
Efficient Synthesis of β-Aryl-γ-lactams and Their Resolution with (S)-Naproxen: Preparation of (R)- and (S)-Baclofen

 and
and
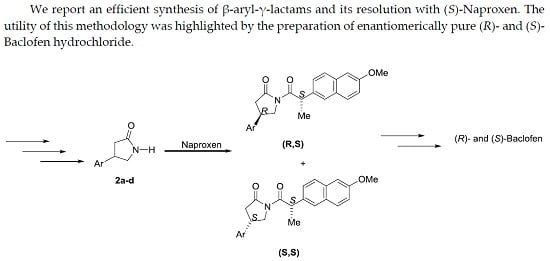
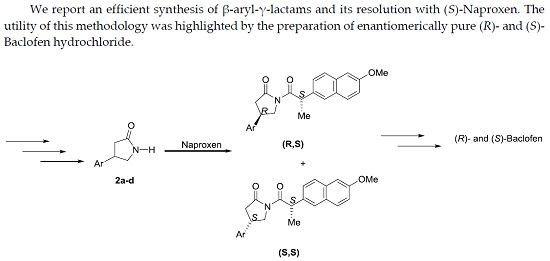
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
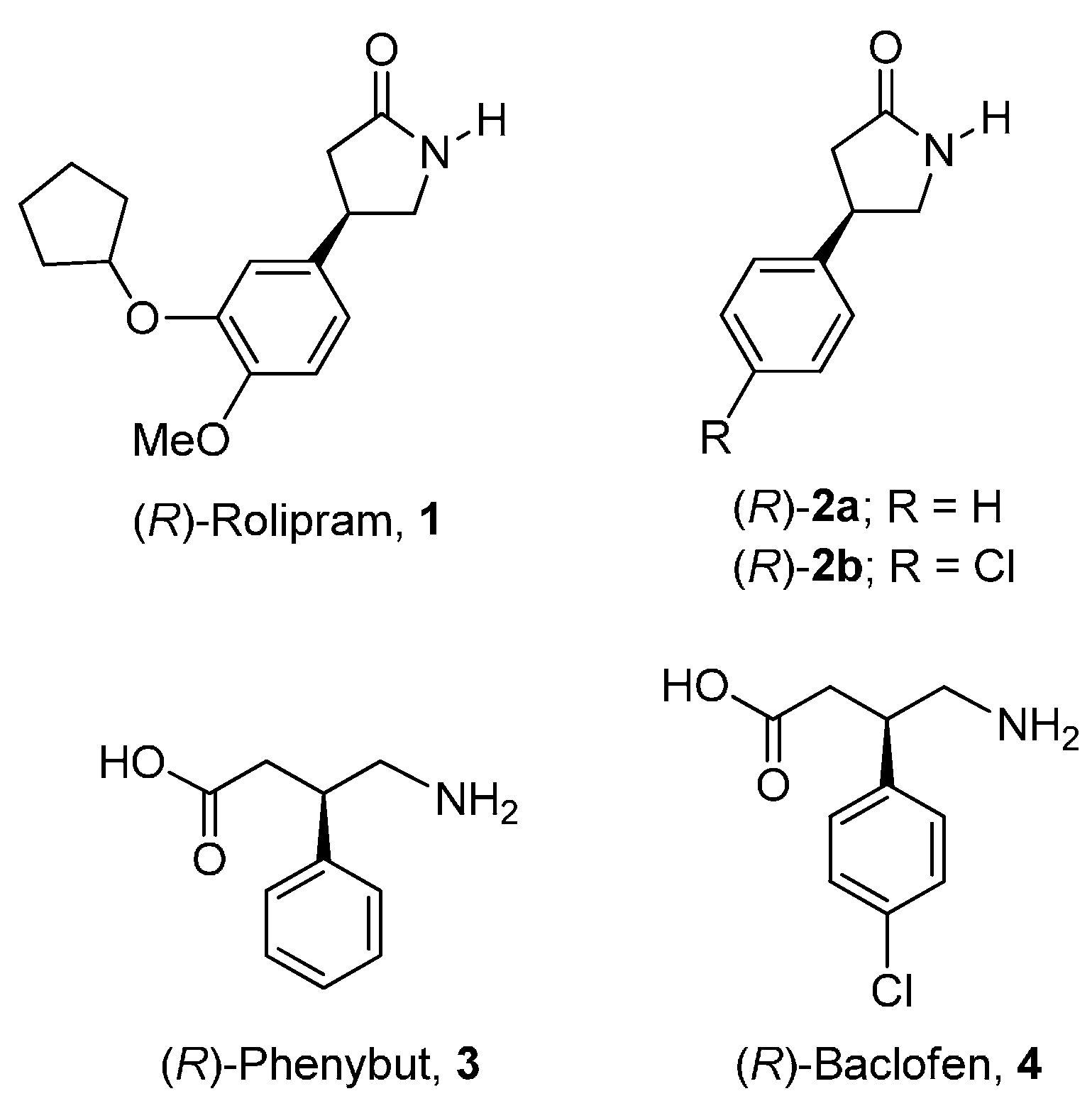
1. Introduction
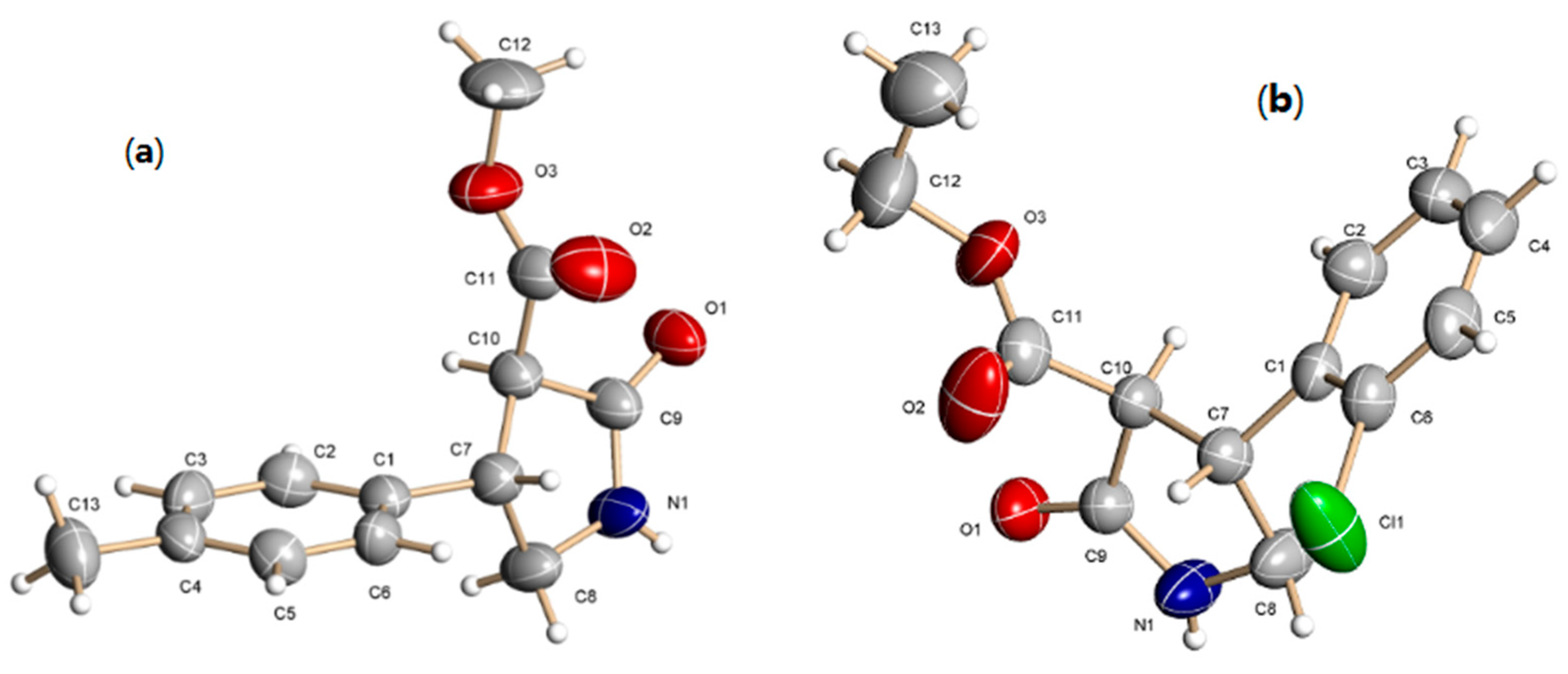
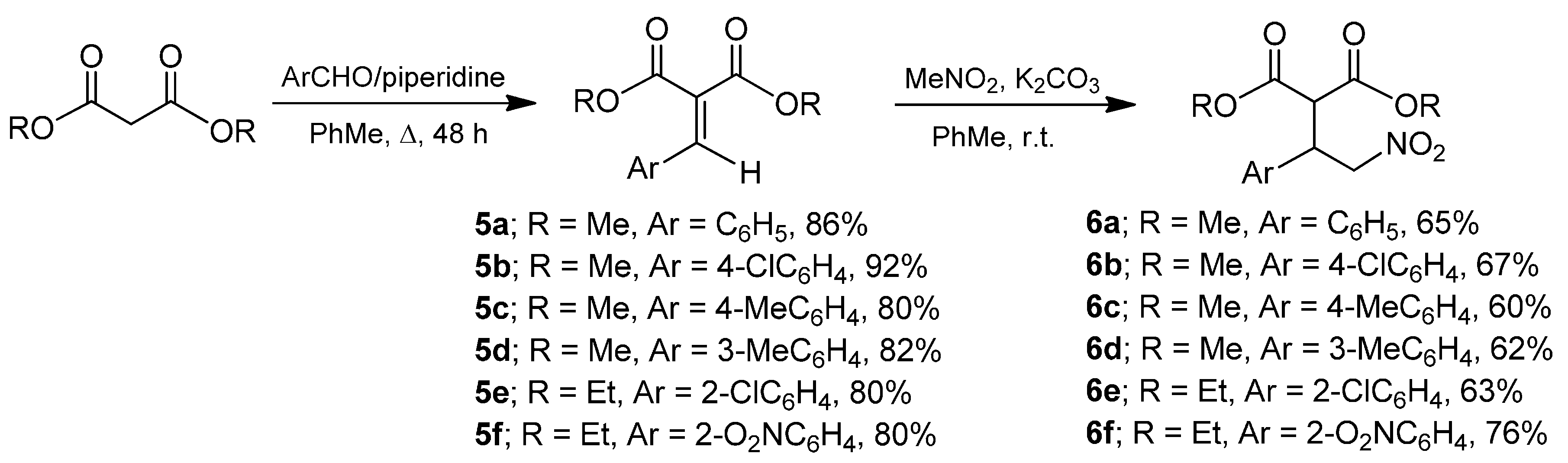
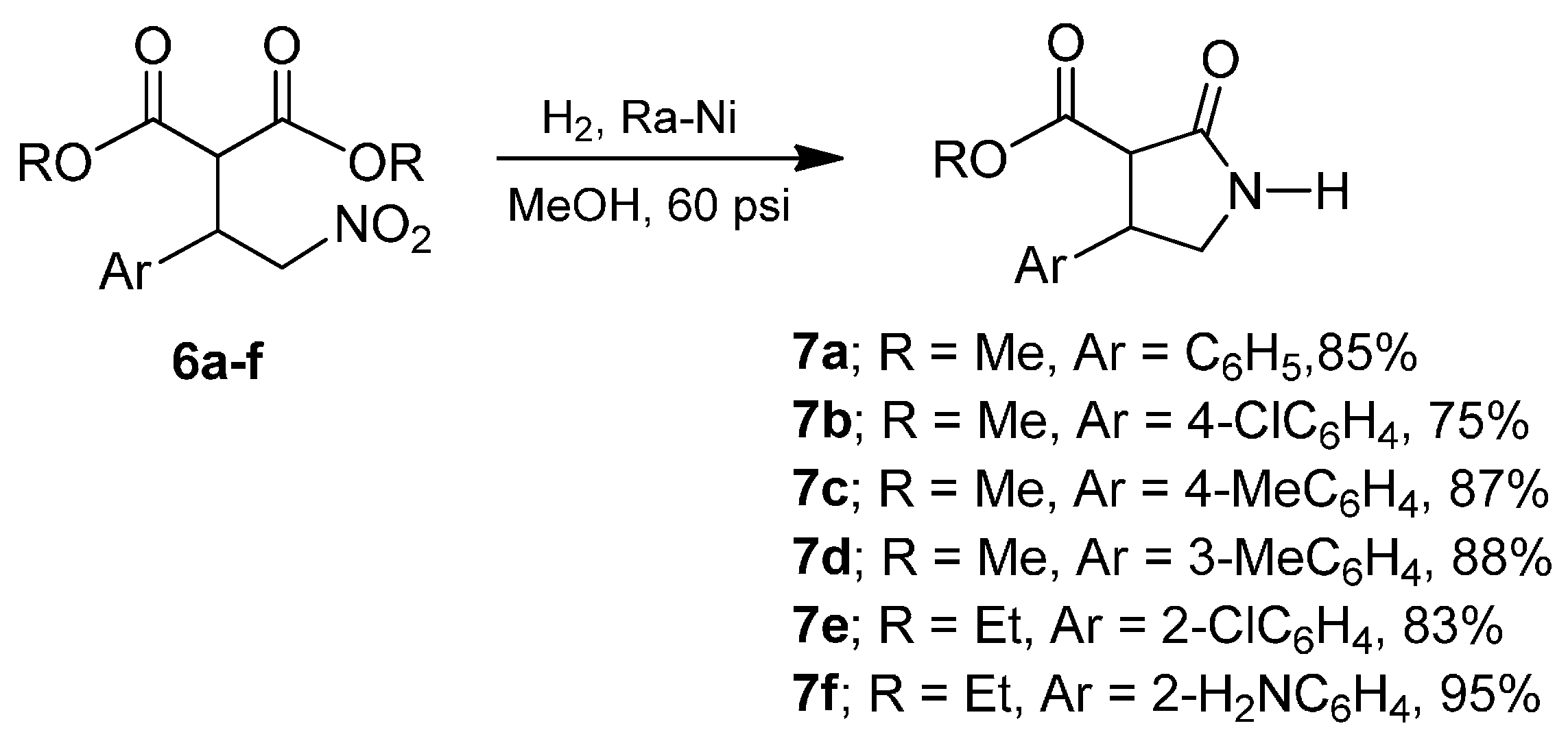
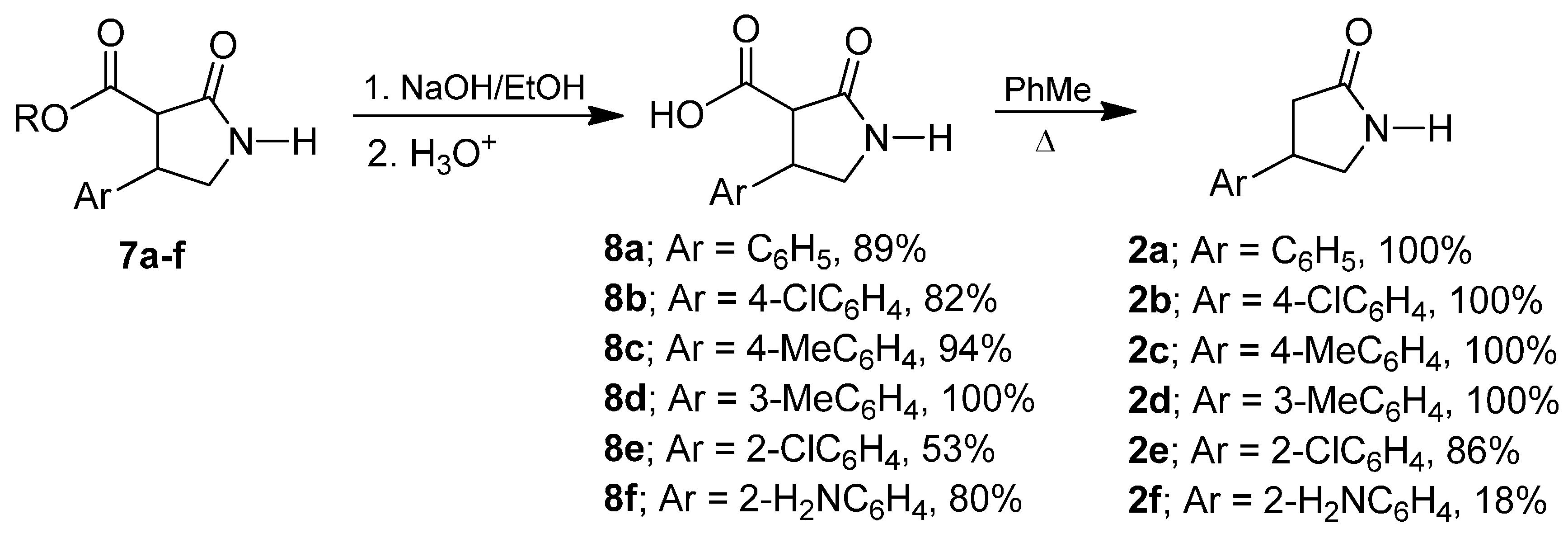
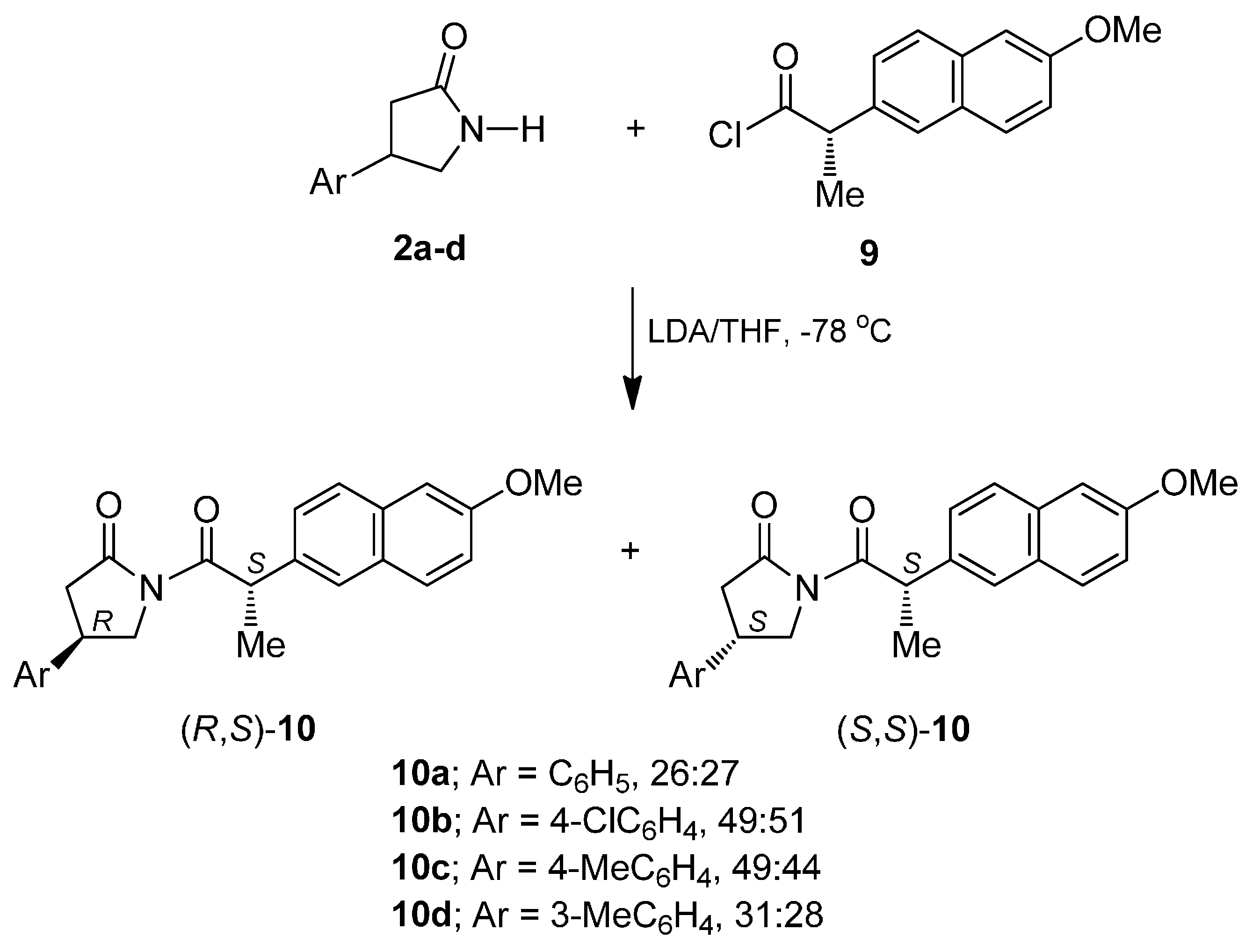
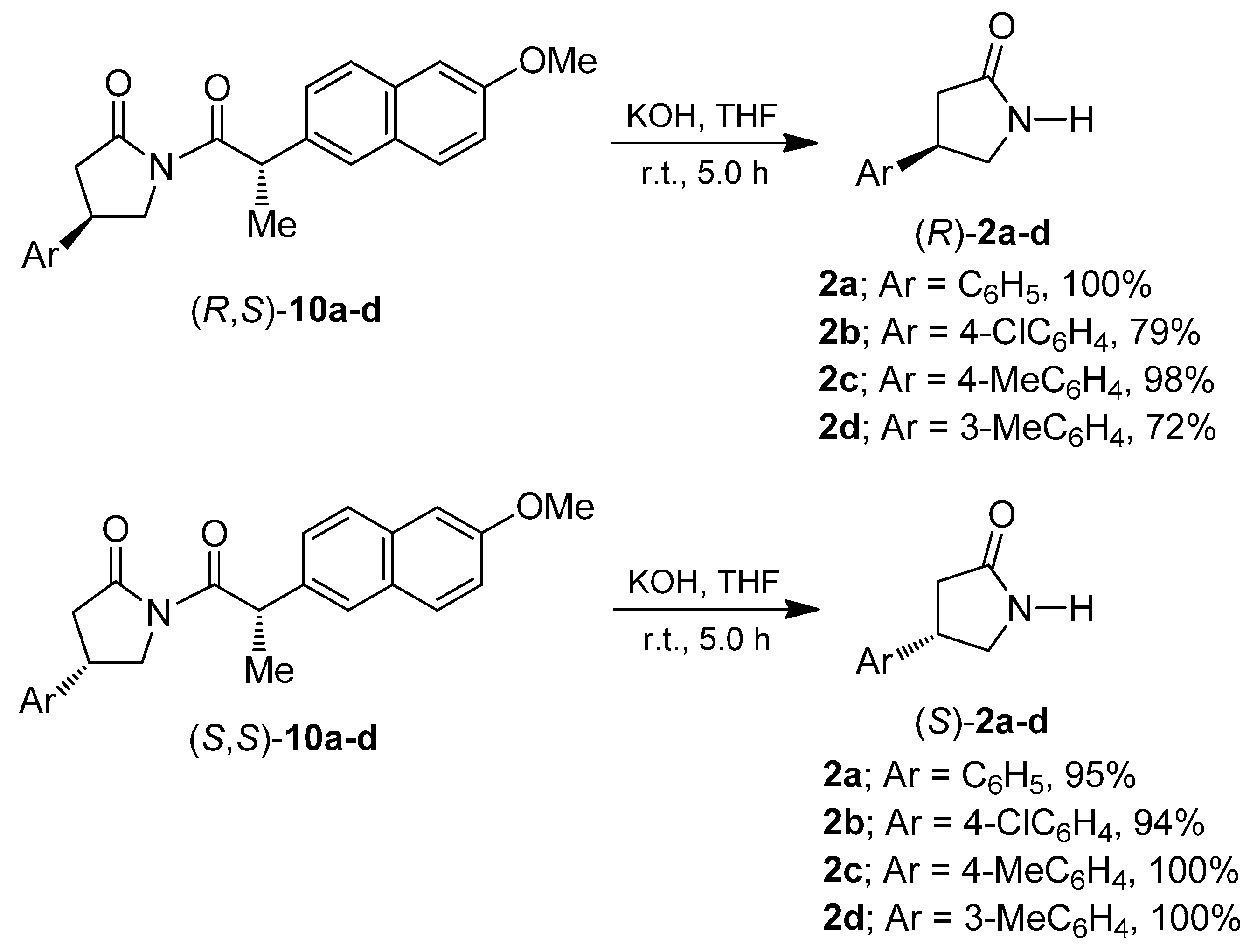
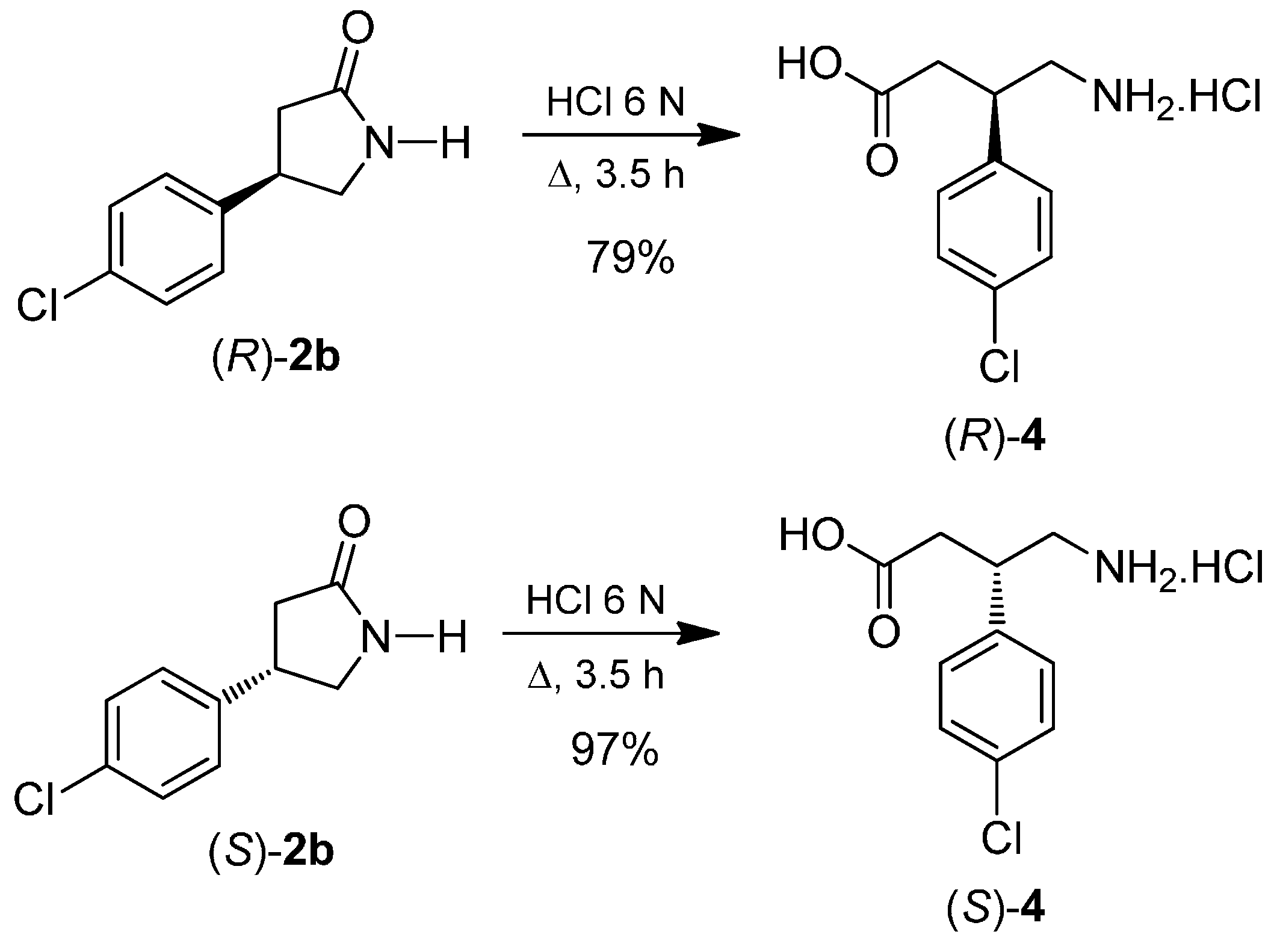
2. Results and Discussion






3. Materials and Methods
3.1. General Comments
3.2. General Procedure for the Preparation of Arylidenemalonates 5a–f
3.2.1. Diethyl 2-(2-Chlorobenzylidene)malonate 5e
3.2.2. Diethyl 2-(2-Nitrobenzyliden)malonate 5f
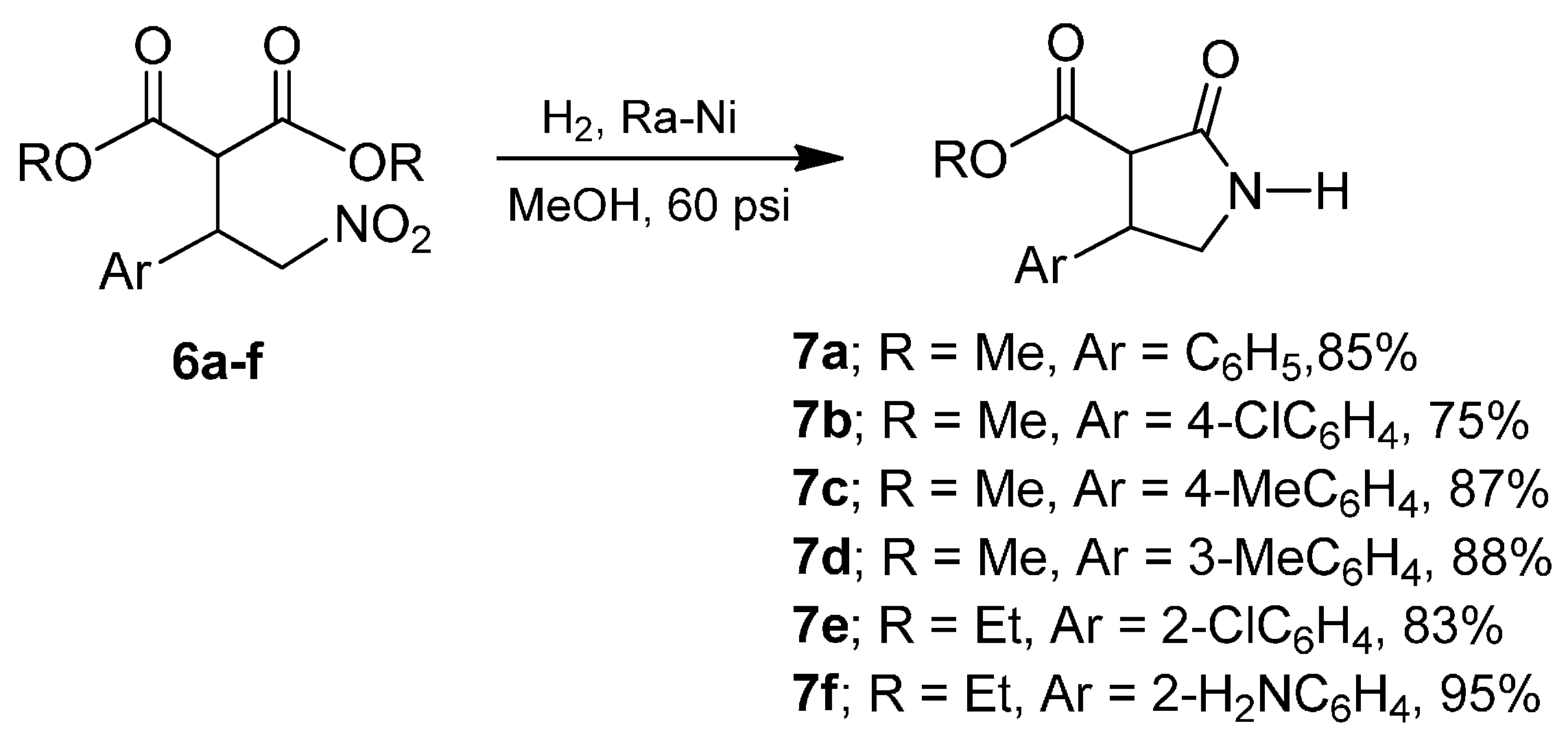
3.3. General Procedure for the Preparation of Nitroderivatives 6a–f
3.4. General Procedure for the Synthesis of the γ-Lactams 7a–f
3.4.1. Ethyl 2-Oxo-4-(4-chlorophenyl)-pyrrolidine-3-carboxylate (±)-7b
3.4.2. Methyl 2-Oxo-4-(4-methylphenyl)-pyrrolidine-3-carboxylate (±)-7c
3.4.3. Methyl 2-Oxo-4-(3-methylphenyl)-pyrrolidine-3-carboxylate (±)-7d
3.4.4. Ethyl 2-Oxo-4-(2-chlorophenyl)-pyrrolidine-3-carboxylate (±)-7e
3.4.5. Ethyl 2-Oxo-4-(2-aminophenyl)-pyrrolidine-3-carboxylate (±)-7f
3.5. General Procedure for The Preparation of the Carboxylic Acids 8a–f
3.5.1. 2-Oxo-4-phenyl-pyrrolidine-3-carboxylic Acid 8a
3.5.2. 2-Oxo-4-(4-chlorophenyl)-pyrrolidine-3-carboxylic Acid 8b
3.5.3. 2-Oxo-4-(4-methylphenyl)-pyrrolidine-3-carboxylic Acid 8c
3.5.4. 2-Oxo-4-(3-methylphenyl)-pyrrolidine-3-carboxylic Acid 8d
3.5.5. 2-Oxo-4-(2-chlorophenyl)-pyrrolidine-3-carboxylic Acid 8e
3.5.6. 2-Oxo-4-(2-aminophenyl)-pyrrolidine-3-carboxylic Acid 8f
3.6. General Procedure of the Synthesis of β-Aryl-γ-lactams (±)-2a–f
3.6.1. 4-(3-Methylphenyl)-pyrrolidin-2-One (±)-2d
3.6.2. 4-(2-Chlorophenyl)-pyrrolidin-2-One (±)-2e
3.6.3. 4-(2-Aminophenyl)-pyrrolidin-2-one (±)-2f
3.7. Synthesis of (S)-Naproxen Acyl Chloride 9
3.8. General Procedures for The Resolution of β-Aryl-γ-lactams (±)-2a–d
3.8.1. (R)-1-((S)-2(6-Methoxynaphth-2-yl)propionyl)-4-phenyl-pyrrolidin-2-one (R,S)-10a and (S)-1-((S)-2(6-methoxynaphth-2-yl)propionyl)-4-phenyl-pyrrolidin-2-one (S,S)-10a
3.8.2. (R)-4-(4-Chlorophenyl)-1-((S)-2-(6-methoxynaphth-2-yl)propionyl)-pyrrolidin-2-one (R,S)-10b and (S)-4-(4-Chlorophenyl)-1-((S)-2-(6-methoxynaphth-2-yl)propionyl)-pyrrolidin-2-one (S,S)-10b
3.8.3. (R)-1-((S)-2-(6-Methoxynaphth-2-yl)propionyl-4-(4-methylphenyl)-pyrrolidin-2-one (R,S)-10c and (S)-1-((S)-2-(6-Methoxynaphth-2-yl)propionyl-4-(4-methylphenyl)-pyrrolidin-2-one (S,S)-10c, (R,S)-10c and (S,S)-10c
3.8.4. (R)-1-((S)-2-(6-Methoxynaphth-2-yl)propionyl)-4-(3-methylphenyl)-pyrrolidin-2-one (R,S)-10d and (S)-1-((S)-2-(6-Methoxynaphth-2-yl)propionyl)-4-(3-methylphenyl)-pyrrolidin-2-one (S,S)-10d
3.9. General Procedure for the Preparation of Enantiomerically-Pure β-Aryl-γ-lactams 2a–d
3.9.1. (R)-4-Phenylpyrrolidin-2-one 2a
3.9.2. (S)-4-Phenylpyrrolidin-2-one 2a
3.9.3 (R)-4-(4-Chlorophenyl)-pyrrolidin-2-one 2b
3.9.4. (S)-4-(4-Chlorophenyl)-pyrrolidin-2-one 2b
3.9.5. (R)-4-(4-Methylphenyl)-pyrrolidin-2-one 2c
3.9.6. (S)-4-(4-Methylphenyl)-pyrrolidin-2-one 2c
3.9.7. (R)-4-(3-Methylphenyl)-pyrrolidin-2-one 2d
3.9.8. (S)-4-(3-Methylphenyl)-pyrrolidin-2-one 2d
3.10. (R)-(−)-Baclofen Hydrochloride 4
3.11. (S)-(+)-Baclofen Hydrochloride 4
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sifferlen, T.; Boller, A.; Chardonneau, A.; Cottreel, E.; Hoecker, J.; Aissaoui, H.; Williams, J.T.; Brotschi, C.; Heidmann, B.; Siegrist, R.; et al. Discovery of substituted lactams as novel dual orexin receptor antagonists. Synthesis, preliminary structure-activity relationship studies and efforts towards improved metabolic stability and pharmacokinetic properties. Part 1. Bioorg. Med. Chem. Lett. 2014, 24, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, G.; Dunn, D.; McKenna, B.A.; Kopec, K.; Chatterjee, S. Lactam and oxazolidinone derived potent 5-hydroxytryptamine 6 receptor antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 2094–2097. [Google Scholar] [CrossRef] [PubMed]
- Vorona, M.; Orlova, N.; Kuznetsov, E.; Vikainis, S.; Liepinshi, E.; Belyakov, S.; Mishnev, A.; Veinberg, G. Method for the preparation of 4-aryl-3-pyrrolin-2-ones and their 5-bromo. Chem. Heterocycl. Compd. 2013, 49, 1118–1127. [Google Scholar] [CrossRef]
- Kanes, S.J.; Tokarczyk, J.; Siegel, S.J.; Bilker, W.; Abel, T.; Kelly, M.P. Rolipram a specific phosphodiesterase 4 inhibitor with potential antipsychotic activity. Neuroscience 2007, 144, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, R-W.; Williams, A.J.; Liao, Z.; Yao, C.; Tortella, F.C.; Dave, J.R. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation. Neurosci. Lett. 2007, 418, 165–169. [Google Scholar]
- Wachtel, H. Potencial antidepressant activity of rolipram and other selective cyclic adenosine 3’,5’-monophosphate phosphodiesterase inhibitors. Neuropharmacology 1983, 22, 267–272. [Google Scholar] [CrossRef]
- Büyüknacar, H.S.; Kumcu, E.K.; Göçmen, C.; Önder, S. Effect of phosphodiesterase type 4 inhibitor rolipram on cyclophosphamide-induced cystitis in rats. Eur. J. Pharmacol. 2008, 586, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Löschmann, P.A.; Northoff, G.H.; Weller, M.; Steinbrecher, A.; Steinbach, J.P.; Lichtenfels, R.; Meyermann, R.; Riethmüller, A.; Fontana, A.; et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat. Med. 1995, 1, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Zvejniece, L.; Vavers, E.; Svalbe, B.; Veinberg, G.; Rizhanova, K.; Liepins, V.; Kalvinsh, I.; Dambrova, M. R-phenibut binds to the α2-δ subunit of voltage-dependent calcium channels and exerts gabapentin-like anti-nociceptive effects. Pharmacol. Biochem. Behav. 2015, 137, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lapin, I. Phenibut (β-Phenyl-GABA): A Tranquilizer and Nootropic Drug. CNS Drug Rev. 2001, 7, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Sharma, S.; Kumar, P.; Deshmukh, H. Therapeutic potential of GABAB receptor ligands in drug addiction, anxiety, depression and other CNS disorders. Pharmacol. Biochem. Behav. 2013, 110, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Zvejniece, L.; Liepinsh, E.; Cirule, H.; Zharkova, O.; Veinberg, G.; Kalvinsh, I. Comparative pharmacological activity of optical isomers of phenibut. Eur. J. Pharmacol. 2008, 583, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Baures, P.W.; Eggleston, D.S.; Erhard, K.F.; Cieslinski, L.B.; Torphy, T.J.F.; Christensen, S.B. The Crystal structure, absolute configuration, and phosphodiesterase-inhibitory activity of (+)-1-(4-bromobenzyl)-4-(3-(cyclopentyloxy)-4-methoxy pheny-1)-pyrrolidin-2-one. J. Med. Chem. 1993, 36, 3274–3277. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.H.; Schmiechen, R.; Brezinski, M.; Seidler, J. Stereospecific binding of the antidepressant rolipram to brain protein structures. Eur. J. Pharmacol. 1986, 127, 105–115. [Google Scholar] [CrossRef]
- Olpe, H.R.; Demiéville, H.; Baltzer, V.; Bencze, W.L.; Koella, W.P.; Wolf, P.; Haas, H.L. The biological activity of d-baclofen (Lipresal®). Eur. J. Pharmacol. 1978, 52, 133–136. [Google Scholar] [CrossRef]
- Xu, F.; Peng, G.; Phan, T.; Dilip, U.; Chen, J.L.; Chernov-Rogan, T.; Zhang, X.; Grindstaff, K.; Annamalai, T.; Koller, K.; et al. Discovery of a novel potent GABAB receptor agonist. Bioorg. Med. Chem. Lett. 2011, 21, 6582–6585. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, M.; Cativiela, C. Stereoselective synthesis of γ-amino acids. Tetrahedron Asymmetry 2007, 18, 3–97. [Google Scholar] [CrossRef]
- Bae, H.Y.; Song, C.E. Unprecedented hydrophobic amplification in noncovalent organocatalysis “on Water”: Hydrophobic chiral squaramide catalyzed michael addition of malonates to nitroalkenes. ACS Catal. 2015, 5, 3613–3619. [Google Scholar] [CrossRef]
- Massolo, E.; Benaglia, M.; Genoni, A.; Annunziata, R.; Celentano, G.; Gaggero, N. Stereoselective reaction of 2-carboxythioesters-1,3-dithiane with nitroalkenes: an organocatalytic strategy for the asymmetric addition of a glyoxylate anion equivalent. Org. Biomol. Chem. 2015, 13, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Powell, R.W., III; Santangelo, S.; Stewart, J.D. Opposite enantioselectivity in the bioreduction of (Z)-β-aryl-β-cyanoacrylates mediated by the tryptophan 116 mutants of old yellow enzyme 1: Synthetic approach to (R)- and (S)-β-aryl-γ-lactams. Adv. Synth. Catal. 2015, 357, 1849–1860. [Google Scholar] [CrossRef]
- Ghislieri, D.; Gilmore, K.; Seeberger, P.H. Chemical assembly systems: layered control for divergent, continuous, multistep syntheses of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Naciuk, F.F.; Vargas, D.Z.; D’Oca, C.R.M.; Moro, C.C.; Russowsky, D. One pot domino reaction accessing γ-nitroesters: synthesis of GABA derivatives. New J. Chem. 2015, 39, 1643–1653. [Google Scholar] [CrossRef]
- Hajra, S.; Aziz, S.M.; Maji, R. Organocatalytic enantioselective conjugate addition of nitromethane to alkylidenemalonates: Asymmetric synthesis of pyrrolidine-3-carboxylic acid derivatives. RSC Adv. 2013, 3, 10185–10188. [Google Scholar] [CrossRef]
- Ooi, T.; Fujioka, S.; Maruoka, K. Highly enantioselective conjugate addition of nitroalkanes to alkylidenemalonates using efficient phase-transfer catalysis of N-spiro chiral ammonium bromides. J. Am. Chem. Soc. 2004, 126, 11790–11791. [Google Scholar] [CrossRef] [PubMed]
- CCDC 1048102 and 1048101 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
- Gruzdev, D.A.; Chulakov, E.N.; Sadretdinova, L.Sh.; Kodess, M.I.; Levit, G.L.; Krasnov, V.P. Synthesis of enantiomers of 3-methyl- and 3-phenyl-3,4-dihydro-2H-[1,4]benzothiazines and their 1,1-dioxides via an acylative kinetic resolution protocol. Tetrahedron Asymmetry 2015, 26, 186–194. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Chulakov, E.N.; Levit, G.L.; Ezhikova, M.A.; Kodess, M.I.; Krasnov, V.P. A comparative study on the acylative kinetic resolution of racemic fluorinated and non-fluorinated 2-methyl-1,2,3,4-tetrahydroquinolines and 3,4-dihydro-3-methyl-2H-[1,4]-benzoxazines. Tetrahedron Asymmetry 2013, 24, 1240–1246. [Google Scholar] [CrossRef]
- Boyd, E.; Coulbeck, E.; Coumbarides, G.S.; Chavda, S.; Dingjan, M.; Eames, J.; Flinn, A.; Motevalli, M.; Northend, J.; Yohannes, Y. Parallel kinetic resolution of racemic oxazolidinones using quasi-enantiomeric active esters. Tetrahedron Asymmetry 2007, 18, 2515–2530. [Google Scholar] [CrossRef]
- Solis, A.; Luna, H.; Pérez, H.I.; Manjarrez, N.; Sánchez, R.; Gutiérrez, A. (S)-Naproxen® as a derivatizing agent to determine enantiomeric excess of cyanohydrins by HPLC. Tetrahedron Lett. 1998, 39, 8759–8762. [Google Scholar] [CrossRef]
- Blazewska, K.; Gajda, T. (S)-Naproxen® and (S)-Ibuprofen® chlorides–convenient chemical derivatizing agents for the determination of the enantiomeric excess of hydroxyl- and amino-phosphonates. Tetrahedron Asymmetry 2002, 13, 671–674. [Google Scholar] [CrossRef]
- Paraskar, A.S.; Sudalai, A. Co-catalyzed reductive cyclization of azido and cyano substituited α,β-unsaturated esters with NaBH4: Enantioselective synthesis of (R)-baclofen and (R)-rolipram. Tetrahedron 2006, 62, 4907–4916. [Google Scholar] [CrossRef]
- Camps, P.; Muñoz-Torrero, D.; Sánchez, L. Synthesis of both enantiomers of baclofen using (R)- and (S)-N-phenylpantolactam as chiral auxiliaries. Tetrahedron Asymmetry 2004, 15, 2039–2044. [Google Scholar] [CrossRef]
- Thakur, V.V.; Nikalje, M.D.; Sudalai, A. Enantioselective synthesis of (R)-(−)-baclofen via Ru(II)-BINAP catalyzed asymmetric hydrogenation. Tetrahedron Asymmetry 2003, 14, 581–586. [Google Scholar] [CrossRef]
- Corey, E.J.; Zhang, F.Y. Enantioselective michael addition of nitromethane to α,β-enones catalyzed by chiral quaternary ammonium salts. A simple synthesis of (R)-baclofen. Org. Lett. 2000, 2, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Langlois, N.; Dahuron, N.; Wang, H.-S. Enantioselective syntheses of (R)-3-phenyl GABA, (R)-baclofen and 4-arylpyrrolidin-2-ones. Tetrahedron 1996, 52, 15117–15126. [Google Scholar] [CrossRef]
- Fallan, C.; Quigley, P.F.; Lam, H.W. Ytterbium-catalyzed conjugate allylation of alkylidene malonates. J. Org. Chem. 2011, 76, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, Y.; Takahashi, K.; Kitazawa, T.; Sakai, N. Indium(III)-catalyzed knoevenagel condensation of aldehydes and activated methylenes using acetic anhydride as a promoter. J. Org. Chem. 2015, 80, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, Y.; Qu, C.; Zhang, L.; Wang, J.; Cui, Y. Catalytic performance of silica supported cinchona alkaloids as heterogeneous catalysts for asymmetric michael reaction. Catal. Lett. 2014, 144, 1681–1688. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Tang, L.; Deng, L. Highly enantioselective conjugate addition of malonate and β-ketoester to nitroalkenes: Asymmetric C-C bond formation with new bifunctional organic catalysts based on cinchona alkaloids. J. Am. Chem. Soc. 2004, 126, 9906–9907. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Chen, D.; Zhang, C.; Zhang, Q.; Cao, B.; Zhao, Z. Mechanosynthesis of γ-nitro dicarbonyl compounds via CaCl2-catalyzed Michael addition. Tetrahedron 2013, 69, 7320–7324. [Google Scholar] [CrossRef]
- Nikonorov, A.A.; Ostroglyadov, E.S.; Vasil’ev, O.S.; Berestovitskaya, B.M. Synthesis and structure of nitroethylpyrrolidone carboxylates. Russ. J. Gen. Chem. 2011, 81, 1681–1690. [Google Scholar] [CrossRef]
- Biswas, K.; Gholap, R.; Srinivas, P.; Kanyal, S.; das Sarma, K. β-Substituted γ-butyrolactams from mucochloric acid: synthesis of (±)-baclofen and other γ-aminobutyric acids and useful building blocks. RSC Adv. 2014, 4, 2538–2545. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Cwiklak, M.; Ostaszewski, R. Studies on the chemoenzymatic synthesis of 3-phenyl-GABA and 4-phenyl-pyrrolid-2-one: The influence of donor of the alkoxy group on enantioselective esterification. Tetrahedron Asymmetry 2013, 24, 427–433. [Google Scholar] [CrossRef]
- Marivet, M.C.; Bourguignon, J.J.; Lugnier, C.; Mann, A.; Stoclet, J.-C.; Wermutht, C.G. Inhibition of cyclic adenosine-3’,5’-monophosphate phosphodiesterase from vascular smooth muscle by rolipram analogues. J. Med. Chem. 1989, 32, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.L.; Poulsen, P.H.; Donslund, B.S.; Morana, F.; Joergensen, K.A. Asymmetric synthesis of γ-nitroesters by an organocatalytic one-pot strategy. Org. Lett. 2012, 14, 1516–1519. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.; Almeida, W.P.; Coelho, F. An efficient synthesis of (R)-(−)-baclofen. Tetrahedron Asymmetry 1999, 10, 2113–2118. [Google Scholar] [CrossRef]
- Yoshifuji, S.; Kaname, M. Stereospecific synthesis of (R)- and (S)-baclofen and (R)- and (S)-PCPGABA[4-amino-2-(4-chlorophenyl)butyric acid] via (R)-and (S)-3-(4-chlorophenyl)pyrrolidines. Chem. Pharm. Bull. 1995, 43, 1302–1306. [Google Scholar] [CrossRef]
- Chênevert, R.; Desjardins, M. Chemoenzymatic synthesis of both enantiomers of baclofen. Tetrahedron Lett. 1991, 32, 4249–4250. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montoya-Balbás, I.J.; Valentín-Guevara, B.; López-Mendoza, E.; Linzaga-Elizalde, I.; Ordoñez, M.; Román-Bravo, P. Efficient Synthesis of β-Aryl-γ-lactams and Their Resolution with (S)-Naproxen: Preparation of (R)- and (S)-Baclofen. Molecules 2015, 20, 22028-22043. https://doi.org/10.3390/molecules201219830
Montoya-Balbás IJ, Valentín-Guevara B, López-Mendoza E, Linzaga-Elizalde I, Ordoñez M, Román-Bravo P. Efficient Synthesis of β-Aryl-γ-lactams and Their Resolution with (S)-Naproxen: Preparation of (R)- and (S)-Baclofen. Molecules. 2015; 20(12):22028-22043. https://doi.org/10.3390/molecules201219830
Chicago/Turabian StyleMontoya-Balbás, Iris J., Berenice Valentín-Guevara, Estefanía López-Mendoza, Irma Linzaga-Elizalde, Mario Ordoñez, and Perla Román-Bravo. 2015. "Efficient Synthesis of β-Aryl-γ-lactams and Their Resolution with (S)-Naproxen: Preparation of (R)- and (S)-Baclofen" Molecules 20, no. 12: 22028-22043. https://doi.org/10.3390/molecules201219830