Characterization of Proanthocyanidins from Parkia biglobosa (Jacq.) G. Don. (Fabaceae) by Flow Injection Analysis — Electrospray Ionization Ion Trap Tandem Mass Spectrometry and Liquid Chromatography/Electrospray Ionization Mass Spectrometry


Abstract
:
1. Introduction
2. Results and Discussion
2.1. FIA-ESI-IT-MSn Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak No. | tR (min) | [M−H]−, m/z | MS2 | MS3 | Proposed Names |
|---|---|---|---|---|---|
| Catechins monomers, esterified-catechin-O-gallate and O-glucuronide monomers (*) | |||||
| 1 | 42.42 | 289 | 137 [M−H−152]− | (epi)catechin | |
| 2 | 24.98 | 305 | 179 [M−H−126]− | (epi)gallocatechin | |
| 3 | 43.33 | 167 [M−H−138]− | |||
| 137 [M−H−168]− | |||||
| 4 | 55.12 | 457 | 331 [M−H−126]− | (epi)gallocatechin-O-gallate | |
| 305 [M−H−152]− | |||||
| 169 [M−H−288]− | |||||
| 5 | 55.77 | 481 | 463 [M−H−18]− | (epi)gallocatechin-O-glucuronide | |
| 305 [M−H−176]− | |||||
| 287 [M−H−176−18]− | |||||
| 313 [M−H−168]− | |||||
| 6 | 55.74 | 617 | 599 [M−H−18]− | 289 [M−H−152−176]− | (epi)catechin-O-gallate-O-glucuronide |
| 465 [M−H−152]− | 271 [M−H−152−176−18]− | ||||
| 463 [M–H−170]− | |||||
| 7 | 55.40 | 633 | 507 [M−H−126]− | 463 [M−H−152−18]− | (epi)gallocatechin-O-gallate-O-glucuronide |
| 481 [M−H−152]− | 305 [M−H−152−176]− | ||||
| 287 [M−H−152−176−18]− | |||||
| Catechins dimers (*) | |||||
| 8 | 36.68 | 577 | 559 [M−H−18]− | (epi)catechin-(epi)catechin | |
| 9 | 46.27 | 451 [M−H−126]− | |||
| 10 | 48.40 | 425 [M−H−152]− | |||
| 11 | 49.89 | 407 [M−H−152−18]− | |||
| 289 [M−H−288]− | |||||
| 12 | 33.21 | 593 | 467 [M−H−126]− | (epi)catechin-(epi)gallocatechin | |
| 13 | 36.67 | 441 [M−H−152]− | |||
| 14 | 39.55 | 289 [M−H−304]− | |||
| 15 | 13.57 | 609 | 483 [M−H−126]− | (epi)gallocatechin-(epi)gallocatechin | |
| 16 | 22.46 | 441 [M−H−168]− | |||
| 305 [M−H−304]− | |||||
| 303 [M−H−306]− | |||||
| Esterified catechin-O-gallate and O-glucuronide dimers – Group I (*) | |||||
| 17 | 50.55 | 729 | 577 [M−H−152]− | (epi)catechin-(epi)catechin-O-gallate | |
| 18 | 54.93 | 559 [M−H−170]− | |||
| 289 [M−H−440]− | |||||
| 287 [M−H−442]− | |||||
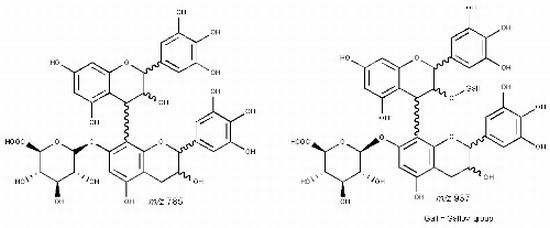
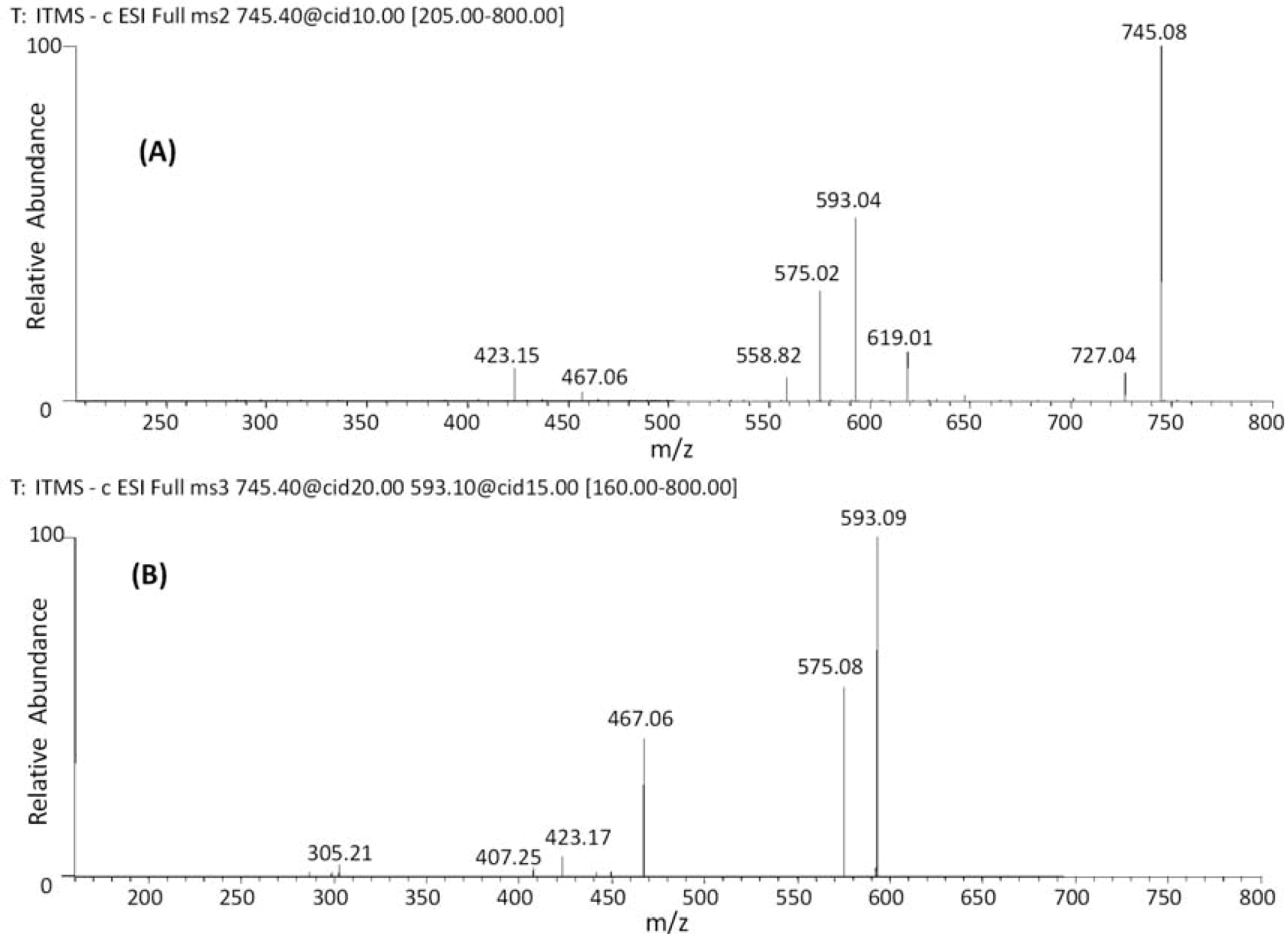
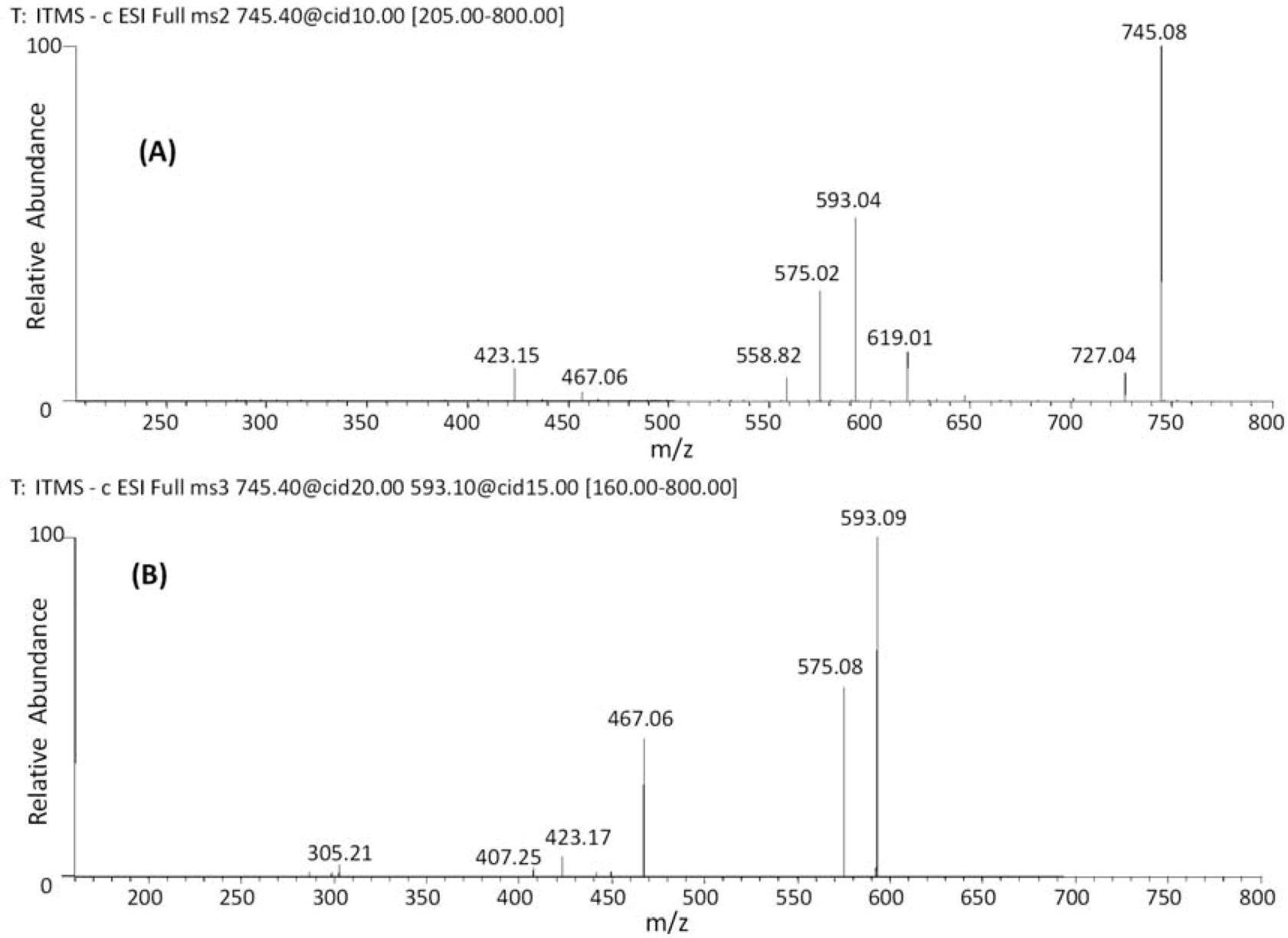
| 19 | 38.09 | 745 | 619 [M−H−126]− | 467 [M–H−152−126]− | (epi)gallocatechin-(epi)catechin-O-gallate |
| 20 | 42.68 | 593 [M−H−152]− | 407 [M–H−152−168−18]− | ||
| 21 | 47.44 | 575 [M−H−152−18]− | |||
| 22 | 49.89 | ||||
| 23 | 29.43 | 761 | 635 [M–H−126]− | 591 [M−H−152−18]− | (epi)gallocatechin-(epi)gallocatechin-O-gallate |
| 24 | 31.65 | 609 [M–H−152]− | 483 [M−H−152−126]− | ||
| 25 | 36.87 | 593 [M–H−168]− | 305 [M−H−152−304]− | ||
| 26 | 38.13 | ||||
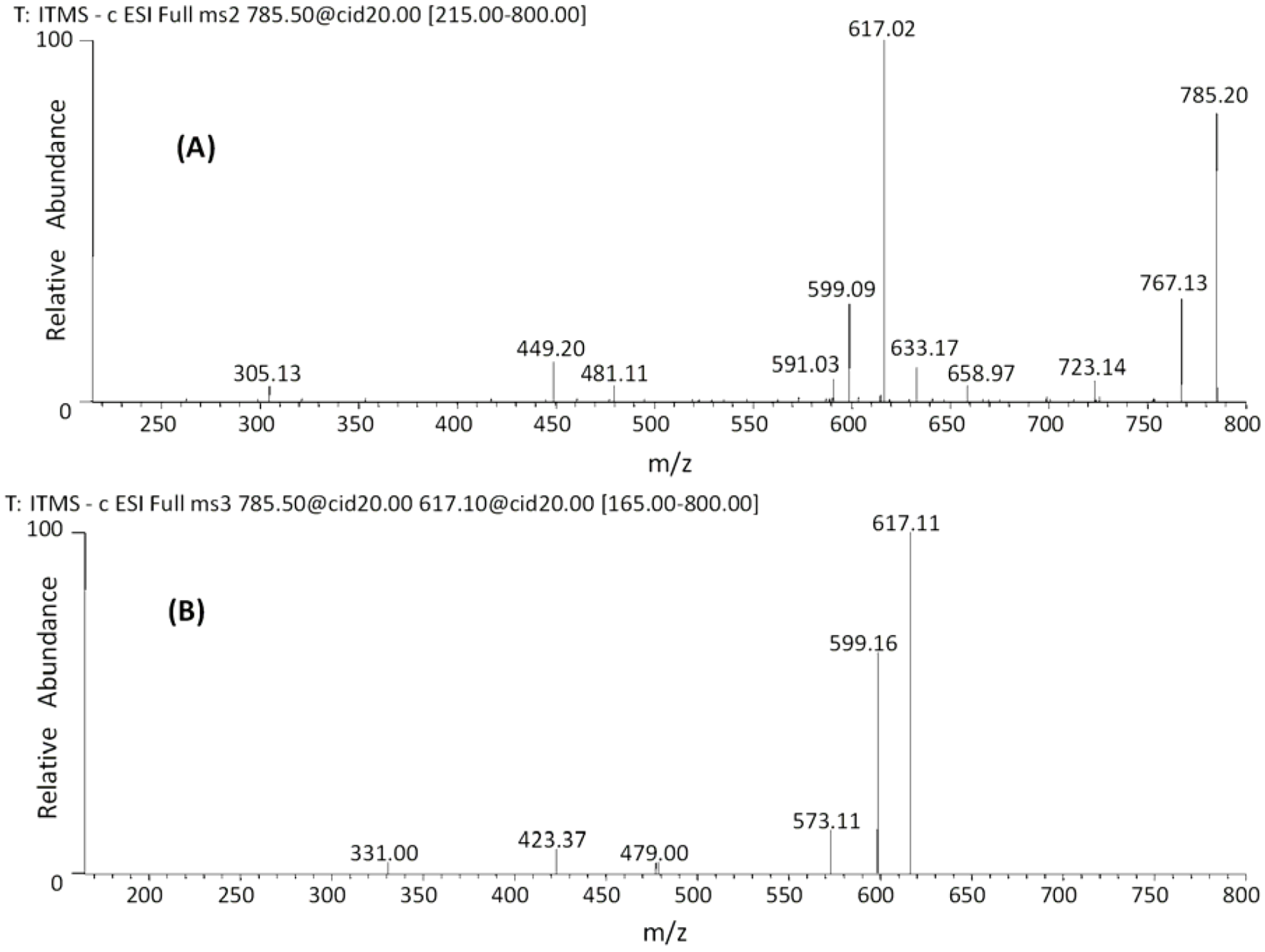
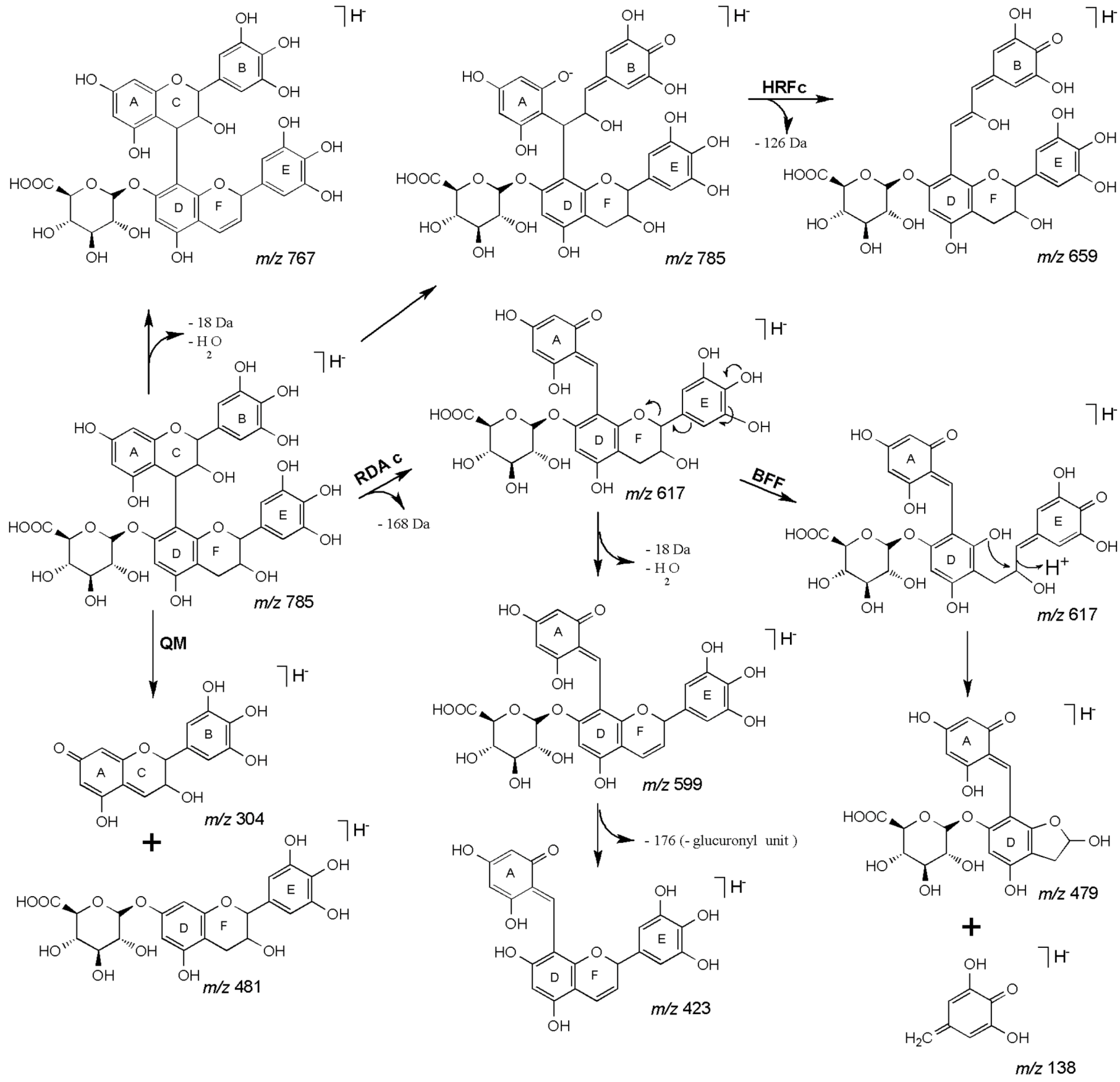
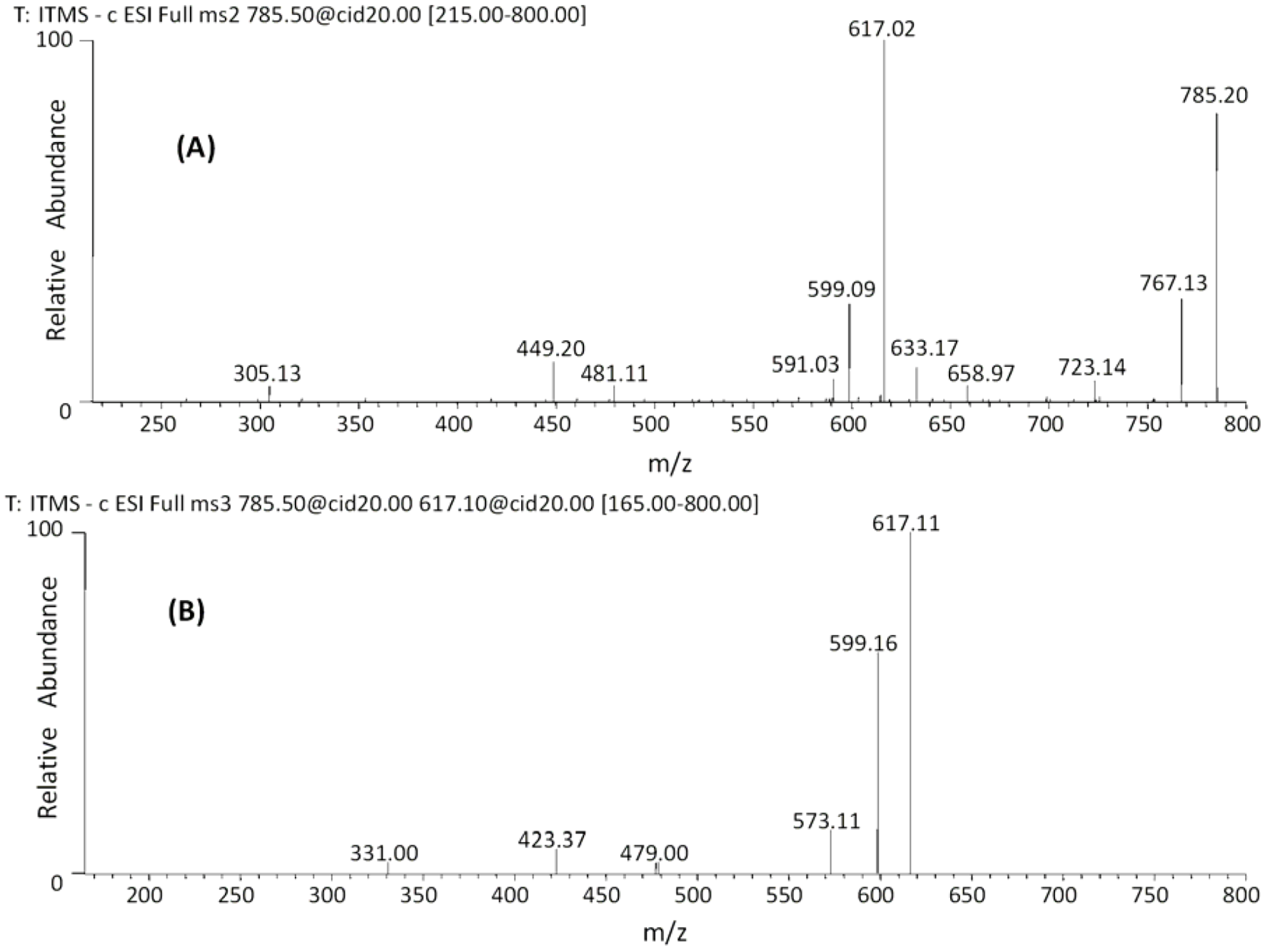
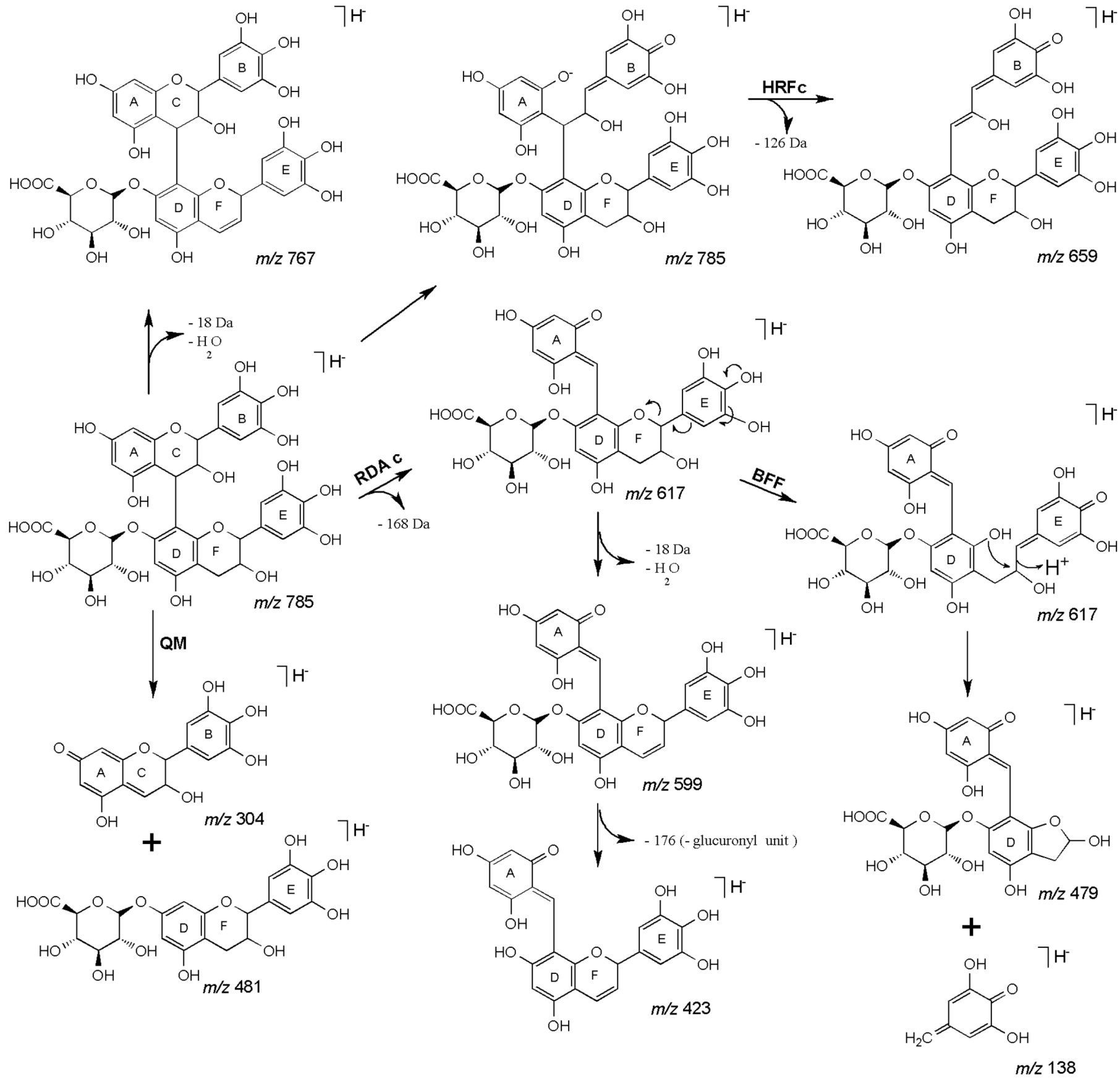
| 27 | 45.18 | 785 | 659 [M−H−126]− | 479 [M−H−168−138]− | (epi)gallocatechin-(epi)gallocatechin-O-glucuronide |
| 28 | 47.48 | 617 [M−H−168]− | 423 [M−H−168−176−18]− | ||
| 591 [M−H−176−18]− | 599 [M−H−168−18]− | ||||
| 481 [M−H−304]− | |||||
| Esterified catechin-O-gallate and O-glucuronide dimers – Group II (*) | |||||
| 29 | 41.69 | 881 | 729 [M−H−152]− | (epi)catechin-O-gallate-(epi)catechin-O-gallate | |
| 30 | 54.71 | 577 [M−H−304]− | or | ||
| 31 | 55.38 | (epi)catechin-O-gallate–(epi)gallocatechin | |||
| 32 | 54.93 | 897 | 745 [M−H−152]− | (epi)catechin-O-gallate-(epi)gallocatechin-O-gallate | |
| 33 | 55.28 | 727 [M−H−170]− | |||
| 34 | 49.44 | 913 | 761 [M–H−152]− | (epi)gallocatechin-O-gallate-(epi)gallocatechin-O-gallate | |
| 35 | 50.62 | 787 [M−H−126]− | |||
| 743 [M−H−170]− | |||||
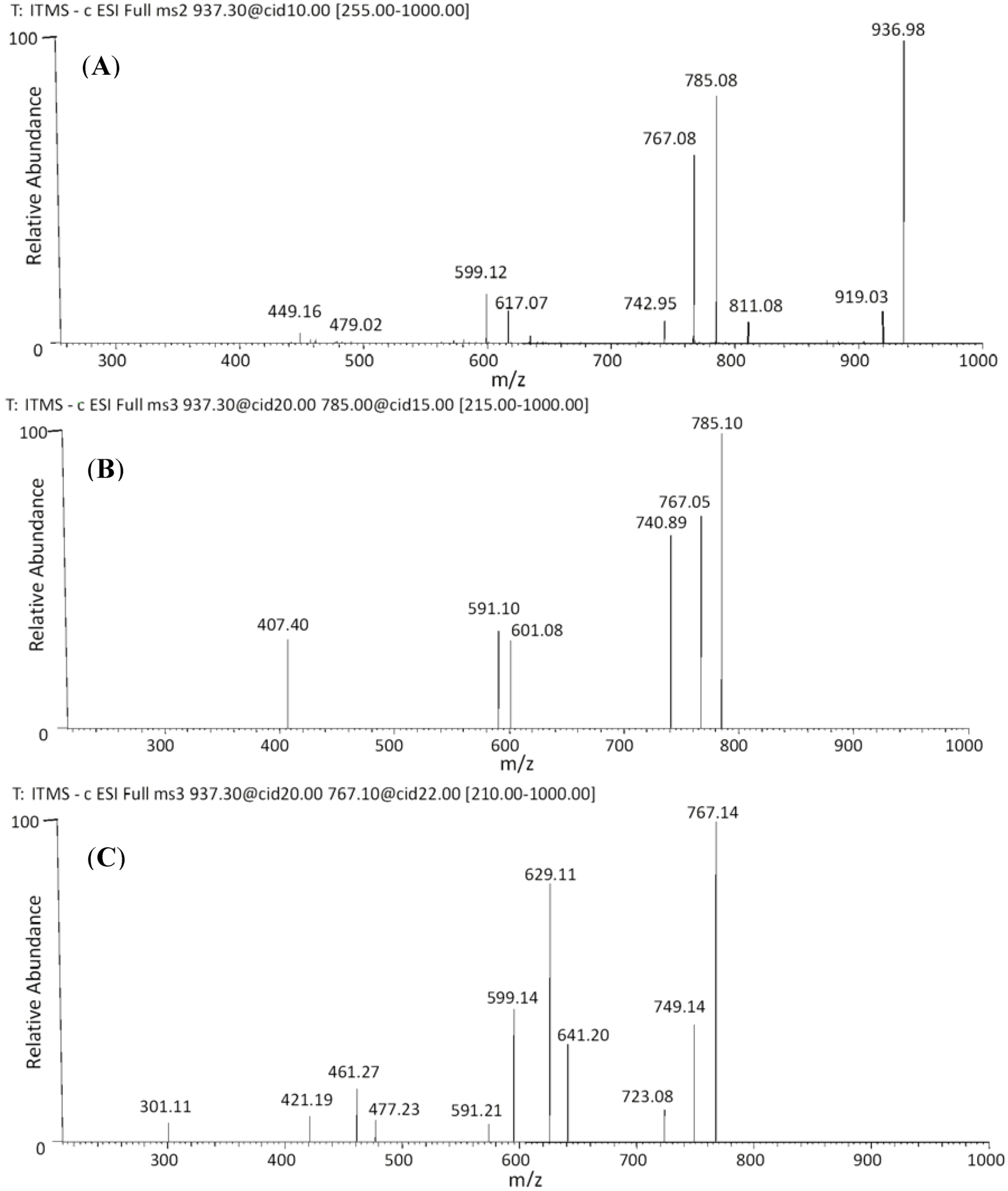
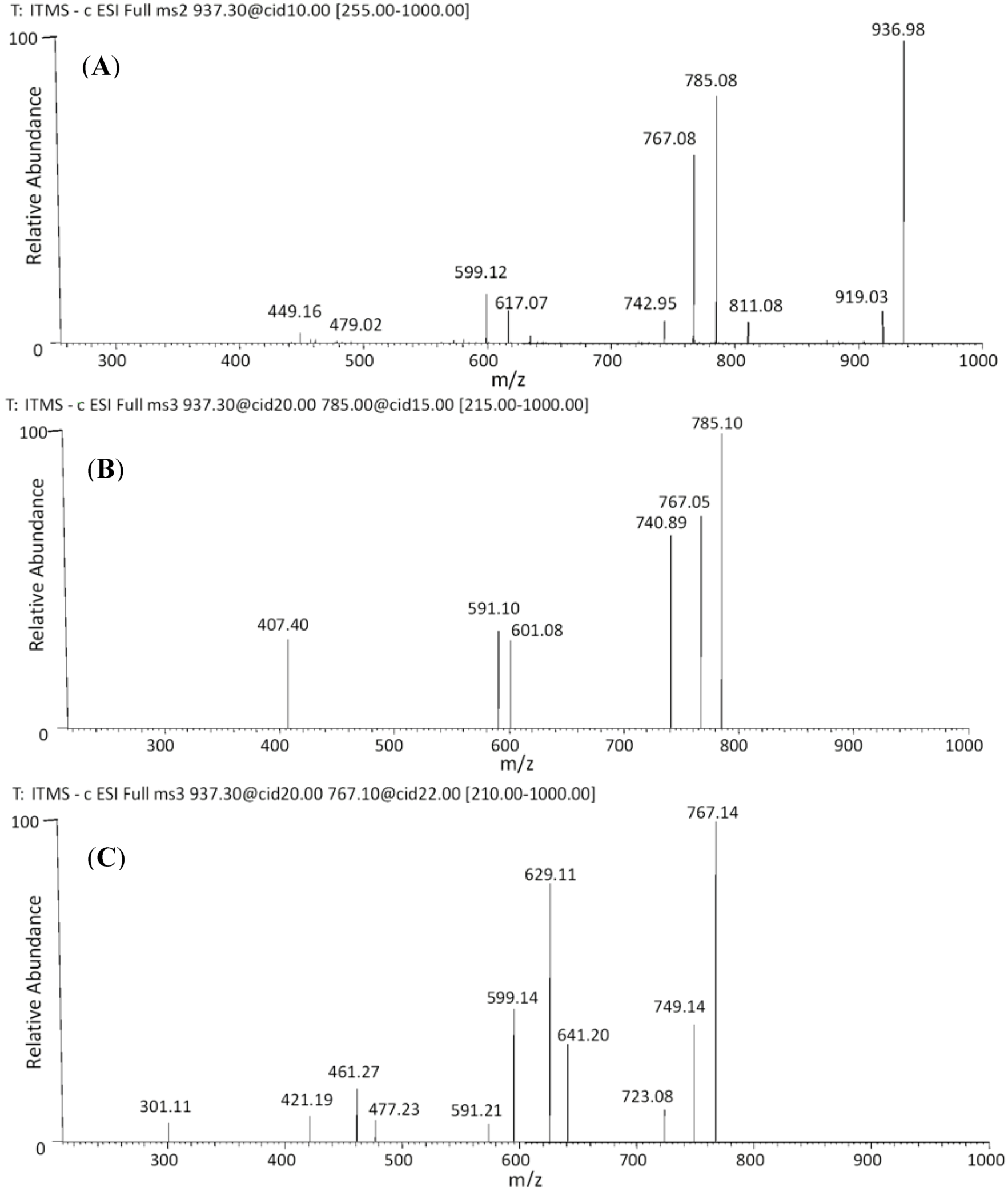
| 36 | 54.88 | 937 | 919 [M−H−18]− | 641 [M−H−170−126]− | (epi)gallocatechin-O-gallate-(epi)gallocatechin-O-glucuronide |
| 37 | 55.27 | 811 [M−H−126]− | 629 [M−H−170−138]− | ||
| 785 [M−H−152]− | 591 [M−H−152−176−18]− | ||||
| 767 [M−H−170]− | 599 [M−H−176−168]− | ||||
| Catechin trimer (*) | |||||
| 38 | 55.54 | 865 | 739 [M−H−126]− | (epi)catechin-(epi)catechin-(epi)catechin | |
| 713 [M−H−152]− | |||||
| 695 [M−H−152−18]− | |||||
| 577 [M−H−288]− | |||||
| 425 [M−H−288−152]− | |||||
| Esterified catechin-O-gallate and O-glucuronide trimers (#) | |||||
| 39 | 54.95 | 1017 | n.a. | n.a. | (epi)catechin-(epi)catechin-(epi)catechin-O-gallate |
| 40 | 40.75 | 1049 | n.a. | n.a. | (epi)gallocatechin-(epi)gallocatechin-(epi)gallocatechin-O-gallate |
| 41 | 45.73 | ||||
| 42 | 54.86 | ||||
| 43 | 55.22 | ||||
| 44 | 46.55 | 1089 | n.a. | n.a. | (epi)gallocatechin-(epi)gallocatechin-(epi)gallocatechin-O-glucuronide |
| 45 | 55.52 | ||||
| Catechin tetramers (#) | |||||
| 46 | 55.25 | 1153 | n.a. | n.a. | (epi)catechin-(epi)catechin-(epi)catechin-(epi)catechin |
| 47 | 55.23 | 1169 | n.a. | n.a. | (epi)catechin-(epi)catechin-(epi)catechin-(epi)gallocatechin |
| 48 | 55.32 | 1185 | n.a. | n.a. | (epi)catechin-(epi)catechin-(epi)gallocatechin-(epi)gallocatechin |
| 49 | 54.83 | 1201 | n.a. | n.a. | (epi)catechin-(epi)gallocatechin-(epi)gallocatechin-(epi)gallocatechin |
| 50 | 54.90 | 1217 | n.a. | n.a. | (epi)gallocatechin-(epi)gallocatechin-(epi)gallocatechin-(epi)gallocatechin |
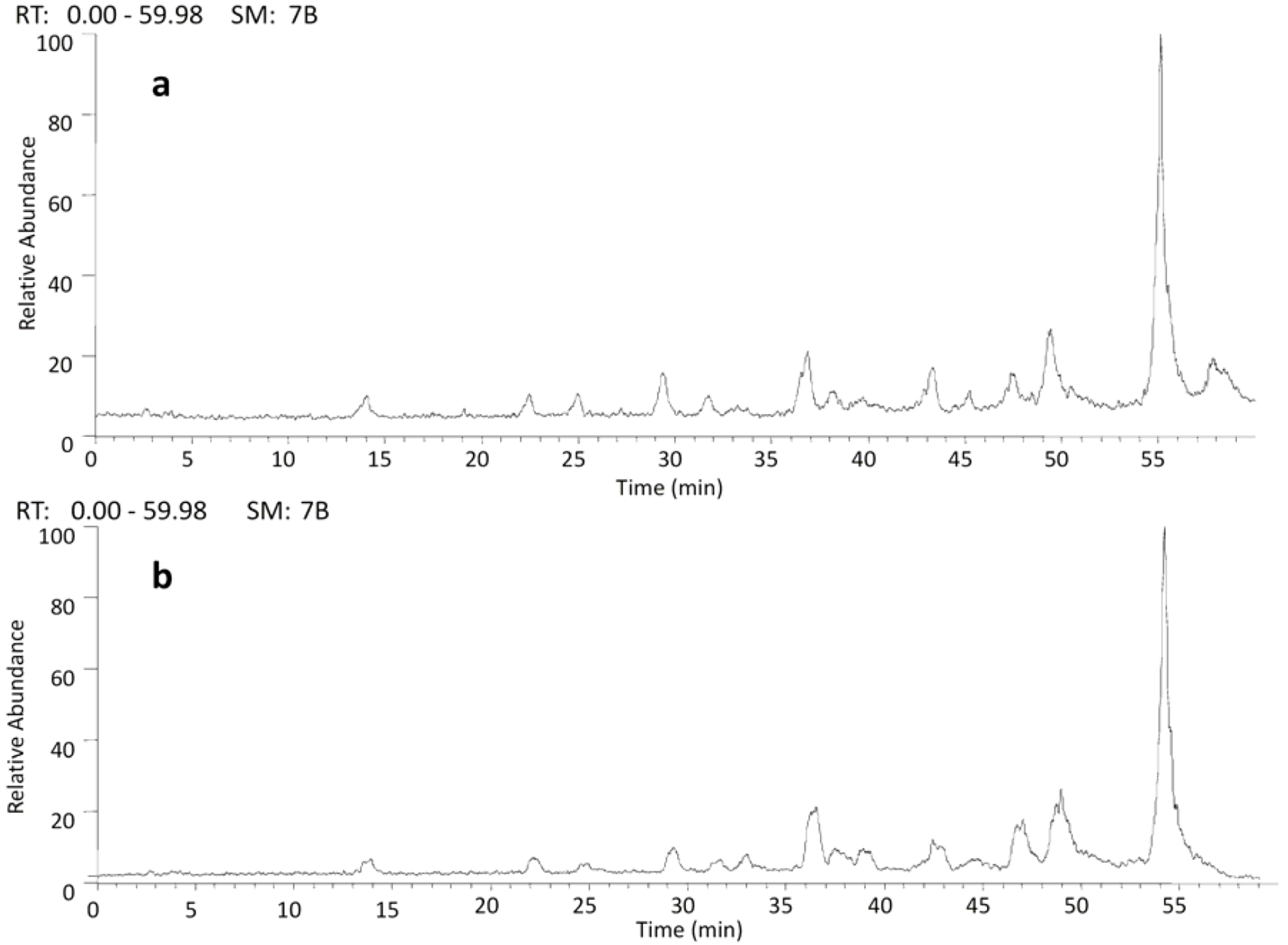
2.2. HPLC/ESI-IT-MS Analysis

3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Sample Preparation
3.4. Mass Spectrometric Analysis
3.4.1. FIA-ESI-IT-MSn
3.4.2. HPLC/ESI-IT-MS
3.5. Antiradical Activity Determination
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Burkill, H.M. The Useful Plants of West Tropical Africa, 2nd ed.; Royal Botanical Gardens: Kew, Richmond, UK, 1995; Volume 3, p. 857. [Google Scholar]
- Hopkins, H.C. The taxonomy, reproductive biology and economic potential of Parkia (Leguminosae: Mimosideae) in Africa and Madagascar. Bot. J. Linn. Soc. 1983, 87, 135–167. [Google Scholar] [CrossRef]
- Dalziel, J.M. The Useful Plants of West Africa; Crown Agents for Overseas Government and Administration: London, UK, 1937; p. 217. [Google Scholar]
- Odunfa, S.A. Dawadawa. In Legume Based Fermented Foods; Reddy, N.R., Pierson, M.D., Salunkhe, D.K., Eds.; CRC Press: New York, NY, USA, 1989; pp. 173–189. [Google Scholar]
- Gan, C.-Y.; Latiff, A.A. Antioxidant Parkia speciosa pod powder as potential functional flour in food application: Physicochemical properties characterization. Food Hydrocolloid. 2011, 25, 1174–1180. [Google Scholar] [CrossRef]
- Ouoba, L.I.I.; Diawara, B.; Jespersen, L.; Jakobsen, M. Antimicrobial activity of Bacillus subtilis and Bacillus pumilus during the fermentation of African locust bean (Parkia biglobosa) for Soumbala production. J. Appl. Microbiol. 2007, 102, 963–970. [Google Scholar]
- Adetutu, A.; Morgan, W.A.; Corcoran, O.; Chimezie, F. Antibacterial activity and in vitro cytotoxicity of extracts and fractions of Parkia biglobosa (Jacq.) Benth. stem bark and Ageratum conyzoides Linn. leaves. Environ. Toxicol. Pharmacol. 2012, 34, 478–483. [Google Scholar]
- Fernandes, H.B.; Silva, F.V.; Passos, F.F.B.; Bezerra, R.D.S.; Chaves, M.H.; Oliveira, F.A.; Oliveira, R.C.M. Gastroprotective effect of the ethanolic extract of Parkia platycephala Benth. leaves against acute gastric lesion models in rodents. Biol. Res. 2010, 43, 451–457. [Google Scholar]
- Assane, M.; Baba, M.R.; Bassene, E.; Sere, A. Antihypertensive action of Parkia biglobosa+ (Jacq) Benth seeds in rat. Darkar Med. 1993, 38, 49–54. [Google Scholar]
- Araujo, M.E.M.; Palma, F.M.S.B.; Moutinho, A.T.; Abreu, P.M. Constituents of Parkia biglobosa. Fitoterapia 1995, 66, 468–469. [Google Scholar]
- Tringali, C.; Spatafora, C.; Longo, O.D. Bioactive constituents of the bark of Parkia biglobosa. Fitoterapia 2000, 71, 118–125. [Google Scholar] [CrossRef]
- Aiyelaagbe, O.O.; Ajaiyeoba, E.O.; Ekundayo, O. Studies on the seed oils of Parkia biglobosa and Parkia bicolor. Plant Foods Hum. Nutr. 1996, 49, 229–233. [Google Scholar] [CrossRef]
- Biswanath, D.; Bikas, C.M.; Sudhan, D.; Biplab, G.; Shiho, A.; Noriko, S.; Yoshihiro, H. Iridoid glucosides from leaves and stem barks of Parkia javanica. J. Asian Nat. Prod. Res. 2009, 11, 229–235. [Google Scholar] [CrossRef]
- Adewoye, R.O.; Ajayi, O.O. Flavonols, flavones and tannins of Parkia clappetoniana. J. Am. Leather Chem. Assoc. 1988, 83, 153–156. [Google Scholar]
- Adewoye, R.O.; Ajayi, O.O. Anthocyanidins of Parkia clappertoniana. J. Soc. Leather Tech. Chem. 1989, 73, 110–121. [Google Scholar]
- Lemmich, E.; Adewunmi, C.O.; Furu, P.; Kristensen, A.; Larsen, L.; Olsen, C.E. 5-Deoxyflavones from Parkia clappertoniana. Phytochemistry 1996, 42, 1011–1013. [Google Scholar] [CrossRef]
- Khanbabaee, K.; van Ree, T. Tannins: Classification and definition. Nat. Prod. Rep. 2001, 18, 641–649. [Google Scholar] [CrossRef]
- Hemingway, R.W.; Karchesy, J.J. Chemistry and Significance of Condensed Tannins; Plenum: New York, NY, USA, 1989. [Google Scholar]
- Rodrigues, C.M.; Rinaldo, D.; dos Santos, L.C.; Montoro, P.; Piacente, S.; Pizza, C.; Hiruma-Lima, C.A.; Souza-Brito, A.R.M.; Vilegas, W. Metabolic fingerprinting using direct flow injection electrospray ionization tandem mass spectrometry for the characterization of proanthocyanidins from the barks of Hancornia speciosa. Rapid Commun. Mass Spectrom. 2007, 21, 1907–1914. [Google Scholar] [CrossRef]
- Porter, L.J. Tannins. In Methods in Plant Biochemistry; Dey, P.M., Harborne, J.B., Eds.; Academic Press: San Diego, CA, USA, 1989; Volume I, pp. 389–420. [Google Scholar]
- Karonen, M.; Loponen, J.; Ossipov, V.; Pihlaja, K. Analysis of procyanidins in pine bark with reversed-phase and normal-phase high-performance liquid chromatography-electrospray ionization mass spectrometry. Anal. Chim. Acta 2004, 522, 105–112. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Zhang, Z.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Liquid chromatographic/electrospray ionization mass spectrometric studies of proanthocyanidins in foods. J. Mass Spectrom. 2003, 38, 1272–1280. [Google Scholar] [CrossRef]
- De Bruyne, T.; Pieters, L.; Deelstra, H.; Vlietinck, A. Condensed vegetable tannins: Biodiversity in structure and biological activities. Biochem. Syst. Ecol. 1999, 27, 445–459. [Google Scholar] [CrossRef]
- Maldonado, P.D.; Rivero-Cruz, I.; Mata, R.; Pedraza-Chaverr, J. Antioxidant activity of A-type proanthocyanidins from Geranium niveum (Geraniaceae). J. Agric. Food Chem. 2005, 53, 1996–2001. [Google Scholar]
- Park, Y.S.; Jeon, M.H.; Hwang, H.J.; Park, M.R.; Lee, S.-H.; Kim, S.G.; Kim, M. Antioxidant activity and analysis of proanthocyanidins from pine (Pinus densiflora) needles. Nutr. Res. Pract. 2011, 5, 281–287. [Google Scholar] [CrossRef]
- Srivastava, R.C.; Husain, M.M.; Hasan, S.K.; Athar, M. Green tea polyphenols and tannic acid act as potent inhibitors of phorbol ester-induced nitric oxide generation in rat hepatocytes independent of their antioxidant properties. Cancer Lett. 2000, 153, 1–5. [Google Scholar] [CrossRef]
- Hiruma-Lima, C.A.; Rodrigues, C.M.; Kushima, H.; Moraes, T.M.; Lolis, S.D.F.; Feitosa, S.B.; Magri, L.P.; Soares, F.R.; Cola, M.M.; Andrade, F.D.P.; et al. The anti-ulcerogenic effects of Curatella americana L. J. Ethnopharmacol. 2009, 121, 425–432. [Google Scholar] [CrossRef]
- Calzada, F.; Cedillo-Rivera, R.; Bye, R.; Mata, R. Geranins C and D, additional new antiprotozoal A-type proanthocyanidins from Geranium niveum. Planta Med. 2001, 67, 677–680. [Google Scholar] [CrossRef]
- Heijmen, F.H.; Du-Pont, J.S.; Middelkoop, E.; Kreis, R.W.; Hoekstra, M. Cross-linking of dermal sheep collagen with tannic acid. J. Biomater. 1997, 18, 749–754. [Google Scholar] [CrossRef]
- Hou, D.-X.; Masuzaki, S.; Hashimoto, F.; Uto, T.; Tanigawa, S.; Fujii, M.; Sakata, Y. Green tea proanthocyanidins inhibit cyclooxygenase-2 expression in LPS-activated mouse macrophages: Molecular mechanisms and structure–activity relationship. Arch. Biochem. Biophys. 2007, 460, 67–74. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Honda, M.; Ikigai, H.; Hara, Y.; Shimamura, T. Inhibitory effects of (−)-Epigallocatechin gallate on the life cycle of human immunodeficiency virus type 1 (HIV-1). Antiviral Res. 2002, 53, 19–34. [Google Scholar] [CrossRef]
- Nakai, M.; Fukui, Y.; Asami, S.; Toyoda-Ono, Y.; Iwashita, T.; Shibata, H.; Mitsunaga, T.; Hashimoto, F.; Kiso, Y. Inhibitory effects of oolong tea polyphenols on pancreatic lipase in vitro. J. Agric. Food Chem. 2005, 53, 4593–4598. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 1–8. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Z.; Lei, Z.; Yang, Y.; Sugiura, N. Comparative study of antioxidant activity and antiproliferative effect of hot water and ethanol extracts from the mushroom Inonotus obliquus. J. Biosci. Bioeng. 2009, 107, 42–48. [Google Scholar]
- Fulcrand, H.; Mané, C.; Preys, S.; Mazerolles, G.; Bouchut, C.; Mazauric, J.P.; Souquet, J.M.; Meudec, E.; Li, Y.; Cole, R.B.; et al. Direct mass spectrometry approaches to characterize polyphenol composition of complex samples. Phytochemistry 2008, 69, 3131–3138. [Google Scholar] [CrossRef]
- Sannomiya, M.; Montoro, P.; Piacente, S.; Pizza, C.; Brito, A.R.M.S.; Vilegas, W. Application of liquid chromatography/electrospray ionization tandem mass spectrometry to the analysis of polyphenolic compounds from an infusion of Byrsonima crassa Niedenzu. Rapid Commun. Mass Spectrom. 2005, 19, 2244–2250. [Google Scholar]
- Friedrich, W.; Eberhardt, A.; Galensa, R. Investigation of proanthocyanidins by HPLC with electrospray ionization mass spectrometry. Eur. Food Res. Technol. 2000, 211, 56–64. [Google Scholar] [CrossRef]
- Li, H.J.; Deinzer, M.L. Tandem mass spectrometry for sequencing proanthocyanidins. Anal. Chem. 2007, 79, 1739–1748. [Google Scholar] [CrossRef]
- Stalmach, A.; Troufflard, S.; Serafini, M.; Crozier, A. Absorption, metabolism and excretion of Choladi green tea flavan-3-ols by humans. Mol. Nutr. Food Res. 2009, 53, S44–S53. [Google Scholar] [CrossRef]
- Shrestha, S.P.; Thompson, J.A.; Wempe, M.F.; Gu, M.; Agarwal, R.; Agarwal, C. Glucuronidation and methylation of procyanidin dimers b2 and 3,3″-Di-O-Galloyl-b2 and corresponding monomers epicatechin and 3-O-galloyl-epicatechin in mouse liver. Pharm Res. 2012, 29, 856–865. [Google Scholar] [CrossRef]
- Kalili, K.M.; de Villiers, A. Off-line comprehensive two-dimensional hydrophilic interaction x reversed phase liquid chromatographic analysis of green tea phenolics. J. Sep. Sci. 2010, 33, 853–863. [Google Scholar] [CrossRef]
- Shui, G.H.; Leong, L.P. Analysis of polyphenolic antioxidants in star fruit using high performance liquid chromatography and mass spectrometry. J. Chromatogr. A 2004, 1022, 67–75. [Google Scholar] [CrossRef]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach; Wiley: New York, NY, USA, 2002; p. 507. [Google Scholar]
- Lazarus, S.A.; Adamson, G.E.; Hammerstone, J.F.; Schmitz, H.H. High-performance liquid chromatography/mass spectrometry analysis of proanthocyanidins in foods and beverages. J. Agric. Food Chem. 1999, 47, 3693–3701. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Rinaldo, D.; Sannomiya, M.; Santos, L.C.; Montoro, P.; Piacente, S.; Pizza, C.; Vilegas, W. High-performance liquid chromatographic separation and identification of polyphenolic compounds from the infusion of Davilla elliptica St. Hill. Phytochem. Anal. 2008, 19, 17–24. [Google Scholar] [CrossRef]
- Monagas, M.; Quintanill-López, J.E.; Gómez-Cordovés, C.; Bartolomé, B.; Lebrón-Aguilar, R. MALDI-ToF MS analysis of plant proanthocyanidins. J. Pharm. Biomed. Anal. 2010, 51, 358–372. [Google Scholar] [CrossRef]
- Srinivasan, P.; Vadhanam, M.V.; Arif, J.M.; Gupta, R.C. A rapid screening assay for antioxidant potential of natural and synthetic agents in vitro. Int. J. Oncol. 2002, 20, 983–986. [Google Scholar]
- Barbosa-Pereira, L.; Angulo, I.; Paseiro-Losada, P.; Cruz, J.M. Phenolic profile and antioxidant properties of a crude extract obtained from a brewery waste stream. Food Res. Int. 2013, in press. [Google Scholar]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Lebensm. Wiss. Technol. 1995, 28, 25–30. [Google Scholar]
- Sample Availability: Samples of the extracts are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tala, V.R.S.; Candida da Silva, V.; Rodrigues, C.M.; Nkengfack, A.E.; Campaner dos Santos, L.; Vilegas, W. Characterization of Proanthocyanidins from Parkia biglobosa (Jacq.) G. Don. (Fabaceae) by Flow Injection Analysis — Electrospray Ionization Ion Trap Tandem Mass Spectrometry and Liquid Chromatography/Electrospray Ionization Mass Spectrometry. Molecules 2013, 18, 2803-2820. https://doi.org/10.3390/molecules18032803
Tala VRS, Candida da Silva V, Rodrigues CM, Nkengfack AE, Campaner dos Santos L, Vilegas W. Characterization of Proanthocyanidins from Parkia biglobosa (Jacq.) G. Don. (Fabaceae) by Flow Injection Analysis — Electrospray Ionization Ion Trap Tandem Mass Spectrometry and Liquid Chromatography/Electrospray Ionization Mass Spectrometry. Molecules. 2013; 18(3):2803-2820. https://doi.org/10.3390/molecules18032803
Chicago/Turabian StyleTala, Viviane Raïssa Sipowo, Viviane Candida da Silva, Clenilson Martins Rodrigues, Augustin Ephrem Nkengfack, Lourdes Campaner dos Santos, and Wagner Vilegas. 2013. "Characterization of Proanthocyanidins from Parkia biglobosa (Jacq.) G. Don. (Fabaceae) by Flow Injection Analysis — Electrospray Ionization Ion Trap Tandem Mass Spectrometry and Liquid Chromatography/Electrospray Ionization Mass Spectrometry" Molecules 18, no. 3: 2803-2820. https://doi.org/10.3390/molecules18032803