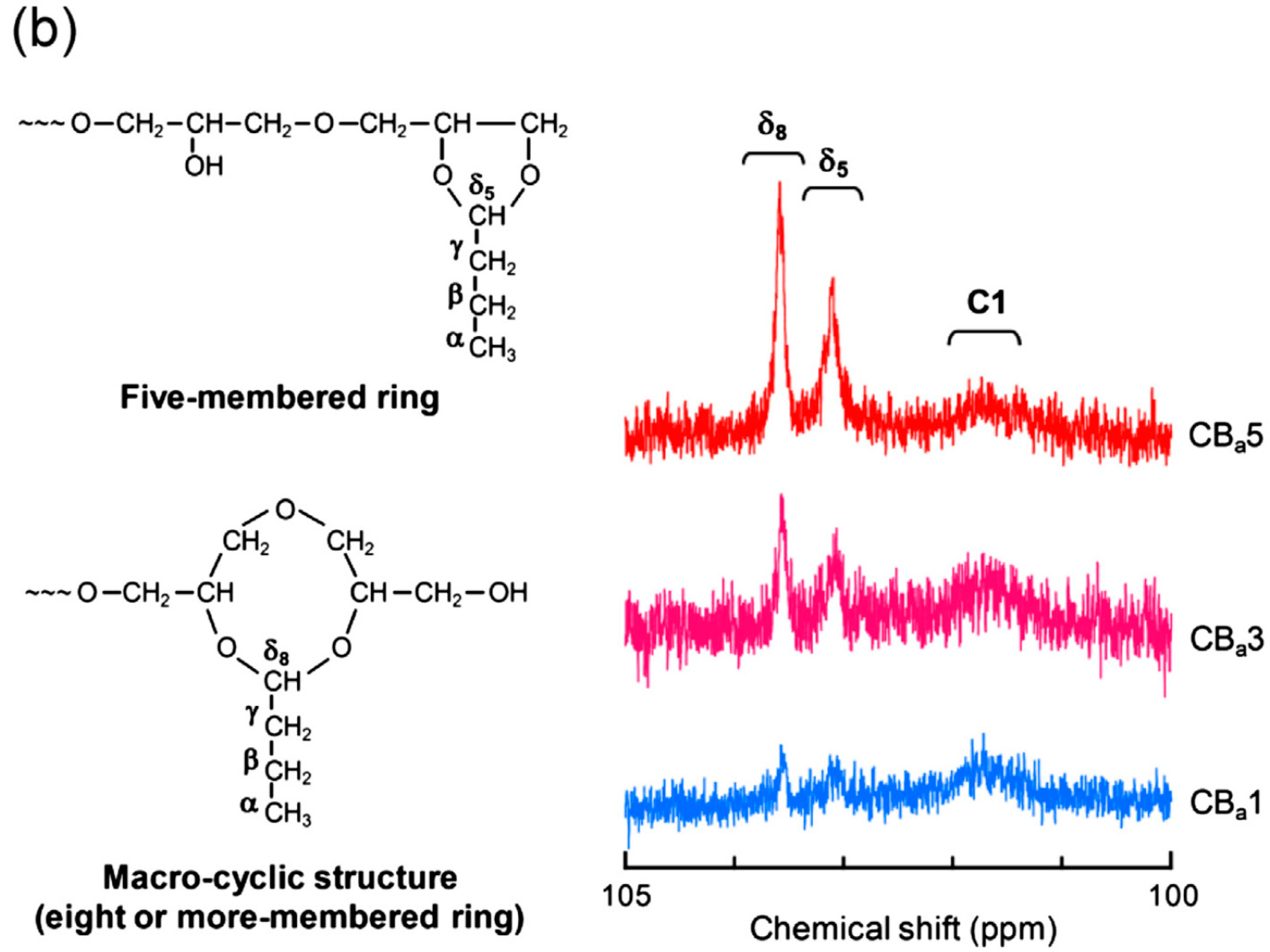
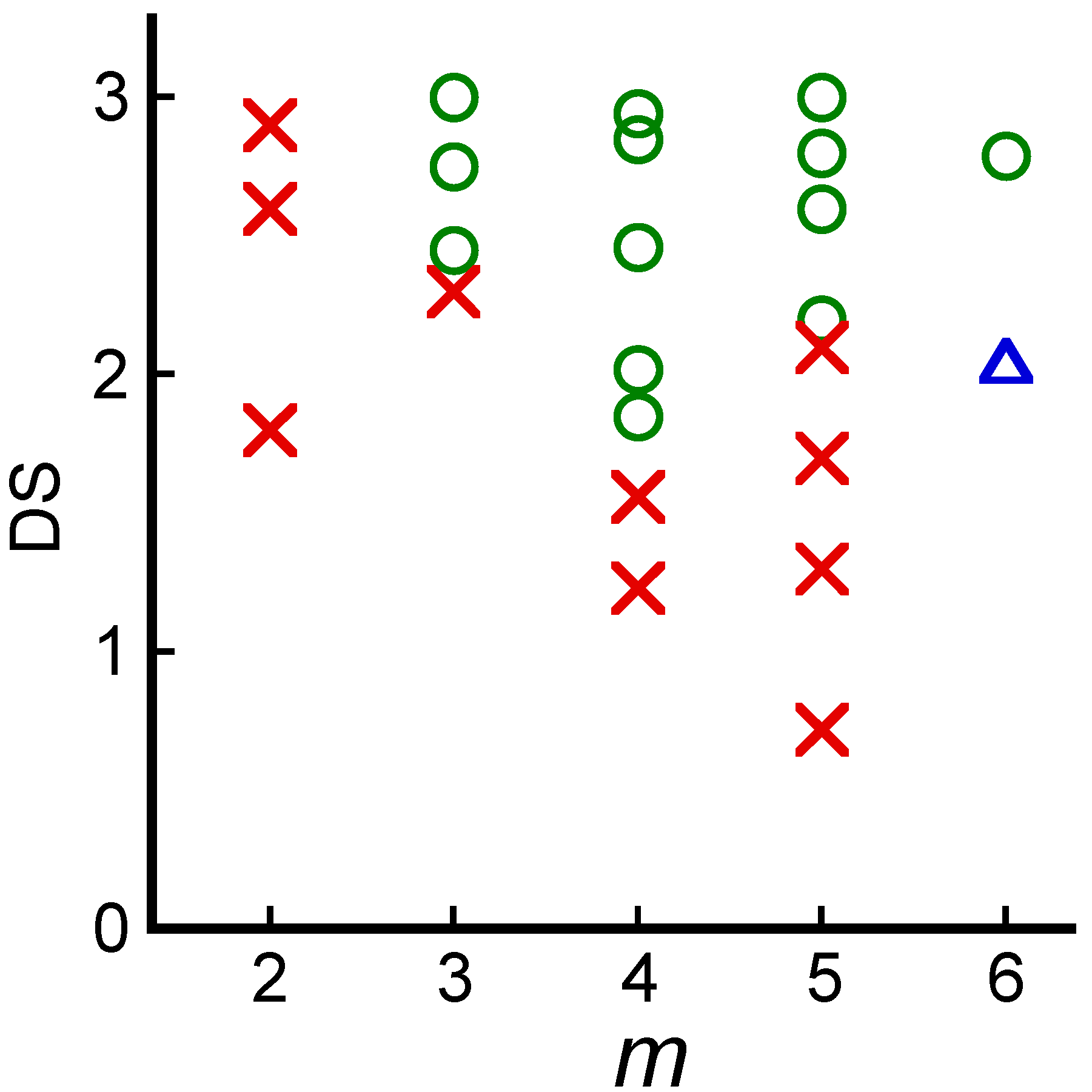
2.2. Systematic Studies of Related Polysaccharides
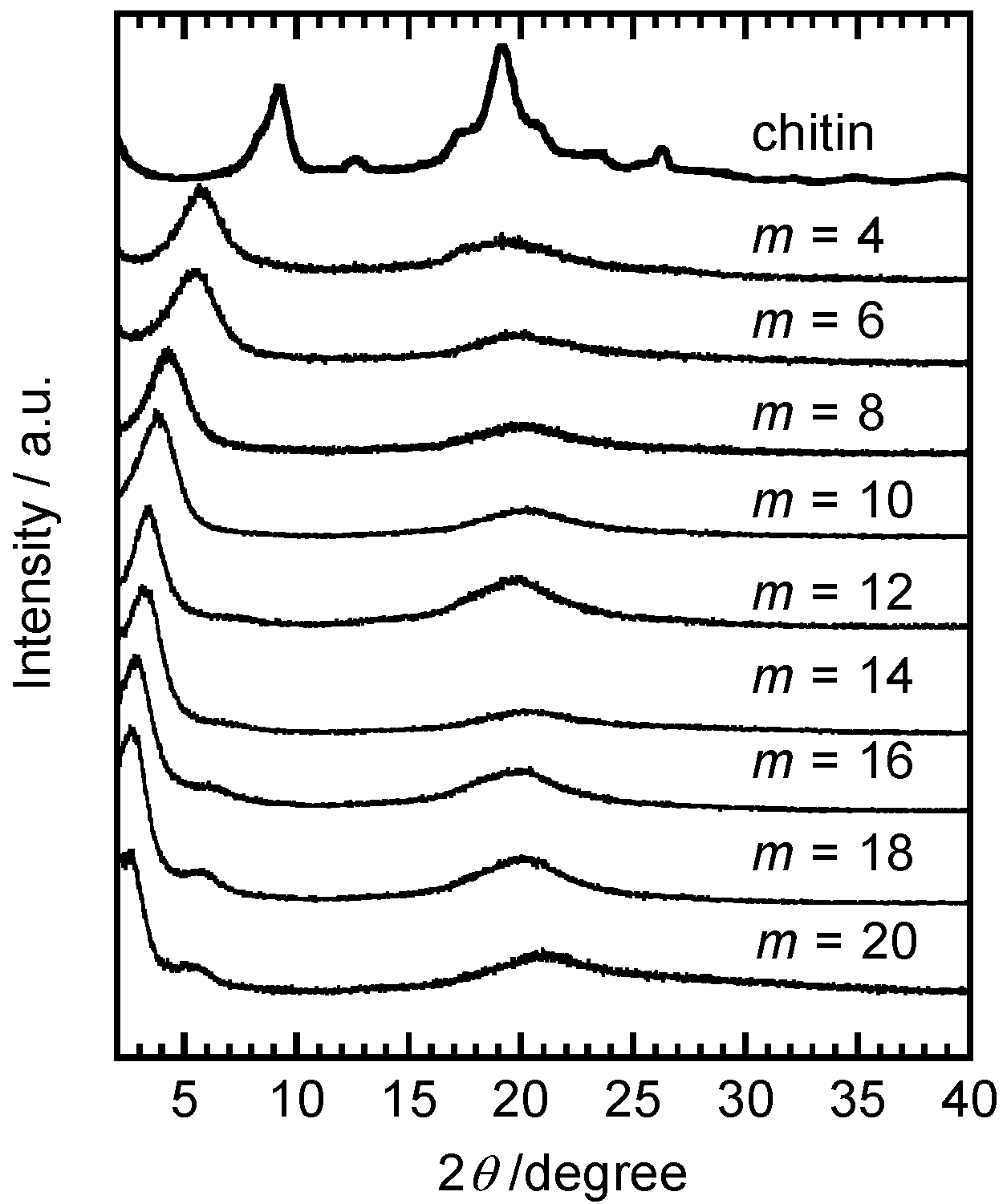

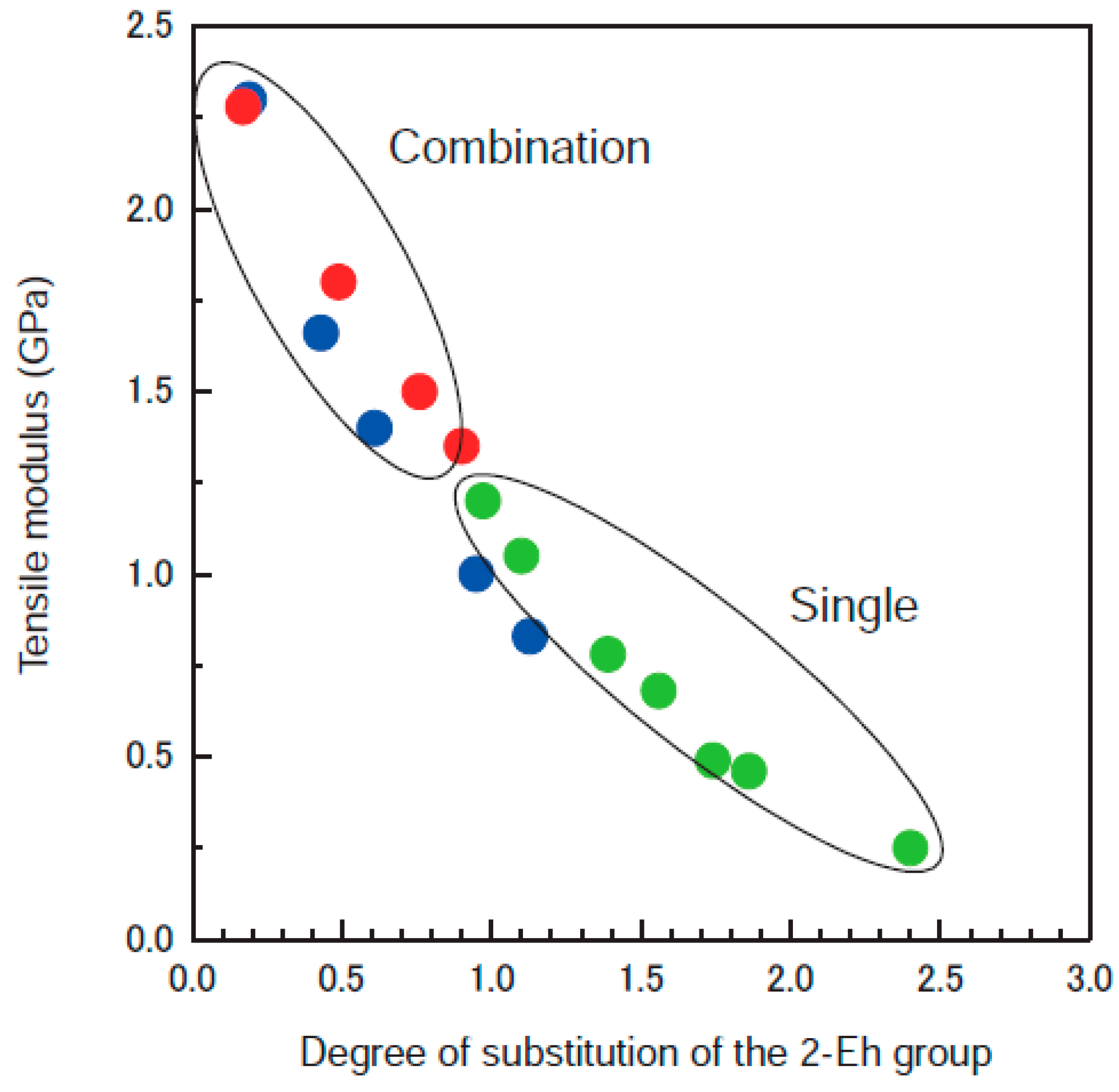
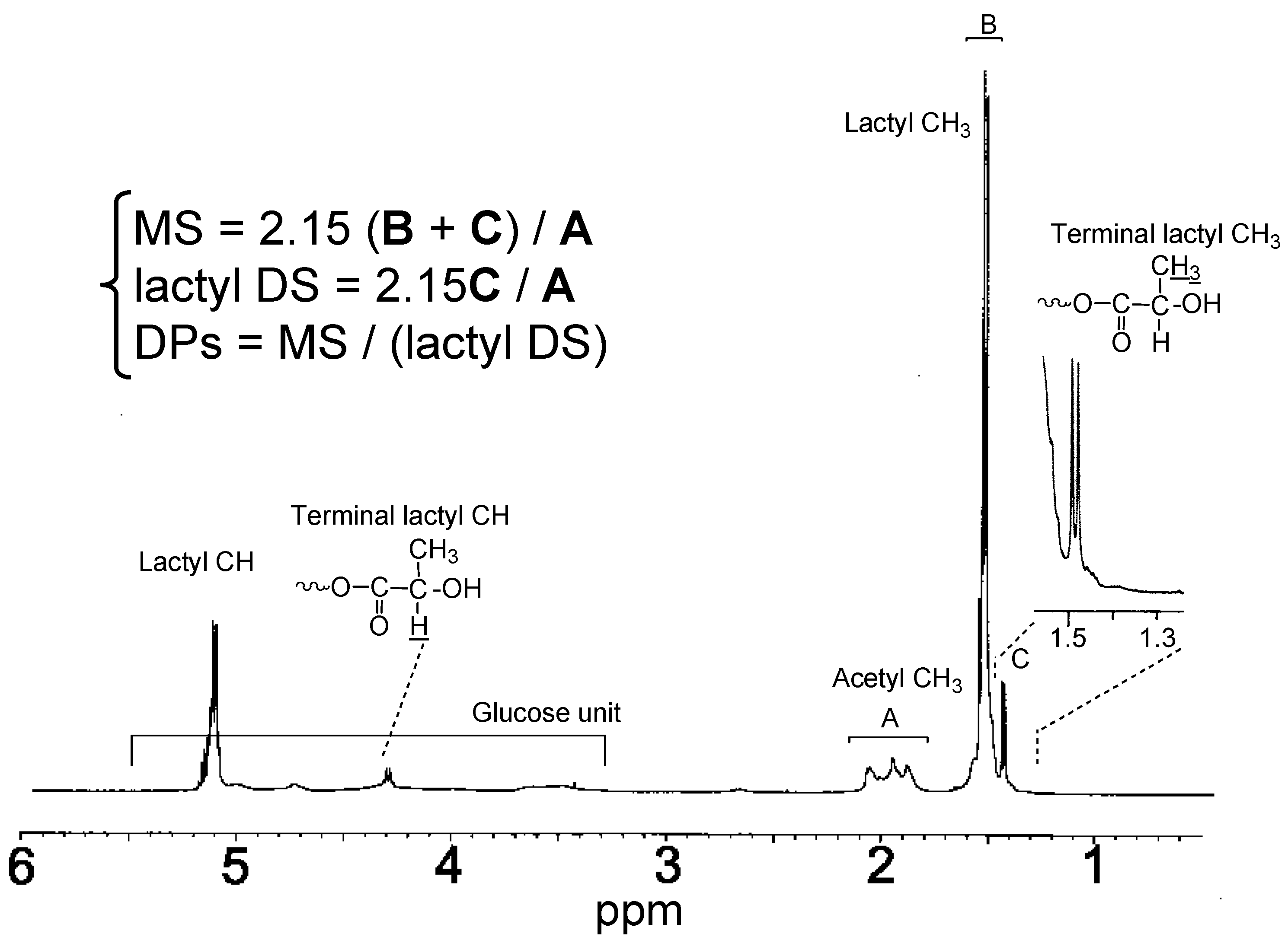
3. Derivatization with Multiple Substituents


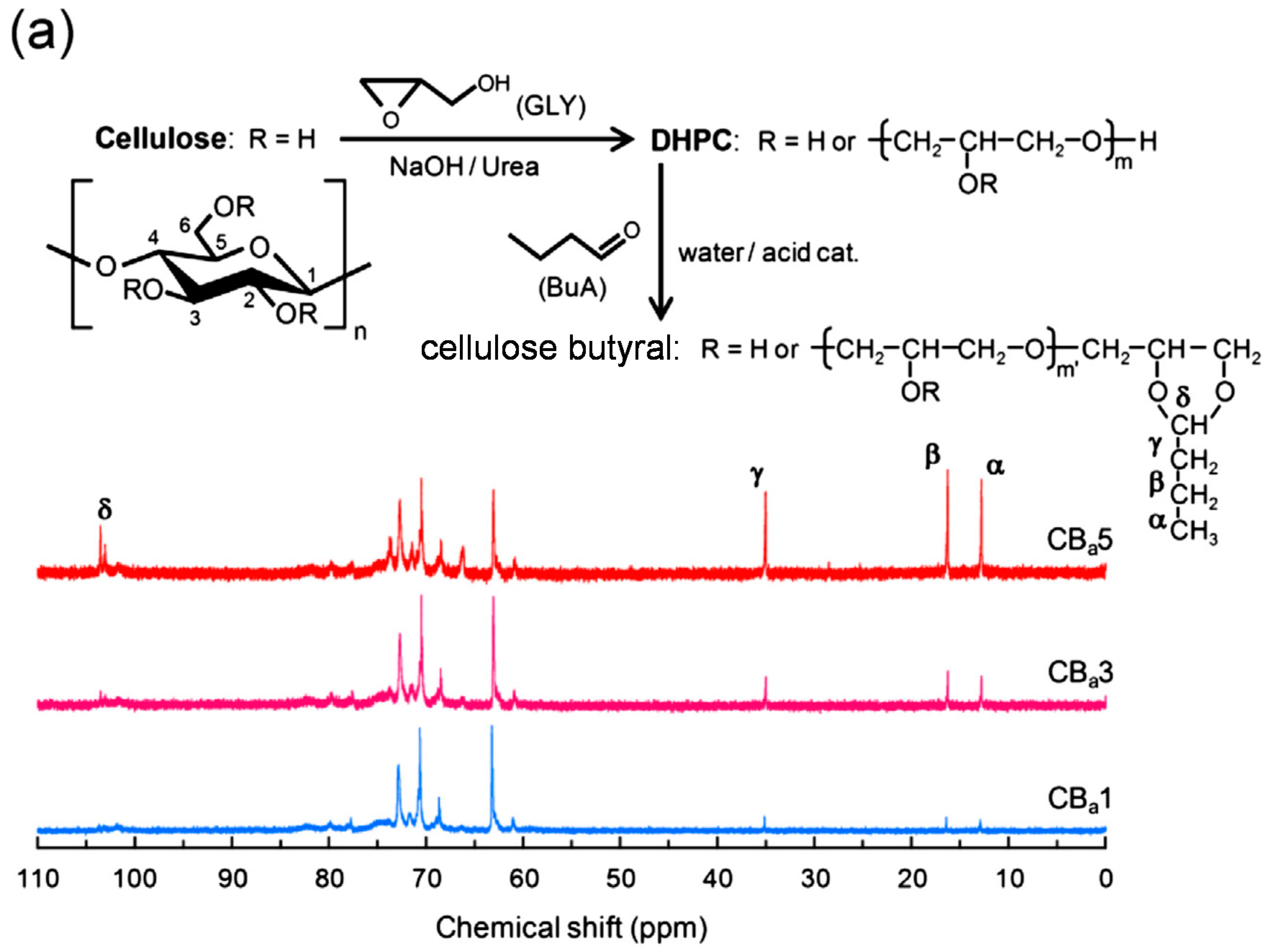

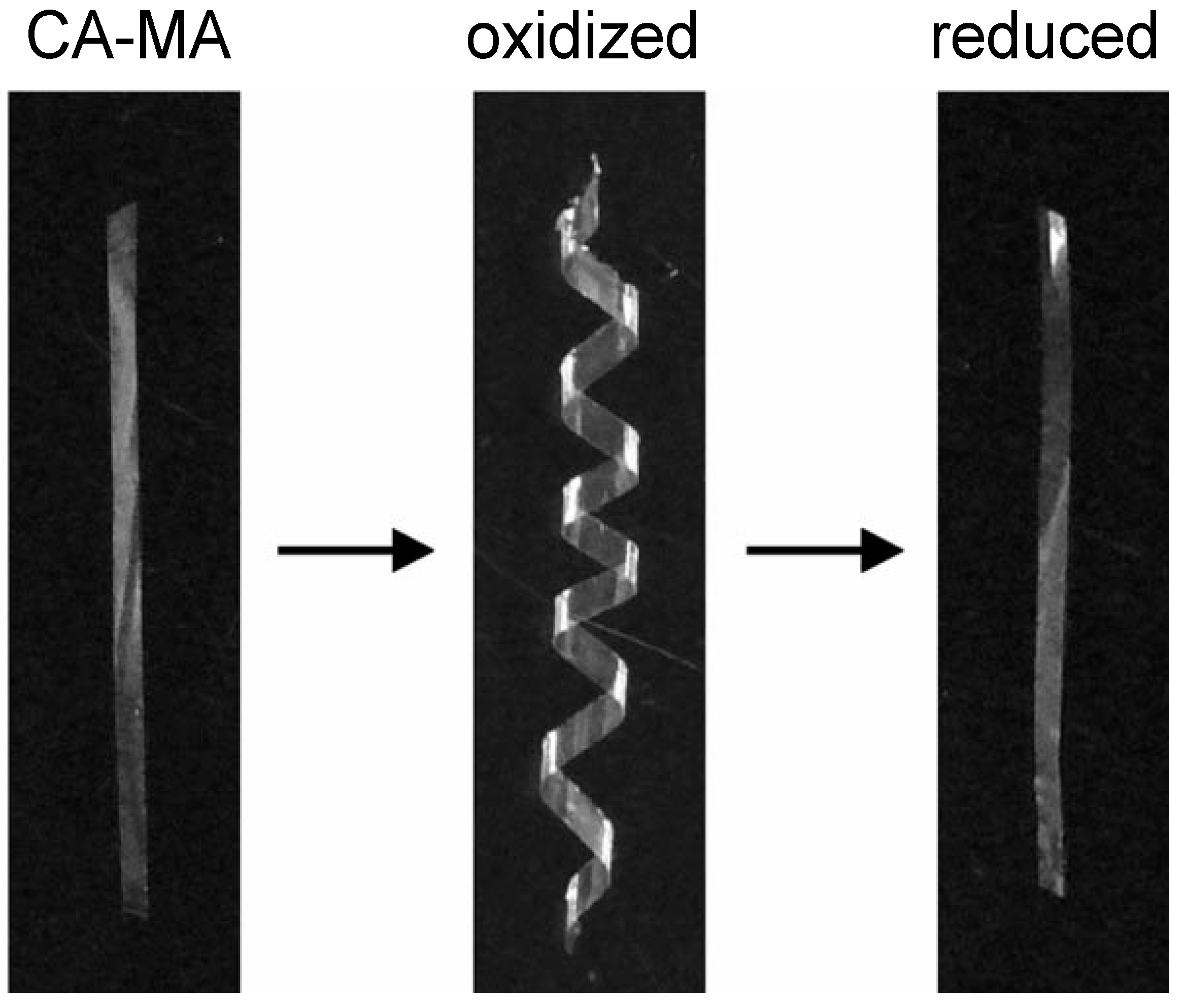
4. Blends of Simple Derivatives with Synthetic Polymers
4.1. Blend with N-Vinyl Pyrrolidone-Based Polymers
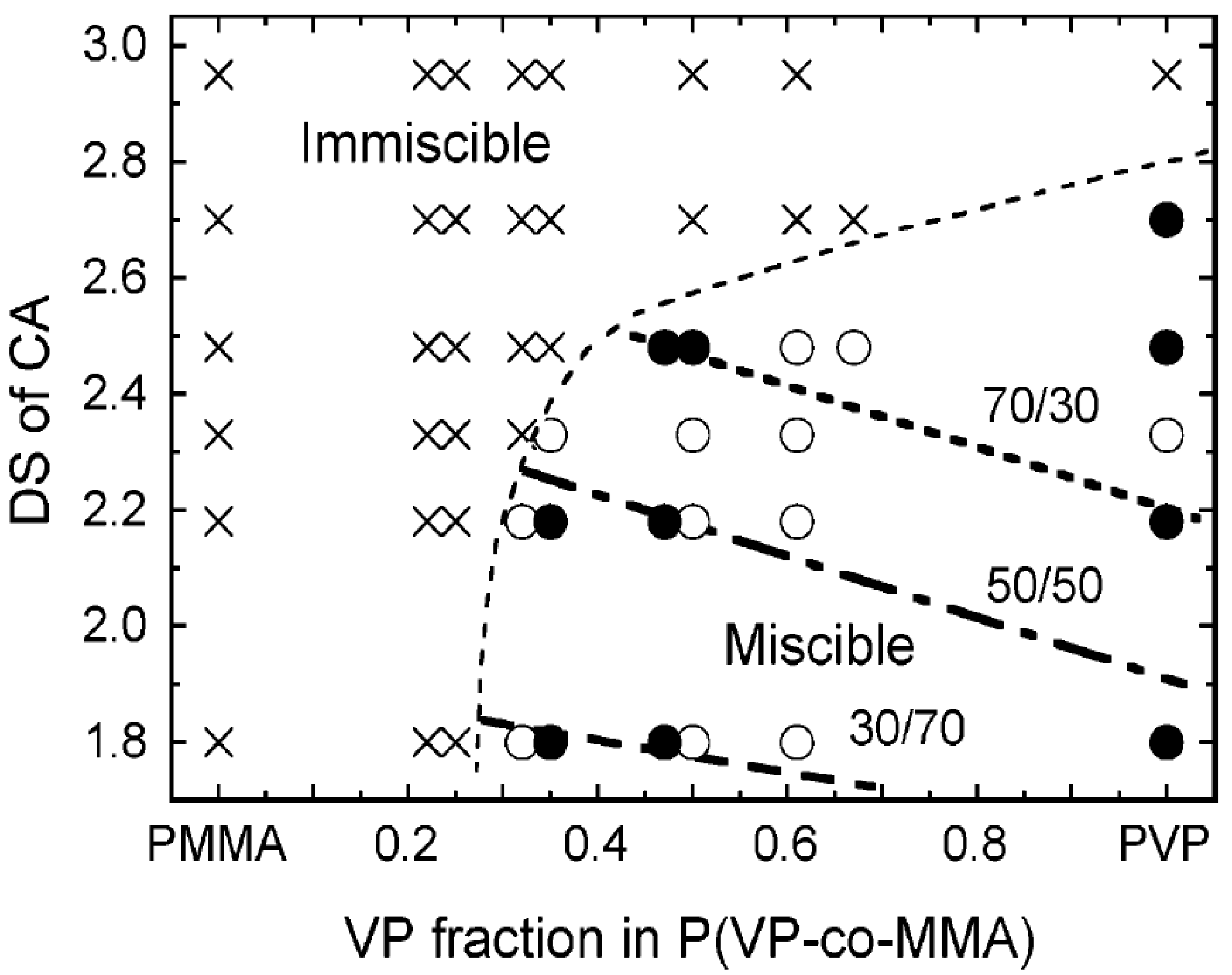
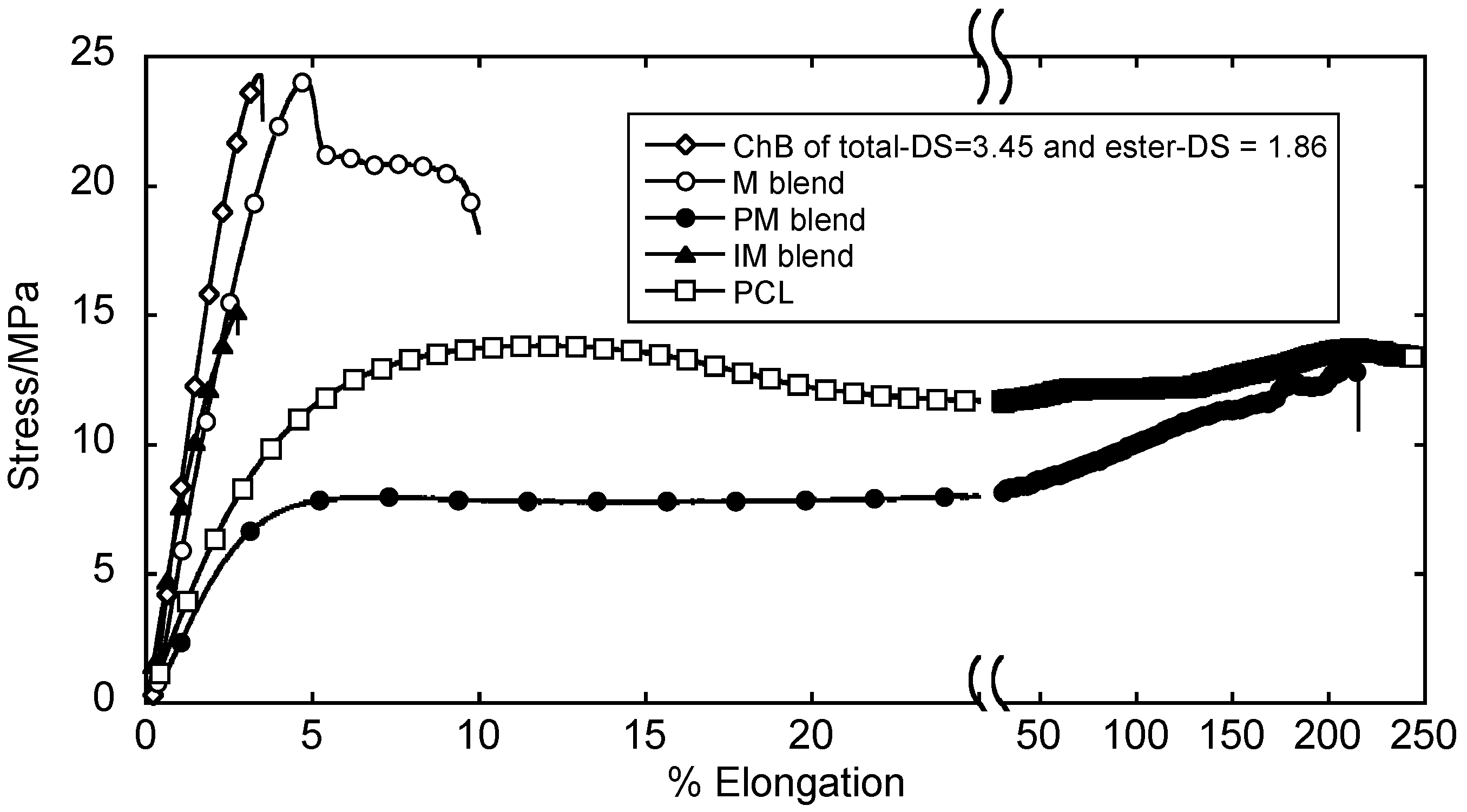
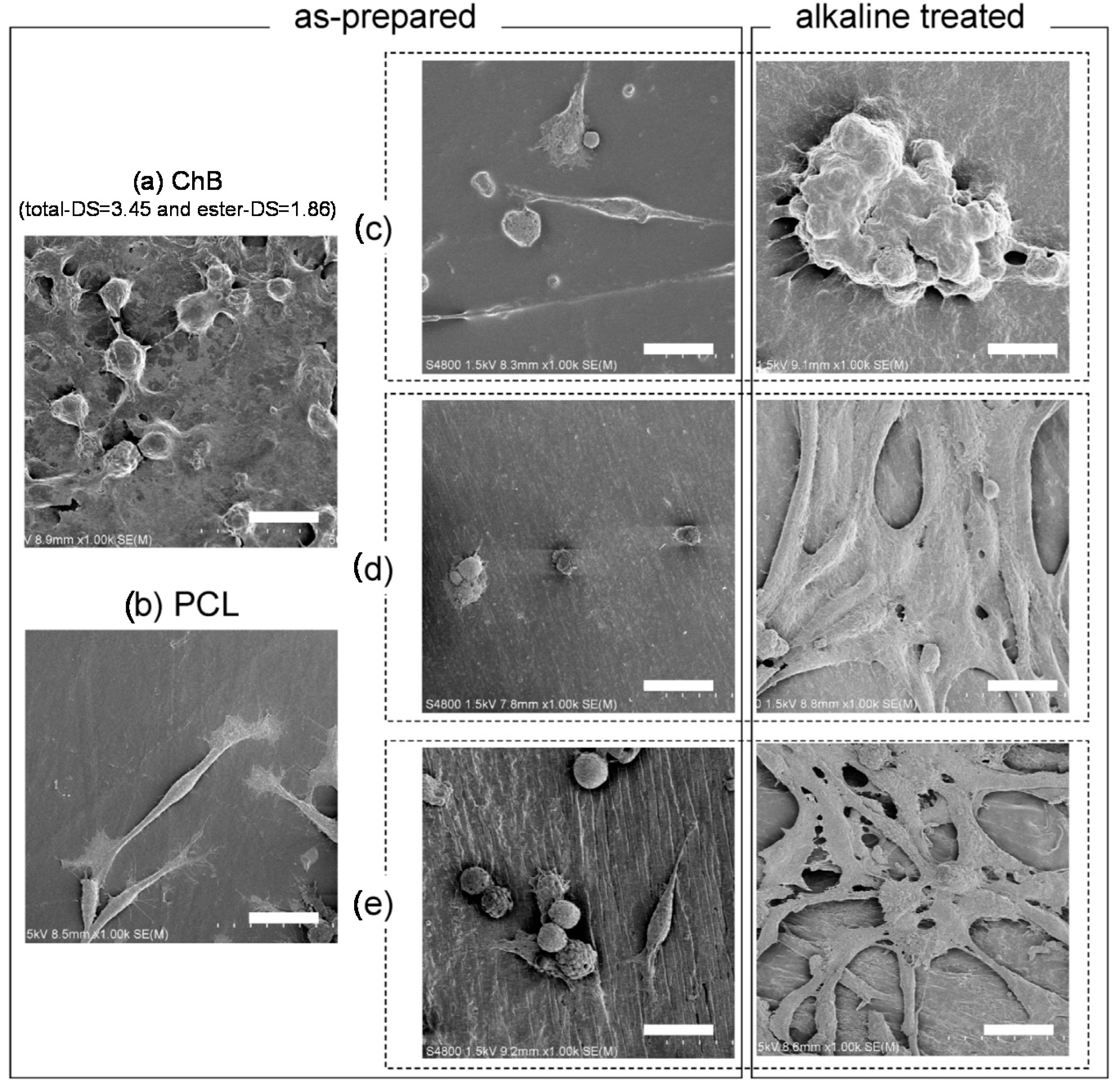
4.2. Blends with Aliphatic Polyesters

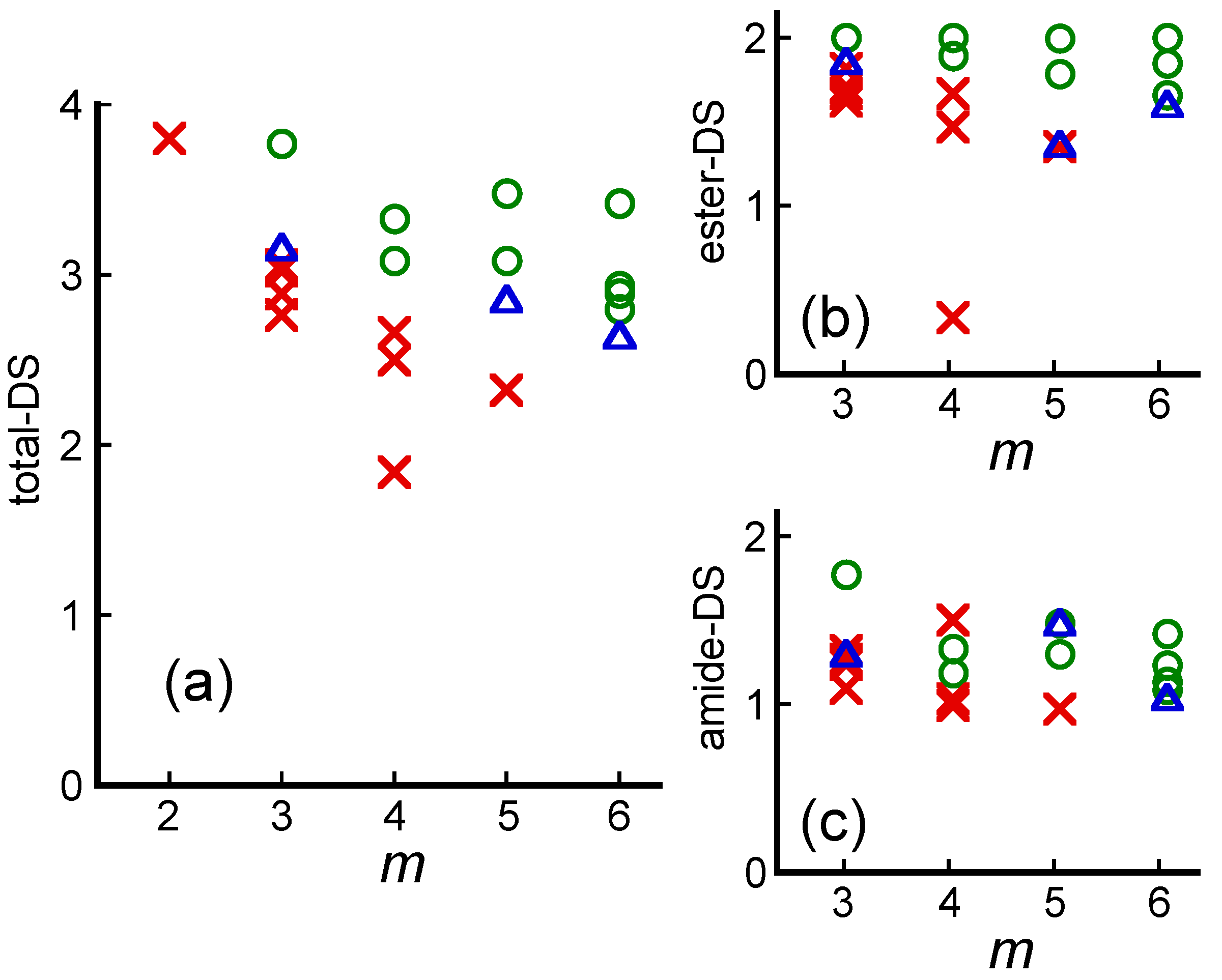


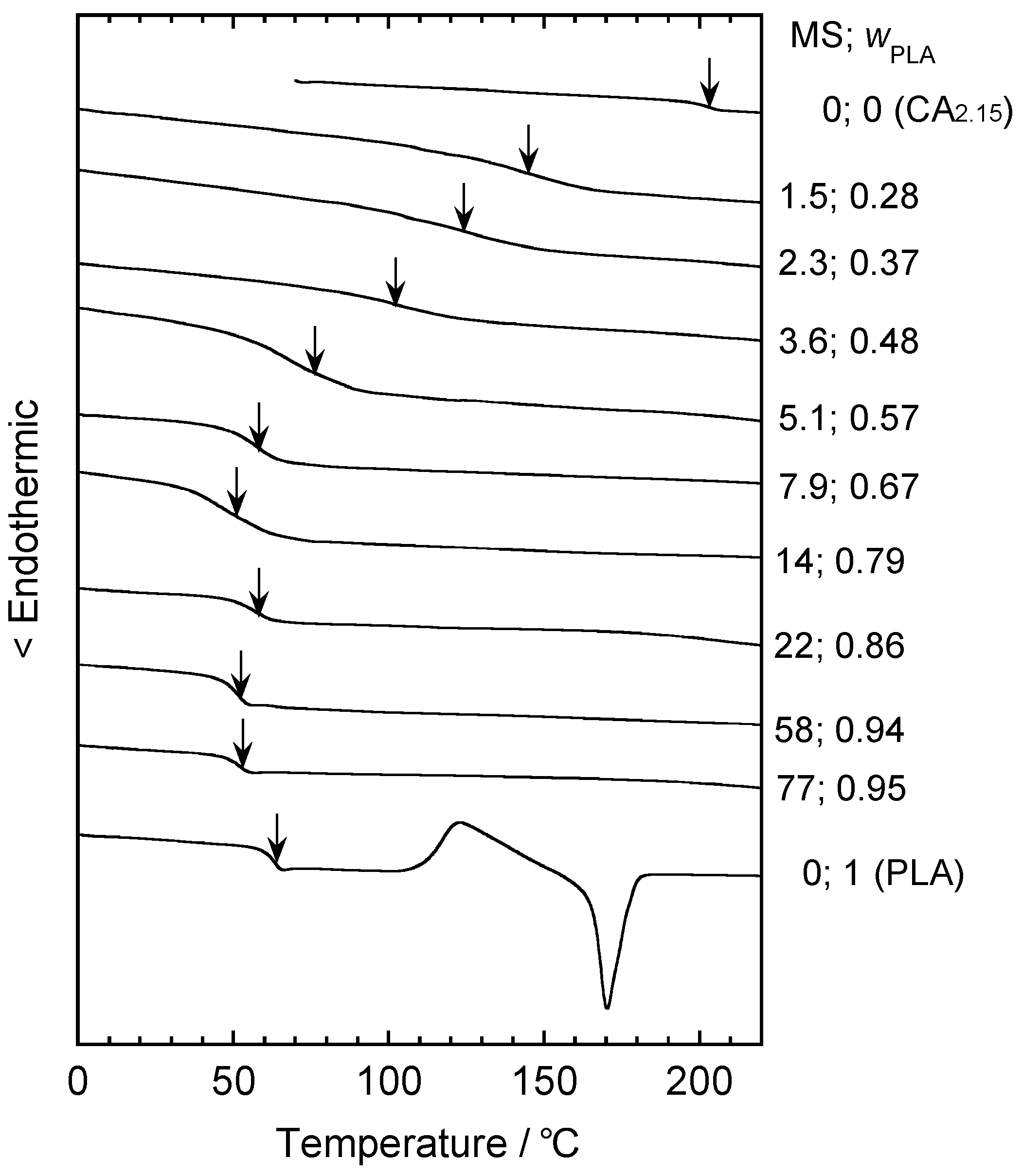
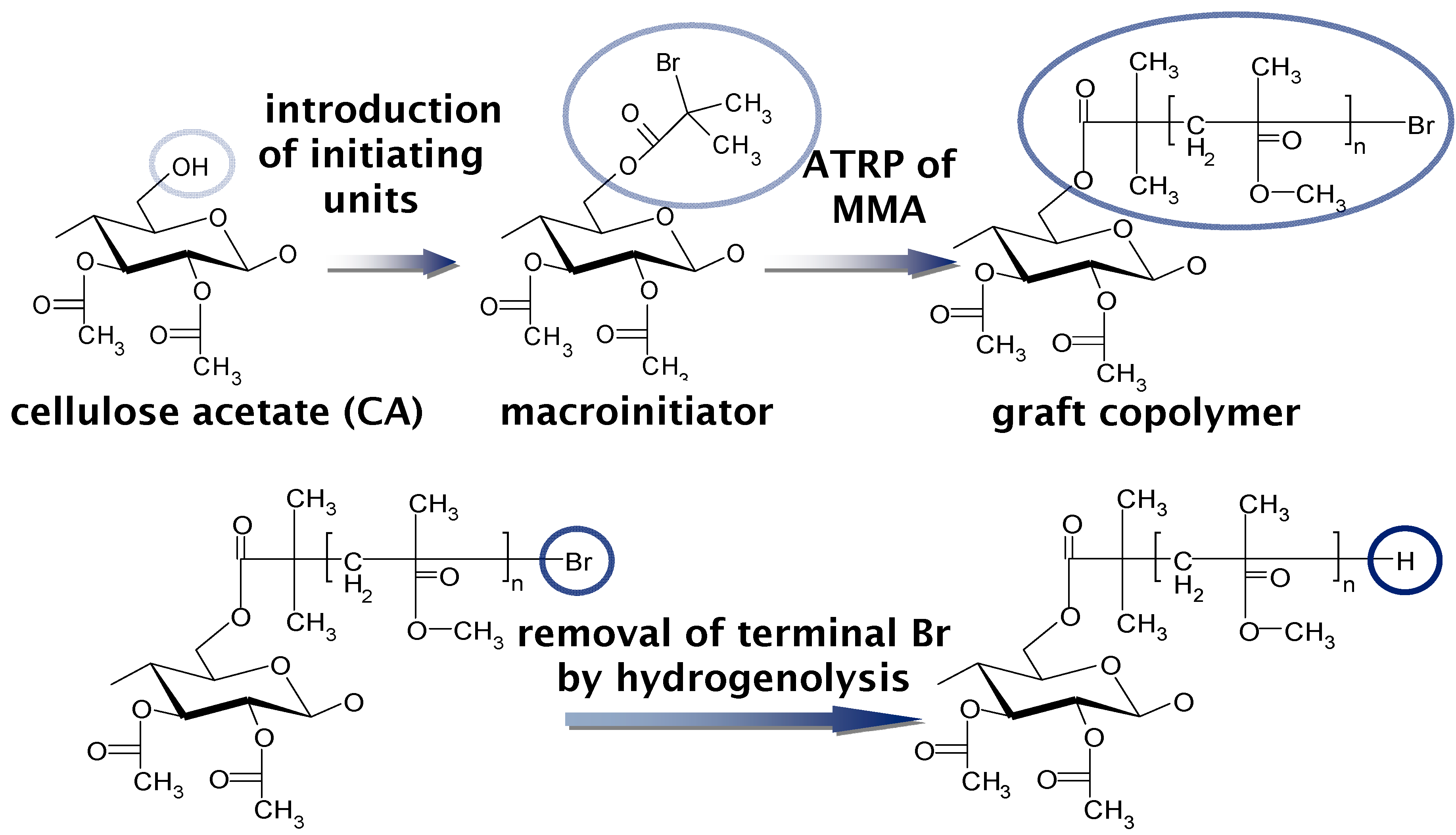
5. Graft Copolymers
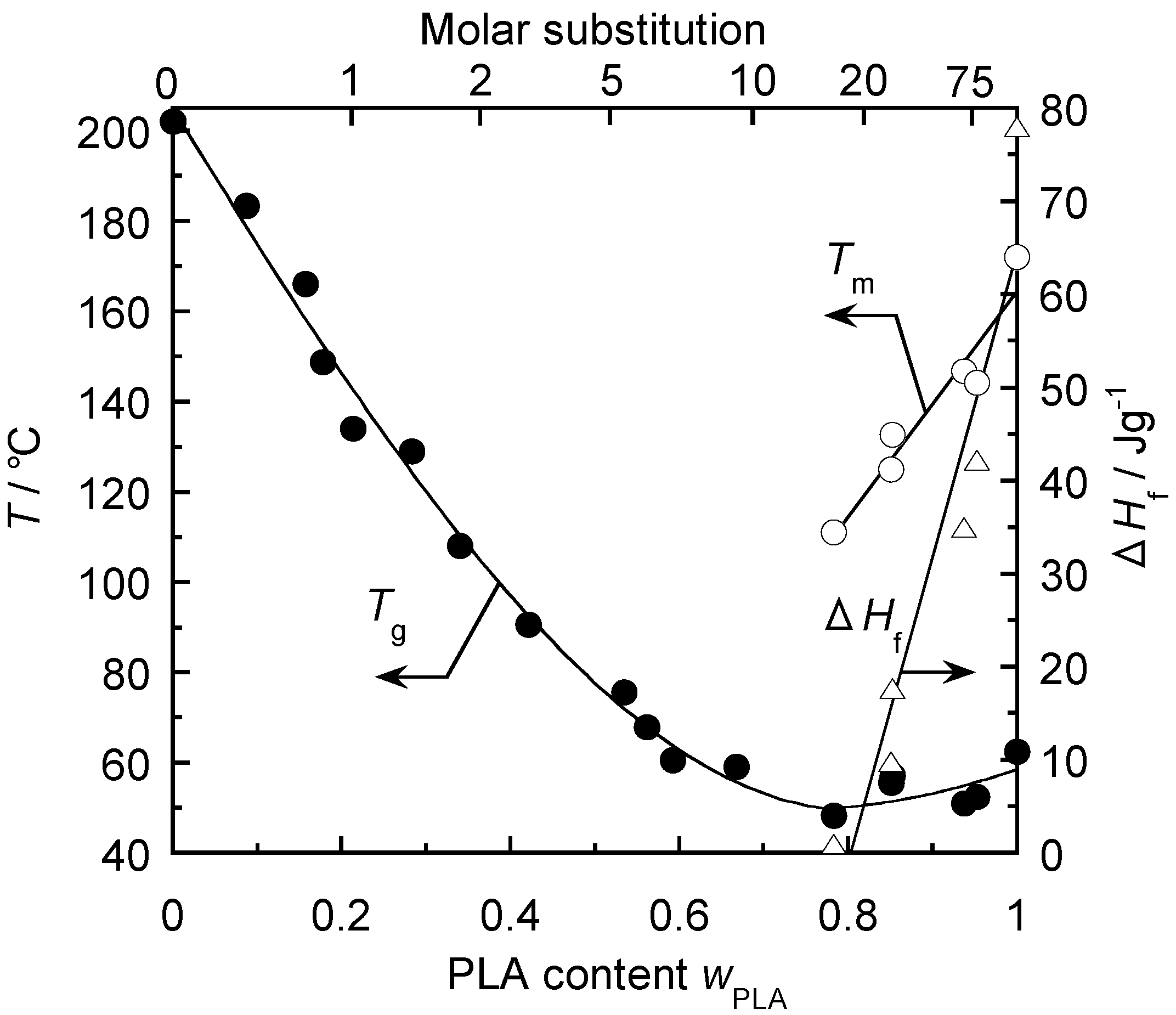
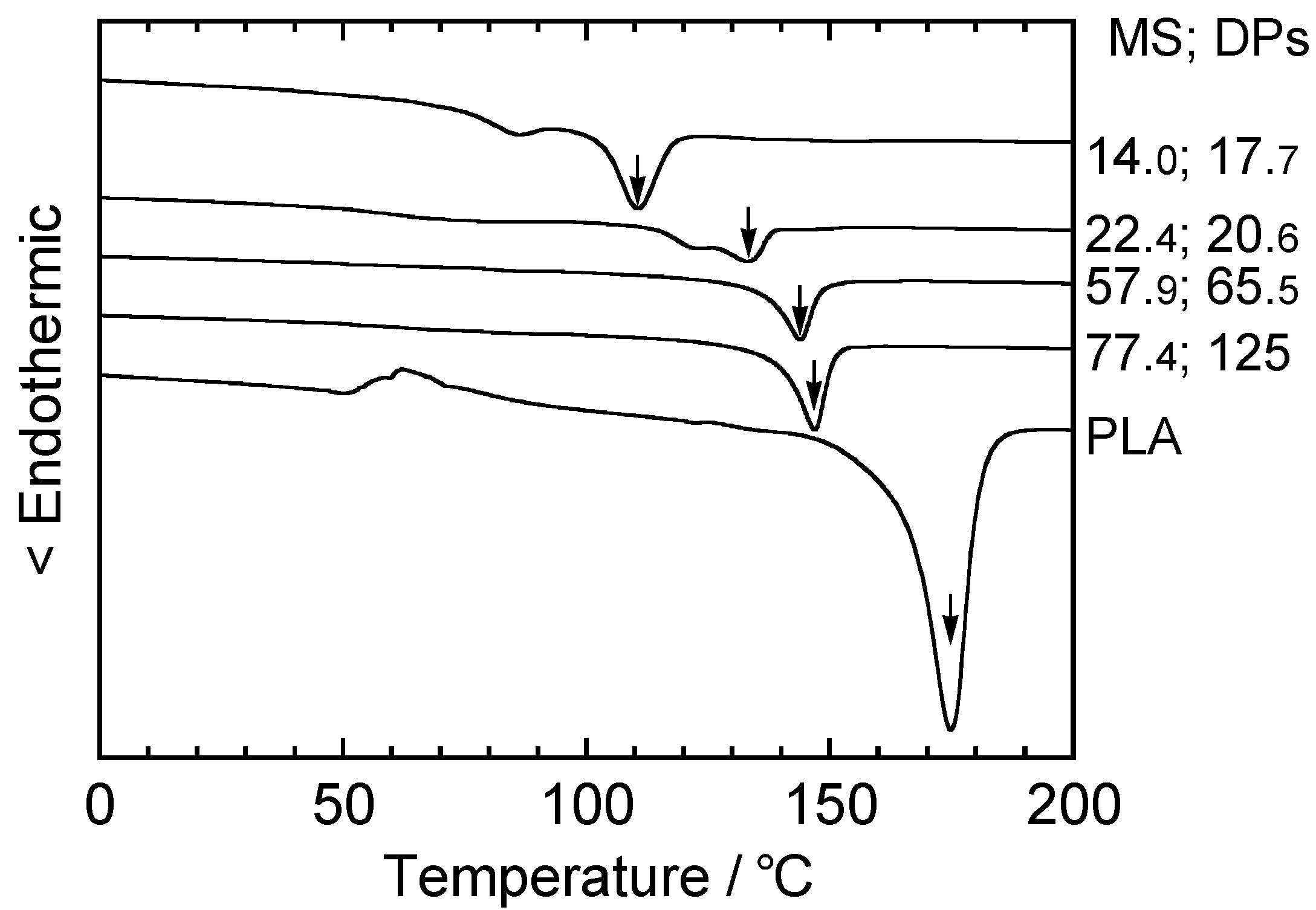
5.1. Synthesis of Biodegradable Cellulosic Graft Copolymers and Formulation of Basic Physical Properties

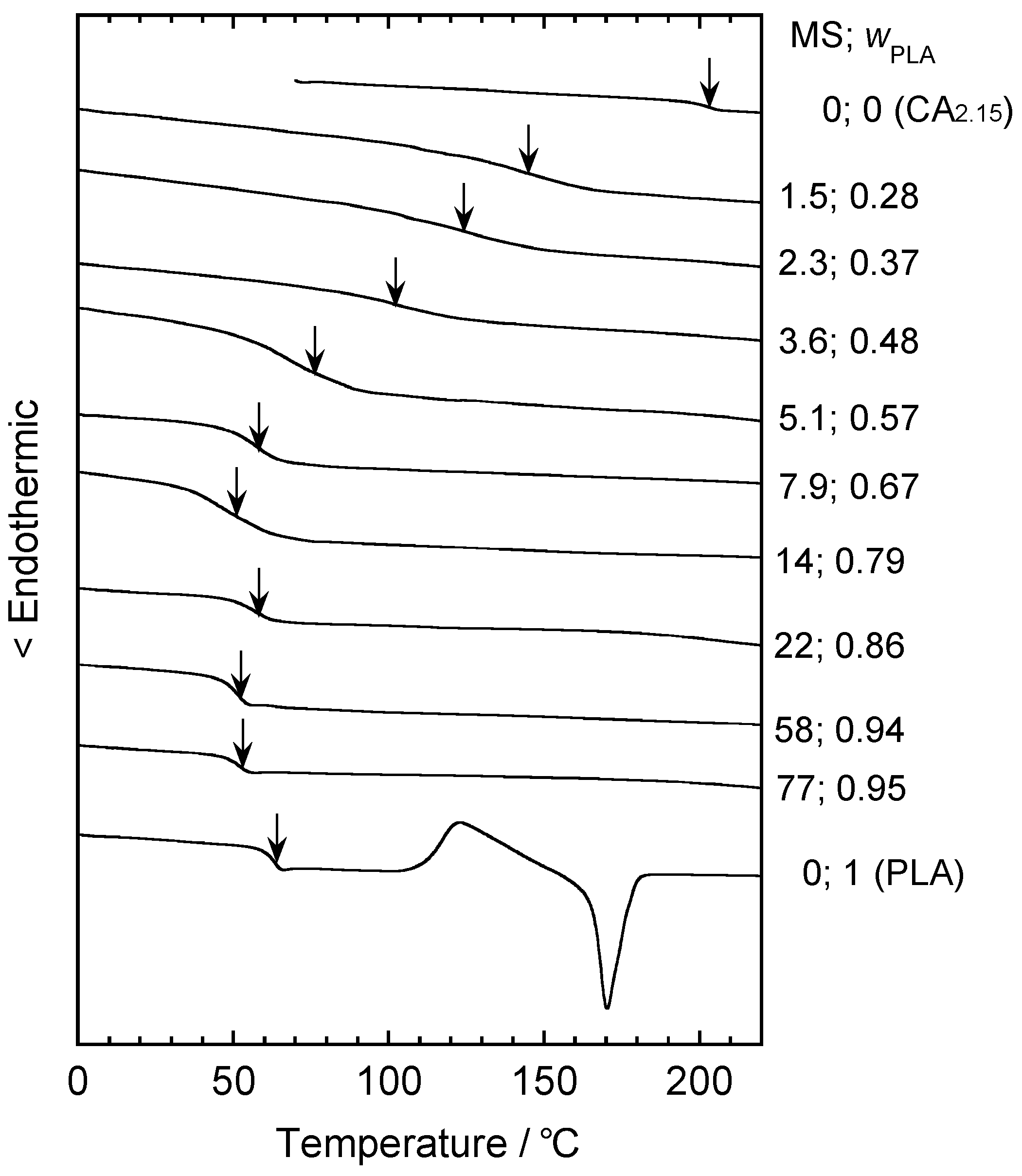
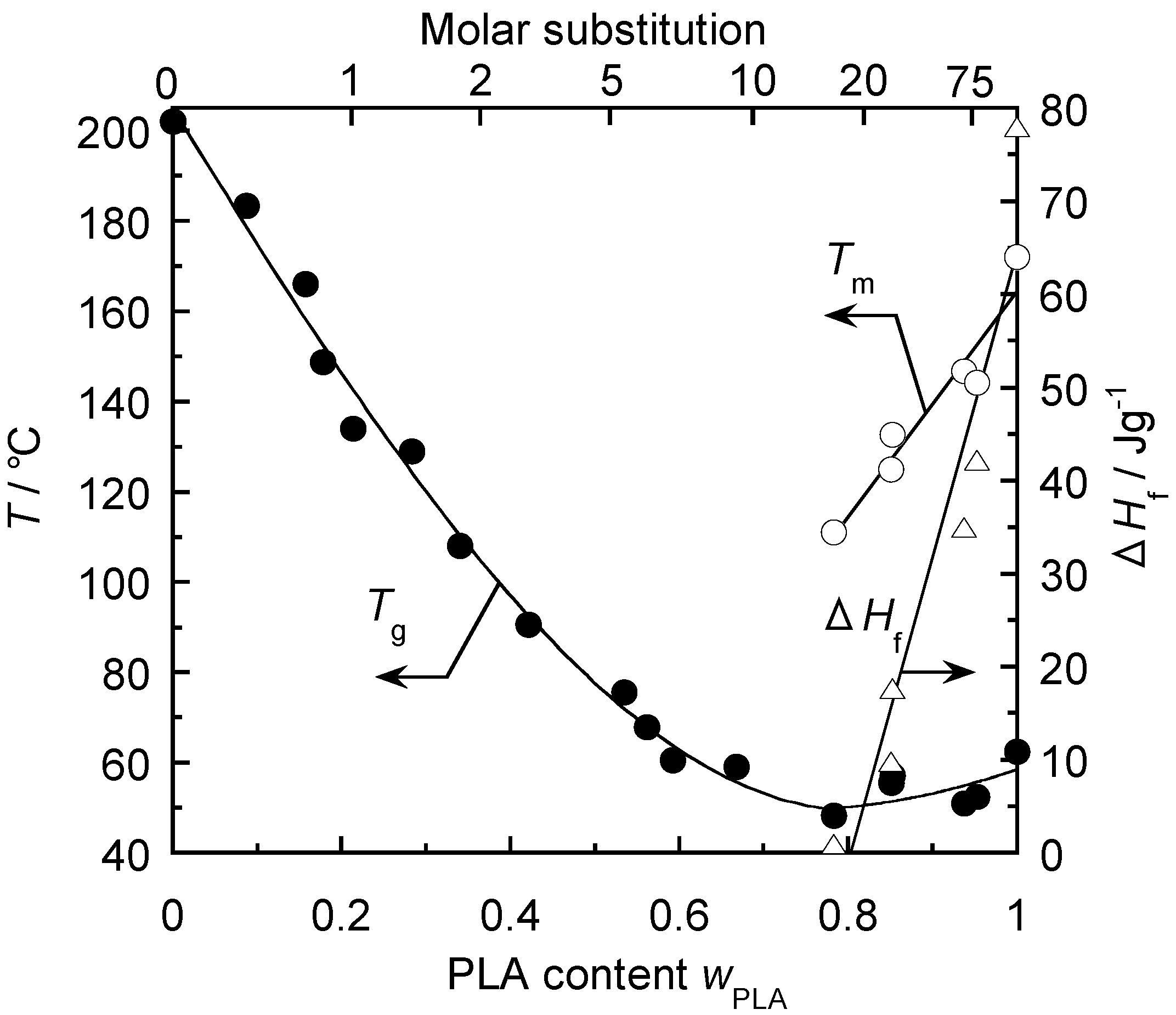
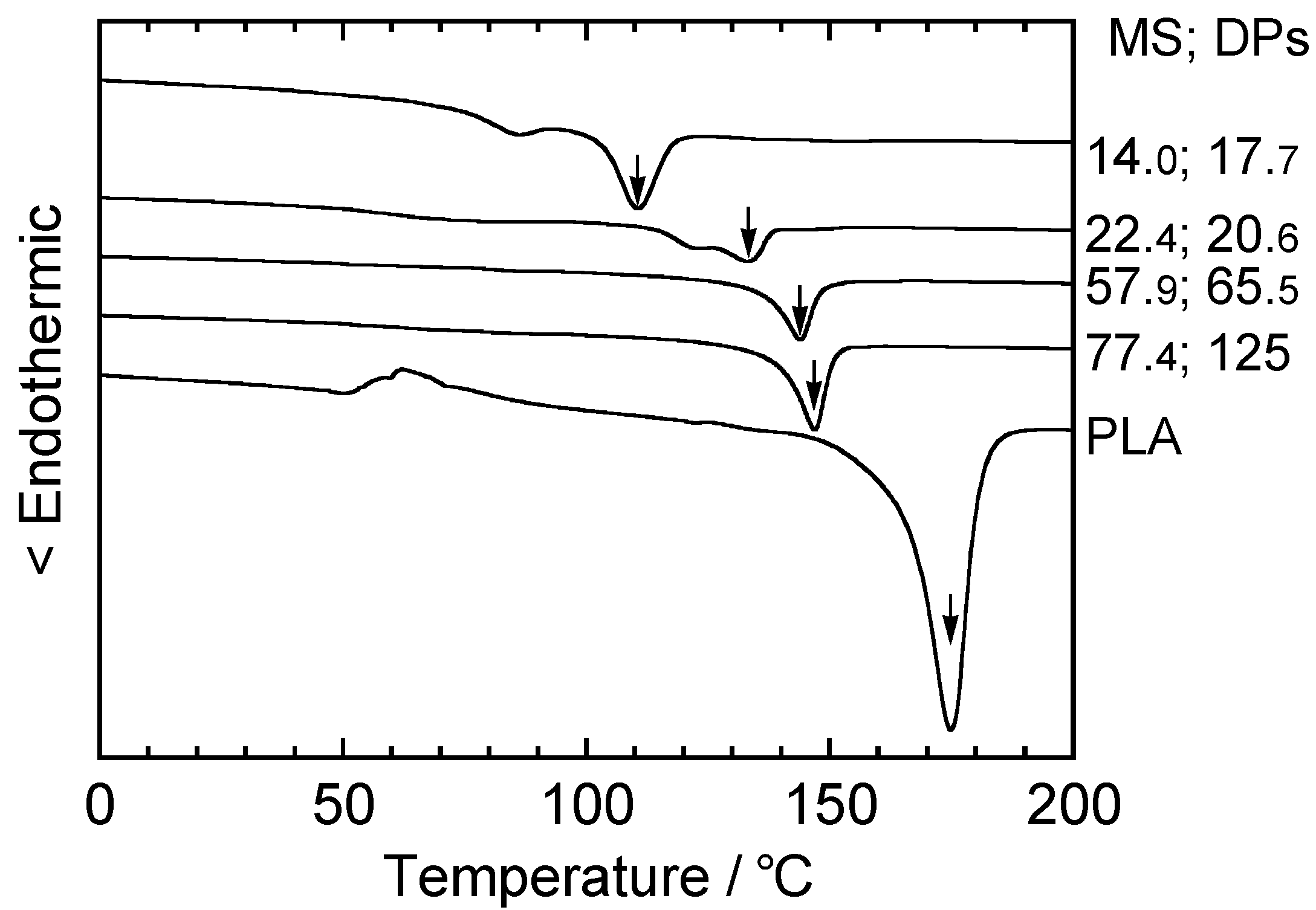
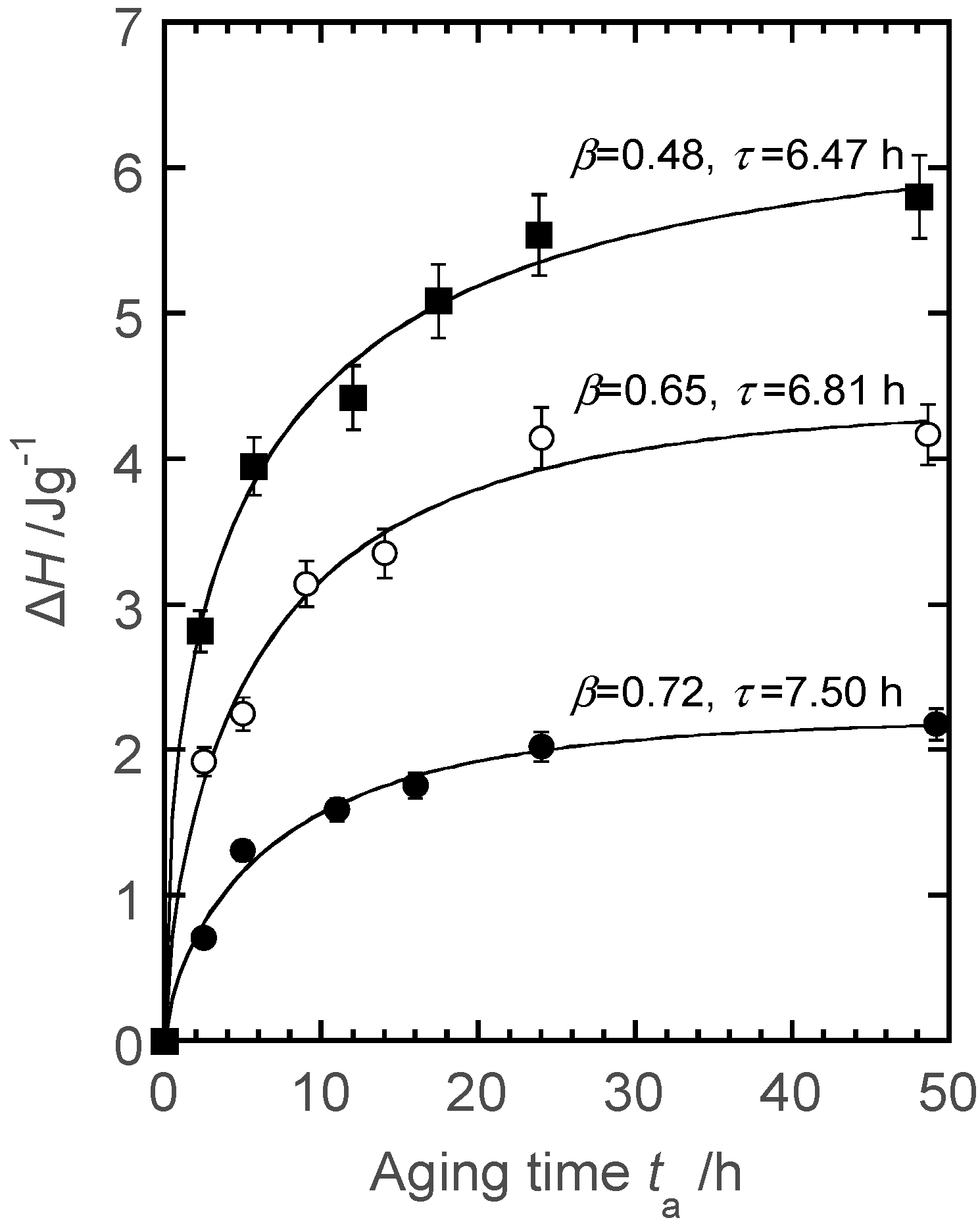
5.2. Thermal Treatment Effect on the Development of Supramolecular Structures
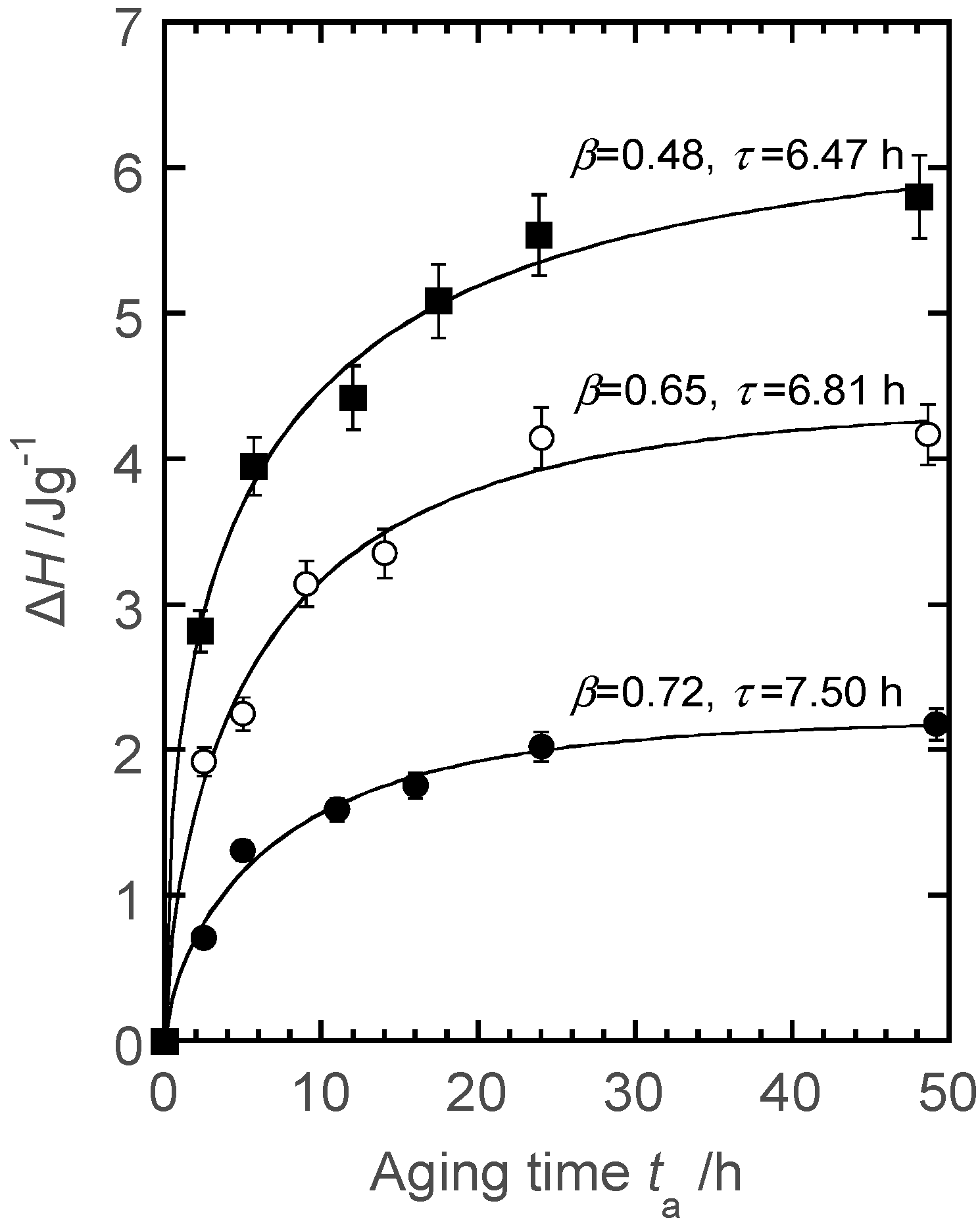

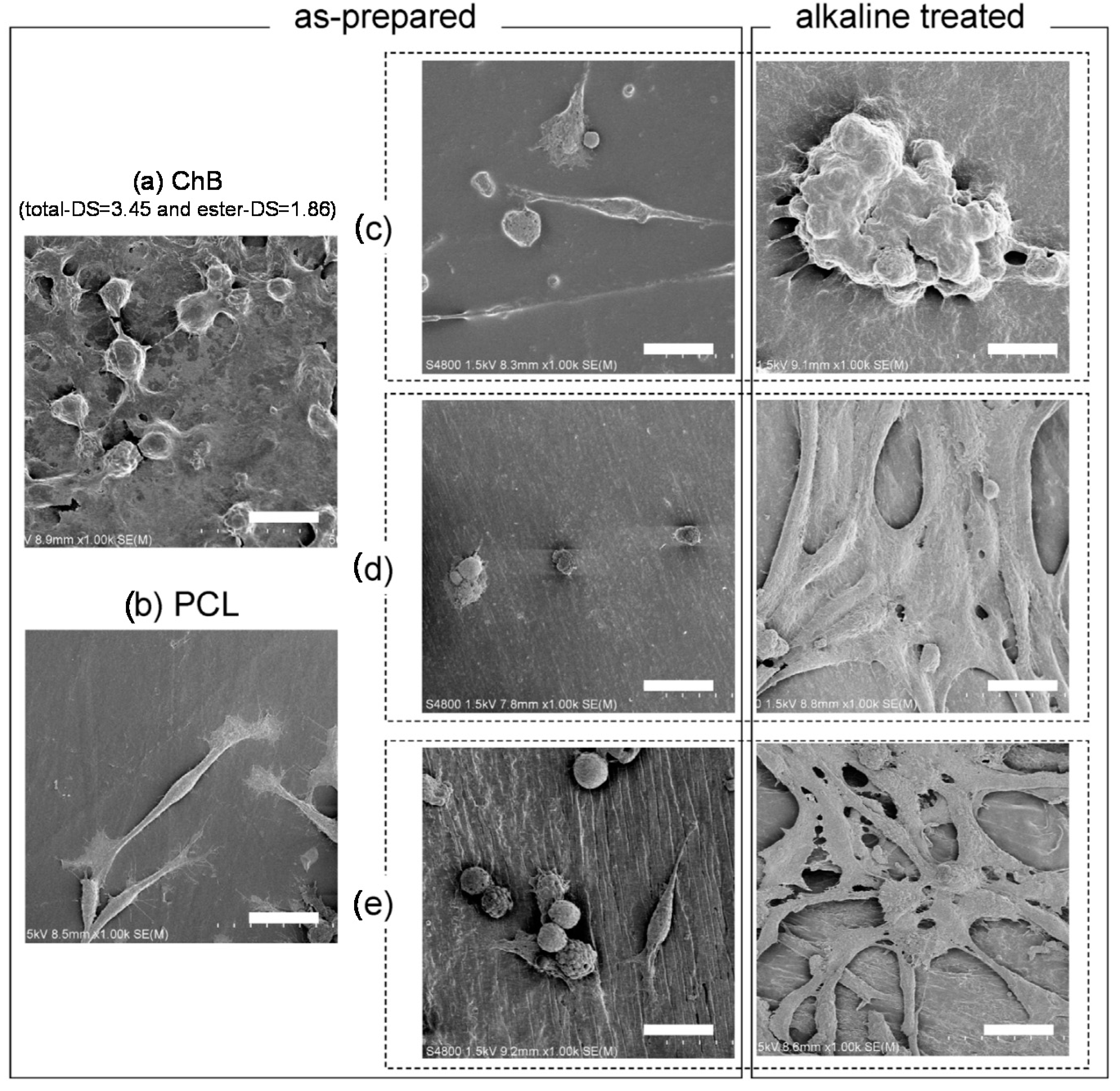
5.3. Enzymatic Hydrolysis and Surface Morphological Characterization

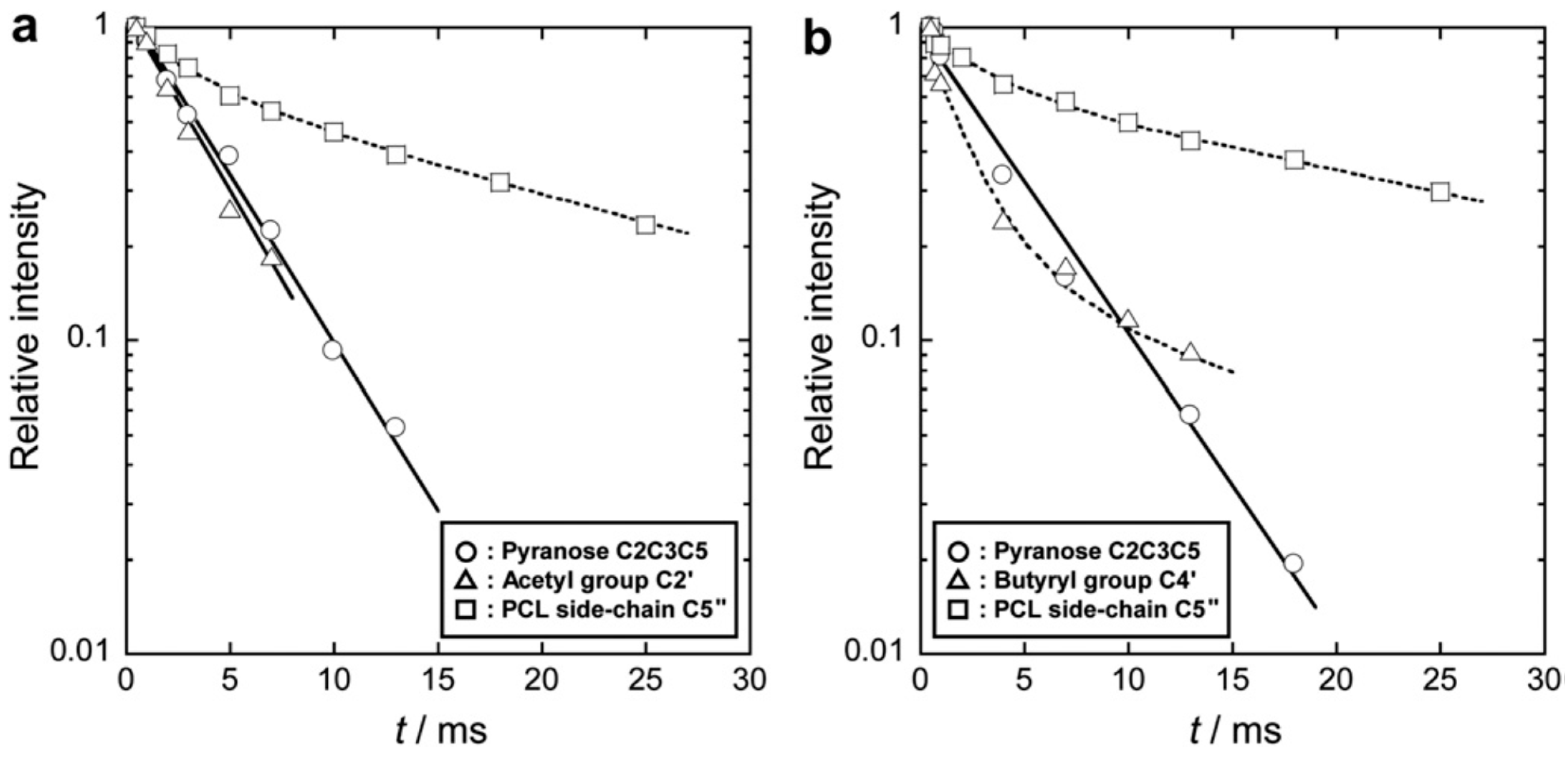
5.4. Molecular and Segmental Dynamics Characterized by Relaxation Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
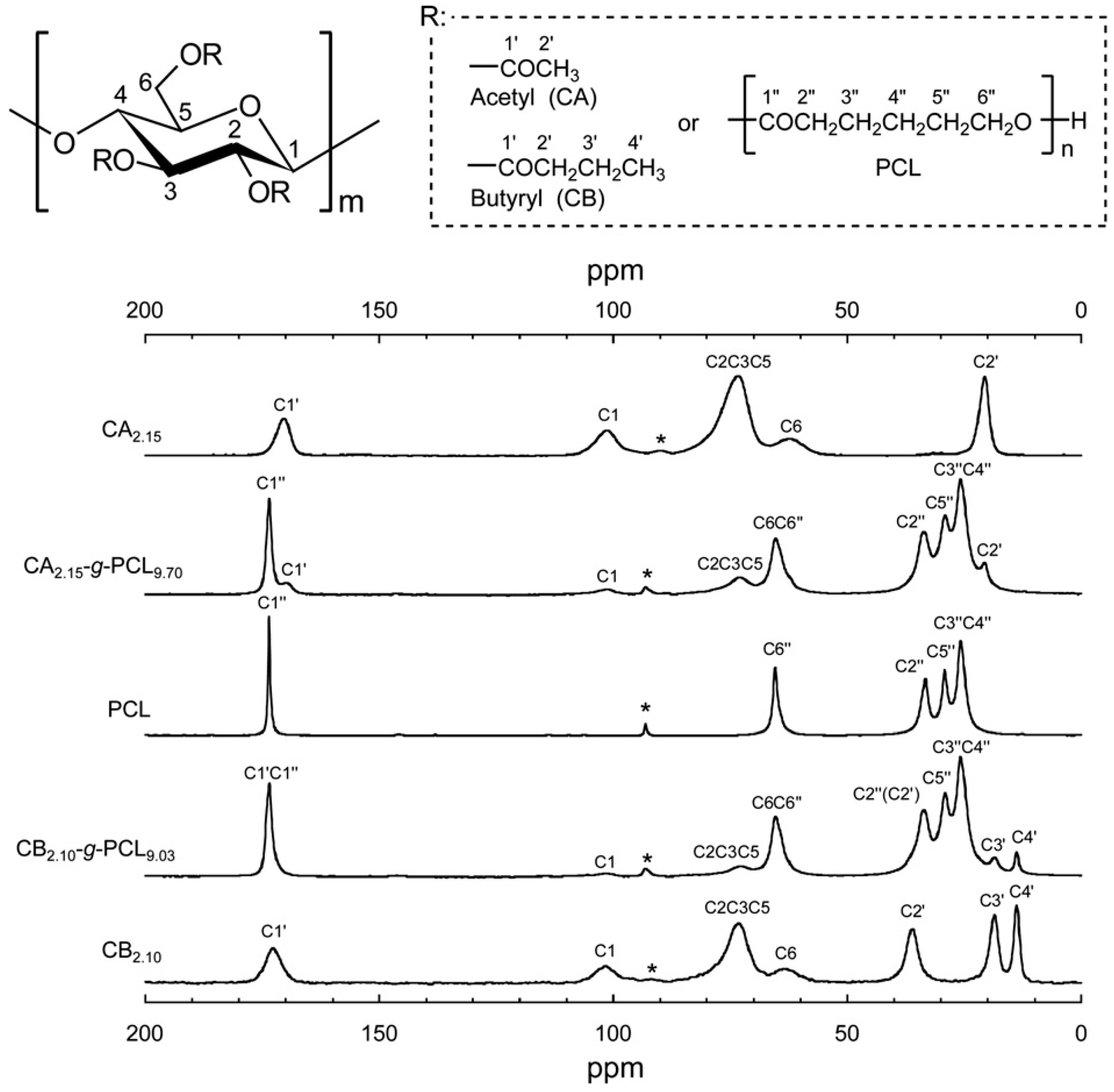
| Samples | /ms | |||
|---|---|---|---|---|
| CA or CB Component | PCL Component | |||
| Pyranose C2C3C5 | Acetyl C2' or Butyryl C4' | C3''C4'' | C5'' | |
| CA2.15 | 13.7 | 13.9 | – | – |
| CA2.15-g-PCL0.27 | 12.9 | 13.3 | 12.8 | 12.3 |
| CA2.15-g-PCL0.87 | 8.46 | 7.87 | 8.02 | 8.30 |
| CA2.15-g-PCL1.30 | 6.99 | 7.11 | 4.72 | 3.72 |
| CA2.15-g-PCL2.50 | 2.79 | 2.74 | 2.20 | 1.71 |
| CA2.15-g-PCL9.70 | 4.05 | 3.79 | 3.01 (a)/24.3 (b) | 3.63 (a)/24.6 (b) |
| CA2.45 | 16.6 | 15.9 | – | – |
| CA2.45-g-PCL0.11 | 14.0 | 14.4 | 13.6 | 13.2 |
| CA2.45-g-PCL0.22 | 11.4 | 11.7 | 9.03 | 7.23 |
| CA2.45-g-PCL1.20 | 6.21 | 5.64 | 4.02 | 4.17 |
| CA2.45-g-PCL2.50 | 3.02 | 2.68 | 2.05 | 2.39 |
| CA2.45-g-PCL9.30 | 7.49 | 8.31 | 4.10 (a)/22.5 (b) | 4.83 (a)/23.3 (b) |
| CA2.98 | 15.7 | 15.4 | – | – |
| CA2.98-g-PCL0.22 | 13.7 | 13.9 | 8.95 | 9.03 |
| CA2.98-g-PCL0.55 | 12.3 | 14.0 | 7.62 | 6.39 |
| CA2.98-g-PCL2.07 | 6.34 | 5.64 | 3.49 | 4.52 |
| CA2.98-g-PCL9.20 | 7.09 | 7.87 | 2.15 (a)/22.0 (b) | 1.75 (a)/21.6 (b) |
| CB2.10 | 9.20 | 8.64 | – | – |
| CB2.10-g-PCL0.16 | 7.45 | 6.73 | 8.58 | 6.42 |
| CB2.10-g-PCL0.60 | 6.58 | 6.23 | 6.47 | 5.23 |
| CB2.10-g-PCL2.33 | 3.24 | 3.11 | 2.48 | 2.69 |
| CB2.10-g-PCL9.03 | 3.94 | 2.02 (a)/18.0 (b) | 3.86 (a)/27.3 (b) | 3.00 (a)/31.1 (b) |
| CB2.50 | 7.60 | 7.90 | – | – |
| CB2.50-g-PCL0.26 | 7.09 | 7.26 | n.d.c) | n.d. |
| CB2.50-g-PCL1.37 | 3.89 | 3.23 | 3.45 | n.d. |
| CB2.50-g-PCL3.49 | 3.26 | 3.50 | 3.96 | 3.55 |
| CB2.50-g-PCL7.42 | 4.82 | 3.20 (a)/18.3 (b) | 3.68 (a)/29.4 (b) | 4.83 (a)/31.6 (b) |
| CB2.93 | 8.26 | 7.60 | – | – |
| CB2.93-g-PCL0.23 | 7.73 | 7.67 | n.d. | n.d. |
| CB2.93-g-PCL0.50 | 5.81 | 5.36 | n.d. | n.d. |
| CB2.93-g-PCL3.58 | 3.25 | 2.80 | 3.64 | 3.00 |
| CB2.93-g-PCL12.6 | n.d. (c) | 3.12 (a)/18.7 (b) | 7.20 (a)/35.0 (b) | 7.29 (a)/38.0 (b) |
| PCL | – | – | 6.29 (a)/60.2 (b) | 7.17 (a)/61.8 (b) |
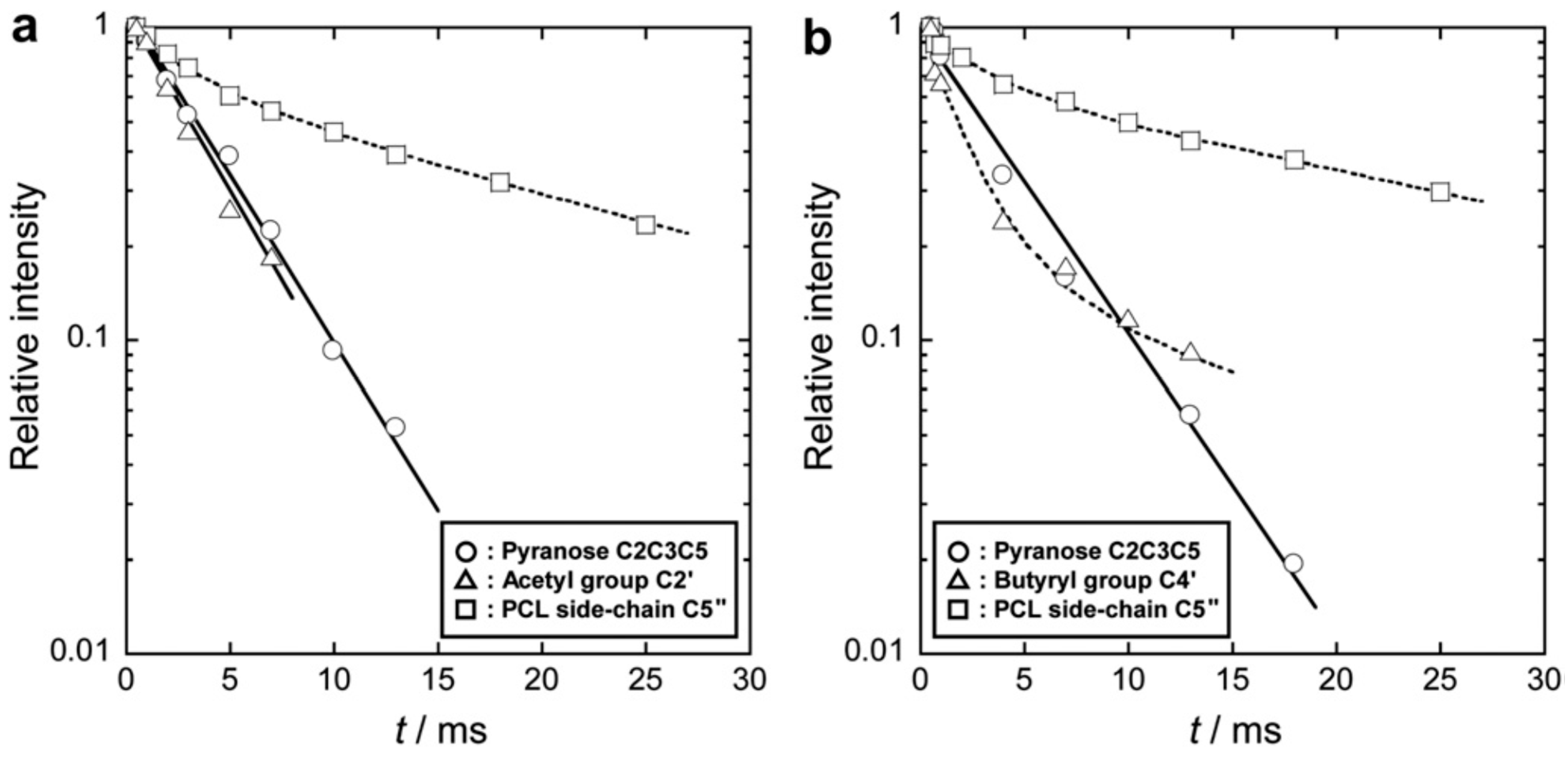
5.5. Extension of Reaction Systems for Graft Copolymerization
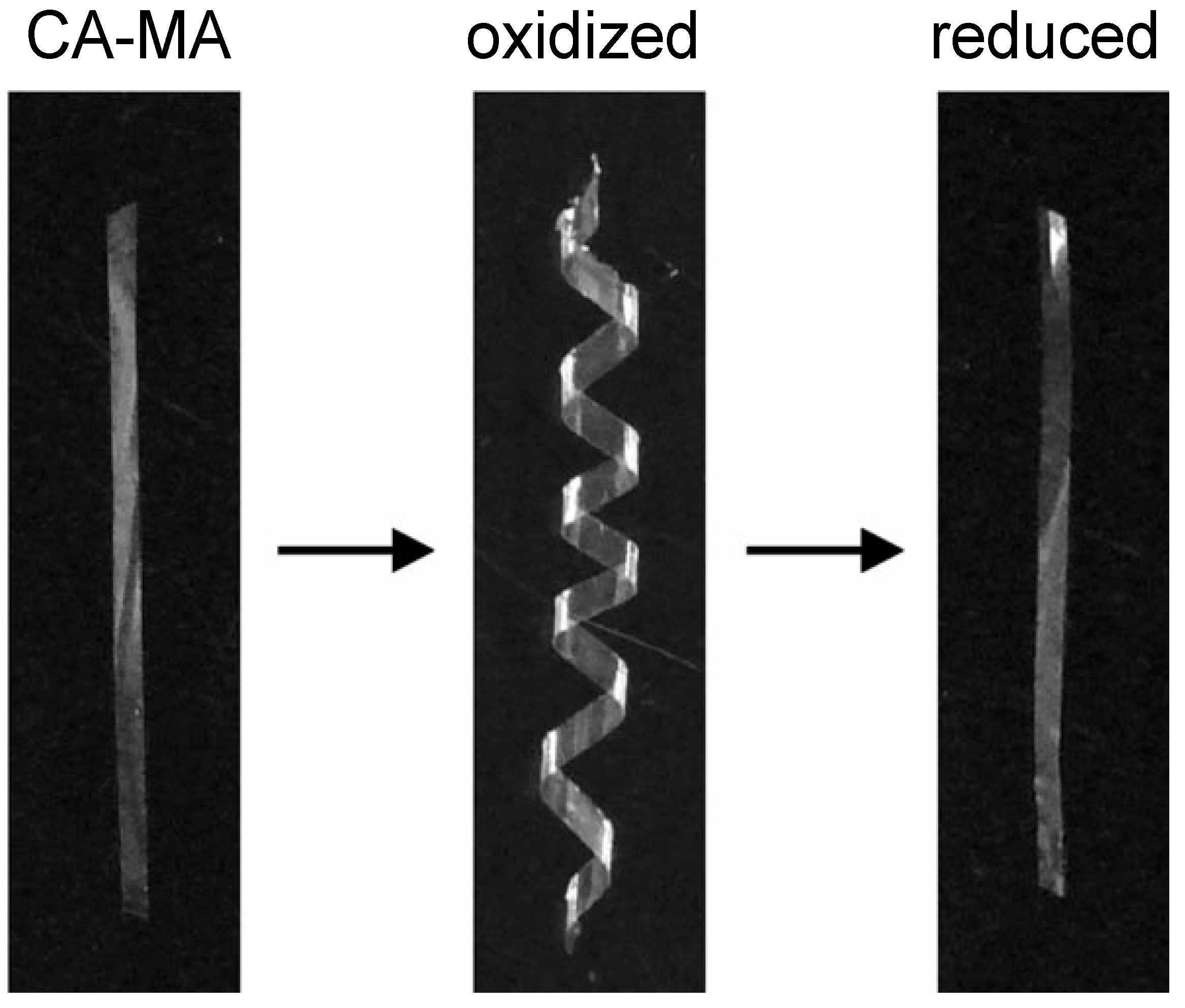
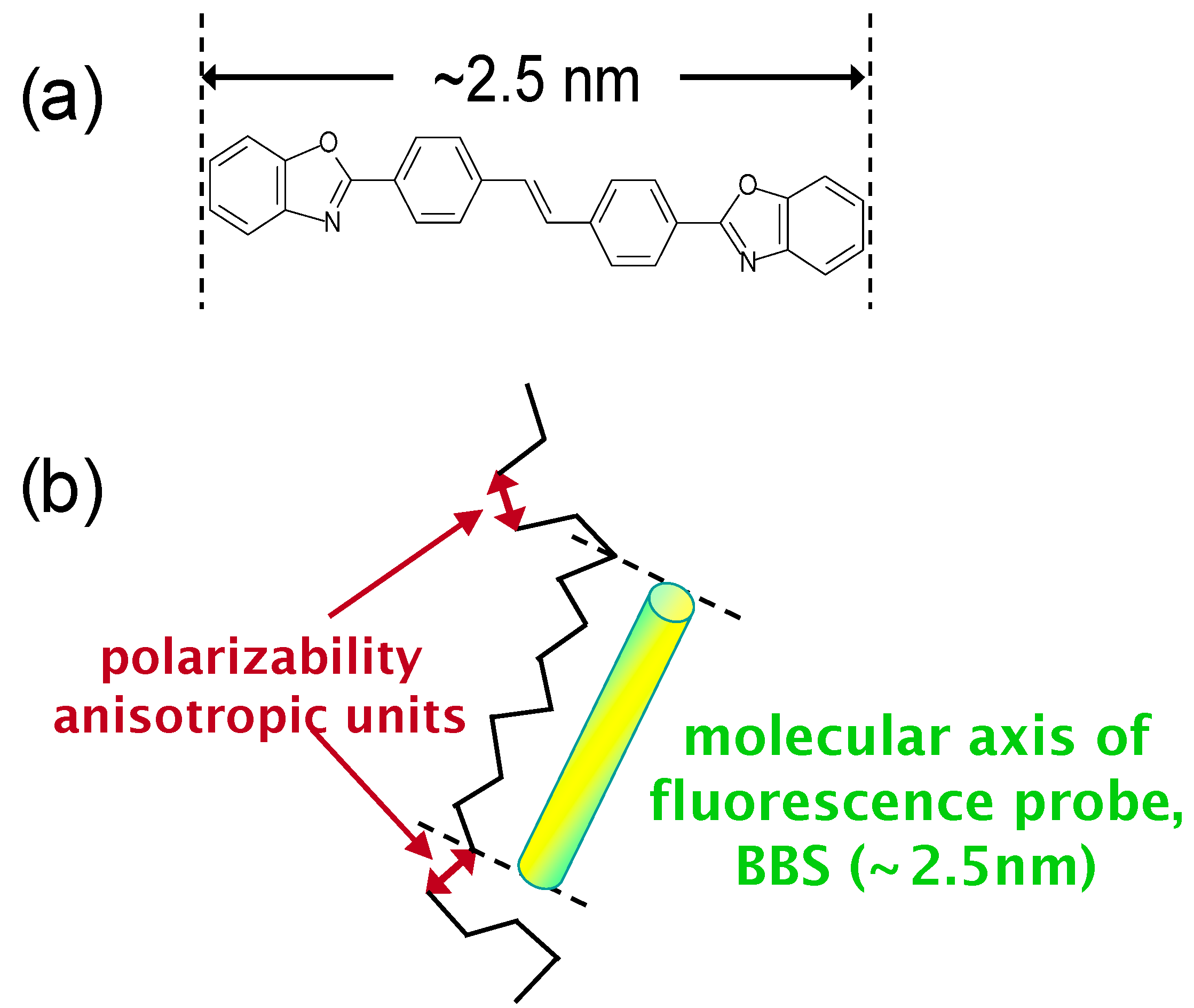
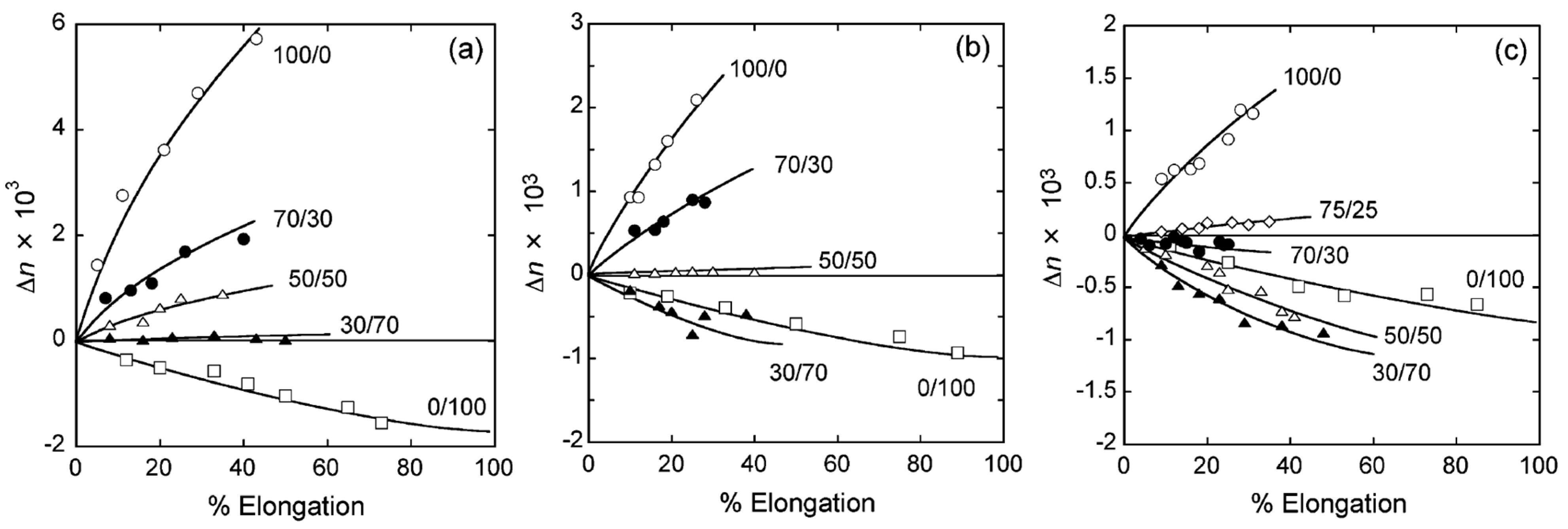
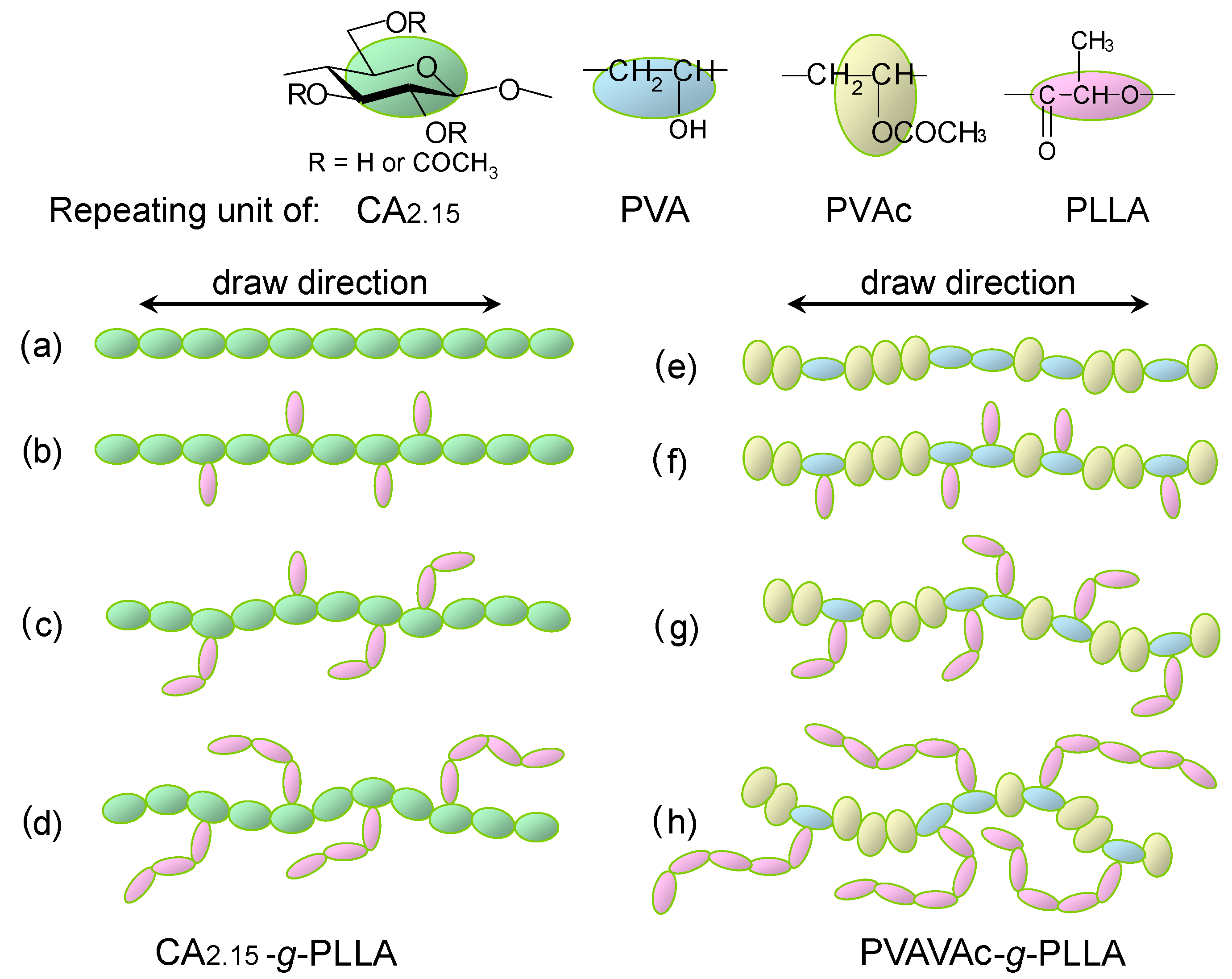
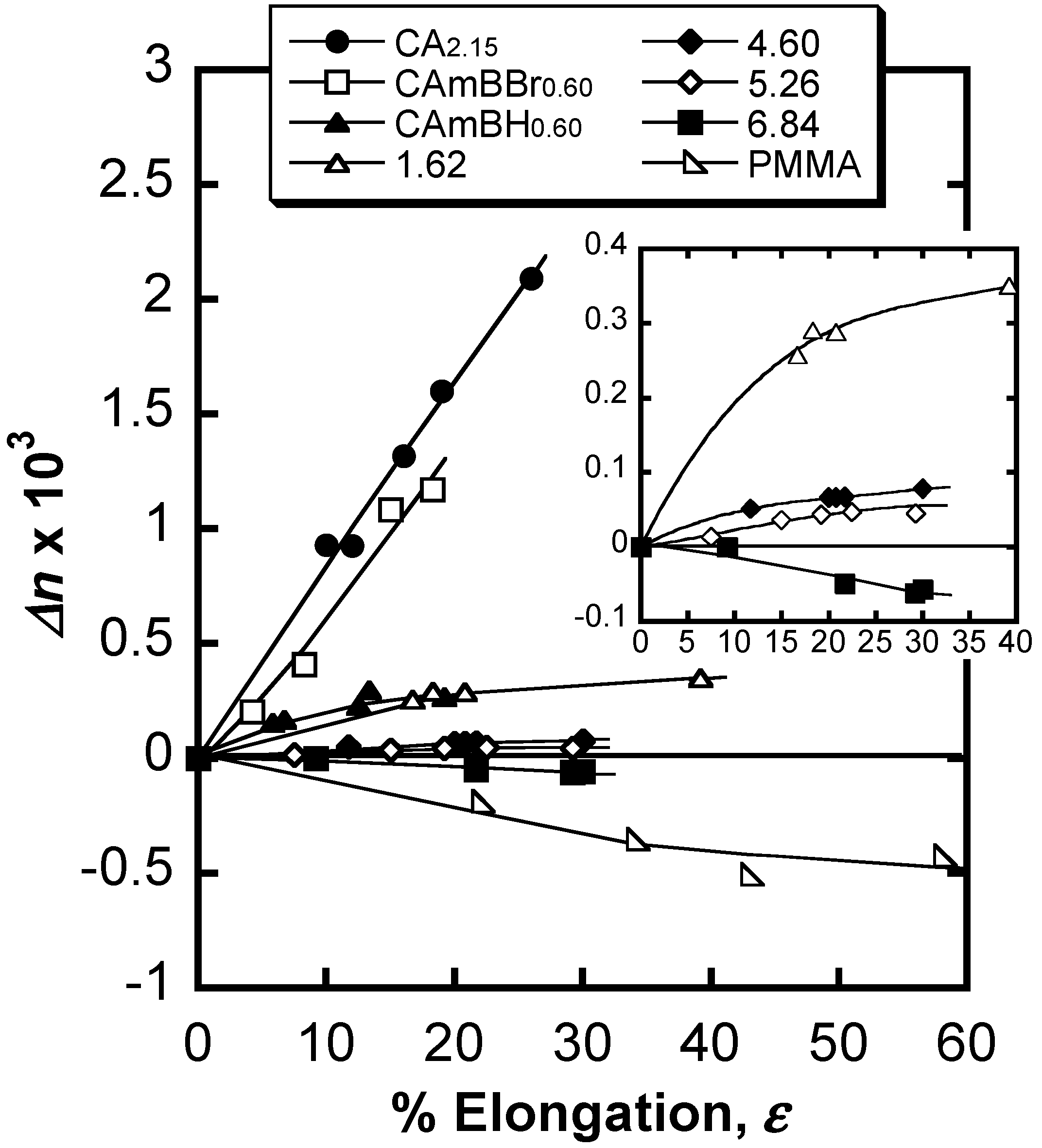
6. Orientation Control
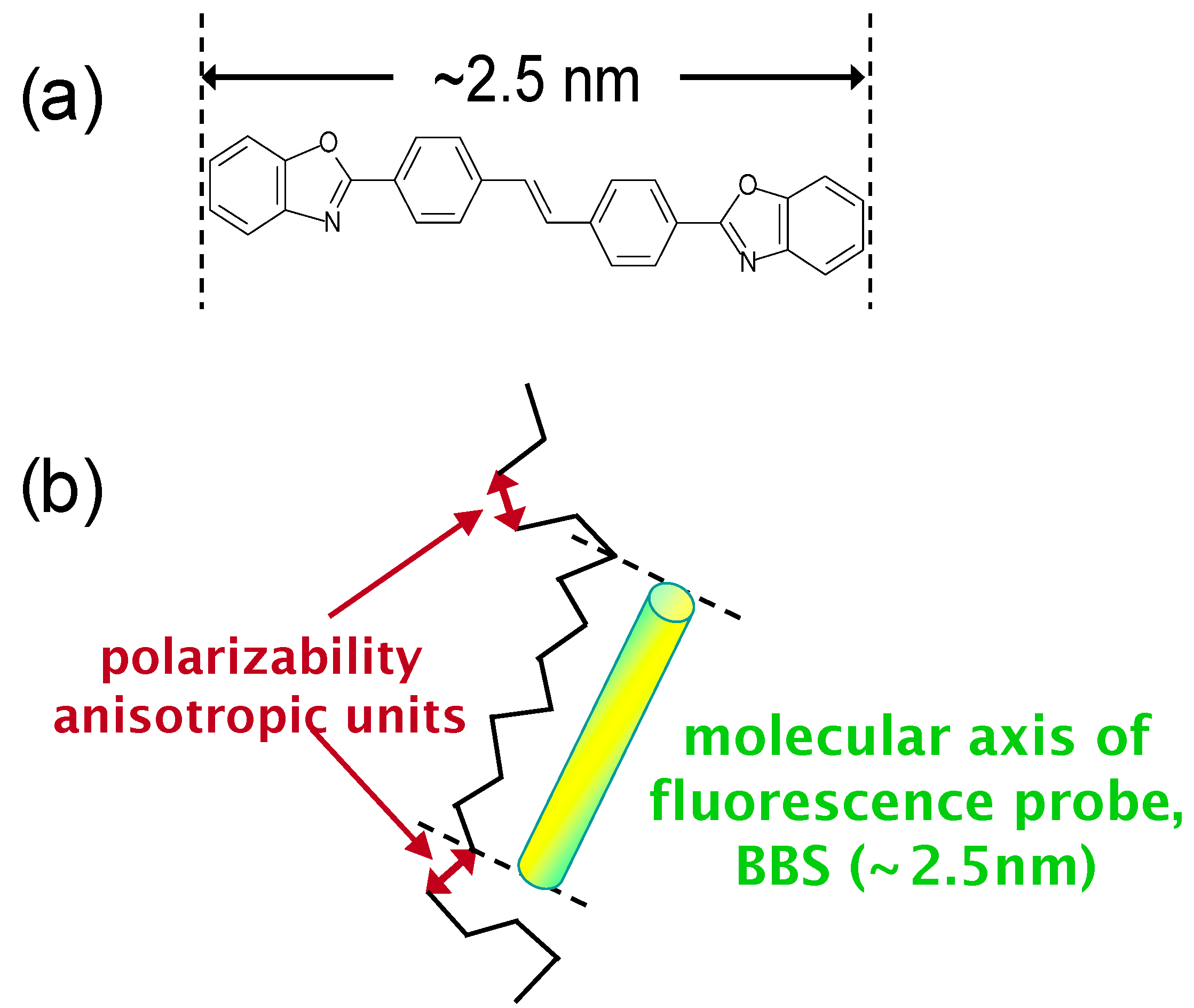
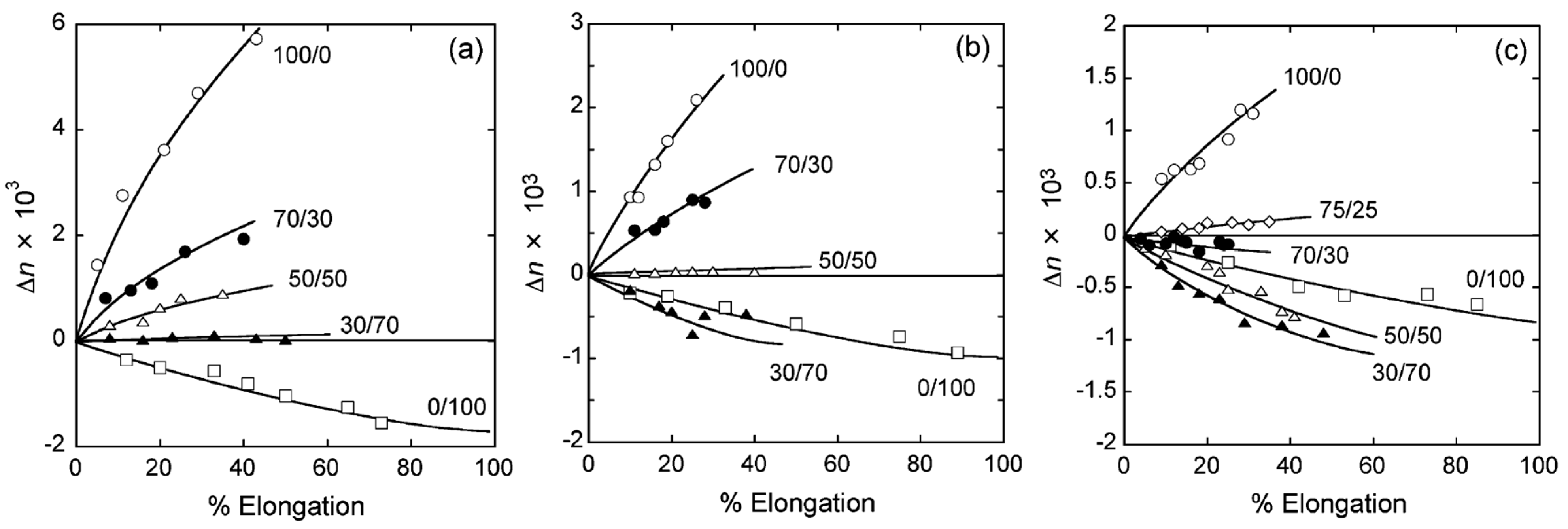
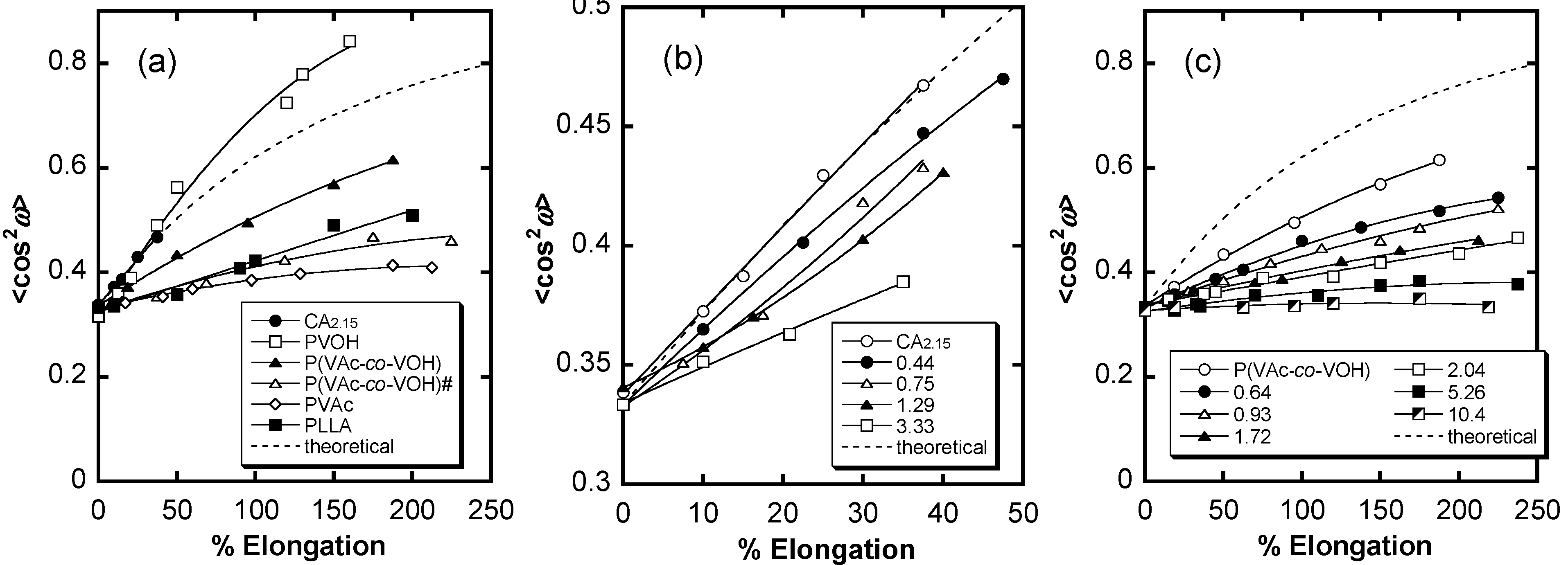
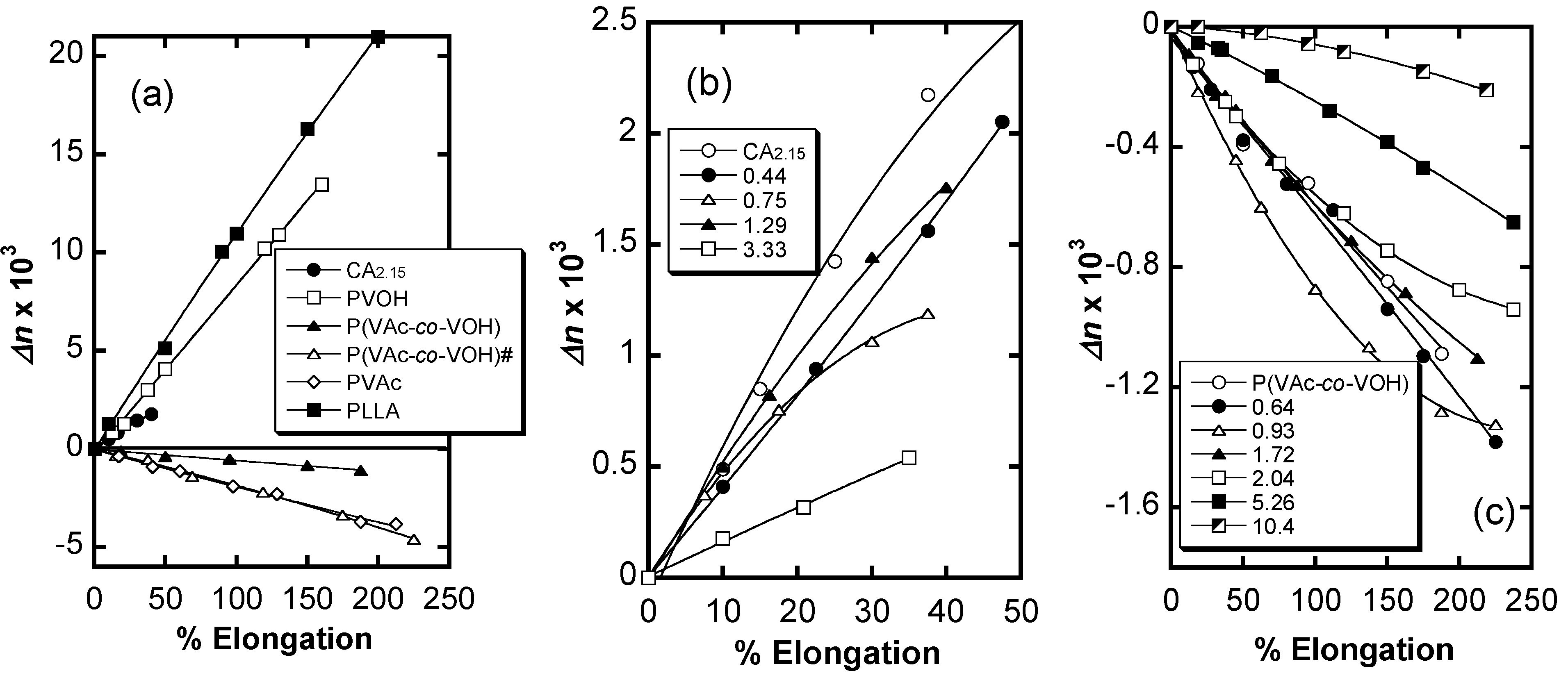



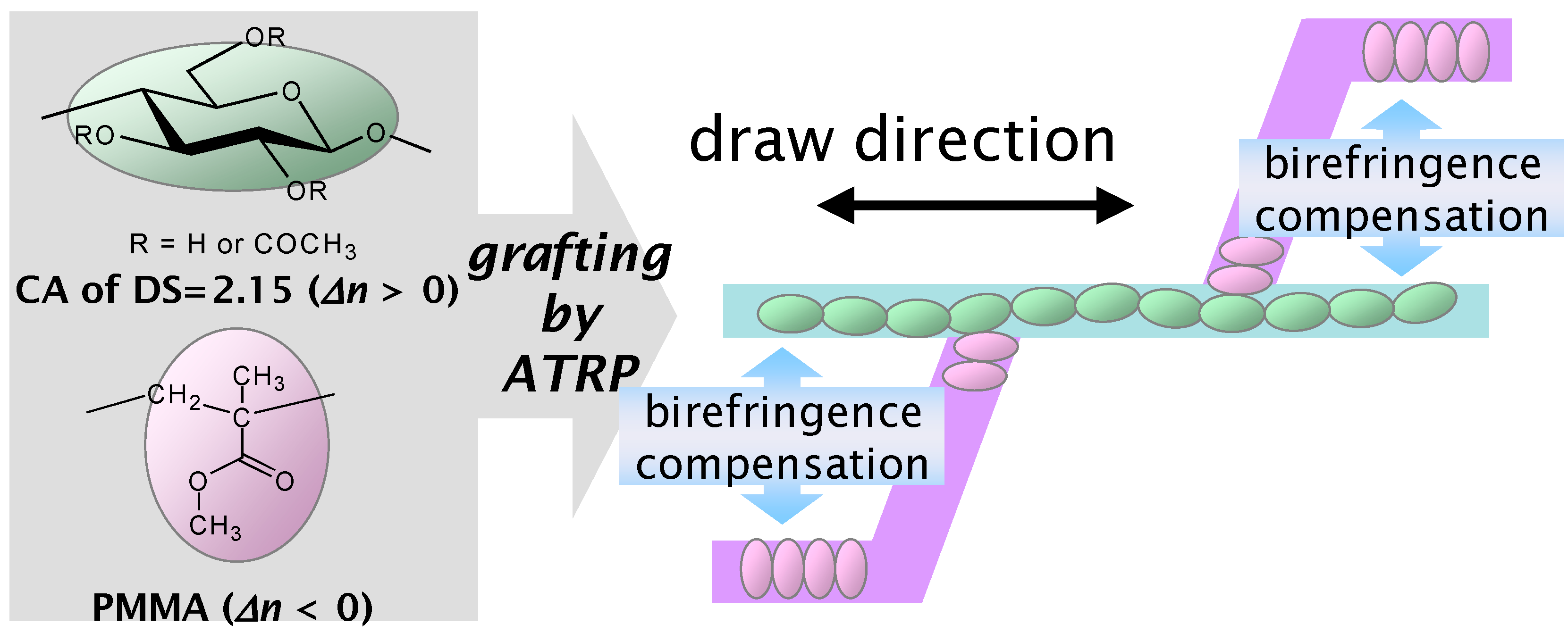
7. Conclusions
Acknowledgments
Abbreviations
| ABS | acrylonitrile butadiene styrene |
| Ac | acetate |
| Acyl-Ch | acyl chitin |
| AFM | atomic force microscopy |
| amide-DS | degree of amide substitution |
| ATR-FTIR | attenuated total reflection FT-IR |
| ATRP | atom-transfer radical polymerization |
| BBS | 4,4'-bis(2-benzoxazolyl)stilbene |
| BL | (R,S)-β-butyrolactone |
| Bu | butyrate |
| CA | cellulose acetate |
| CB | cellulose butyrate |
| CD | curdlan |
| CDA | cellulose diacetate |
| Cell | cellulose |
| CE | cellulose ester |
| ChB | chitin butyrate |
| CL | ε-caprolactone |
| CMC | tri-O-carboxymethyl cellulose |
| CP | cellulose propionate |
| CP/MAS | cross-polarization magic angle-spinning |
| De | decanoate |
| DHPC | O-(2,3-dihydroxypropyl) cellulose |
| DMA | dynamic mechanical analysis |
| DMAc | N,N-dimethylacetamide |
| DMSO | dimethyl sulfoxide |
| Δn | birefringence |
| DPs | average degree of polymerization of side chains |
| DS | degree of substitution |
| DSC | differential scanning calorimetry |
| EC | ethyl cellulose |
| 2-Eh | 2-ethylhexanoyl |
| ester-DS | degree of ester substitution |
| FE-SEM | field emission scanning electron microscope |
| GM | konjac glucomannan |
| He | hexanoate |
| IM | immiscible |
| La | laurate |
| LC | liquid crystalline |
| LCD | liquid crystal display |
| m | side chain length, where m is defined as the number of the carbon atoms forming the side chain skeleton |
| M | miscible |
| M(t) | magnetization intensity observed as a function of the spin-locking time t |
| MA | mercaptoacetic acid |
| MMA | methyl methacrylate |
| bis-MPA | bis(methylol)-2-methylpropanamide |
| MS | molar substitution |
| Oc | octanoate |
| oxyalkanoyl DS | degree of oxyalkanoyl substitution |
| P(VP-co-MMA) | poly(N-vinyl pyrrolidone-co-methyl methacrylate) |
| PAA | 3-pentadecylphenoxy acetic acid |
| PCL | poly(ε-caprolactone) |
| PHA | poly(hydroxy alkanoate) |
| PLA | poly(lactic acid) |
| PLLA | poly(l-lactide) |
| PM | partially miscible |
| PMMA | poly(methyl methacrylate) |
| POM | polarized optical microscope |
| Pr | propionate |
| PVAVAc | poly(vinyl acetate-co-vinyl alcohol) |
| PVB | poly(vinyl butyral) |
| PVP | poly(N-vinyl pyrrolidone) |
| T1ρH | 1H spin-lattice relaxation time in the rotating frame |
| TAC | triacetyl cellulose |
| Tg | glass transition temperature |
| total-DS | total degree of subsitution |
| Va | valerate |
| VAc | vinyl acetate |
| VL | δ-valerolactone |
| VP | N-vinyl pyrrolidone |
| WAXD | wide-angle X-ray diffractometry |
| XG | xyloglucan |
| XylC6N3 | di-O-(6-azidohexanoyl)-xylan |
Conflicts of Interest
References
- Edgar, K.J.; Buchanan, C.M.; Debenham, J.S.; Rundquist, P.A.; Seiler, B.D.; Shelton, M.C.; Tindall, D. Advances in cellulose ester performance and application. Prog. Polym. Sci. 2001, 26, 1605–1688. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar]
- Nishio, Y. Material functionalization of cellulose and related polysaccharides via diverse microcompositions. Adv. Polym. Sci. 2006, 205, 97–151. [Google Scholar]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindstrom, T.; Ankerfors, M.; Gray, D.G.; Dorris, A. Nanocelluloses: A new family of nature-based materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef]
- Eichhorn, S.J. Cellulose nanowhiskers: Promising materials for advanced applications. Soft Matter 2011, 7, 303–315. [Google Scholar] [CrossRef]
- Heinze, T.; Liebert, T. Unconventional methods in cellulose fictionalization. Prog. Polym. Sci. 2001, 26, 1689–1762. [Google Scholar] [CrossRef]
- Roy, D.; Semsarilar, M.; Guthrie, J.T.; Perrier, S. Cellulose modification by polymer grafting: A review. Chem. Soc. Rev. 2009, 38, 2046–2064. [Google Scholar] [CrossRef] [PubMed]
- Carlmark, A.; Larsson, E.; Malmström, E. Grafting of cellulose by ring-opening polymerization—A review. Eur. Polym. J. 2012, 48, 1646–1659. [Google Scholar] [CrossRef]
- Habibi, Y. Key advances in the chemical modification of nanocelluloses. Chem. Soc. Rev. 2014, 43, 1519–1542. [Google Scholar] [CrossRef] [PubMed]
- Malm, C.; Mench, J.; Kendall, D.; Hiatt, G. Aliphatic acid esters of cellulose: Properties. Ind. Eng. Chem. 1951, 43, 688–691. [Google Scholar] [CrossRef]
- Morooka, T.; Norimoto, M.; Yamada, T.; Shiraishi, N. Dielectric properties of cellulose acylates. J. Appl. Polym. Sci. 1984, 29, 3981–3990. [Google Scholar] [CrossRef]
- Sealey, J.; Sarmaranayake, G.; Todd, J.; Glasser, W.G. Novel cellulose derivatives. IV. Preparation and thermal analysis of waxy esters of cellulose. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1613–1620. [Google Scholar] [CrossRef]
- Vaca-Garcia, C.; Borredon, M.; Gaseta, A. Determination of the degree of substitution (DS) of mixed cellulose esters by elemental analysis. Cellulose 2001, 8, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Glasser, W.G. Prospects for future applications of cellulose acetate. Macromol. Symp. 2004, 208, 371–394. [Google Scholar] [CrossRef]
- Vaca-Garcia, C.; Gozzelino, G.; Glasser, W.G.; Borredon, M. Dynamic mechanical thermal analysis transitions of partially and fully substituted cellulose fatty esters. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 281–288. [Google Scholar] [CrossRef]
- Fukuda, T.; Sugiura, M.; Takada, A.; Sato, T.; Miyamoto, T. Characteristics of cellulosic thermotropics. Bull. Inst. Chem. Res. Kyoto Univ. 1991, 69, 211–218. [Google Scholar]
- Flory, P.J. Molecular theory of liquid crystals. Adv. Polym. Sci. 1984, 59, 1–36. [Google Scholar]
- Teramoto, Y.; Miyata, T.; Nishio, Y. Dual Mesomorphic assemblage of chitin normal acylates and rapid enthalpy relaxation of their side chains. Biomacromolecules 2006, 7, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Plate, N.; Shbaev, V.; Petrukhin, B.; Zubov, Y.; Kargin, V. Structure of crystalline polymers with unbranched long side chains. J. Polym. Sci. Part A Polym. Chem. 1971, 9, 2291–2298. [Google Scholar] [CrossRef]
- Fundador, N.; Enomoto-Rogers, Y.; Takemura, A.; Iwata, T. Syntheses and characterization of xylan esters. Polymer 2012, 53, 3885–3893. [Google Scholar] [CrossRef]
- Enomoto-Rogers, Y.; Ohmomo, Y.; Takemura, A.; Iwata, T. Syntheses of glucomannan esters and their thermal andmechanical properties. Carbohydr. Polym. 2014, 101, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Marubayashi, H.; Yukinaka, K.; Enomoto-Rogers, Y.; Takemura, A.; Iwata, T. Curdlan ester derivatives: Synthesis, structure, and properties. Carbohydr. Polym. 2014, 103, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Sawai, D.; Nozoe, Y.; Yoshitani, T.; Tsukada, Y. Curdlan ester derivatives: Synthesis, structure, and properties. FUJIFILM Res. Dev. 2012, 57, 55–58. [Google Scholar]
- Glasser, W.G.; Sarmaranayake, G.; Durmay, M.; Dave, V. Novel cellulose derivatives. III. Thermal analysis of mixed esters with butyric and hexanoic acid. J. Polym. Sci. Part B Polym. Phys. 1995, 33, 2045–2054. [Google Scholar] [CrossRef]
- Kano, H.; Aranishi, Y.; Maeda, Y.; Nishio, Y. Chemical modification of cellulose for melt spinning process. In Proceedings of the International Symposium on Fiber Science and Technology (ISF2014), Tokyo, Japan, 1 October 2014; pp. G1–G36.
- Iji, M.; Moon, S.; Tanaka, S. Hydrophobic, mechanical and thermal characteristics of thermoplastic cellulose diacetate bonded with cardanol from cashew nutshell. Polym. J. 2011, 43, 738–741. [Google Scholar] [CrossRef]
- Yao, K.; Okoshi, M.; Kawashima, M.; Yamanoi, K. The development of flame-resistant bio-based plastic from inedible wood material. Fuji Xerox Tech. Rep. 2013, 22, 96–103. (In Japanese) [Google Scholar]
- Shibakami, M.; Tsubouchi, G.; Hayashi, M. Thermoplasticization of euglenoid β-1,3-glucans by mixedesterification. Carbohydr. Polym. 2014, 105, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Teramoto, Y.; Nishio, Y. Synthesis of O-(2,3-dihydroxypropyl) cellulose in NaOH/urea aqueous solution: As a precursor for introducing “necklace-like” structure. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3590–3597. [Google Scholar] [CrossRef]
- Chang, C.; Teramoto, Y.; Nishio, Y. High performance films of cellulose butyral derivative having a necklace-like annular structure in the side chains. Polymer 2014, 55, 3944–3950. [Google Scholar] [CrossRef] [Green Version]
- Culter, R.; Kleinlein, B. Effect of the hydroxyl content and molecular weight of polyvinyl butyral on tape properties. J. Eur. Ceram. Soc. 2009, 29, 3211–3218. [Google Scholar] [CrossRef]
- Jeong, H.; Rooney, M.; David, D.; MacKnight, W.J.; Karasz, F.E.; Kajiyama, T. Miscibility of polyvinyl butyral/nylon 6 blends. Polymer 2000, 41, 6003–6013. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.; Liu, S.; Liu, Y.; Xu, X.; Chen, X.; Chu, B.; Guo, X.; Xu, J.; Cheng, H.; et al. Dynamic self-assembly induced rapid dissolution of cellulose at low temperatures. Macromolecules 2008, 41, 9345–9351. [Google Scholar] [CrossRef]
- Aoki, D.; Teramoto, Y.; Nishio, Y. SH-containing cellulose acetate derivatives: Preparation and characterization as a shape memory-recovery material. Biomacromolecules 2007, 8, 3749–3757. [Google Scholar] [CrossRef] [PubMed]
- Utracki, L.A. Polymer Alloys and Blends; Hanser Gardner Publications: Munich, Germany, 1990. [Google Scholar]
- Miyashita, Y.; Suzuki, T.; Nishio, Y. Miscibility of cellulose acetate with vinyl polymers. Cellulose 2002, 9, 215–223. [Google Scholar] [CrossRef]
- Ohno, T.; Yoshizawa, S.; Miyashita, Y.; Nishio, Y. Interaction and scale of mixing in cellulose acetate/poly(N-vinyl pyrrolidone-co-vinyl acetate) blends. Cellulose 2005, 12, 281–291. [Google Scholar] [CrossRef]
- Ohno, T.; Nishio, Y. Cellulose alkyl ester/vinyl polymer blends: Effects of butyryl substitution and intramolecular copolymer composition on the miscibility. Cellulose 2006, 13, 245–259. [Google Scholar] [CrossRef]
- Ohno, T.; Nishio, Y. Estimation of miscibility and interaction for cellulose acetate and butyrate blends with N-vinylpyrrolidone copolymers. Macromol. Chem. Phys. 2007, 208, 622–634. [Google Scholar] [CrossRef]
- Ohno, T.; Nishio, Y. Molecular orientation and optical anisotropy in drawn films of miscible blends composed of cellulose acetate and poly(N-vinylpyrrolidone-co-methyl methacrylate). Macromolecules 2007, 40, 3468–3476. [Google Scholar] [CrossRef]
- Sugimura, K.; Teramoto, Y.; Nishio, Y. Cellulose propionate/poly(N-vinyl pyrrolidone-co-vinyl acetate) blends: Dependence of the miscibility on propionyl DS and copolymer composition. Cellulose 2013, 20, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Sugimura, K.; Teramoto, Y.; Nishio, Y. Blend miscibility of cellulose propionate with poly(N-vinyl pyrrolidone-co-methyl methacrylate). Carbohydr. Polym. 2013, 98, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, Y.; Matsuda, K.; Miyashita, Y.; Kimura, N.; Suzuki, H. Blends of poly(ε-caprolactone) with cellulose alkyl esters: Effect of the alkyl side-chain length and degree of substitution on miscibility. Cellulose 1997, 4, 131–145. [Google Scholar] [CrossRef]
- Kusumi, R.; Inoue, Y.; Shirakawa, M.; Miyashita, Y.; Nishio, Y. Cellulose alkyl ester/poly(ε-caprolactone) blends: Characterization of miscibility and crystallization behaviour. Cellulose 2008, 15, 1–16. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kawahara, M.; Teramoto, Y.; Nishio, Y. Synthesis of acyl chitin derivatives and miscibility characterization of their blends with poly(ε-caprolactone). Carbohydr. Polym. 2010, 79, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Hashiwaki, H.; Teramoto, Y.; Nishio, Y. Fabrication of thermoplastic ductile films of chitin butyrate/poly(ε-caprolactone) blends and their cytocompatibility. Carbohydr. Polym. 2014, 114, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Kondo, K.; Ueno, M.; Ito, K.; Kawahara, S.; Isono, Y.; Suzuki, J.; Matsushita, Y. Preparation and morphology of model graft copolymers of the A3B2 type with different graft junction points. Polym. J. 2001, 33, 732–740. [Google Scholar] [CrossRef]
- Buchanan, C.M.; Gardner, R.M.; Komarek, R.J. Aerobic biodegradation of cellulose acetate. J. Appl. Polym. Sci. 1993, 47, 1709–1719. [Google Scholar] [CrossRef]
- Sakai, K.; Yamauchi, T.; Nakasu, F.; Ohe, T. Biodegradation of cellulose acetate by Neisseria sicca. Biosci. Biotechnol. Biochem. 1996, 60, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Ajioka, M.; Enomoto, K.; Suzuki, K.; Yamaguchi, A. Basic properties of polylactic acid produced by the direct condensation polymerization of lactic acid. Bull. Chem. Soc. Jpn. 1995, 68, 2125–2131. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Kreiser-Saunders, I.; Boettcher, C. Polylactones: 31. Sn(II)octoate-initiated polymerization of l-lactide: A mechanistic study. Polymer 1995, 36, 1253–1259. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hagiwara, N.; Shiraishi, N. Thermoplasticization of cellulose acetates by grafting of cyclic esters. Cellulose 1999, 6, 193–212. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Yoshida, T.; Hatakeyama, T. The effect of side chain association on thermal and viscoelastic properties of celluloses acetate based polycaprolactones. J. Therm. Anal. Calorim. 2000, 59, 157–168. [Google Scholar] [CrossRef]
- Teramoto, Y.; Yoshioka, M.; Shiraishi, N.; Nishio, Y. Plasticization of cellulose diacetate by graft copolymerization of epsilon-caprolactone and lactic acid. J. Appl. Polym. Sci. 2002, 84, 2621–2628. [Google Scholar] [CrossRef]
- Teramoto, Y.; Nishio, Y. Cellulose diacetate-graft-poly(lactic acid)s: Synthesis of wide-ranging compositions and their thermal and mechanical properties. Polymer 2003, 44, 2701–2709. [Google Scholar] [CrossRef]
- Teramoto, Y.; Ama, S.; Higeshiro, T.; Nishio, Y. Cellulose acetate-graft-poly(hydroxyalkanoate)s: Synthesis and dependence of the thermal properties on copolymer composition. Macromol. Chem. Phys. 2004, 205, 1904–1915. [Google Scholar] [CrossRef]
- Reimschuessel, H. On the glass transition temperature of comblike polymers: Effects of side chain length and backbone chain structure. J. Polym. Sci. Part A Polym. Chem. 1979, 17, 2447–2457. [Google Scholar] [CrossRef]
- Teramoto, Y.; Nishio, Y. Biodegradable cellulose diacetate-graft-poly(l-lactide)s: Thermal treatment effect on the development of supramolecular structures. Biomacromolecules 2004, 5, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, Y.; Nishio, Y. Biodegradable cellulose diacetate-graft-poly(l-lactide)s: Enzymatic hydrolysis behavior and surface morphological characterization. Biomacromolecules 2004, 5, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, R.; Teramoto, Y.; Nishio, Y. Structural characterization of poly(ε-caprolactone)-grafted cellulose acetate and butyrate by solid-state 13C-NMR, dynamic mechanical, and dielectric relaxation analyses. Polymer 2011, 52, 5912–5921. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.M.; Yang, S.J. Studying the miscibility and thermal behavior of polybenzoxazine/poly(ε-caprolactone) blends using DSC, DMA, and solid state 13C-NMR spectroscopy. Polymer 2005, 46, 8068–8078. [Google Scholar] [CrossRef]
- Kusumi, R.; Teramoto, Y.; Nishio, Y. Crystallization behavior of poly(ε-caprolactone) grafted onto cellulose alkyl esters: Effects of copolymer composition and intercomponent miscibility. Macromol. Chem. Phys. 2008, 209, 2135–2146. [Google Scholar] [CrossRef]
- Mayumi, A.; Kitaoka, T.; Wariishi, H. Partial substitution of cellulose by ring-opening esterification of cyclic esters in a homogeneous system. J. Appl. Polym. Sci. 2006, 102, 4358–4364. [Google Scholar] [CrossRef]
- Vidéki, B.; Klébert, S.; Pukánszky, B. External and internal plasticization of cellulose acetate with caprolactone: Structure and properties. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 873–883. [Google Scholar] [CrossRef]
- Yuan, W.; Yuan, J.; Zhang, F.; Xie, X. Syntheses, characterization, and in vitro degradation of ethyl cellulose-graft-poly(ε-caprolactone)-block-poly(l-lactide) coplymers by sequential ring-opening polymerization. Biomacromolecules 2007, 8, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, J.; Lv, Y.; Yu, J.; Wu, J.; Zhang, J.; He, J. Thermoplastic cellulose-graft-poly(l-lactide) copolymers homogeneously synthesized in an ionic liquid with 4-dimethylaminopyridine catalyst. Biomacromolecules 2009, 10, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Lönnberg, H.; Zhou, Q.; Brumer, H.; Teeri, T.; Malmström, E.; Hult, A. Grafting of cellulose fibers with poly(ε-caprolactone) and poly(l-lactic acid) via ring-opening polymerization. Biomacromolecules 2006, 7, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Habibi, Y.; Goffin, A.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(ε-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 18, 5002–5010. [Google Scholar] [CrossRef]
- Enomoto-Rogers, Y.; Iwata, T. Synthesis of xylan-graft-poly(l-lactide) copolymers via click chemistry and their thermal properties. Carbohydr. Polym. 2012, 87, 1933–1940. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Masuzawa, K. Birefringence control for binary blends of cellulose acetate propionate and poly(vinyl acetate). Eur. Polym. J. 2007, 43, 3277–3282. [Google Scholar] [CrossRef]
- Nakayama, H.; Fukagawa, N.; Nishiura, Y.; Yasuda, T.; Ito, T.; Mihayashi, K. Development of low-retardation TAC film for protection films of LCD’s polarizer. J. Photopolym. Sci. Technol. 2006, 19, 169–173. [Google Scholar] [CrossRef]
- Unohara, T.; Teramoto, Y.; Nishio, Y. Molecular orientation and optical anisotropy in drawn films of cellulose diacetate-graft-PLLA: Comparative investigation with poly(vinyl acetate-co-vinyl alcohol)-graft-PLLA. Cellulose 2011, 18, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, H.; Teramoto, Y.; Nishio, Y. Orientation and birefringence compensation of trunk and graft chains in drawn films of cellulose acetate-graft-PMMA synthesized by ATRP. Macromolecules 2013, 46, 3074–3083. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teramoto, Y. Functional Thermoplastic Materials from Derivatives of Cellulose and Related Structural Polysaccharides. Molecules 2015, 20, 5487-5527. https://doi.org/10.3390/molecules20045487
Teramoto Y. Functional Thermoplastic Materials from Derivatives of Cellulose and Related Structural Polysaccharides. Molecules. 2015; 20(4):5487-5527. https://doi.org/10.3390/molecules20045487
Chicago/Turabian StyleTeramoto, Yoshikuni. 2015. "Functional Thermoplastic Materials from Derivatives of Cellulose and Related Structural Polysaccharides" Molecules 20, no. 4: 5487-5527. https://doi.org/10.3390/molecules20045487