Induction of Cell Death Mechanisms and Apoptosis by Nanosecond Pulsed Electric Fields (nsPEFs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Therapies for Hallmarks of Cancer
1.2. Physical Treatments that Target Whole Tumors
2. Results and Discussion
2.1. NsPEFs Induce Nanopores in Plasma Membranes
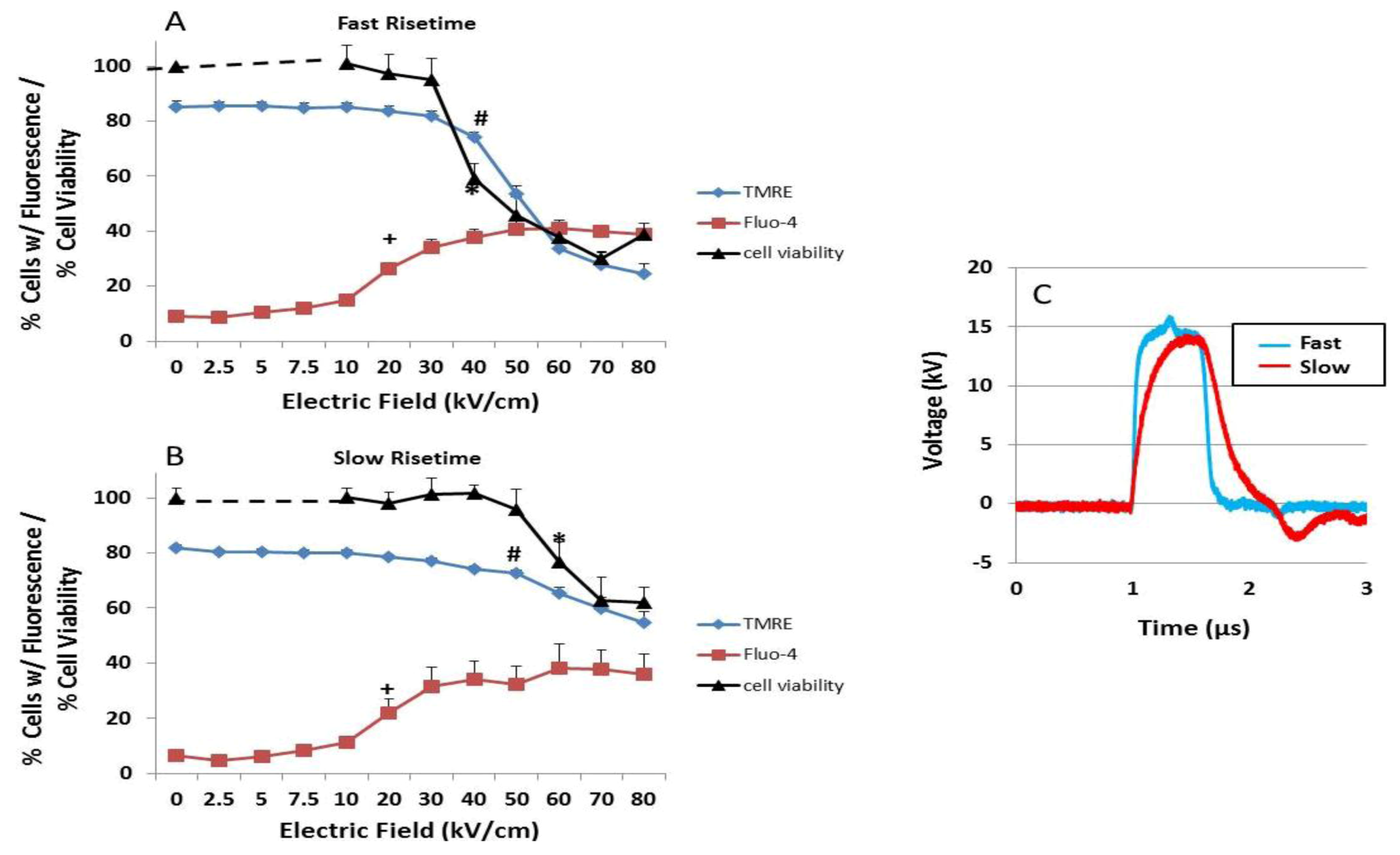
2.2. Transient Features in nsPEFs Differentially Modulate Intracellular Functions


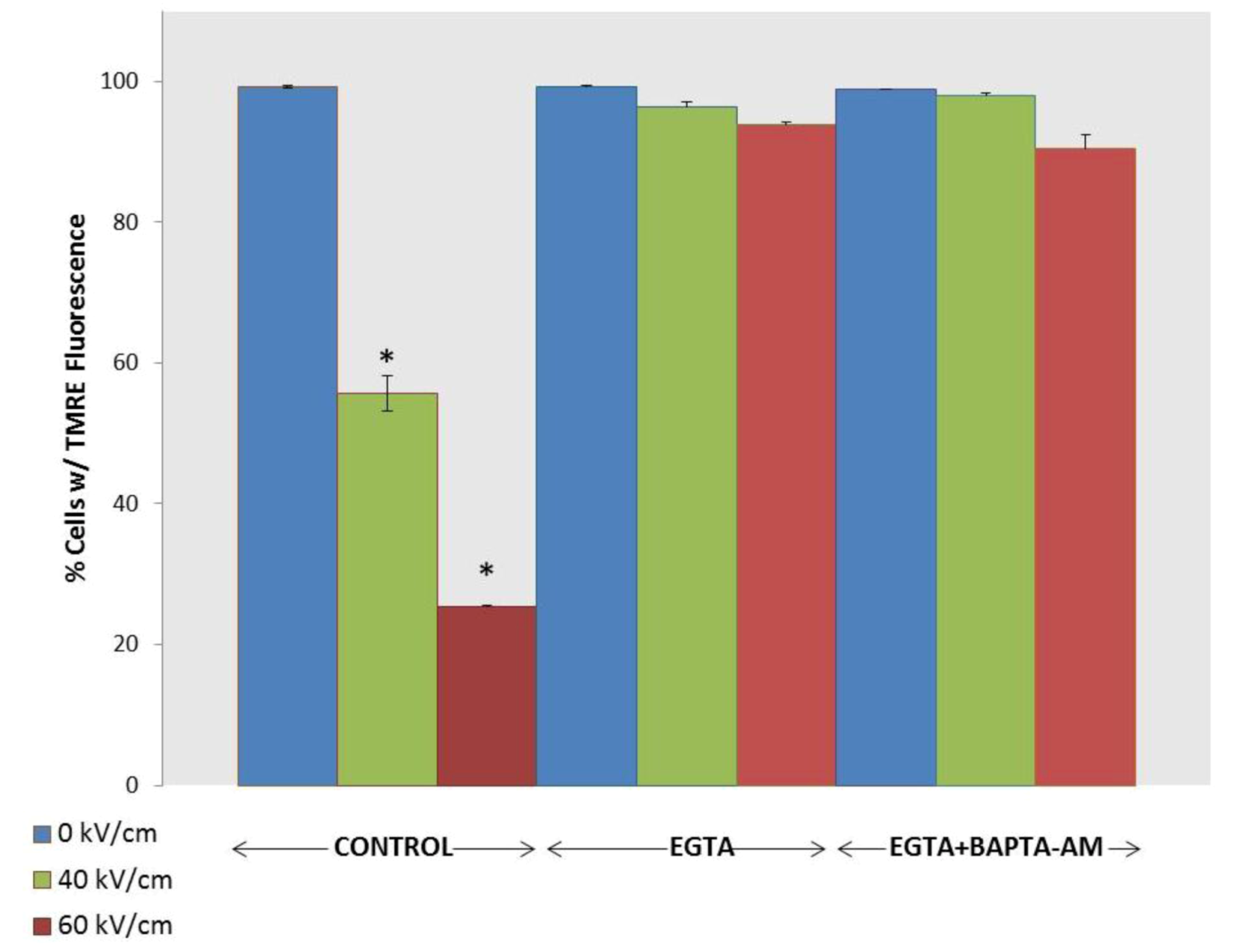
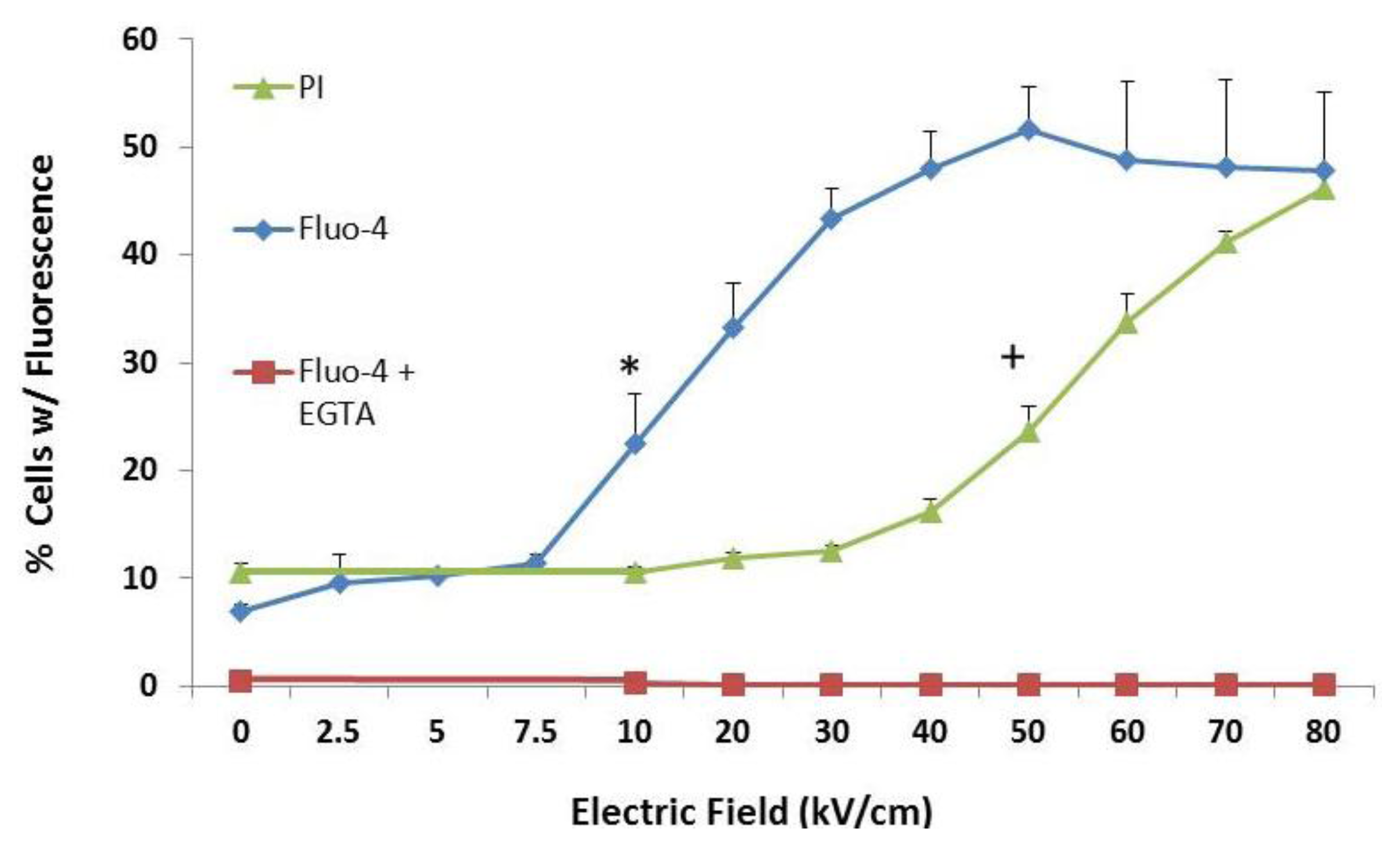
2.3. NsPEF-Induced Ca2+ Mobilization and Cell Viability

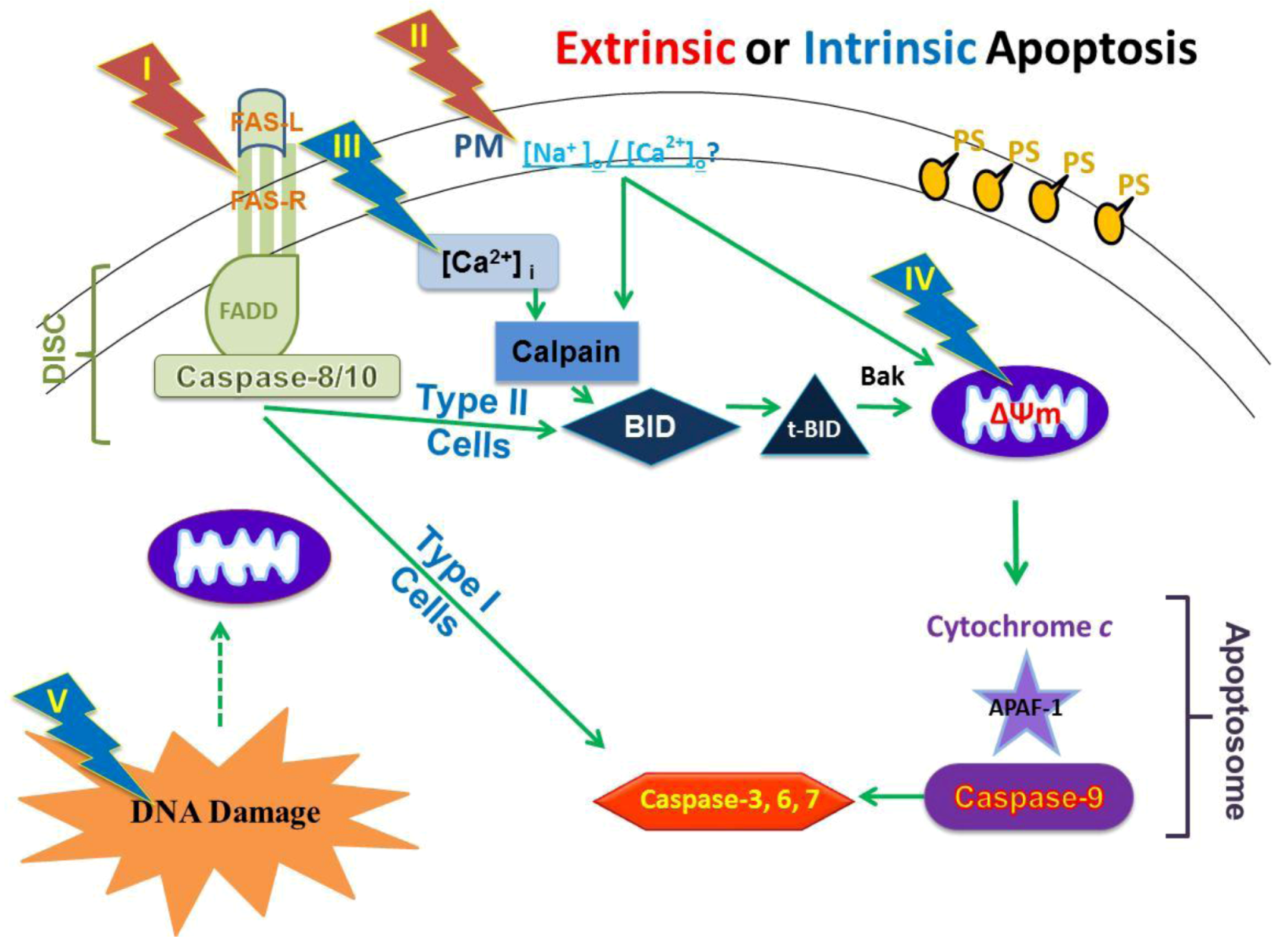
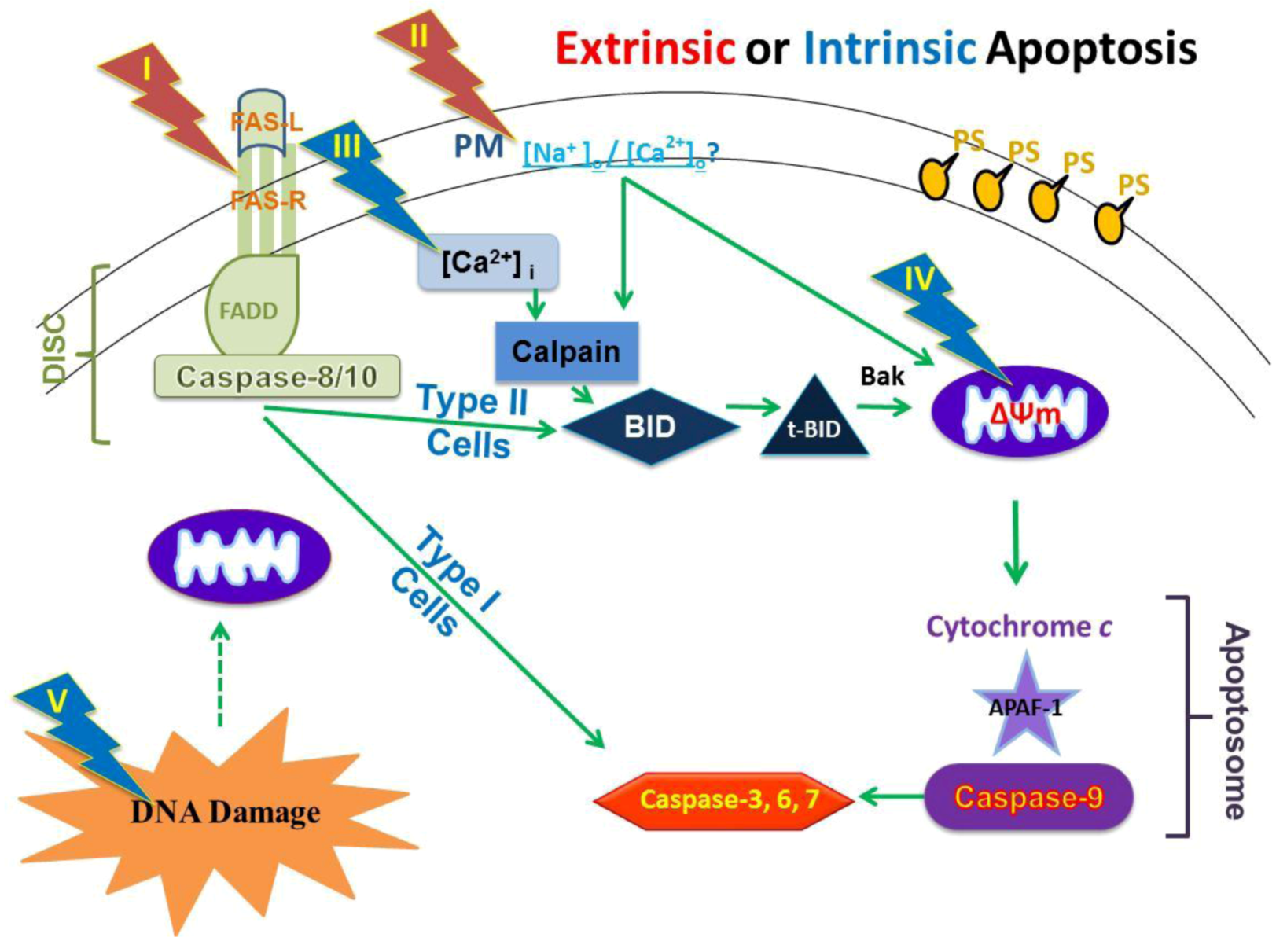
2.4. Possible Cellular Targets and Cell Death Pathways for nsPEFs
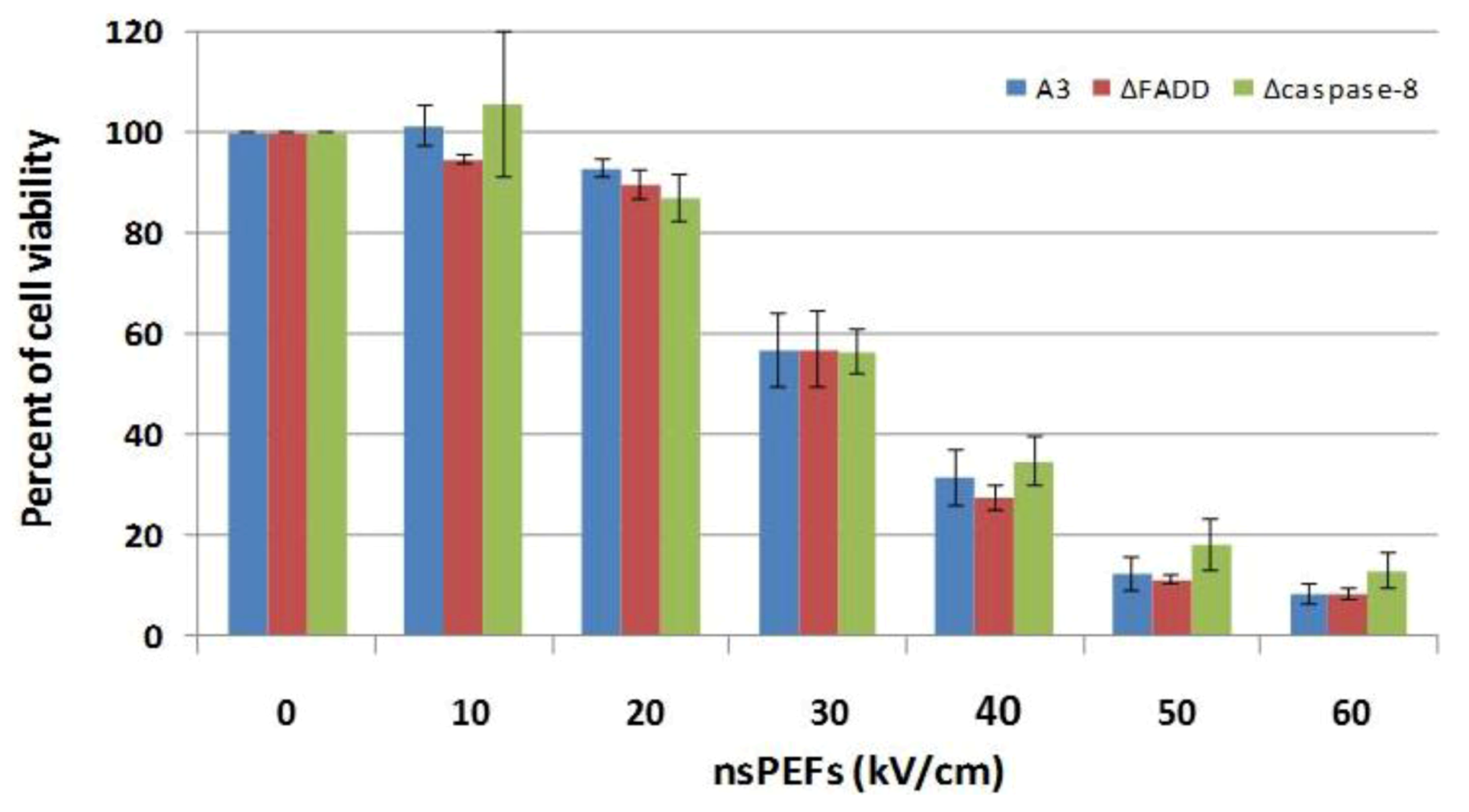
2.5. NsPEFs Do Not Induce Cell Death Through the Extrinsic Apoptosis Pathway in Jurkat Cells
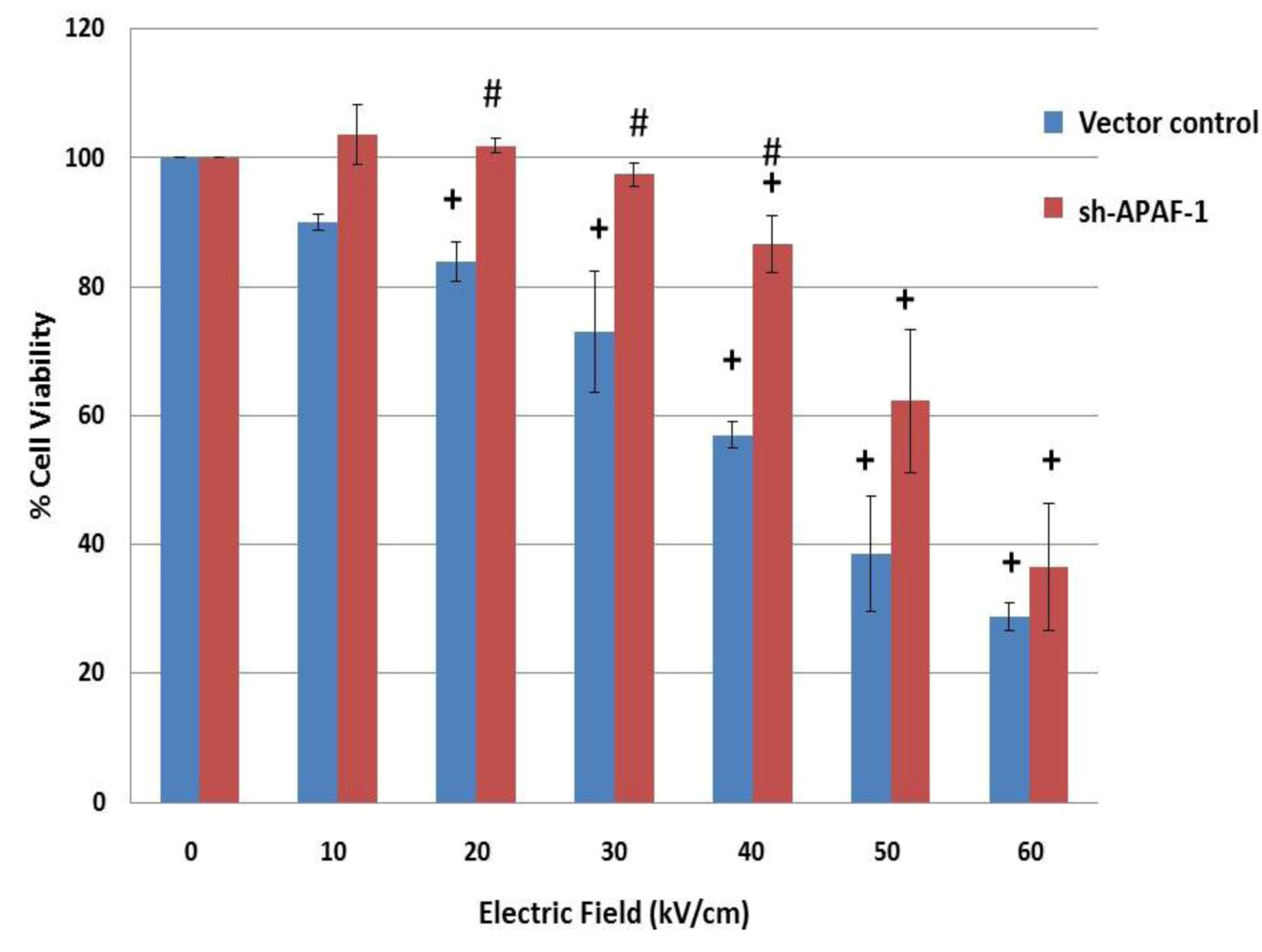
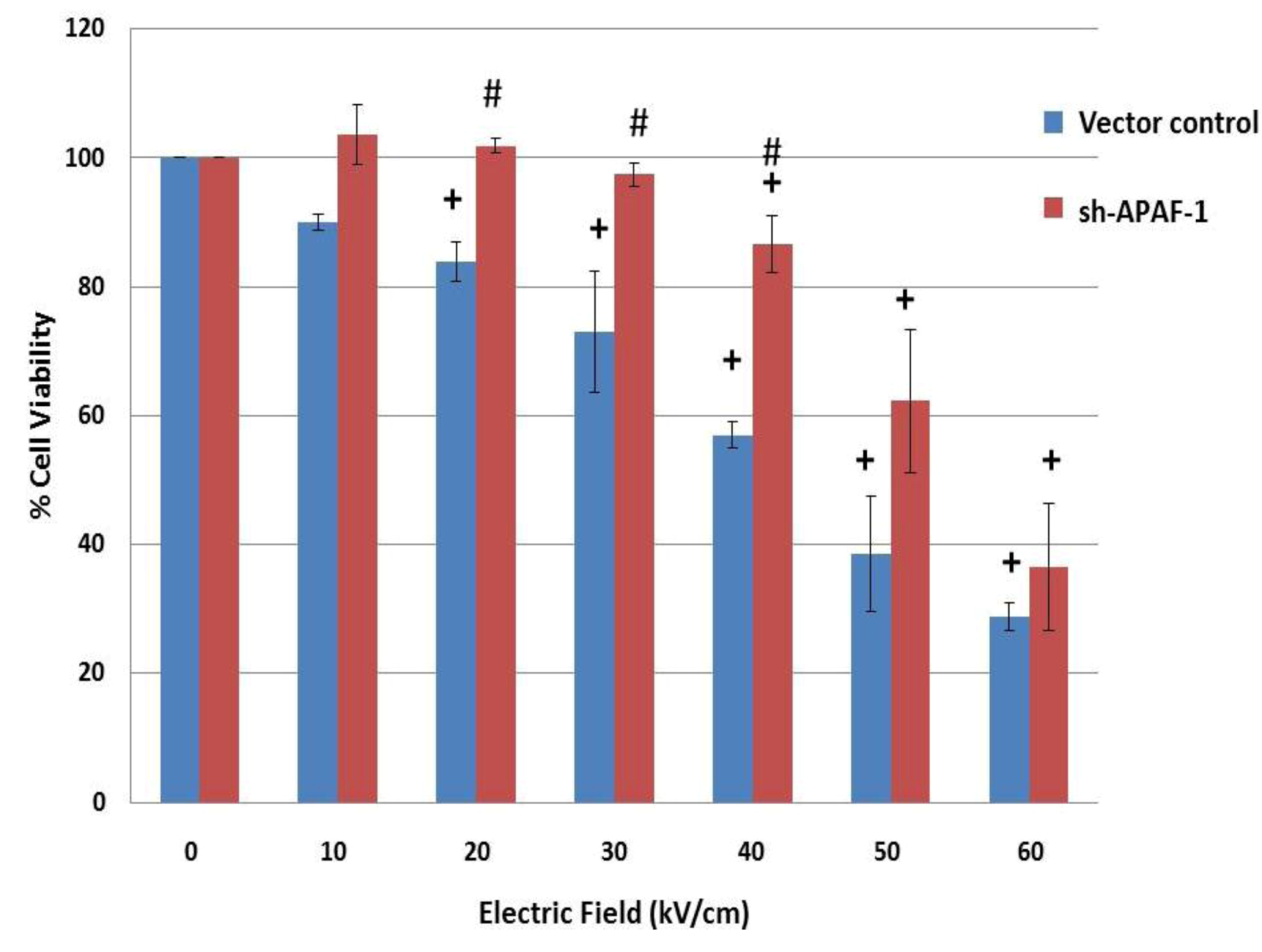
2.6. NsPEFs Induce Intrinsic Caspase-Dependent at Lower Electric Fields (20-40 kV/cm) and Caspase-Independent Cell Death at Higher Electric Fields (≥ 50 kV/cm) in Jurkat Cells
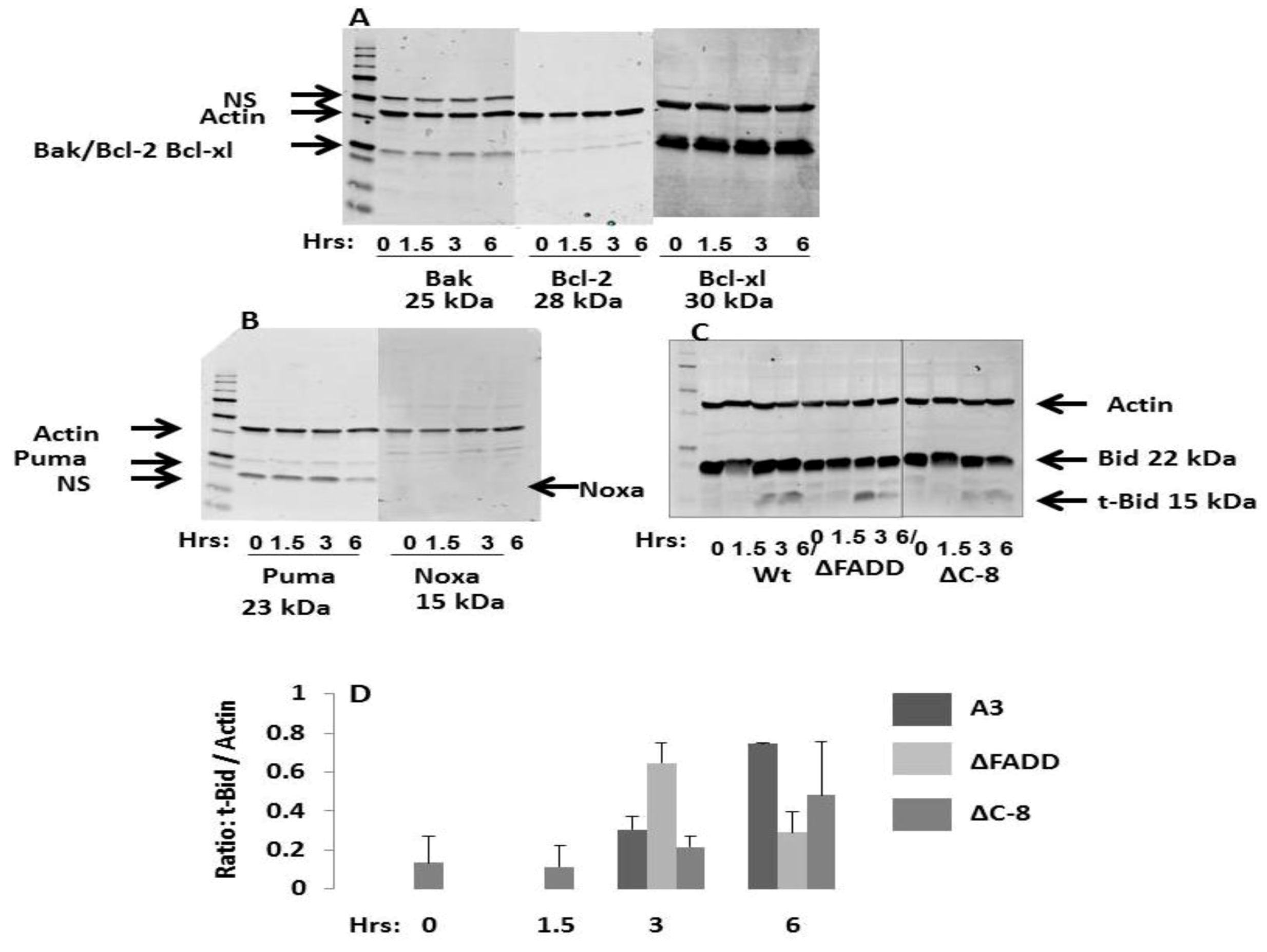
2.7. NsPEFs Activate Bid but Do Not Alter Levels of Other Endogenous Bcl-2 Family Proteins
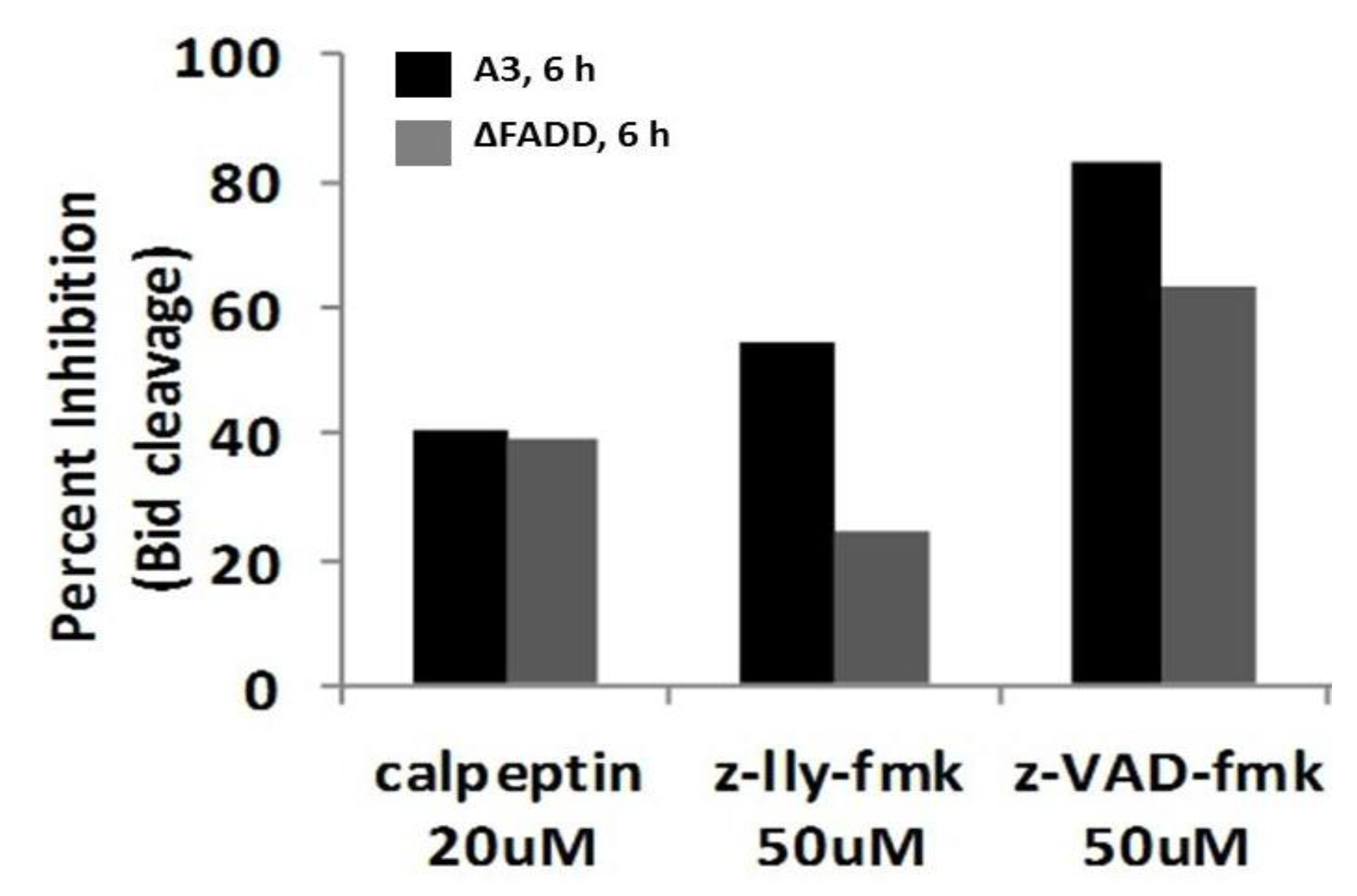
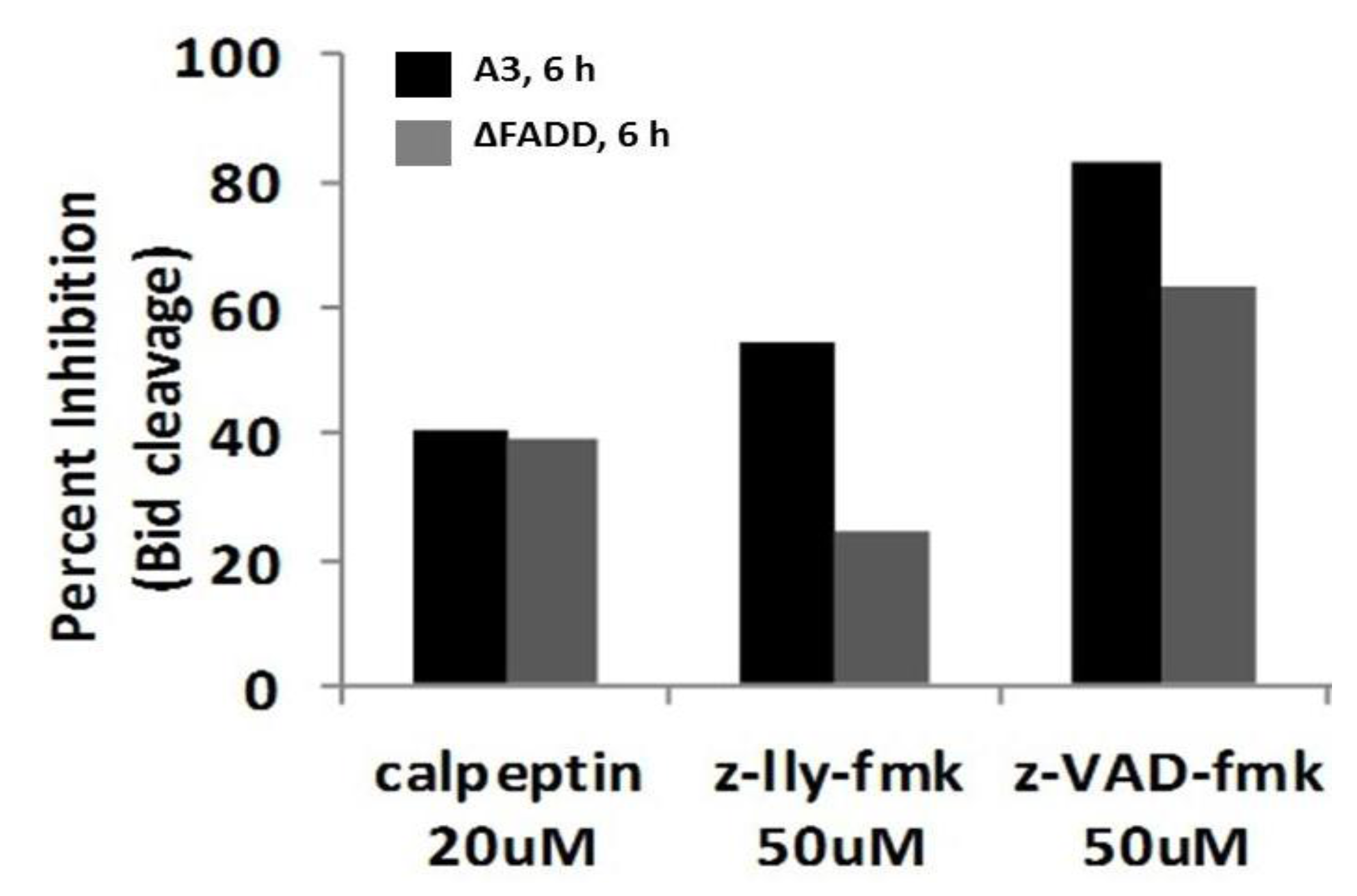
2.8. Bid Cleavage Is Sensitive to Inhibition of Caspases and Calpains in E4 Squamous Carcinoma and Jurkat Clones
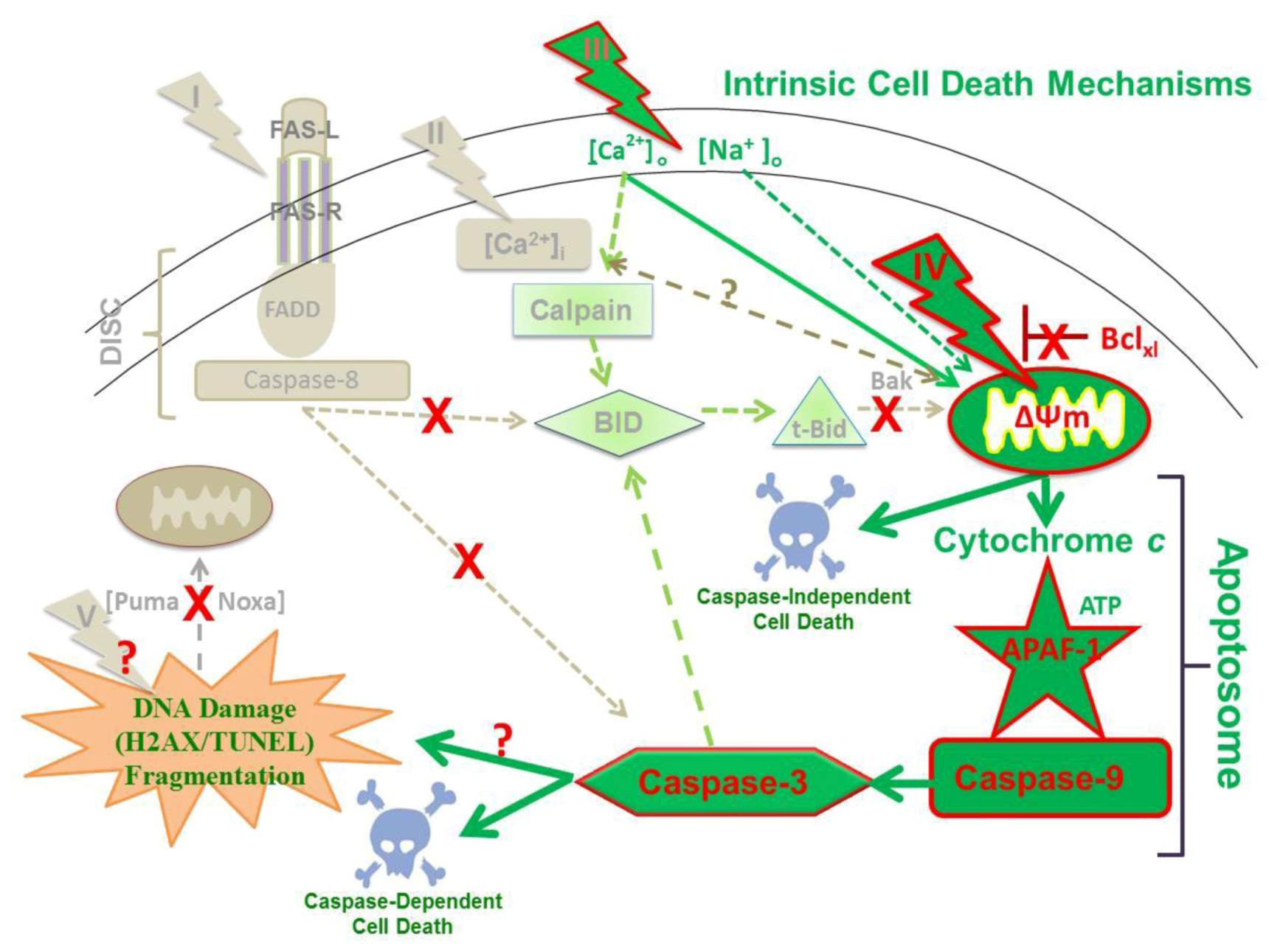
2.9. Hypothesized Cell Targets and Cell Death Pathways Activated by nsPEFs
3. Experimental Section
3.1. Cell Culture
3.2. Treatment of Cells with nsPEFs
3.3. Determination of Propidium Iodide (PI) Uptake
3.4. Flow Cytometry Analysis of Ca2+ and Δψm
3.5. Determination of Cell Viability
3.6. Flow Cytometry Analysis of Cytochrome c Release
3.7. Determination of Bcl-2 Family Proteins by Immunoblot Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Loges, S.; Schmidt, T.; Carmeliet, P. Mechanisms of Resistance to Anti-Angiogenic Therapy and Development of Third-Generation Anti-Angiogenic Drug Candidates. Genes Cancer 2010, 1, 12–25. [Google Scholar] [CrossRef]
- Mir, L.M.; Orlowski, S.; Belehradek, J.; Paoletti, C. Electrochemotherapy potentiation of antitumour effect of bleomycin by local electric pulses. Eur. J. Cancer 1991, 27, 68–72. [Google Scholar] [CrossRef]
- Sersa, G.; Cemazar, M.; Miklavcic, D. Antitumor effectiveness of electrochemotherapy with cis-diamminedichloroplatinum(II) in mice. Cancer Res. 1995, 55, 3450–3455. [Google Scholar]
- Goto, T.; Nishi, T.; Tmura, T.; Dev, S.B.; Takeshima, H.; Kochi, M.; Yoshizato, K.; Kuratsu, K.; Sakata, T.; Hofmann, G.A.; Ushio, Y. Highly efficient electro-gene therapy of solid tumor by using an expression plasmid for t herpes simplex virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA 2000, 97, 354–359. [Google Scholar] [CrossRef]
- Li, S.; Zhang, X.; Xia, X. Regression of Tumor Growth and Induction of Long-Term Antitumor Memory by Interleukin 12 Electro-Gene Therapy. J. Natl. Cancer Inst. 2002, 94, 762–768. [Google Scholar] [CrossRef]
- Davalos, R.V.; Mir, I.L.; Rubinsky, B. Tissue ablation with irreversible electroporation. Ann. Biomed. Eng. 2005, 33, 223–231. [Google Scholar] [CrossRef]
- Beebe, S.J.; Fox, P.M.; Rec, L.J.; Somers, K.; Stark, R.H.; Schoenbach, K.H. Nanosecond pulsed electric field (nsPEF) effects on cells and tissues: apoptosis induction and tumor growth inhibition. IEEE Trans. Plasma Sci. 2002, 30, 286–292. [Google Scholar] [CrossRef]
- Nuccitelli, R.; Pliquett, U.; Chen, X.; Ford, W.; Swanson, J.R.; Beebe, S.J.; Kolb, J.F.; Schoenbach, K.H. Nanosecond pulsed electric fields cause melanomas to self-destruct. Biochem. Biophys. Res. Commun. 2006, 343, 351–360. [Google Scholar] [CrossRef]
- Garon, E.B.; Sawcer, D.; Vernier, P.T.; Tang, T.; Sun, Y.; Marcu, L.; Gundersen, M.A.; Koeffler, H.P. In vitro and in vivo evaluation and a case report of intense nanosecond pulsed electric field as a local therapy for human malignancies. Int. J. Cancer 2007, 121, 675–682. [Google Scholar] [CrossRef]
- Chen, X.; Kolb, J.F.; Swanson, R.J.; Schoenbach, K.H.; Beebe, S.J. Apoptosis initiation and angiogenesis inhibition: Melanoma targets for nanosecond pulsed electric fields. Pigment. Cell Melanoma Res. 2010, 23, 554–563. [Google Scholar] [CrossRef]
- Chen, X.; Zhuang, J.; Kolb, J.F.; Schoenbach, K.H.; Beebe, S.J. Long term survival of mice with hepatocellular carcinoma after pulse power ablation with nanosecond pulsed electric fields. Technol. Cancer Res. Treat. 2012, 11, 83–93. [Google Scholar]
- Tiong, L.; Maddern, G.J. Systematic review and meta-analysis of survival and disease recurrence after radiofrequency ablation for hepatocellular carcinoma. Br. J. Surg. 2011, 98, 1210–1224. [Google Scholar] [CrossRef]
- Sersa, G.; Cemazar, M.; Snoj, M. Electrochemotherapy of solid tumors—Preclinical and clinical experience. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 728–731. [Google Scholar]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; Heller, R. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar]
- Heller, R.; Shirley, S.; Guo, S.; Donate, A.; Heller, L. Electroporation based gene therapy—From the bench to the bedside. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 736–738. [Google Scholar]
- Neal, R.E., 2nd; Rossmeis, J.H., Jr.; Garcia, P.A.; Lanz, O.I.; Henao-Guerrero, N.; Davalos, R.V. Successful treatment of a large soft tissue sarcoma with irreversible electroporation. J. Clin. Oncol. 2011, 29, 372–377. [Google Scholar] [CrossRef]
- Kingham, T.P.; Karkar, A.M.; D'Angelica, M.I.; Allen, P.J.; Dematteo, R.P.; Getrajdman, G.I.; Sofocleous, C.T.; Solomon, S.B.; Jarnagin, W.R.; Fong, Y. Ablation of Perivascular Hepatic Malignant Tumors with Irreversible Electroporation. J. Am. Coll. Surg. 2012, 215, 379–387. [Google Scholar]
- Stewart, D.A.; Gowrishankar, T.R.; Weaver, J.C. Transport lattice approach to describing electroporation: use of local asymptotic model. IEEE Transact. Plasma Sci. 2004, 32, 1696–1708. [Google Scholar]
- Gowrishankar, T.R.; Esser, A.T.; Vasilkoski, Z.; Smith, K.C.; Weaver, J.C. Microdosimetry for conventional and supra-electroporation in cells with organelles. Biochem. Biophys. Res. Commun. 2006, 341, 1266–1276. [Google Scholar]
- Beebe, S.J.; Fox, P.M.; Rec, L.J.; Willis, E.L.; Schoenbach, K.H. Nanosecond, high-intensity pulsed electric fields induce apoptosis in human cells. FASEB J. 2003, 17, 1493–1495. [Google Scholar]
- Vernier, P.T.; Aimin, L.; Marcu, L.; Craft, C.M.; Gundersen, M.A. Ultrashort pulsed electric fields induce membrane phospholipid translocation and caspase activation: Differential sensitivities of Jurkat T lymphoblasts and rat glioma C6 cells. IEEE Trans. Dielectr. Electr. Insul. 2003, 10, 795–809. [Google Scholar]
- Beebe, S.J.; Blackmore, P.F.; White, J.; Joshi, R.P.; Schoenbach, K.H. Nanosecond pulsed electric fields modulate cell function through intracellular signal transduction mechanisms. Physiol. Meas. 2004, 25, 1077–1093. [Google Scholar]
- Hall, E.H.; Schoenbach, K.H.; Beebe, S.J. Nanosecond pulsed electric fields induce apoptosis in p53-wildtype and p53-null HCT116 colon carcinoma cells. Apoptosis 2007, 12, 1721–1731. [Google Scholar] [CrossRef]
- Ford, W.E.; Ren, W.; Blackmore, P.F.; Schoenbach, K.H.; Beebe, S.J. Nanosecond pulsed electric fields stimulate apoptosis without release of pro-apoptotic factors from mitochondria in B16f10 melanoma. Arch. Biochem. Biophys 2010, 497, 82–89. [Google Scholar]
- Ren, W.; Beebe, S.J. An apoptosis targeted stimulus with nanosecond pulsed electric fields (nsPEFs) in E4 squamous cell carcinoma. Apoptosis 2011, 16, 382–393. [Google Scholar] [CrossRef]
- Ren, W.; Sain, N.M.; Beebe, S.J. Nanosecond pulsed electric fields (nsPEFs) activate intrinsic caspase-dependent and caspase-independent cell death in Jurkat cells. Biochem Biophys. Res. Commun. 2012, 421, 808–812. [Google Scholar]
- Schoenbach, K.H.; Beebe, S.J.; Buescher, E.S. Intracellular effect of ultrashort electrical pulses. Bioelectromagnetics 2001, 22, 440–448. [Google Scholar] [CrossRef]
- Stacey, M.; Stickley, J.; Fox, P.; Statler, V.; Schoenbach, K.H.; Beebe, S.J.; Buescher, S. Differential effects in cells exposed to ultra-short, high intensity electric fields: Cell survival, DNA damage, and cell cycle analysis. Mutat. Res. 2003, 542, 65–75. [Google Scholar] [CrossRef]
- Stacey, M.; Fox, P.; Buescher, S.; Kolb, J. Nanosecond pulsed electric field induced cytoskeleton, nuclear membrane and telomere damage adversely impact cell survival. Bioelectrochemistry 2011, 82, 131–134. [Google Scholar] [CrossRef]
- Beebe, S.J.; White, J.; Blackmore, P.F.; Deng, Y.; Somers, K.; Schoenbach, K.H. Diverse effects of nanosecond pulsed electric fields on cells and tissues. DNA Cell Biol. 2003, 22, 785–796. [Google Scholar] [CrossRef]
- Vernier, P.T.; Sun, Y.; Marcu, L.; Salemi, S.; Craft, C.M.; Gundersen, M.A. Calcium bursts induced by nanosecond electric pulses. Biochem. Biophys. Res. Commun. 2003, 310, 286–295. [Google Scholar] [CrossRef]
- White, J.A.; Blackmore, P.F.; Schoenbach, K.H.; Beebe, S.J. Stimulation of capacitative Ca2+ entry in HL-60 cells by nanosecond pulsed electric fields. J. Biol. Chem. 2004, 279, 22964–22972. [Google Scholar]
- Buescher, E.S.; Smith, R.R.; Schoenbach, K.H. Submicrosecond intense pulsed electric field effects on intracellular free Ca2+: mechanisms and effects. IEEE Trans. Plasma Sci. 2004, 32, 1563–1572. [Google Scholar] [CrossRef]
- Zhang, J.; Blackmore, P.F.; Hargrave, B.Y.; Xiao, S.; Beebe, S.J.; Schoenbach, K.H. Nanosecond pulse electric field (nanopulse): A novel non-ligand agonist for platelet activation. Arch. Biochem. Biophys. 2008, 471, 240–248. [Google Scholar]
- Vernier, P.T. Mitochondrial membrane permeabilization with nanosecond electric pulses. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 743–745. [Google Scholar]
- Batista Napotnik, T.; Wu, Y.H.; Gundersen, M.A.; Miklavčič, D.; Vernier, P.T. Nanosecond electric pulses cause mitochondrial membrane permeabilization in Jurkat cells. Bioelectromagnetics 2012, 33, 257–264. [Google Scholar]
- Shawgo, M.E.; Shelton, S.N.; Robertson, J.D. Caspase-mediated Bak activation and cytochrome c release during intrinsic apoptotic cell death in Jurkat cells. J. Biol. Chem. 2008, 283, 35532–35538. [Google Scholar]
- Shelton, S.N.; Shawgo, M.E.; Robertson, J.D. Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J. Biol. Chem. 2009, 284, 11247–11255. [Google Scholar]
- Shelton, S.N.; Dillard, C.D.; Robertson, J.D. Activation of caspase-9, but not caspase-2 or caspase-8, is essential for heat-induced apoptosis in Jurkat cells. J. Biol. Chem. 2010, 285, 40525–40533. [Google Scholar]
- Deng, J.; Schoenbach, K.H.; Buescher, E.S.; Hair, P.; Fox, P.M.; Beebe, S.J. The effects of intense submicrosecond electrical pulses on cells. Biophys. J. 2003, 84, 2709–2714. [Google Scholar] [CrossRef]
- Tekle, E.; Oubrahim, H.; Dzekunov, S.M.; Kolb, J.F.; Schoenbach, K.H.; Chock, P.B. Selective field effects on intracellular vacuoles and vesicle membranes with nanosecond electric pulses. Biophys. J. 2005, 89, 274–284. [Google Scholar]
- Pakhomov, A.G.; Bowman, A.M.; Ibey, B.L.; Andre, F.M.; Pakhomova, O.N.; Schoenbach, K.H. Lipid nanopores can form a stable, ion channel-like conduction pathway in cell membrane. Biochem Biophys. Res. Commun. 2009, 385, 181–186. [Google Scholar] [CrossRef]
- Bowman, A.M.; Nesin, O.M.; Pakhomova, O.N.; Pakhomov, A.G. Analysis of plasma membrane integrity by fluorescent detection of Tl(+) uptake. J. Membr. Biol. 2010, 236, 15–26. [Google Scholar] [CrossRef]
- Creighton, T.E. Proteins: Structures and Molecular Properties; WH Freeman: New York, NY, USA, 1993. [Google Scholar]
- Tieleman, D.P. The molecular basis of electroporation. BMC Biochem. 2004, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Tarek, M. Membrane electroporation: a molecular dynamics simulation. Biophys. J. 2005, 88, 4045–4053. [Google Scholar]
- Vernier, P.T.; Ziegler, M.J.; Sun, Y.; Chang, W.V.; Gundersen, M.A.; Tieleman, D.P. Nanopore formation and phosphatidylserine externalization in a phospholipid bilayer at high transmembrane potential. J. Am. Chem. Soc. 2006, 128, 6288–6289. [Google Scholar]
- Levine, Z.A.; Vernier, P.T. Life cycle of an electropore: field-dependent and field-independent steps in pore creation and annihilation. J. Membr. Biol. 2010, 236, 27–36. [Google Scholar] [CrossRef]
- Sugar, I.P.; Neumann, E. Stochastic model for electric field-induced membrane pores. Electroporation Biophys. Chem. 1984, 19, 211–225. [Google Scholar] [CrossRef]
- Weaver, J.C. Electroporation of biological membranes from multicellular to nano scales. IEEE Trans. Dielect. Electr. Insul. 2003, 10, 754–768. [Google Scholar] [CrossRef]
- Delemotte, L.; Tarek, M. Molecular dynamics simulations of lipid membrane electroporation. J. Membr. Biol. 2012, 245, 531–543. [Google Scholar] [CrossRef]
- Beebe, S.J.; Chen, Y.-J.; Sain, N.M.; Schoenbach, K.H.; Xiao, S. Transient features in nanosecond pulsed electric fields differentially modulate mitochondria and viability. PloS One 2012, 7, e51349. [Google Scholar]
- Jacobson, J.; Duchen, M.R. Mitochondrial oxidative stress and cell death in astrocytes—Requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 2002, 115, 1175–1188. [Google Scholar]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Pakhomova, O.N.; Khorokhorina, V.A.; Bowman, A.M.; Rodaitė-Riševičienė, R.; Saulis, G.; Xiao, S.; Pakhomov, A.G. Oxidative effects of nanosecond pulsed electric field exposure in cells and cell-free media. Arch. Biochem. Biophys. 2012, 527, 55–64. [Google Scholar]
- Leung, A.W.; Halestrap, A. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta. 2008, 1777, 946–952. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta. 2009, 1787, 1402–1415. [Google Scholar] [CrossRef]
- Wang, S.; Chen, J.; Chen, M.T.; Vernier, P.T.; Gundersen, M.A.; Valderrábano, M. Cardiac myocyte excitation by ultrashort high-field pulses. Biophys. J. 2009, 96, 1640–1648. [Google Scholar]
- Morotomi-Yano, K.; Akiyama, H.; Yano, K.I. Nanosecond pulsed electric fields activate AMP-activated protein kinase: Implications for Ca2+-mediated activation of cellular signaling. Biochem. Biophys. Res. Commun. 2012, 428, 371–375. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and Ca2+: From cell signaling to cell death. J. Physiol. 2002, 529, 57–68. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef]
- Ichas, F.; Mazat, J.P. From Ca2+ signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta. 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Schoenbach, K.H.; Joshi, R.P.; Stark, R.H.; Dobbs, F.C.; Beebe, S.J. Bacterial decontamination of liquids with pulsed electric fields. IEEE Trans. Dielectr. Electr. Insul. 2000, 7, 637–645. [Google Scholar] [CrossRef]
- Lawrence, C.P.; Chow, S.C. FADD deficiency sensitizes Jurkat T cells to TNF-alpha-dependent necrosis during activation-induced cell death. FEBS Lett. 2005, 579, 6465–6472. [Google Scholar] [CrossRef]
- Vonarbourg, C.; Stolzenberg, M.C.; Hölzelova, E.; Fischer, A.; Deist, F.L.; Rieux-Laucat, F. Differential sensitivity of Jurkat and primary T cells to caspase-independent cell death triggered upon Fas stimulation. Eur. J. Immunol. 2002, 32, 2376–2384. [Google Scholar] [CrossRef]
- Kroemer, G.; Martin, S.J. Caspase-independent cell death. Nat. Med. 2005, 11, 725–730. [Google Scholar] [CrossRef]
- Proskuryakov, S.Y.; Gabai, V.L. Mechanisms of tumor cell necrosis. Curr. Pharm. Des. 2010, 16, 56–68. [Google Scholar] [CrossRef]
- Nuccitelli, R.; Chen, X.; Pakhomov, A.G.; Baldwin, W.H.; Sheikh, S.; Pomicter, J.L.; Ren, W.; Osgood, C.; Swanson, R.J.; Kolb, J.F.; Beebe, S.J.; Schoenbach, K.H. A new pulsed electric field therapy for melanoma disrupts the tumor's blood supply and causes complete remission without recurrence. Int. J. Cancer 2009, 125, 438–445. [Google Scholar] [CrossRef]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.; Gottlieb, R.A. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 2001, 276, 30724–30728. [Google Scholar]
- Mandic, A.; Viktorsson, K.; Strandberg, L.; Heiden, T.; Hansson, J.; Linder, S.; Shoshan, M.C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol. Cell. Biol. 2002, 22, 3003–3013. [Google Scholar]
- Dejean, L.M.; Martinez-Caballero, S.; Kinnally, K.W. Is MAC the knife that cuts cytochrome c from mitochondria during apoptosis? Cell Death Differ. 2006, 13, 1387–1395. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Tornero, D.; Posadas, I.; Ceña, V. Bcl-x(L) blocks a mitochondrial inner membrane channel and prevents Ca2+ overload-mediated cell death. PLoS One 2011, 6, e20423. [Google Scholar] [CrossRef]
- Peixoto, P.M.; Lue, J.K.; Ryu, S.Y.; Wroble, B.N.; Sible, J.C.; Kinnally, K.W. Mitochondrial Apoptosis-Induced Channel MAC) Function Triggers a Bax/Bak-Dependent Bystander Effect. Am. J. Pathol. 2011, 178, 48–54. [Google Scholar] [CrossRef]
- Hair, P.S.; Schoenbach, K.H.; Buescher, E.S. Sub-microsecond, intense pulsed electric field applications to cells show specificity of effects. Bioelectrochemistry 2003, 61, 65–72. [Google Scholar] [CrossRef]
- Ibey, B.L.; Pakhomov, A.G.; Gregory, B.W.; Khorokhorina, V.A.; Roth, C.C.; Rassokhin, M.A.; Bernhard, J.A.; Wilmink, G.J.; Pakhomova, O.N. Selective cytotoxicity of intense nanosecond-duration electric pulses in mammalian cells. Biochim. Biophys. Acta. 2010, 1800, 1210–1219. [Google Scholar] [CrossRef]
- Yang, W.; Wu, Y.H.; Yin, D.; Koeffler, H.P.; Sawcer, D.E.; Vernier, P.T.; Gundersen, M.A. Differential sensitivities of malignant and normal skin cells to nanosecond pulsed electric fields. Technol. Cancer Res. Treat. 2011, 10, 281–286. [Google Scholar]
- Nuccitelli, R.; Tran, K.; Athos, B.; Kreis, M.; Nuccitelli, P.; Chang, K.S.; Epstein, E.H., Jr.; Tang, J.Y. Nanoelectroablation therapy for murine basal cell carcinoma. Biochem Biophys. Res. Commun. 2012, 424, 446–450. [Google Scholar] [CrossRef]
- Yin, D.; Yang, W.G.; Weissberg, J.; Goff, C.B.; Chen, W.; Kuwayama, Y.; Leiter, A.; Xing, H.; Meixel, A.; Gaut, D.; Kirkbir, F.; Sawcer, D.; Vernier, P.T.; Said, J.W.; Gundersen, M.A.; Koeffler, H.P.; Burinsky, B. Cutaneous papilloma and squamous cell carcinoma therapy utilizing nanosecond pulsed electric fields (nsPEF). PLoS One 2012, 7, e43891. [Google Scholar]
- Nuccitelli, R.; Huynh, J.; Lui, K.; Wood, R.; Kreis, M.; Athos, B.; Nuccitelli, P. Nanoelectroablation of human pancreatic carcinoma in a murine xenograft model without recurrence. Int. J. Cancer 2012, 132, 1933–1939. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beebe, S.J.; Sain, N.M.; Ren, W. Induction of Cell Death Mechanisms and Apoptosis by Nanosecond Pulsed Electric Fields (nsPEFs). Cells 2013, 2, 136-162. https://doi.org/10.3390/cells2010136
Beebe SJ, Sain NM, Ren W. Induction of Cell Death Mechanisms and Apoptosis by Nanosecond Pulsed Electric Fields (nsPEFs). Cells. 2013; 2(1):136-162. https://doi.org/10.3390/cells2010136
Chicago/Turabian StyleBeebe, Stephen J., Nova M. Sain, and Wei Ren. 2013. "Induction of Cell Death Mechanisms and Apoptosis by Nanosecond Pulsed Electric Fields (nsPEFs)" Cells 2, no. 1: 136-162. https://doi.org/10.3390/cells2010136