Synthesis, X-ray Diffraction, Thermogravimetric and DFT Analyses of Pyrimidine Derivatives

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
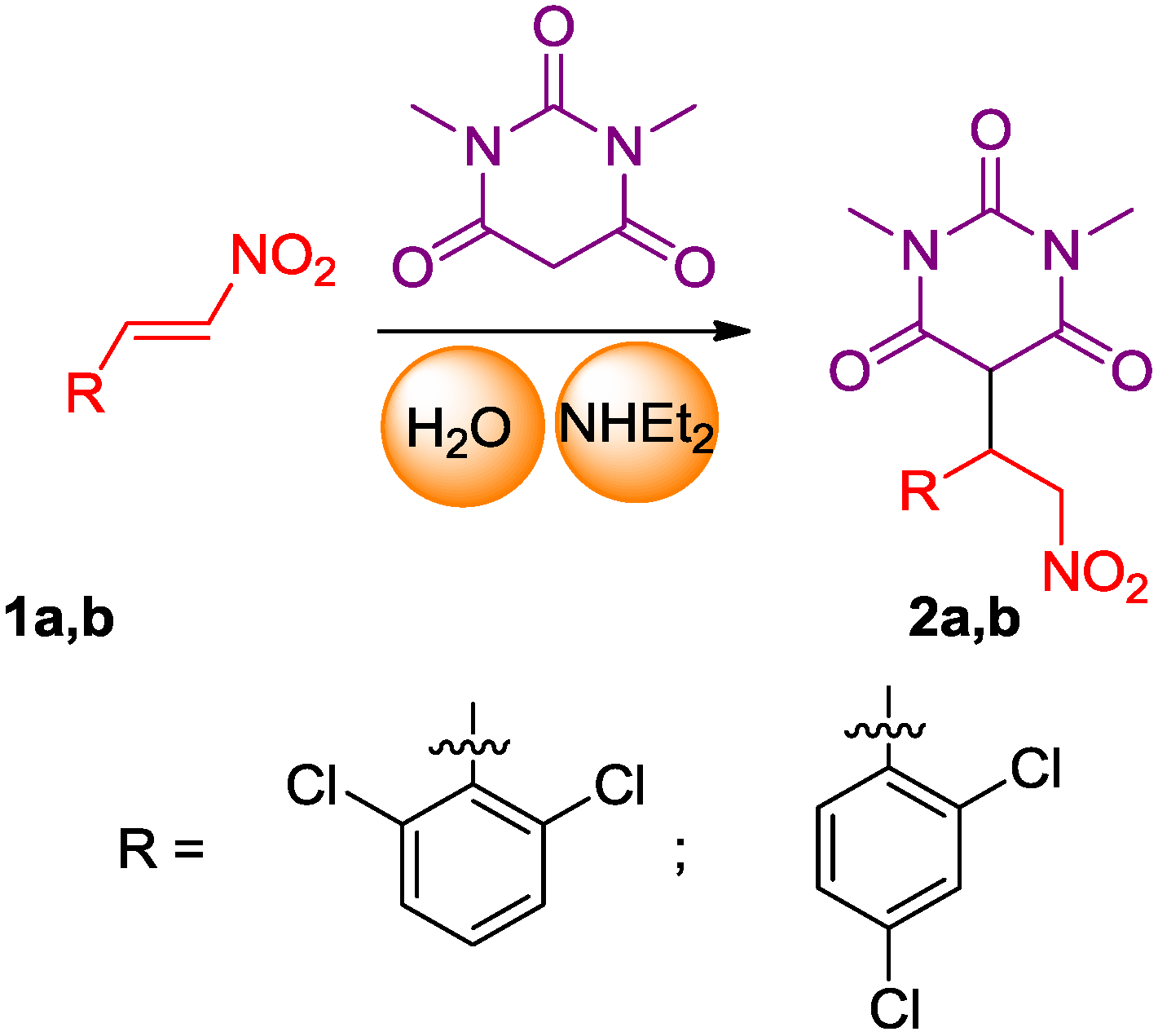
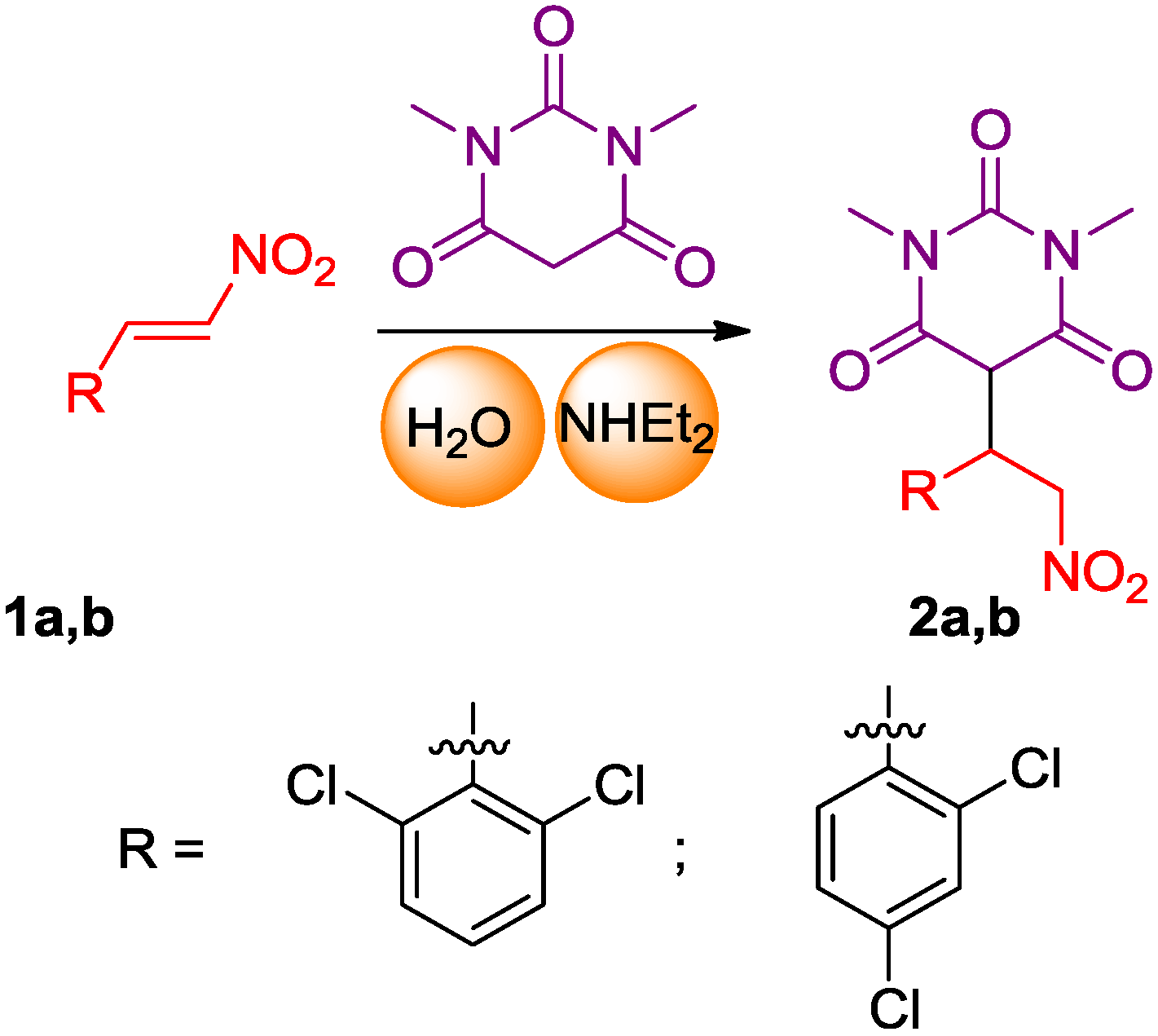
2.1. Synthesis of the Pyrimidine Derivatives
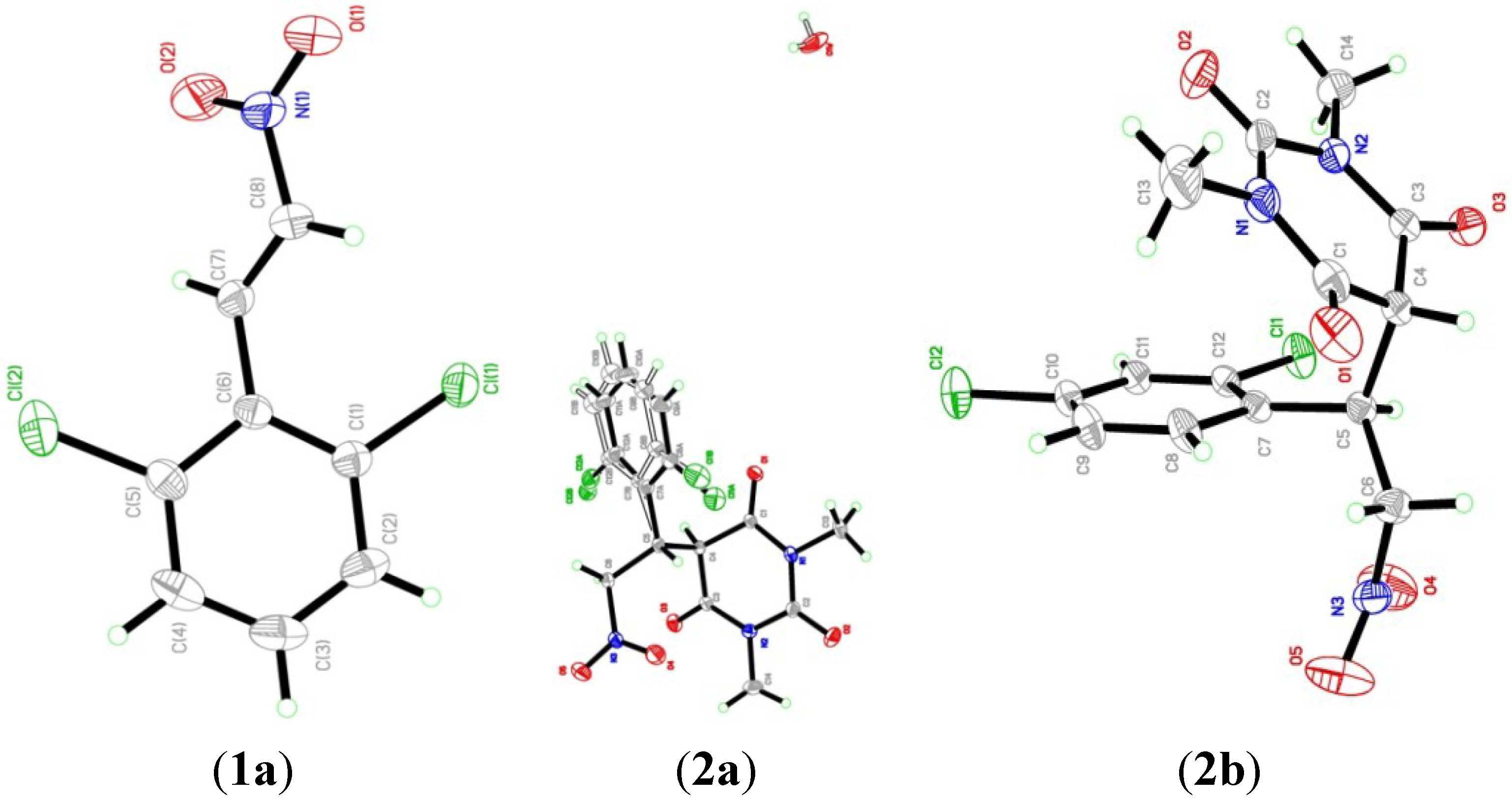
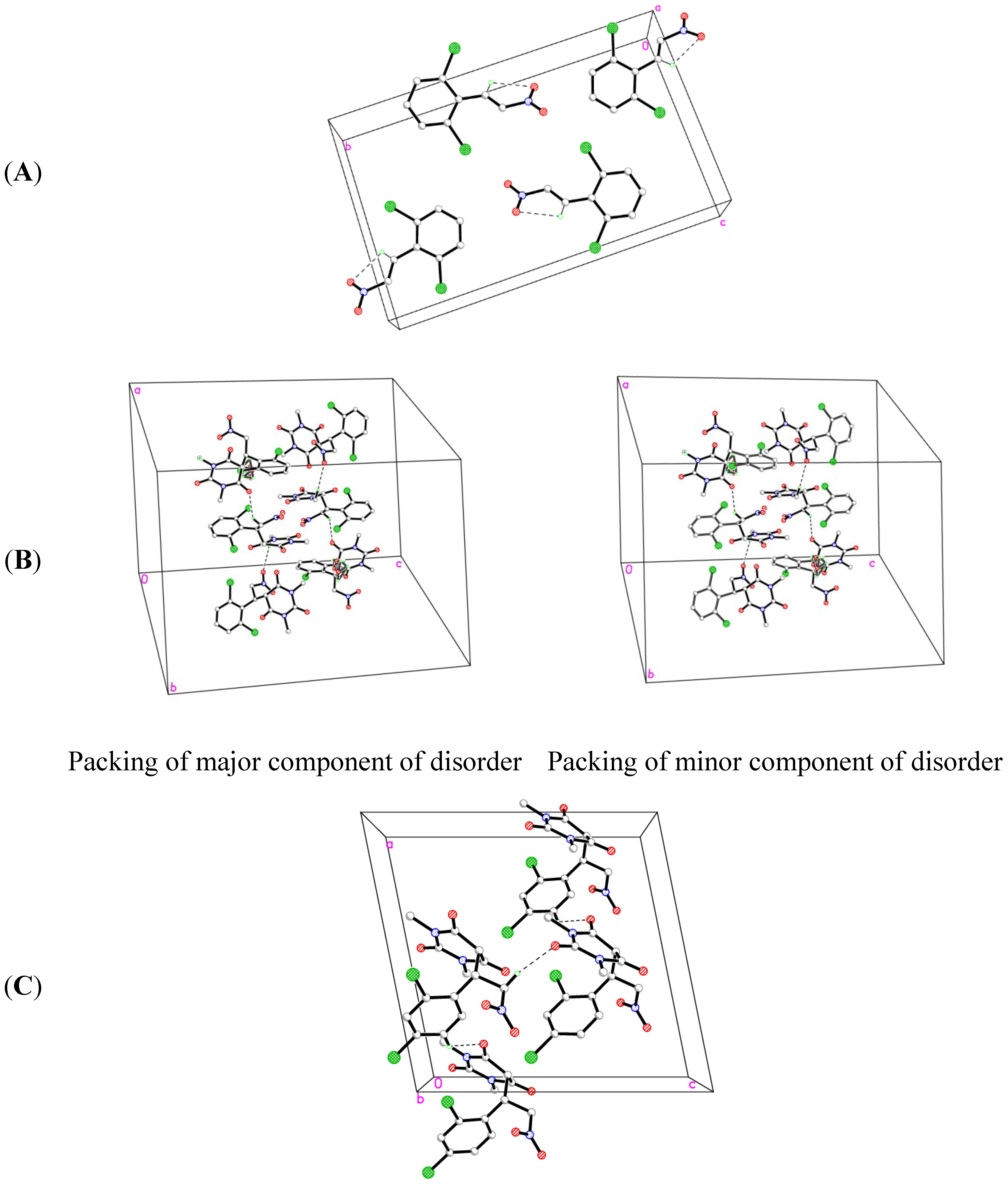
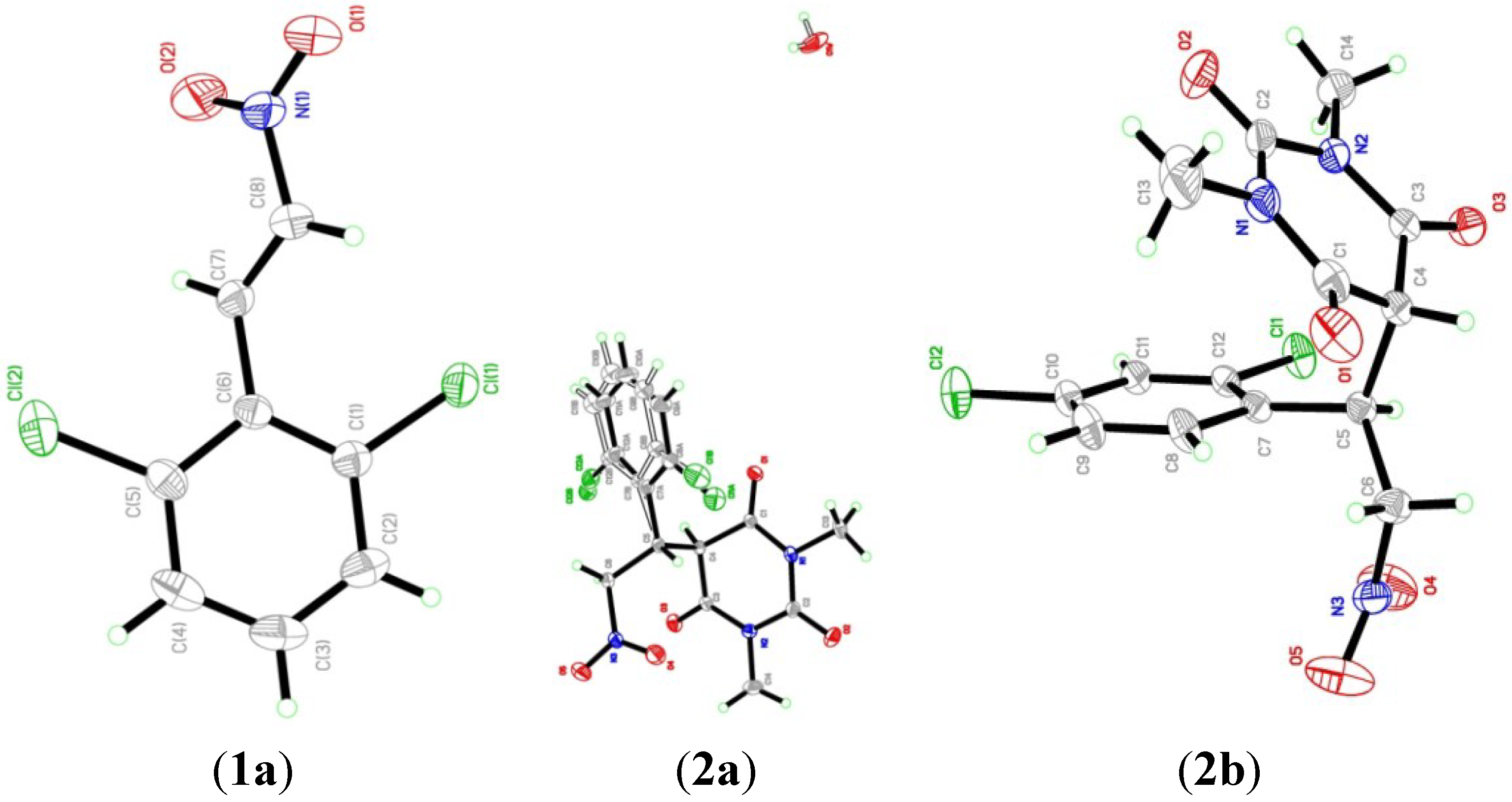
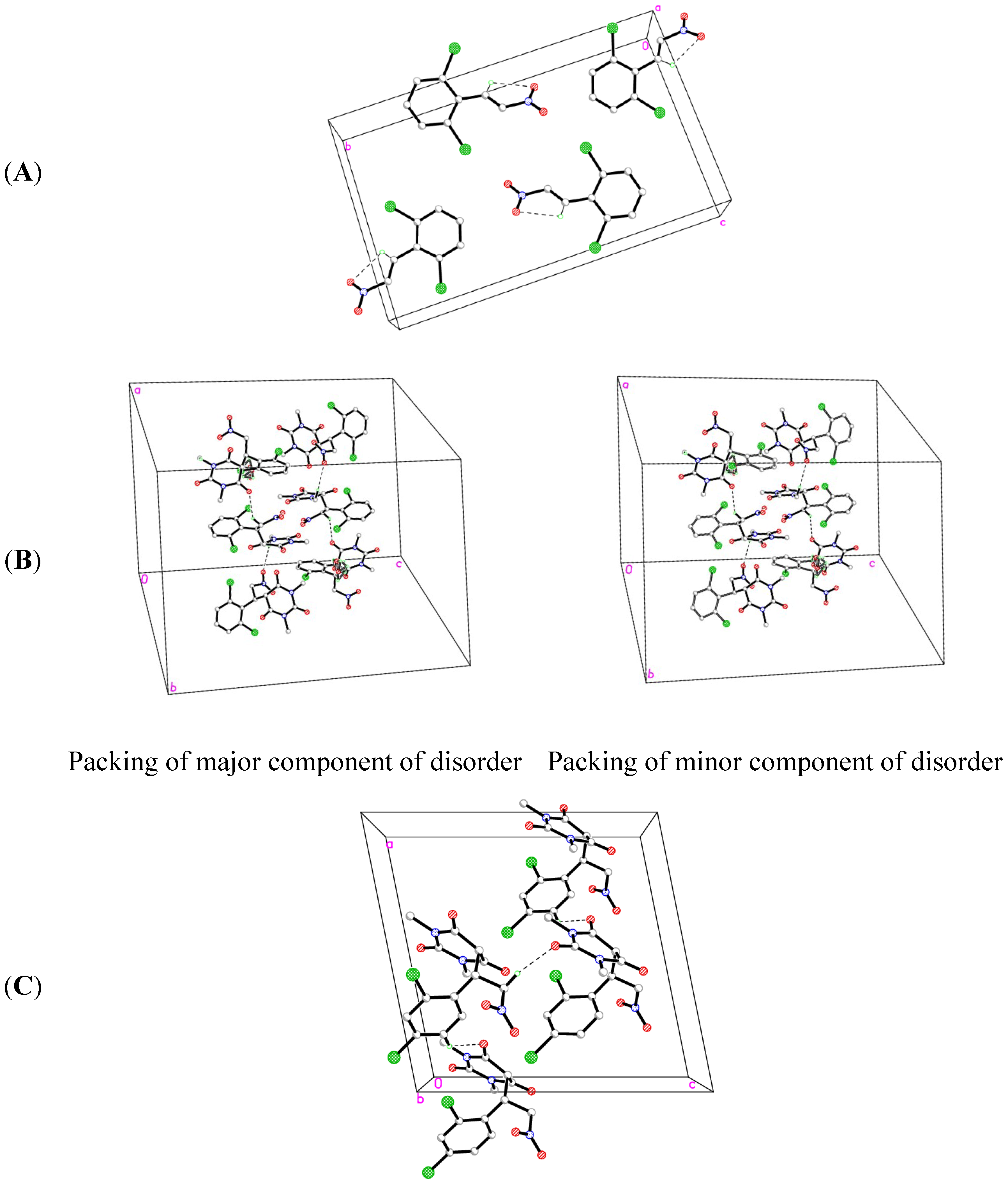
2.2. X-ray Crystal Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Compound 1a | Compound 2a | Compound 2b |
|---|---|---|---|
| Empirical formula | C8H5Cl2NO2 | C14H13Cl2N3O5·H2O | C14H13Cl2N3O5 |
| Formula weight | 218.03 | 391.02 | 374.17 |
| Temperature | 293(2) K | 293(2) K | 293(2) K |
| Wavelength (Mo Kα radiation, λ) | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | monoclinic | trigonal | Monoclinic |
| Space group | P 21/c | R-3 | Cc |
| Unit cell dimensions | a = 3.8395 (1) Å, α = 90.00° b = 19.8653 (7) Å β = 90.6258 (10)° c = 11.9619 (4) Å, γ = 90.00° | a = 18.0177(7) Å, α = 90.00° b = 18.0177(7) Å, β = 90.00° c = 26.6558(9) Å, γ = 120.00° | a = 12.9432 (4) Å, α = 90.00° b = 9.3084 (4) Å, β = 101.3572 (16)° c = 13.4650 (5) Å, γ = 90.00° |
| Volume | 912.31 (5)Å3 | 7494.1(6) Å3 | 1590.51 (11) Å3 |
| Z | 4 | 6 | 4 |
| Density (calculated) | 1.587 Mg/m3 | 1.535 Mg/m3 | 1.563 Mg/m3 |
| Absorption coefficient | 0.67 mm−1 | 0.424 mm−1 | 0.439 mm−1 |
| F(000) | 440 | 3564 | 768 |
| Crystal size | 0.58 × 0.35 × 0.27 mm | 0.33 × 0.21 × 0.09 mm | 0.57 × 0.35 × 0.26 mm |
| Theta range for data collection | 2.7 to 30.5°. | 2.3 to 30.6°. | 2.7 to 30.6°. |
| Index ranges | −5 ≤ h ≤ 5, −28 ≤ k ≤ 28, −17 ≤ l ≤ 17 | −25 ≤h ≤ 25, −25 ≤ k ≤ 25, −38 ≤ l ≤ 38 | −18 ≤ h ≤ 18, −13 ≤ k ≤ 13, −19 ≤ l ≤ 19 |
| Reflections collected/ unique | 41303/2404 [R(int) = 0.042] | 107278/5130 [R(int) = 0.059] | 35961/4754 [R(int) = 0.024] |
| Completeness to theta = 30.57° | 99.8% | 99.6% | 99.6% |
| Absorption correction | multi-scan | multi-scan | multi-scan |
| Refinement | method Full-matrix least-squares on F2 | method Full-matrix least-squares on F2 | method Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 | 1.04 | 1.03 | 1.27 |
| Largest diff. peak and hole | 0.28 and −0.22 e.Å−3 | 0.72 and −0.26 e.Å−3 | 0.22 and −0.25 e.Å−3 |
| Bond | Experimental | Calculated |
| Cl1-C1 | 1.7320 (13) | 1.7314 |
| Cl2-C5 | 1.7295 (14) | 1.7405 |
| O1-N1 | 1.222 (2) | 1.2370 |
| O2-N1 | 1.211 (2) | 1.2372 |
| N1-C8 | 1.4563 (18) | 1.4511 |
| Atom Angle | Experimental | Calculated |
| O1-N1-O2 | 124.37 (14) | 125.1111 |
| O1-N1-C8 | 115.73 (14) | 115.6492 |
| O2-N1-C8 | 119.90 (14) | 119.2397 |
| Cl1-C1-C2 | 117.62 (11) | 124.7209 |
| Cl1-C1-C6 | 120.34 (9) | 113.6358 |
| Cl2-C5-C4 | 118.18 (11) | 114.3525 |
| Cl2-C5-C6 | 119.58 (10) | 123.4094 |
| N1-C8-C7 | 120.57 (13) | 119.8515 |
| D-H···A | D-H | H···A | D···A | D-H···A |
|---|---|---|---|---|
| C7-H7A···O2 | 0.9300 | 2.4000 | 2.7323 (18) | 101.00 |
| Bond | Experimental | Calculated |
| Cl1A-C8A | 1.738 (4) | 1.7277 |
| Cl1B-C8B | 1.721 (14) | NC |
| Cl2A-C12A | 1.751 (6) | 1.7365 |
| Cl2B-C12B | 1.715 (14) | NC |
| O1-C1 | 1.204 (2) | 1.2278 |
| O2-C2 | 1.205 (2) | 1.2325 |
| O3-C3 | 1.212 (2) | 1.2276 |
| O4-N3 | 1.196 (3) | 1.2362 |
| O5-N3 | 1.215 (3) | 1.2378 |
| O1W-O1W i | 0.969 (19) | NC |
| O1W-O1W ii | 0.97 (2) | NC |
| N1-C1 | 1.378 (2) | 1.3855 |
| N1-C13 | 1.473 (3) | 1.4513 |
| N1-C2 | 1.382 (2) | 1.3724 |
| N2-C2 | 1.387 (3) | 1.3925 |
| N2-C14 | 1.463 (3) | 1.4515 |
| N2-C3 | 1.370 (2) | 1.3881 |
| N3-C6 | 1.500 (2) | 1.4962 |
| Atom Angle | Experimental | Calculated |
| O1W ii-O1W-O1W i | 60.0 (18) | NC |
| C1-N1-C2 | 124.36 (15) | 122.9525 |
| C2-N1-C13 | 117.61 (15) | 117.6313 |
| C1-N1-C13 | 117.39 (15) | 117.0595 |
| C2-N2-C3 | 124.74 (15) | 123.0628 |
| C3-N2-C14 | 117.62 (17) | 116.9594 |
| C2-N2-C14 | 117.50 (16) | 117.4505 |
| O4-N3-C6 | 121.03 (18) | 117.2445 |
| O4-N3-O5 | 123.45 (19) | 125.7819 |
| O5-N3-C6 | 115.53 (16) | 116.9697 |
| O1-C1-N1 | 121.69 (16) | 123.7575 |
| O1-C1-C4 | 121.91 (15) | 122.9936 |
| N1-C1-C4 | 116.24 (15) | 113.2488 |
| O2-C2-N2 | 120.68 (18) | 123.3871 |
| N1-C2-N2 | 117.42 (14) | 116.2983 |
| O2-C2-N1 | 121.9 (2) | 121.7454 |
| O3-C3-C4 | 121.55 (15) | 123.7560 |
| O3-C3-N2 | 121.13 (15) | 121.7802 |
| N2-C3-C4 | 117.24 (14) | 112.8565 |
| N3-C6-C5 | 112.43 (14) | 111.9825 |
| Cl1A-C8A-C7A | 119.7 (3) | 124.0885 |
| Cl1A-C8A-C9A | 117.4 (3) | 114.4806 |
| Cl1B-C8B-C9B | 116.7 (10) | NC |
| Cl1B-C8B-C7B | 121.7 (9) | NC |
| Cl2A-C12A-C7A | 120.8 (3) | 122.7008 |
| Cl2A-C12A-C11A | 114.7 (4) | 115.2511 |
| Cl2B-C12B-C7B | 122.5 (8) | NC |
| Cl2B-C12B-C11B | 114.4 (10) | NC |
| D-H···A | D-H | H···A | D···A | D-H···A |
|---|---|---|---|---|
| O1W-H2W1···O1W iii | 0.9100 | 2.1000 | 2.859 (6) | 140.00 |
| O1W-H2W1···O1W iv | 0.9100 | 1.8500 | 2.689 (6) | 152.00 |
| O1W-H2W1···O1W v | 0.9100 | 2.0500 | 2.689 (7) | 125.00 |
| C5-H5A···O4 | 0.9800 | 2.2700 | 2.741 (3) | 108.00 |
| C6-H6A···O1 vi | 0.9700 | 2.5300 | 3.337 (3) | 141.00 |
| C6-H6B···Cl2A | 0.9700 | 2.6800 | 3.125 (4) | 109.00 |
| C6-H6B···O3 | 0.9700 | 2.5800 | 2.979 (2) | 105.00 |
| C13-H13A···O2 | 0.9600 | 2.2900 | 2.716 (3) | 106.00 |
| C13-H13C···O5 vii | 0.9600 | 2.5100 | 3.425 (3) | 159.00 |
| C14-H14B···O2 | 0.9600 | 2.2400 | 2.690 (4) | 108.00 |
| Bond | Experimental | Calculated |
| Cl1-C12 | 1.7310 (11) | 1.7247 |
| Cl2-C10 | 1.7338 (13) | 1.7195 |
| O1-C1 | 1.2101 (19) | 1.2276 |
| O2-C2 | 1.219 (2) | 1.2325 |
| O3-C3 | 1.2148 (16) | 1.2278 |
| O4-N3 | 1.212 (2) | 1.2362 |
| O5-N3 | 1.204 (3) | 1.2378 |
| N1-C1 | 1.376 (2) | 1.3881 |
| N1-C2 | 1.386 (2) | 1.3925 |
| N1-C13 | 1.469 (2) | 1.4515 |
| N2-C2 | 1.3947 (19) | 1.4071 |
| N2-C3 | 1.3661 (16) | 1.3855 |
| N2-C14 | 1.471 (2) | 1.4513 |
| N3-C6 | 1.496 (2) | 1.4962 |
| Atom Angle | Experimental | Calculated |
| C1-N1-C2 | 124.73 (12) | 122.7743 |
| C1-N1-C13 | 117.37 (15) | 117.4505 |
| C2-N1-C13 | 117.89 (15) | 117.0006 |
| C2-N2-C3 | 124.33 (11) | 122.9525 |
| C2-N2-C14 | 117.89 (12) | 117.0593 |
| C3-N2-C14 | 117.64 (12) | 117.6313 |
| O4-N3-O5 | 122.4 (2) | 125.7819 |
| O4-N3-C6 | 118.79 (15) | 116.9697 |
| O5-N3-C6 | 118.86 (17) | 117.2445 |
| O1-C1-N1 | 122.31 (14) | 123.3871 |
| O1-C1-C4 | 120.81 (13) | 123.7560 |
| N1-C1-C4 | 116.80 (12) | 112.8565 |
| O2-C2-N1 | 121.70 (15) | 121.7802 |
| O2-C2-N2 | 120.54 (14) | 121.7454 |
| N1-C2-N2 | 117.74 (12) | 116.2983 |
| O3-C3-N2 | 121.68 (12) | 123.7575 |
| O3-C3-C4 | 121.06 (11) | 122.9936 |
| N2-C3-C4 | 117.18 (10) | 113.2488 |
| N3-C6-C5 | 109.74 (11) | 111.9825 |
| Cl2-C10-C9 | 118.98 (10) | 119.8634 |
| Cl2-C10-C11 | 119.27 (9) | 119.9545 |
| Cl1-C12-C7 | 121.01 (8) | 123.6597 |
| Cl1-C12-C11 | 116.44 (9) | 115.4596 |
| D-H···A | D-H | H···A | D···A | D-H···A |
|---|---|---|---|---|
| C6-H6A···O2 i | 0.9700 | 2.3900 | 3.222 (2) | 144.00 |
| C6-H6B···O1 | 0.9700 | 2.5500 | 3.118 (2) | 117.00 |
| C9-H9A···O3 ii | 0.9300 | 2.5100 | 3.2290 (18) | 134.00 |
| C13-H13B···O2 | 0.9600 | 2.3100 | 2.727 (3) | 105.00 |
| C14-H14C···O2 | 0.9600 | 2.2700 | 2.715 (2) | 107.00 |
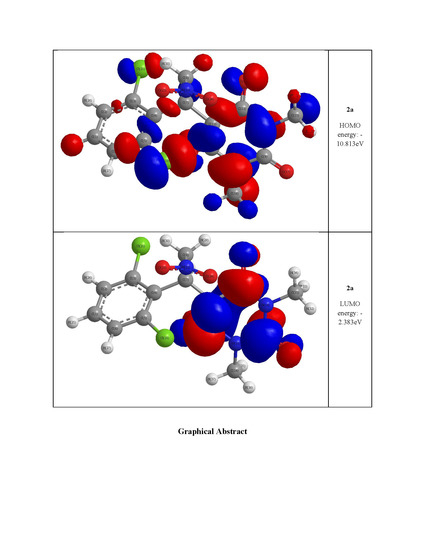
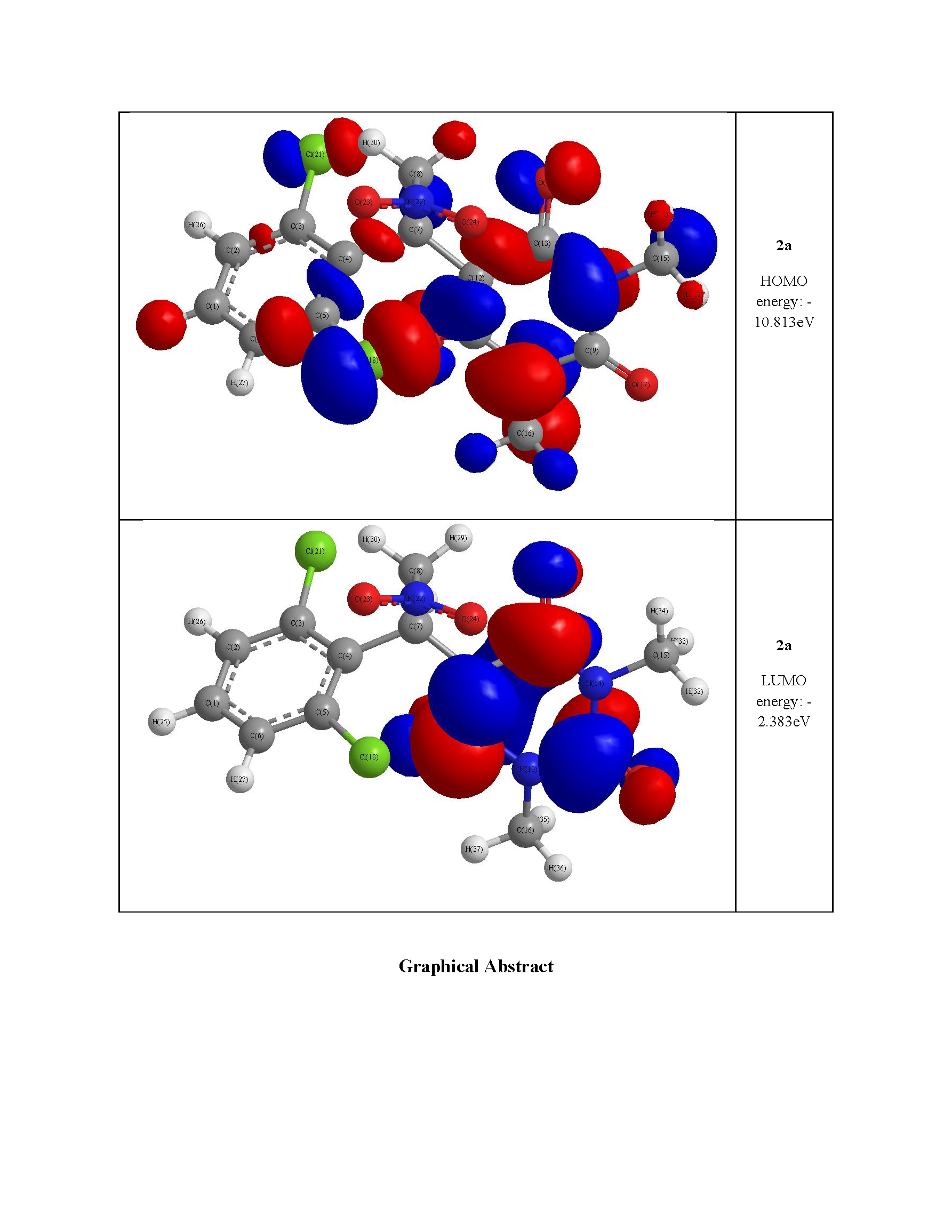
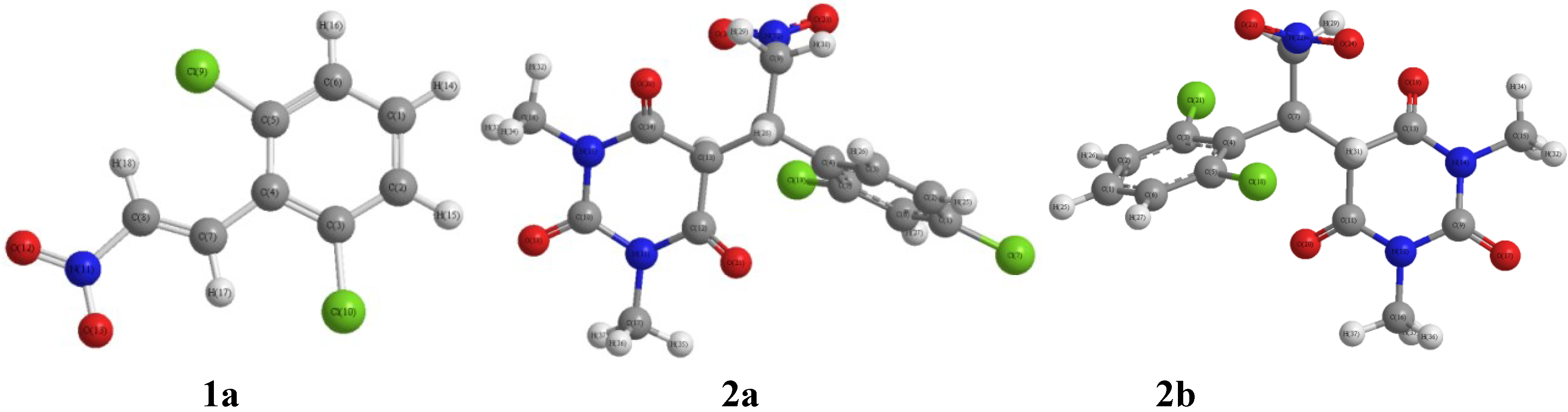
2.3. Optimized Molecular Geometry
| Parameter | 1a | 2a | 2b |
|---|---|---|---|
| Heat of Formation (kcal/mol) | 22.075 | −100.903 | −99.472 |
| Total Energy (kcal/mol) | −896424.216 | −1244859.443 | −102495.469 |
| Dipole (Debye) | 8.281 | 4.116 | 3.848 |
| HOMO energy (eV) | −10.019 | −10.575 | −10.813 |
| LUMO energy (eV) | −4.320 | −2.278 | −2.383 |
| HOMO–LUMO energy gap (eV) | 5.699 | 8.297 | 8.430 |

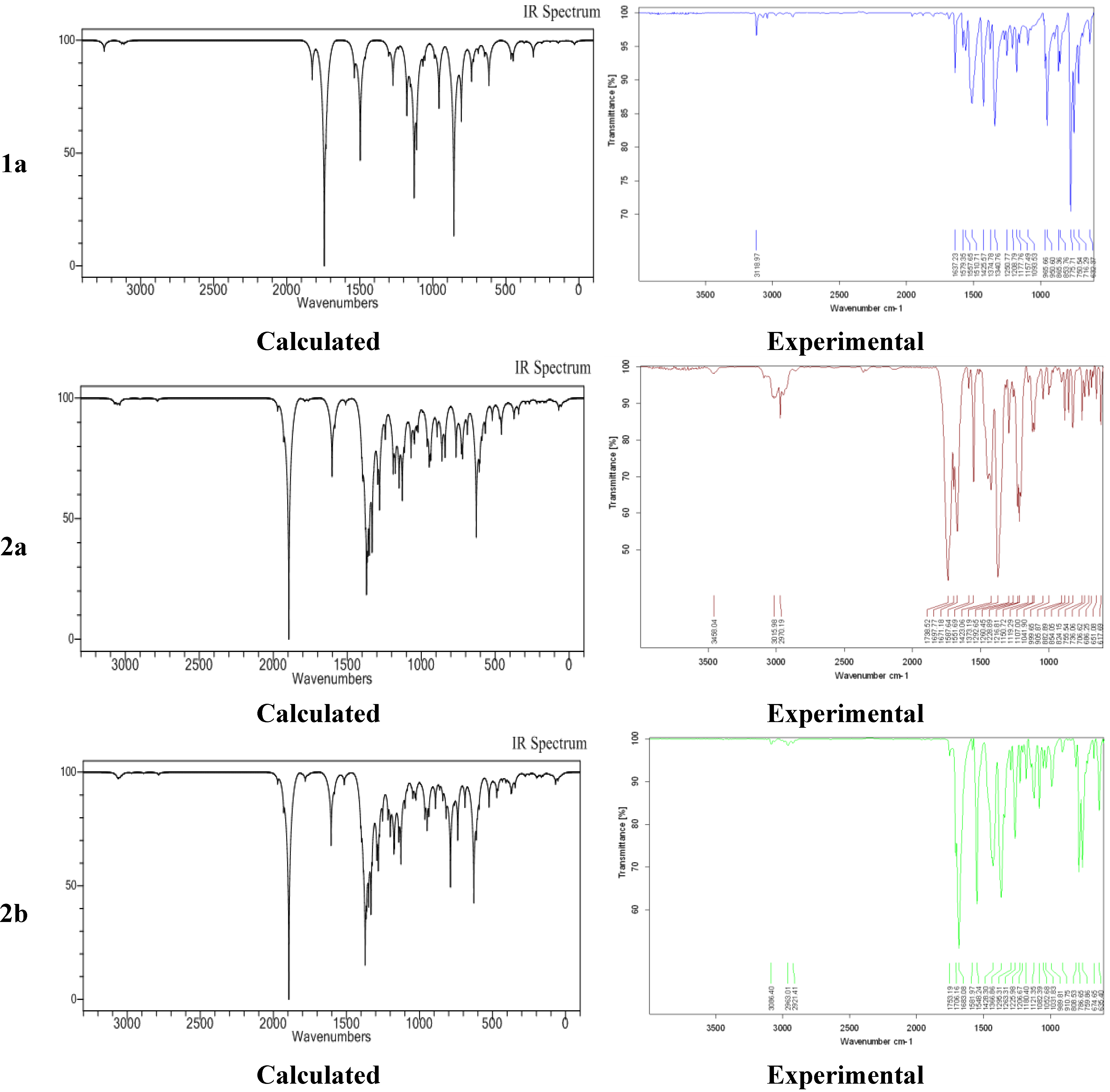
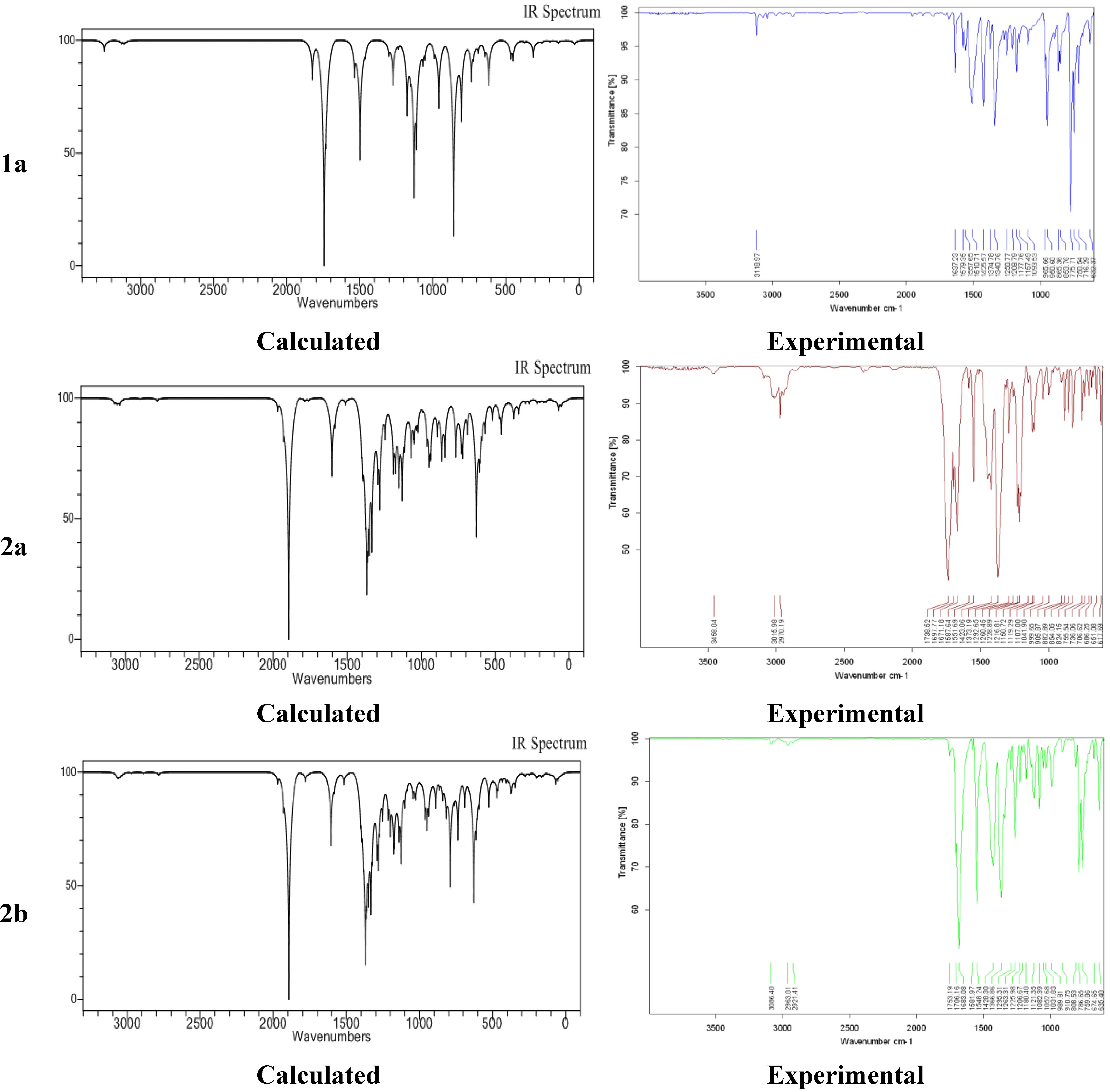
2.4. Experimental and Calculated IR Vibrations
2.5. Geometric Parameters
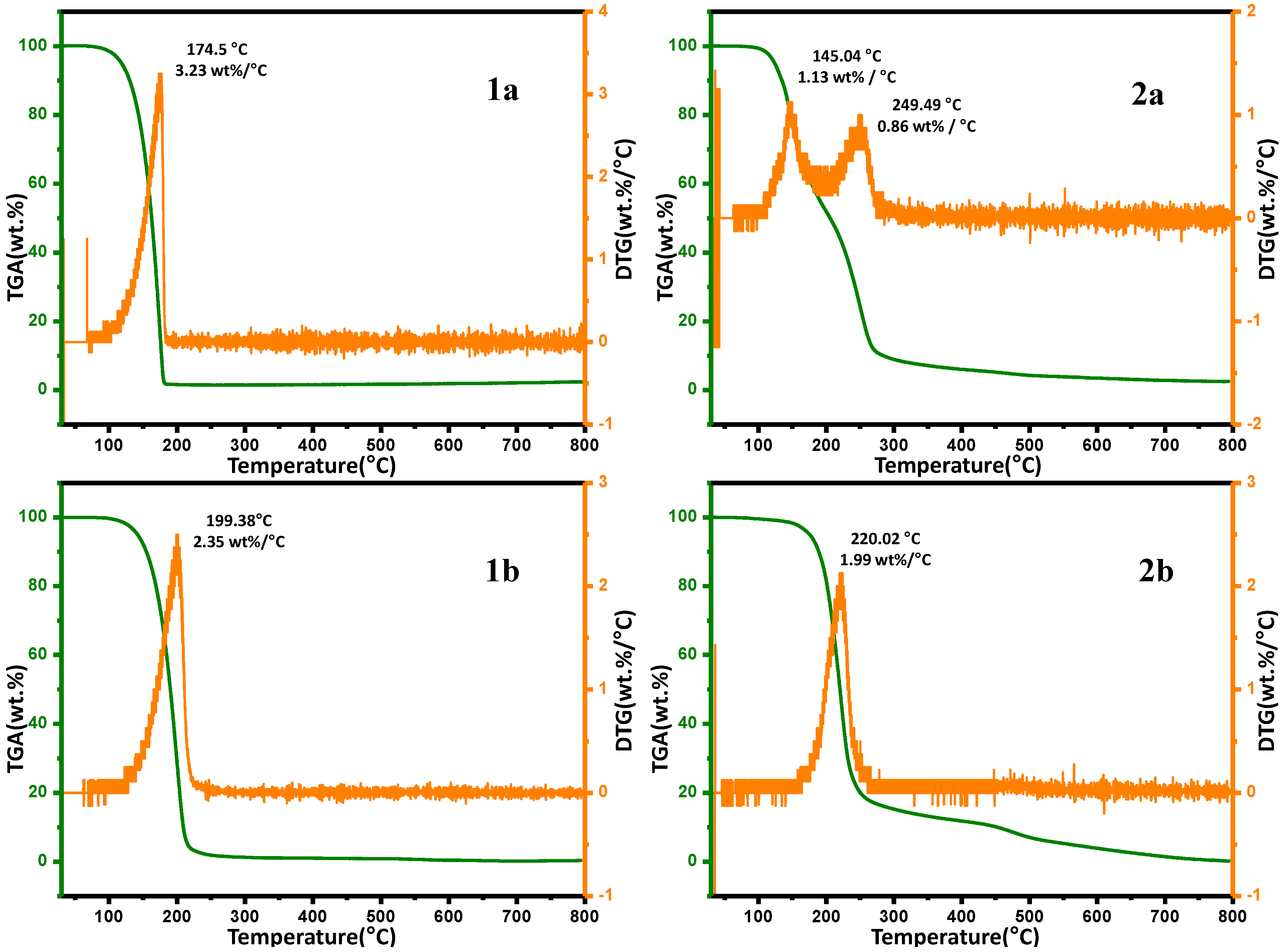
2.6. Thermal Gravimetric Analysis (TGA)
| Temperature | Weight Loss (%) | |||
|---|---|---|---|---|
| 1a | 2a | 1b | 2b | |
| 100 | 0.33 | 0.63 | 1.42 | 0.44 |
| 200 | 70.85 | 48.06 | 98.37 | 18.45 |
| 300 | 98.72 | 91.07 | 98.46 | 84.73 |
| 400 | 98.98 | 93.95 | 98.40 | 88.19 |
| 500 | 99.11 | 95.67 | 98.29 | 92.93 |
| 600 | 99.56 | 96.47 | 98.08 | 96.10 |
| 700 | 99.77 | 97.14 | 97.87 | 98.52 |
| 800 | 99.66 | 97.47 | 97.55 | 99.81 |
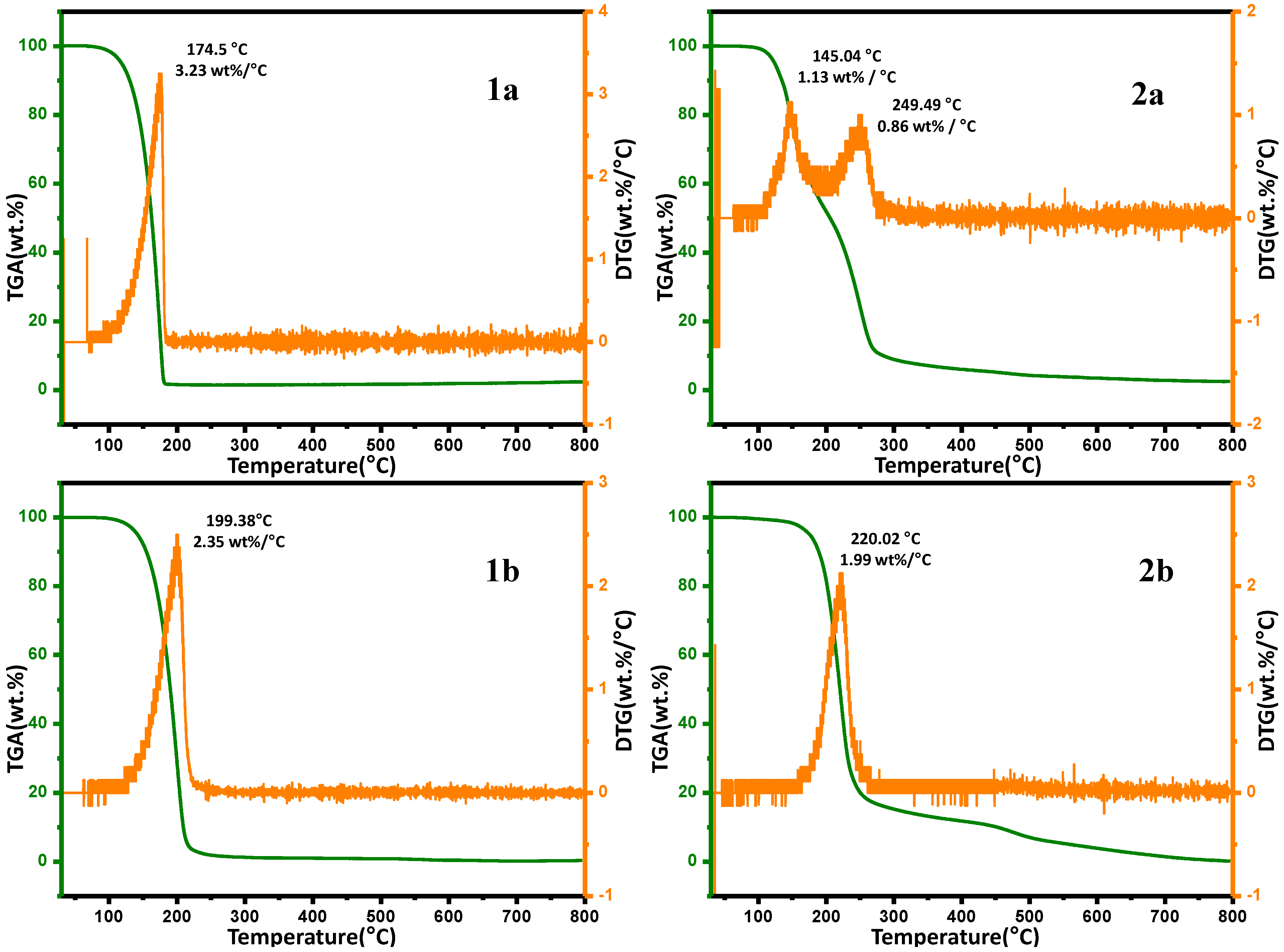
3. Experimental Section
3.1. General
3.2. 5-(1-(2,6-Dichlorophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (2a)
3.3. 5-(1-(2,4-Dichlorophenyl)-2-nitroethyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (2b)
3.4. DFT Calculations
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rastogi, V.K.; Mittal, H.P.; Sharma, Y.C.; Sharma, S.N. Spectroscopy of Biological Molecules; Hester, R.E., Girling, R.B., Eds.; Royal Society of Chemistry: London, UK, 1991; pp. 403–404. [Google Scholar]
- Bandekar, J.; Zundel, G. Normal coordinate analysis treatment on uracil in solid state. Spectrochim. Acta A 1983, 39, 343–355. [Google Scholar]
- Aruna, S.; Shanmugam, G. Vibrational assignments of six-membered heterocyclic compounds: Normal vibrations of 6-amino uracil and 6-amino 2-thio uracil. Spectrochim. Acta A 1985, 41, 531–536. [Google Scholar] [CrossRef]
- Portalone, G.; Ballirano, P.; Maras, A. The crystal structure of 3-methyluracil from X-ray powder diffraction data. J. Mol. Struct. 2002, 608, 35–39. [Google Scholar] [CrossRef]
- Grosmaire, L.; Delabre, J.L. Vibrational spectra of 6-methyluracil, 6-methyl-2-thiouracil and their deuterated analogues. J. Mol. Struct. 2012, 1011, 42–49. [Google Scholar] [CrossRef]
- Yaniv, M.; Folk, W.R. The nucleotide sequences of the two glutamine transfer ribonucleic acids from Escherichia coli. J. Biol. Chem. 1975, 250, 3243–3253. [Google Scholar] [PubMed]
- Amir, M.; Javed, S.A.; Kumar, H. Pyrimidine as antiinflammatory agent: A review. Indian J. Pharm. Sci. 2007, 69, 337–343. [Google Scholar] [CrossRef]
- Çırak, Ç.; Sert, Y.; Ucun, F. Experimental and computational study on molecular structure and vibrational analysis of a modified biomolecule: 5-Bromo-2'-deoxyuridine. Spectrochim. Acta A 2012, 92, 406–414. [Google Scholar] [CrossRef]
- Demirbaş, N.; Ug̵urluog̵lu, R.; Demirbaş, A. Synthesis of 3-alkyl(aryl)-4-alkylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-ones and 3-alkyl-4-alkylamino-4,5-dihydro-1H-1,2,4-triazol-5-ones as antitumor agents. Bioorg. Med. Chem. 2002, 10, 3717–3723. [Google Scholar]
- Elinson, M.N.; Vereshchagin, A.N.; Stepanov, N.O.; Zaimovskaya, T.A.; Merkulova, V.M.; Nikishin, G.I. The first example of the cascade assembly of a spirocyclopropane structure: Direct transformation of benzylidenemalononitriles and N,N-dialkylbarbituric acids into substituted 2-aryl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitriles. Tetrahedron Lett. 2010, 51, 428–431. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Javaid, S.; Yousuf, S.; Choudhary, M.I. Zwitterionic pyrimidinium adducts as antioxidants with therapeutic potential as nitric oxide scavenger. Eur. J. Med. Chem. 2014, 84, 146–154. [Google Scholar] [CrossRef]
- Ray, R.; Krishna, S.H.; Sharma, J.D.; Limaye, S.N. Variations in the Physico-Chemical Parameters of Some 5a and 5b Substituted Barbiturate Derivatives. Asian J. Chem. 2007, 19, 2497–2501. [Google Scholar]
- Brunton, L.L.; Lazo, J.S.; Lazo, P.; Keith, L. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th ed.; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Fillaut, J.L.; de Los Rios, I.; Masi, D.; Romerosa, A.; Zanobini, F.; Peruzzini, M. Synthesis and Structural Characterization of (Carbene)ruthenium Complexes Binding Nucleobases. Eur. J. Inorg. Chem. 2002, 935–942. [Google Scholar]
- Temiz-Arpaci, O.; Ozdemir, A.; Yalcin, I.; Yildiz, I.; Aki-Sener, E.; Altanlar, N. Synthesis and antimicrobial activity of some 5-[2-(morpholin-4-yl)acetamido] and/or 5-[2-(4-substituted piperazin-1-yl)acetamido]-2-(p-substituted phenyl)benzoxazoles. Arch. Pharm. 2005, 338, 105–111. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Phenobarbital for the treatment of epilepsy in the 21st century: A critical review. Epilepsia 2004, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Al-Najjar, H.J.; Barakat, A.; Al-Majid, A.M.; Mabkhot, Y.N.; Weber, M.; Ghabbour, H.A.; Fun, H.-K. A greener and efficient approach to Michael addition of barbituric acid to nitroalkene in aqueous diethylamine mediu. Molecules 2014, 19, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Al-Majid, A.M.; Barakat, A.; Al-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H-K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013, 14, 23762–23773. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; Al-Ghamdi, A.M.; Mabkhot, Y.N.; Siddiqui, M.R.; Ghabbour, H.A.; Fun, H-K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to dimedone-barbituric acid derivatives. Chem. Cent. J. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H-K. An efficient and green procedure for synthesis of rhodanine derivatives by Aldol-thia-Michael protocol using aqueous diethylamine. RSC Adv. 2014, 4, 4909–4916. [Google Scholar] [CrossRef]
- Barakat, A.; Al Majid, A.M.A.; Islam, M.S.; Al-Othman, Z.A. Highly enantioselective Friedel-Crafts alkylations of indoles with α,β-unsaturated ketones under Cu(II)-simple oxazoline-imidazoline catalysts. Tetrahedron 2013, 69, 5185–5192. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Islam, M.S. Barakat; Elkahatany, N.; Yousuf, S.S.; Choudhary, M.I. Tandem Knoevenagel-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-Dimedone derivatives. Arab. J. Chem. 2014. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.A.; Al-Othman, Z.A.; Barakat, A. Highly Enantioselective Friedel-Crafts Alkylation of Indole with electron deficient trans-β-Nitroalkenes under simple Zn(II)-Oxazoline-Imidazoline Catalysts. Tetrahedron: Asymmetry 2014, 25, 245–251. [Google Scholar] [CrossRef]
- Bruker, APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
- Sample Availability: Samples of the compounds 1a,b and 2a,b are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakat, A.; Al-Najjar, H.J.; Al-Majid, A.M.; Adil, S.F.; Ali, M.; Masand, V.H.; Ghabbour, H.A.; Fun, H.-K. Synthesis, X-ray Diffraction, Thermogravimetric and DFT Analyses of Pyrimidine Derivatives. Molecules 2014, 19, 17187-17201. https://doi.org/10.3390/molecules191117187
Barakat A, Al-Najjar HJ, Al-Majid AM, Adil SF, Ali M, Masand VH, Ghabbour HA, Fun H-K. Synthesis, X-ray Diffraction, Thermogravimetric and DFT Analyses of Pyrimidine Derivatives. Molecules. 2014; 19(11):17187-17201. https://doi.org/10.3390/molecules191117187
Chicago/Turabian StyleBarakat, Assem, Hany J. Al-Najjar, Abdullah M. Al-Majid, Syed F. Adil, Mohamed Ali, Vijay H. Masand, Hazem A Ghabbour, and Hoong-Kun Fun. 2014. "Synthesis, X-ray Diffraction, Thermogravimetric and DFT Analyses of Pyrimidine Derivatives" Molecules 19, no. 11: 17187-17201. https://doi.org/10.3390/molecules191117187