
Potential Coagulation Factor-Driven Pro-Inflammatory Responses in Ovarian Cancer Tissues Associated with Insufficient O2 and Plasma Supply

Abstract
:
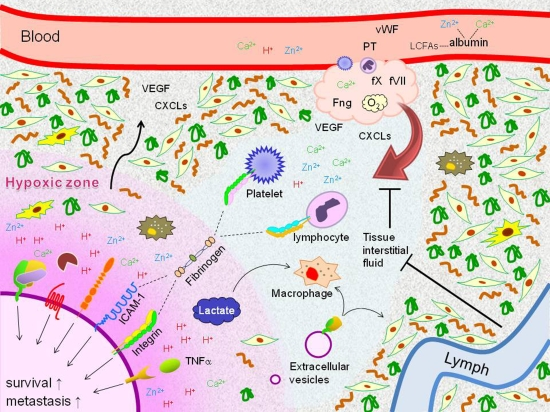{kind=link}
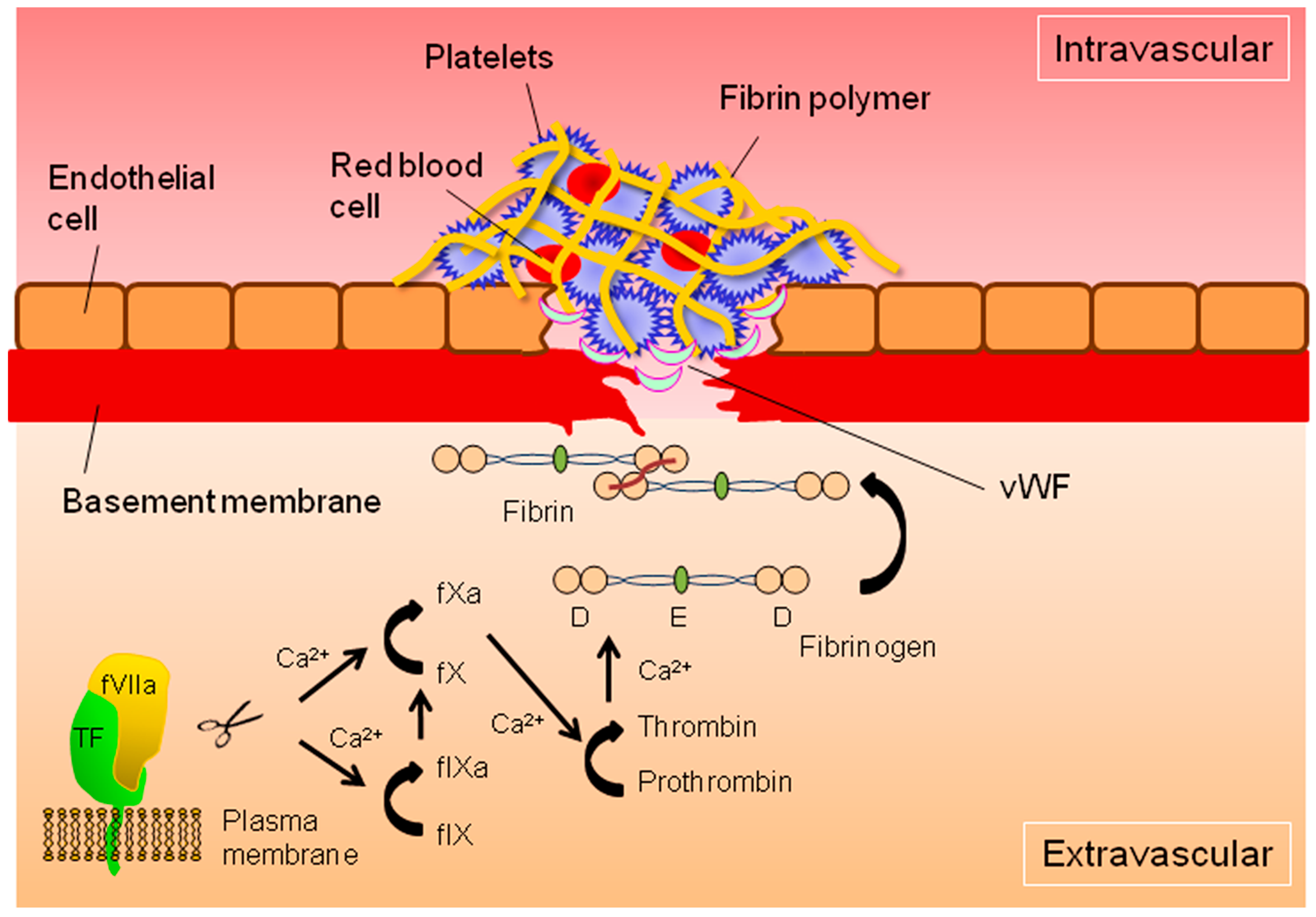{kind=link}
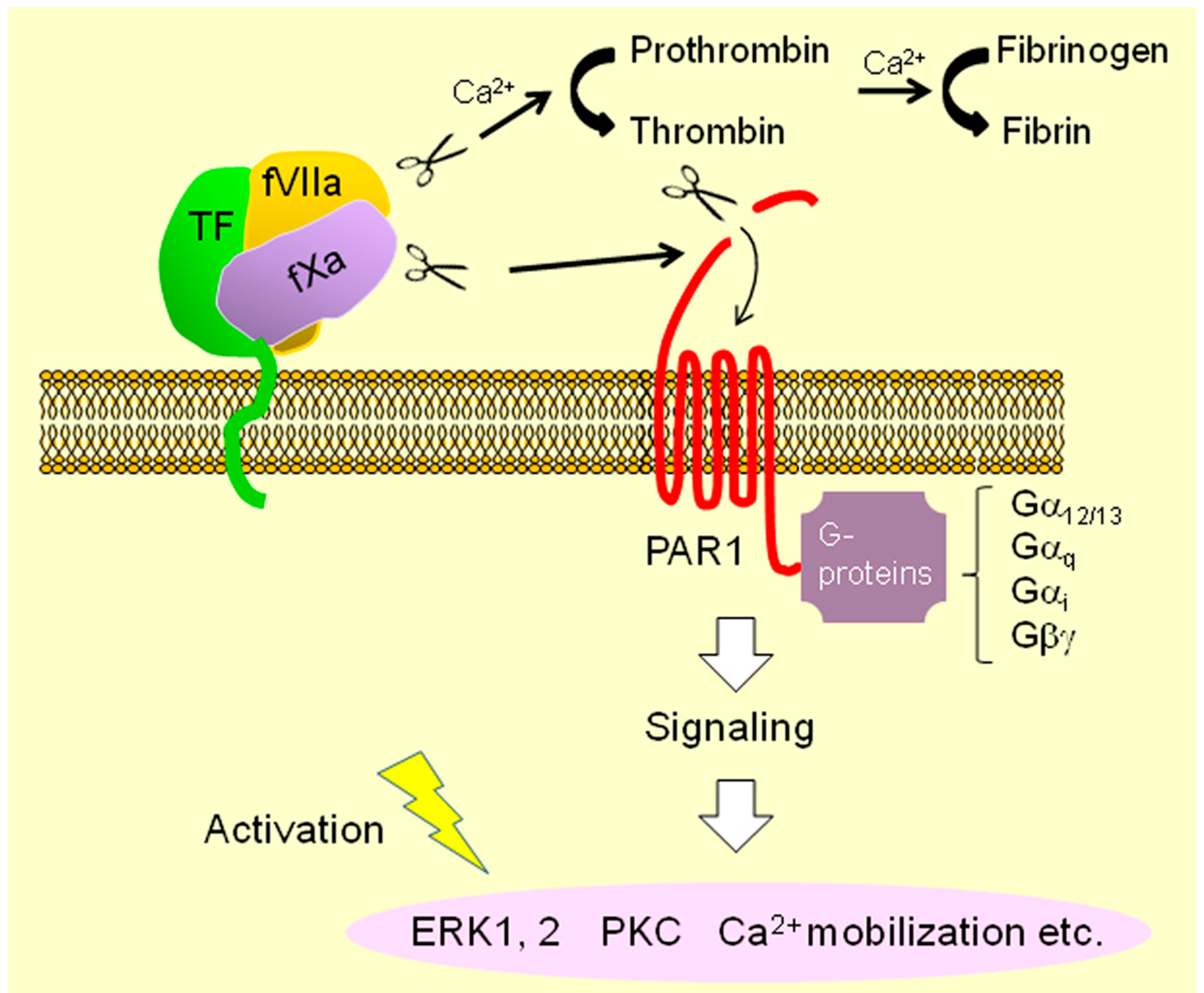{kind=link}
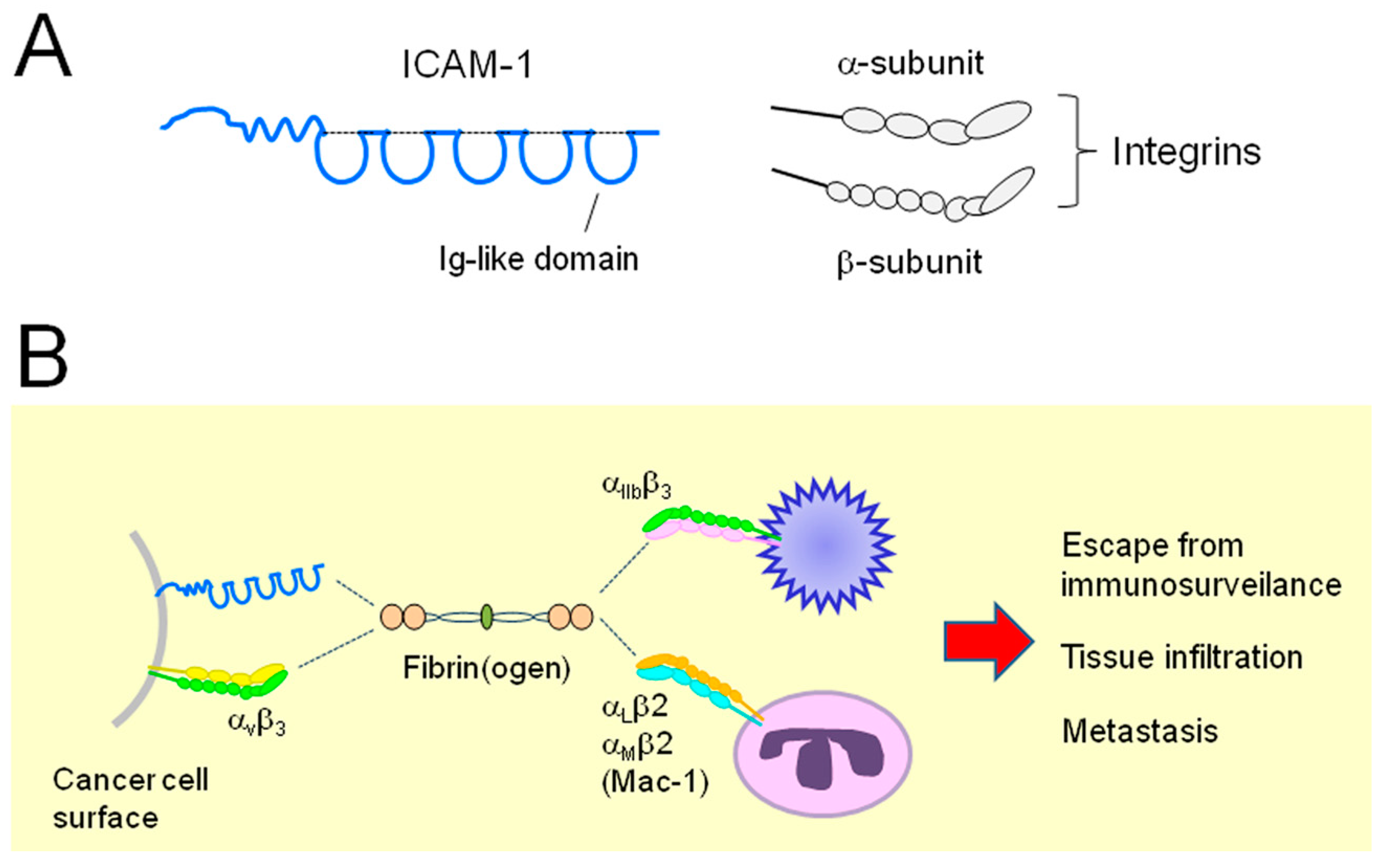{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Potential Relationship between Blood Coagulation and Inflammatory Factors in EOC Tissue: Overview of Current Knowledge
2.1. TF-fVIIa-Dependent Phenotypes of EOC Cells
2.2. TF-fVIIa Pathway and Inflammation in Cancer Tissue
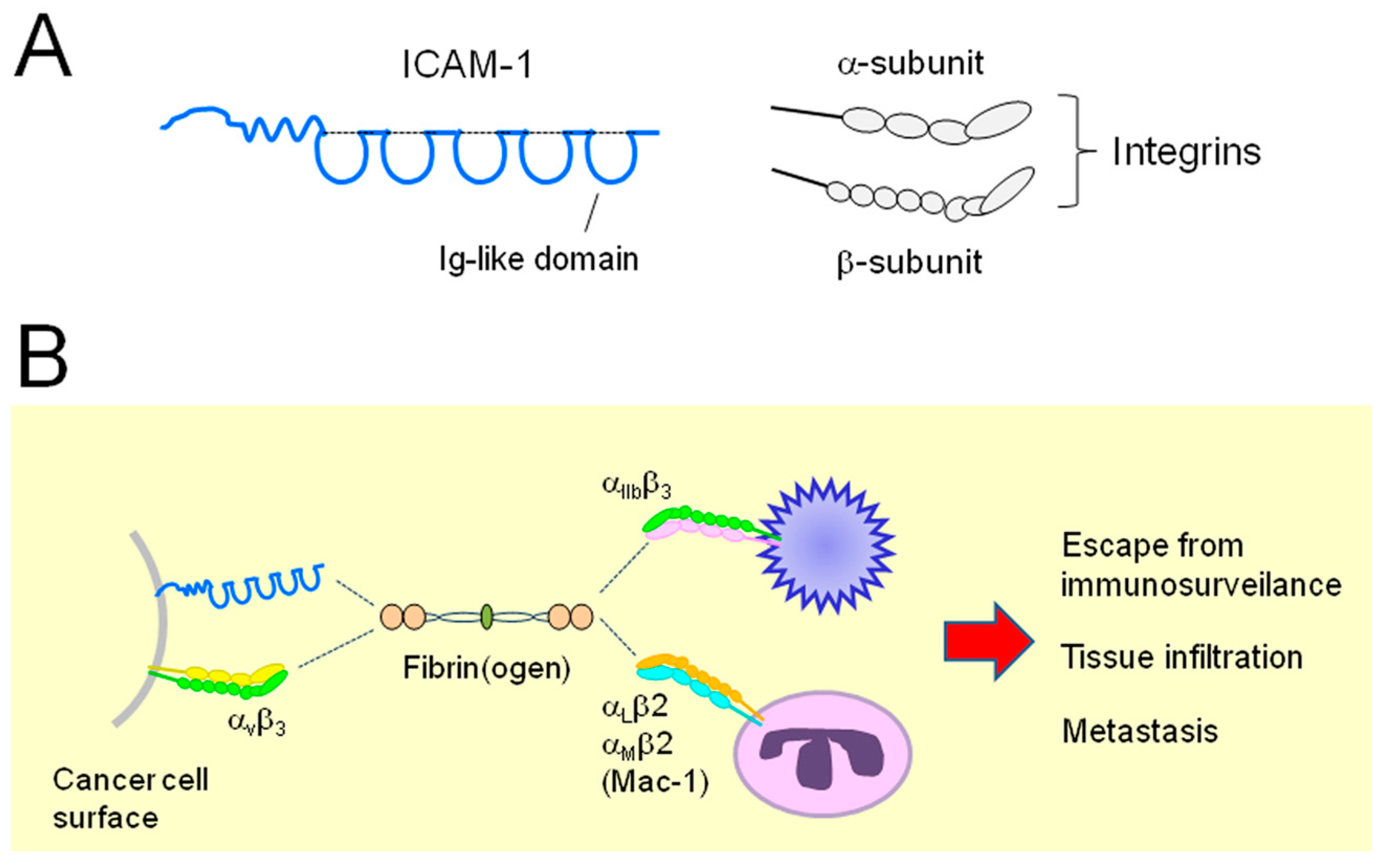
2.3. Relationship between ICAM-1 and EOC
2.4. Relationship between Integrins and EOC
3. Potential TF-fVIIa-Driven Inflammatory Responses within Hypoxic EOC Tissue
3.1. Potential TF Regulation
3.1.1. Regulation by Protein Disulfide Isomerase and Phosphatidylserine: Effect of Hypoxia and Intra-Tumoral pH Level
3.1.2. Lipid-Mediated Regulation
3.1.3. Potential Regulation by MicroRNAs
3.1.4. Potential Involvement of TFPIs
3.1.5. Possible Involvement of Integrins
3.1.6. Potential Involvement of Matriptase and Metalloproteinases
3.2. Implications Regarding ICAM-1
3.2.1. Potential Inflammatory Responses under Deprivation of Both O2 and Serum
3.2.2. Relationship to Intra-Tumoral Albumin Level
4. Potential Roles of Metal Ions in Coagulation Factor-Driven Inflammatory Responses
4.1. Potential Involvement of Zinc Ion
4.2. Potential Involvement of Calcium Ion
5. Potential Coagulation Factor-Driven Immune Responses within EOC Tissues Insufficiently Supplied with O2 and Plasma Components
6. Clinical Implications
7. Summary and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kurman, R.J.; Shih, I.-M. The origin and pathogenesis of epitherial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Ovarian Cancer National Alliance. Statistics (webpage on the Internet). Washington, DC: Ovarian Cancer Research Fund Alliance. 2017. Available online: https://ocrfa.org (accessed on 31 January 2017).
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-M.; Chuang, C.-M.; Wang, M.-L.; Yang, Y.-P.; Chuang, J.-H.; Yang, M.-J.; Yen, M.-S.; Chiou, S.-H.; Chang, C.-C. Gene set—Based integrative analysis revealing two distinct functional regulation patterns in four common subtypes of epithelial ovarian cancer. Int. J. Mol. Sci. 2016, 17, 1272. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.P.; Miller, R.E.; Kaye, S.B. New perspectives on molecular targeted therapy in ovarian clear cell carcinoma. Br. J. Cancer 2013, 108, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. The molecular basis of blood coagulation. Cell 1988, 53, 505–518. [Google Scholar] [CrossRef]
- Van den Berg, Y.W.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. The relationship between tissue factor and cancer progression: Insights from bench and bedside. Blood 2012, 119, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Tissue-factor factor VII complex as a key regulator of ovarian cancer phenotypes. Biomark. Cancer 2015, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aberg, M.; Siegbahn, A. Tissue factor non-coagulant signaling—Molecular mechanisms and biological consequences with a focus on cell migration and apoptosis. J. Thromb. Haemost. 2013, 11, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nawarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumors, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, E.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.N.; Afshar-Kharghan, V.; Sood, A.K. Platelet effects on ovarian cancer. Semin. Oncol. 2014, 41, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, M.; Benencia, F. Toll-like receptors in ovarian cancer as targets for immunotherapies. Front. Immunol. 2014, 5, 341. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; McNeish, I.A. Clear cell carcinoma of ovary and uterus. Curr. Oncol. Rep. 2013, 15, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Akassogloy, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene 2013, 32, 4057–4063. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Swartz, M.A. Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Regulation of the tissue factor gene. FASEB J. 1995, 9, 883–889. [Google Scholar] [PubMed]
- Rong, Y.; Post, D.E.; Pieper, R.O.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005, 65, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Yokota, N.; Koizume, S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Ruf, W.; Sakuma, Y.; Tsuchiya, E.; Miyagi, Y. Self-production of tissue factor-coagulation factor VII complex by ovarian cancer cells. Br. J. Cancer 2009, 101, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Ito, S.; Yoshioka, Y.; Kanayama, T.; Nakamura, Y.; Yoshihara, M.; Yamada, R.; Ochiya, T.; Ruf, W.; Miyagi, E.; et al. High level secretion of tissue factor-rich extracellular vesicles from ovarian cancer cells mediated by filamin-A and protease activated receptors. Thromb. Haemost. 2016, 115, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.Q.; Lima, L.G.; Goncalves, N.P.; De Souza, M.R.; Leal, A.C.; Demasi, M.A.A.; Sogayar, M.C.; Carneiro-Lobo, T.C. Hypoxia regulates the expression of tissue factor pathway signaling elements in a rat glioma model. Oncol. Lett. 2016, 12, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Jin, M.-S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Piao, J.-H.; Asai, A.; Yoshida, A.; Tsuchiya, E.; Ruf, W.; et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 2006, 66, 9453–9460. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Ito, S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Sakuma, Y.; Osaka, H.; Takanao, Y.; Ruf, W.; Miyagi, Y. HIF2α-Sp1 interaction mediates a deacetylation-dependent FVII-gene activation under hypoxic conditions in ovarian cancer cells. Nucleic Acid Res. 2012, 40, 5389–5401. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Lipid droplets: A key cellular organelle associated with cancer cell survival under normoxia and hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Ito, S.; Nakamura, Y.; Yoshihara, M.; Furuya, M.; Yamada, R.; Miyagi, E.; Hirahara, F.; Takano, Y.; Miyagi, Y. Lipid starvation and hypoxia synergistically activates ICAM1 and multiple genes in an Sp1-dependent manner to promote the growth of ovarian cancer. Mol. Cancer 2015, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.; Crowley, D.; Smyth, P.; O’Toole, S.; Spillane, C.; Martin, C.; Gallagher, M.; Canney, A.; Norris, L.; Conlon, N.; et al. Platelet adhesion and degranulation induce pro-survival and pro-angiogenic signaling in ovarian cancer cells. PLoS ONE 2011, 6, e26125. [Google Scholar] [CrossRef] [PubMed]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, T.; Wang, R.; Cheng, Z.; Xu, H.; Weiping, L.; Wang, Y.; Wang, X. Tissue factor-factor VIIa complex induces epithelial ovarian cancer cell invasion and metastasis through a monocytes-dependent mechanism. Int. J. Gynecol. Cancer 2011, 21, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Claussen, C.; Rausch, A.-V.; Lezius, S.; Amirkhosravi, A.; Davila, M.; Francis, J.L.; Hisada, Y.M.; Mackman, N.; Bokemeyer, C.; Schmalfeldt, B.; et al. Microvesicle-associated tissue factor procoagulant activity for the preoperative diagnosis of ovarian cancer. Thromb. Res. 2016, 141, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, N.; Miyagi, E.; Nomura, A.; Morita, E.; Ino, Y.; Ohtake, N.; Miyagi, Y.; Hirahara, F.; Hirano, H. Secretome-based identification of TFPI2, a novel serum biomarker for detection of ovarian clear cell carcinoma. J. Proteome Res. 2013, 12, 4340–4350. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, N.; Kobayashi, H.; Yonemoto, N.; Masuishi, Y.; Ino, Y.; Shigetomi, H.; Furukawa, N.; Ohtake, N.; Miyagi, Y.; Hirahara, F.; et al. Clinical significance of tissue factor pathway inhibitor 2, a serum biomarker candidate for ovarian clear cell carcinoma. PLoS ONE 2016, 11, e0165609. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Rothmeier, A.S.; Graf, C. Targeting clotting proteins in cancer therapy—Progress and challenges. Thromb. Res. 2016, 140, S1–S7. [Google Scholar] [CrossRef]
- Tsao, B.P.; Fair, D.S.; Curtiss, L.K.; Edgington, T.S. Monocytes can be induced by lipopolysaccharide-triggered T lymphocytes to express functional factor VII/VIIa protease activity. J. Exp. Med. 1984, 159, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A., Jr.; Allen, C.L.; Stone, O.L.; Fair, D.S. Human alveolar macrophages synthesize factor VII in vitro. Possible role in intestinal lung disease. J. Clin. Investig. 1985, 75, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Yokota, N.; Carneiro-Lobo, T.; Kitano, M.; Schaffer, M.; Anderson, G.M.; Muller, B.M.; Esmon, C.T.; Ruf, W. Endothelial protein C receptor function in murine and human breast cancer development. PLoS ONE 2013, 8, e61071. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Ricks, T.K.; Trejo, J. Protease-activated receptor signaling, endocytic sorting and dysregulation in cancer. J. Cell Sci. 2007, 120, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Biggerstaff, J.P.; Seth, N.; Amirkhosravi, A.; Amaya, M.; Fogarty, S.; Meyer, T.V.; Siddiqui, M.F.; Francis, J.L. Soluble fibrin augments platelet/tumor cell adherence in vitro and in vivo, and enhance experimental metastasis. Clin. Exp. Metastasis 1999, 17, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Bottsford-Miller, J.; Choi, H.-J.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Haemmerle, M.; Nick, A.M.; Pradeep, S.; Zand, B.; Previs, R.A. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin. Cancer Res. 2015, 21, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.; Cooke, N.; Kenny, D. Living in shear: Platelets protect cancer cells from shear induced damage. Clin. Exp. Metastasis 2014, 31, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, X. Platelets are associated with xenograft tumor growth and the clinical malignancy of ovarian cancer through an angiogenesis-dependent mechanism. Mol. Med. Rep. 2015, 11, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ozdemir, T.; Chung, C.-Y.; Robertson, G.P.; Dong, C. Sequential binding of avb3 and ICAM-1 determines fibrin-mediated melanoma capture and stable adhesion to CD11b/CD18 on neutrophils. J. Immunol. 2011, 186, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Biggerstaff, J.P.; Weidow, B.; Vidosh, J.; Dexheimer, J.; Patel, S.; Patel, P. Soluble fibrin inhibits monocyte adherence and cytotoxicity against tumor cells: Implications for cancer metastasis. Thromb. J. 2006, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Tsakadze, N.L.; Zhao, Z.; D’Souza, S.E. Interaction of intercellular adhesion molecule-1 with fibrinogen. Trends Cardiovasc. Med. 2002, 12, 101–108. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Liu, K.-J.; Hu, G.-L.; Liang, W.-J. Preoperative platelet/lymphocyte ratio is a superior prognostic factor compared to other systemic inflammatory response markers in ovarian cancer patients. Tumor Biol. 2015, 36, 8831–8837. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef]
- Wong, C.W.; Dye, D.E.; Coombe, D.R. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 340296. [Google Scholar] [CrossRef] [PubMed]
- Marlin, S.D.; Springer, T.A. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 1987, 51, 813–819. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; deFougerolles, A.R.; Stacker, S.A.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. ICAM-1 (CD54): A counter receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Hum, X.; Wohler, J.E.; Dugger, K.J.; Bamum, S.R. β2-integrins in demyelinating disease: Not adhering to the paradigm. J. Leukoc. Biol. 2010, 87, 397–403. [Google Scholar]
- Roche, Y.; Pasquire, D.; Rambeaud, J.-J.; Seigneurin, D.; Duperray, A. Fibrinogen mediates bladder cancer cell migration in an ICAM-1-dependent pathway. Thromb. Haemost. 2003, 89, 1089–1097. [Google Scholar] [PubMed]
- Strell, C.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Exp. Cell Res. 2010, 316, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-F.; Chen, M.-K.; Hsieh, Y.-S.; Chung, T.-T.; Hsieh, Y.-H.; Lin, C.-W.; Su, J.-L.; Tsai, M.-H.; Tang, C.-H. Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM1) expression and cell motility in oral cancer cells. J. Biol. Chem. 2010, 285, 29808–29816. [Google Scholar] [CrossRef] [PubMed]
- Rosette, C.; Roth, R.B.; Oeth, P.; Braun, A.; Kammerer, S.; Ekblom, J.; Denissenko, M.F. Role of ICAM1 invasion of human breast cancer cells. Carcinogenesis 2005, 26, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, K.; Mikuta-Pietrasik, J.; Catar, R.; Dworacki, G.; Winckiewics, M.; Frydrychowics, M.; Dragun, D.; Staniszewski, R.; Jorres, A.; Witowski, J. Oxidative stress-dependent increase in ICAM-1 expression promotes adhesion of colorectal and pancreatic cancers to the senescent peritoneal mesothelium. Int. J. Cancer 2010, 127, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Huang, J.; Wang, L.; Jia, Y.; Dillon, D.A.; Zurakowski, D.; Mao, H.; Moses, M.A.; Auguste, D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14710–14715. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Dineen, S.P.; Toombs, J.E.; Carbon, J.G.; Smith, C.W.; Brekken, R.A.; Barnet, C.C., Jr. Tumor-derived intercellular adhesion molecule-1 mediates tumor-associated leukocyte infiltration in orthotopic pancreatic xenografts. Exp. Biol. Med. 2010, 235, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.M.; Cummings, M.; Purdie, D.; Chenevix-Trench, G. Reduced expression of intercellular adhesion molecule-1 in ovarian adenocarcinomas. Br. J. Cancer 2001, 85, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- De Groote, M.L.; Kazemier, H.G.; Huisman, C.; van der Gun, B.T.F.; Faas, M.M.; Rots, M.G. Upregulation of endogenous ICAM-1 reduces ovarian cancer cell growth in the absence of immune cells. Int. J. Cancer 2014, 134, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Taverna, P.; Karpf, A.R.; Griffiths, A. Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics 2015, 10, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Matte, I.; lane, D.; Laplante, C.; Rancourt, C.; Piche, A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar] [PubMed]
- Cai, G.; Ma, X.; Zou, W.; Huang, Y.; Zhang, J.; Wang, D.; Chen, B. Prediction value of intercellular adhesion molecule-1 gene polymorphysms for epithelial ovarian cancer risk, clinical features, and prognosis. Gene 2014, 546, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Ghazy, A.A.; El-Etreby, N.M. relevance of HLA-DP/DQ and ICAM-1 SNPs among ovarian cancer patients. Front. Immunol. 2016, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Yang, Y.; Cui, L.; Yang, J.; Li, X.; Yang, Y.; Duan, H. Bisdemethoxycurcumin inhibits ovarian cancer via deducing oxidative stress mediated MMPs expressions. Sci. Rep. 2016, 6, 28773. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Roggiani, F.; Mezzanzanica, D.; Rea, K.; Tomassetti, A. Guidance of signaling activations by cadherins and integrins in epithelial ovarian cancer cells. Int. J. Mol. Sci. 2016, 17, 1387. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-Met by fibronectin and α5β1-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Chiang, C.-Y.; White, E.A.; Schryver, E.M.; Habis, M.; Romero, I.L.; Ladanyi, A.; Penicka, C.V.; George, J.; Matlin, K.; et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J. Clin. Investig. 2014, 124, 4614–4628. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Ozhegov, E.; van den Berg, Y.W.; Aronow, B.J.; Franco, R.S.; Palascak, M.B.; Fallon, J.T.; Ruf, W.; Versteeg, H.H.; Bogdanov, V.Y. Splice variants of tissue factor promote monocyte-endothelial interactions by triggering the expression of cell adhesion molecules via integrin-mediated signaling. J. Thromb. Haemost. 2011, 9, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, M.; Suzuki, T.; Suzuki, M.; Tanaka, R.; Ito, E.; Saito, T. Statin-mediated reduction of osteopontin expression induces apoptosis and cell growth arrest in ovarian clear cell carcinoma. Oncol. Rep. 2011, 25, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Riley, C.; Oliva, K.; Stutt, E.; Rice, G.E.; Quinn, M.A. Integrin-linked kinase expression increases with ovarian tumor grade and is sustained by peritoneal tumour fluid. J. Pathol. 2003, 201, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.P.B.; Naish, J.H.; Parker, G.J.M.; Waterton, J.C.; Watson, Y.; Jayson, G.C.; Buonaccorsi, G.A.; Cheung, S.; Buckley, D.L.; McGrath, D.M.; et al. Preliminary study of oxygen-enhanced longitudinal relaxation in MRI: A potential novel biomarker of oxygenation changes in solid tumors. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp, M.P.; Winner, K.K.; Davies, S.; Muller, C.; Zhang, Y.; Hoffman, R.M.; Shirinifard, A.; Moses, M.; Jiang, Y.; Wilson, B.S. Ovarian tumor attachment, invasion, and vascularization reflect unique microenvironments in the peritoneum: Insights from xenograft and mathematical models. Front. Oncol. 2013, 3, 97. [Google Scholar] [CrossRef] [PubMed]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, L.M.; O’Toole, S.A.; Spillane, C.D.; Martin, C.M.; Gallagher, M.F.; Stordal, B.; Blackshields, G.; Sheils, O.; O’Leary, J.J. Identifying novel hypoxia-associated markers of chemoresistance in ovarian cancer. BMC Cancer 2015, 15, 547. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.F.; malm, S.W.; Pandey, R.; Laughren, C.; Cui, H.; Roe, D.; Chambers, S.K. Evaluation of a hypoxia regulated gene panel in ovarian cancer. Cancer Microenviron. 2015, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contribution to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Versteeg, H.H.; Kerver, M.; Chen, V.M.; Mueller, B.M.; Hogg, P.J.; Ruf, W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13932–13937. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.V.M.; Pendurthi, U.R. Regulation of tissue factor coagulant activity on cell surface. J. Thromb. Haemast. 2012, 10, 2242–2253. [Google Scholar] [CrossRef] [PubMed]
- Langer, F.; Ruf, W. Synthesis of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb. Haemost. 2014, 111, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Bendapudi, P.; Sharda, A.; Chen, V.; Bellido-Martin, L.; Jasuja, R.; Furie, B.C.; Flaumenhaft, R.; Furie, B. Extracellular thiol isomerases and their role in thrombus formation. Antioxid. Redox Signal. 2016, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bekandam, R.H.; Flaumenhaft, R. Inhibition of protein disulfide isomerase in thrombosis. Basic Clin. Pharmacol. Toxicol. 2016, 119, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Santo, N.D.; Ehrisman, J. Research perspective: Potential role of nitazoxanide in ovarian cancer treatment. Old drug, new purpose? Cancers 2013, 5, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.K.; Wang, H.; Yim, A.M.; Naour, F.L.; Brichory, F.; Jang, J.H.; Zhao, R.; Puravs, E.; Tra, J.; Michael, C.W.; et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 2003, 278, 7607–7616. [Google Scholar] [CrossRef] [PubMed]
- Choschzick, M.; Oosterwijk, E.; Muller, V.; Woelber, L.; Simon, R.; Moch, H.; Tennstedt, P. Overexpression of carbonic anhydrase IX (CAIX) is an independent unfavorable prognostic marker in endometrioid ovarian cancer. Virchows Arch. 2011, 459, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Martin, S.; Moss, R.; Durrant, L.; Deen, S. Co-expression of VEGF and CA9 in ovarian high-grade serous carcinoma and relationship to survival. Virchows Arch. 2012, 461, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, C.; Araos, J.; Villalobos, N.R.; Westermeier, F.; Salomon, C.; Beltran, A.R.; Ramirez, M.A.; Gutierrez, J.; Pardo, F.; Leiva, A.; et al. Modulation of intracellular pH in human ovarian cancer. Curr. Mol. Med. 2016, 16, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Wiig, H. Tumor interstitial fluid formation, characterization, and clinical implications. Front. Oncol. 2015, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Narayan, M. pH dependence of the isomerase activity of protein disulfide isomerase: Insights into its functional relevance. Protein J. 2008, 27, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, L.W.; Hirst, T.R.; Freedman, R.B. pH-dependence of the dithiol-oxidizing activity of DsbA (a periplastic protein thiol: Disulphide oxidoreductase) and protein disulphide-isomerase: Studies with a novel simple peptide substrate. Biochem. J. 1996, 315, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Uehara, T.; Nomura, Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J. Biol. Chem. 2000, 275, 10388–10393. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, B.S.; Horsman, M.R.; Vorum, H.; Honore, B.; Overgaard, J.; Alsner, J. Proteins upregulated by mild and severe hypoxia in squamous cell carcinomas in vitro identified by proteomics. Radiother. Oncol. 2009, 92, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Dorfleutner, A.; Ruf, W. Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood 2003, 102, 3998–4005. [Google Scholar] [CrossRef] [PubMed]
- Pampalakis, G.; Politi, A.-L.; Papanastasiou, A.; Sotiropoulou, G. Distinct cholesterogenic and lipidogenic gene expression patterns in ovarian cancer—A new pool of biomarkers. Genes Cancer 2015, 6, 472–479. [Google Scholar] [PubMed]
- Goldman, A.R.; Bitler, B.G.; Schug, Z.; Conejo-Garcia, J.R.; Zhang, R.; Speicher, D.W. The primary effect of the proteome of ARID1A-mutated ovarian clear cell carcinoma is downregulation of the mevalonate pathway at the post-transcriptional level. Mol. Cell Proteom. 2016, 15, 3348–3360. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, N.; Morin, P.J. MicroRNAs in ovarian carcinomas. Endocr. Relat. Cancer 2010, 17, F77–F89. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weithauser, A.; Tabaraie, T.; Steffens, D.; Krankel, N.; Witkowski, M.; Stratmann, B.; Tschoepe, D.; Landmesser, U.; Rauch-Kroehnert, U. Micro-RNA-126 reduces the blood thrombogenicity in diabetes mellitus via targeting tissue factor. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, H.; Ren, J.; Geng, Q.; Song, J.; Lee, C.; Cao, C.; Zhang, J.; Xu, N. MicroRNA-223 inhibits tissue factor expression in vascular endothelial cells. Atherosclerosis 2014, 237, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Liu, J.; He, F.; Xu, W.; Yao, K. Hypoxia-induced deregulation of miR-126 and its regulative effect on VEGF and MMP-9 expression. Int. J. Med. Sci. 2014, 11, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Kojonazarov, B.; Elgheznawy, A.; Popp, R.; Dahal, B.K.; Bohm, M.; Pullamsetti, S.S.; Ghofrani, H.-A.; Godecke, A.; Jungmann, A.; et al. miR-223-IGF-IR signaling in hypoxia- and load-induced right-ventricular failure: A novel therapeutic approach. Cardiovasc. Res. 2016, 111, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.P.; Elley, P.E.; Maroney, S.A.; Mast, A.E. Biology of tissue factor pathway inhibitor. Blood 2014, 123, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.Y.; Tinholt, M.; Stavik, B.; Dahm, A.E.A.; Kanse, S.; Jin, Y.; Seidl, S.; Sahlberg, K.K.; Iversen, N.; Skretting, G.; et al. Effect of hypoxia on tissue factor pathway inhibitor expression in breast cancer. J. Thromb. Haemost. 2016, 14, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Stavik, B.; Espada, S.; Cui, X.Y.; Iversen, N.; Holm, S.; Mowinkel, M.-C.; Halvorsen, B.; Skretting, G.; Sandset, P.M. EPAS1/HIF-2 alpha-mediated downregulation of tissue factor pathway inhibitor leads to a pro-thrombotic potential in endothelial cells. Biochim. Biophys. Acta 2016, 1862, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.; Harkness, T. Development, maintenance, and reversal of multiple drug resistance: At the crossroads of TFPI1, ABC transporters, and HIF1α. Cancers 2015, 7, 2063–2082. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, F.A.; Norris, L.; O’Toole, S.; Mohamed, B.M.; Langhe, R.; O’Leary, J.; Gleeson, N. Tumor expression of tissue factor and tissue factor pathway inhibitor in ovarian cancer-relationship with venous thrombosis risk. Thromb. Res. 2013, 132, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Sotiriou, C.; Cockman, M.E.; Ratcliffe, P.J.; Maxwell, P.; Liu, E.; Harris, A.L. Gene array of VHL mutation and hypoxia shows novel hypoxia-induced genes and that cyclin D1 is a VHL target gene. Br. J. Cancer 2004, 90, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Osada, R.; Horiuchi, A.; Kikuchi, N.; Yoshida, J.; Hayashi, A.; Ota, M.; Katsuyama, Y.; Mellio, G.; Konishi, I. Expression of hypoxia-inducible factor 1α, hypoxia-inducible factor 2α, and von Hippel-Lindau protein in epithelial ovarian neoplasms and allelic loss of von Hippel-Lindau gene: Nuclear expression of hypoxia-inducible factor 1a is an independent prognostic factor in ovarian carcinoma. Hum. Pathol. 2007, 38, 1310–1320. [Google Scholar] [PubMed]
- Yamamoto, S.; Tsuda, H.; Suzuki, K.; Takano, M.; Tamai, S.; Matsubara, O. An allelotype analysis indicating the presence of two distinct ovarian clear-cell carcinogenic pathways: Endometriosis-associated pathway vs. clear-cell adenocarcinoma-associated pathway. Virchows Arch. 2009, 455, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Ellery, P.E.; Wood, J.P.; Ferrel, J.P.; Bonesho, C.E.; Mast, A.E. Caveolae optimize tissue factor-factor VIIa inhibitory activity of cell-surface-associated tissue factor pathway inhibitor. Biochem. J. 2012, 443, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, D.J.; Jack, G.G.; Page, K.L.; Tetztoff, T.A.; Hall, C.L.; Mast, A.E. Localization of tissue factor pathway inhibitor to lipid rafts is not required for inhibition of factor VIIa/tissue factor activity. Thromb. Haemost. 2003, 89, 65–73. [Google Scholar] [PubMed]
- Koike, T.; Kimura, N.; Miyazaki, K.; Yabuta, T.; Kumamoto, K.; Takenoshita, S.; Chen, J.; Kobayashi, M.; Hosokawa, M.; Taniguchi, A.; et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc. Natl. Acad. Sci. USA 2004, 101, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.E.; Park, H.M.; Chung, J.; Lee, C.H.; Park, H.A. Hypoxia-inducible factor-1α mediates oral squamous cell carcinoma invasion via upregulation of α5 integrin and fibronectin. Biochem. Biophys. Res. Commun. 2010, 393, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.-H.; Lee, Y.; Zhao, X.-F.; Park, Y.-K.; Lee, J.W.; Kim, S. ZEB2-Sp1 cooperation induces invasion by upregulating cadherin-11 and integrin α5 expression. Carcinogenesis 2014, 35, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrin. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Petersen, H.H.; Ahamed, J.; Felding-Habermann, B.; Takada, Y.; Mueller, B.M.; Ruf, W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008, 111, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, F.; Miyagi, Y.; Miyagi, E.; Yasumitsu, H.; Koshikawa, N.; Nagashima, Y.; Kitamura, H.; Minaguchi, H.; Umeda, M.; Miyazaki, K. Trypsinogen expression in human ovarian carcinoma. Int. J. Cancer 1995, 63, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Koshikawa, N.; Yasumitsu, H.; Miyazaki, K. Trypsin stimulates integrin α5β1-dependent adhesion to fibronectin and proliferatio of human gastric carcinoma cells through activation of proteinase-activated receptor-2. J. Biol. Chem. 2000, 275, 4592–4598. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.M.; Szabo, R.; Lee, M.; Kirchhofer, D.; Craik, C.S.; Bugge, T.H.; Camerer, E. Matriptase activation connects tissue factor-dependent coagulation initiation to epithelial proteolysis and signaling. Blood 2016, 127, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Oberst, M.D.; Johnson, M.D.; Dickson, R.B.; Lin, C.-Y.; Singh, B.; Stewart, M.; Williams, A.; Al-Nafussi, A.; Smyth, J.F.; Gabra, H.; et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: Correlation with clinical outcome and tumor clinicopathological parameters. Clin. Cancer Res. 2002, 8, 1101–1107. [Google Scholar] [PubMed]
- Tanimoto, H.; Shigemasa, K.; Tian, X.; Gu, L.; Beard, J.B.; Sawasaki, T.; O’Brien, T.J. Transmemebrane serine protease TADG-15 (ST14/Matriptase/MT-SP1): Expression and prognostic value in ovarian cancer. Br. J. Cancer 2005, 92, 278–283. [Google Scholar] [PubMed]
- Jin, J.-S.; Hsieh, D.-S.; Loh, S.-H.; Chen, A.; Yao, C.-W.; Yen, C.-Y. Increasing expression of serine protease matriptase in ovarian tumors: Tissue microarray analysis of immunostaining score with clinicopathological parameters. Mod. Pathol. 2006, 19, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jiang, Z.; Chen, X.; Xue, L.; Mao, X.; Ruan, G.; Song, Y.; Mustea, A. Decreasing the ratio of matriptase/HAI-1 by downregulation of matriptase as a potential adjuvant therapy in ovarian cancer. Mol. Med. Rep. 2016, 14, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.; Heiden, M.; Seitz, R.; Salge-Bartels, U. Inducible expression of tissue factor in small cell-lung cancer: Impact on morphology and matrix metalloproteinase secretion. J. Cancer Res. Clin. Oncol. 2012, 138, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Schmalfeldt, B.; Prechtel, D.; Härting, K.; Späthe, K.; Rutke, S.; Konik, E.; Fridman, R.; Berger, U.; Schmitt, M.; Kuhn, W.; et al. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin. Cancer Res. 2001, 7, 2396–2404. [Google Scholar] [PubMed]
- Sebastiano, M.; Momi, S.; Falcinelli, E.; Bury, L.; Hoylaerts, M.F.; Gresele, P. A novel mechanism regulating human platelet activation by MMP-2-mediated PAR1 biased signaling. Blood 2017, 129, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Covic, L.; Sevigny, L.M.; Kaneider, N.C.; Lazarides, K.; Azabdaftari, G.; Sharifi, S.; Kuliopulos, A. Targeting a metalloprotease-PAR1 signaling system with cell-penetrating pepducins inhibits angiogenesis, ascites, and progression of ovarian cancer. Mol. Cancer Ther. 2008, 7, 2746–2757. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.-C.; Zhang, T.; Li, W.-P. Thrombin promotes epithelial ovarian cancer cell invasion by inducing epithelial-mesenchymal transition. J. Gynecol. Oncol. 2013, 24, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, K.; Gu, Y.; Feng, B.; Ren, G.; Zhang, L.; wang, Y.; Nie, Y.; Fan, D. Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem. Biophys. Res. Commun. 2011, 415, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Diverse mechanisms of Sp1-dependent transcriptional regulation potentially involved in the adaptive response of cancer cells to oxygen-deficient conditions. Cancers 2016, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Torres-Collado, A.X.; Kisiel, W.; Iruela-Arispe, M.L.; Rodriguez-Manzaneque, J.C. ADAMTS1 interacts with, cleaves, and modifies the extracellular location of the matrix inhibitor tissue factor pathway inhibitor-2. J. Biol. Chem. 2006, 281, 17827–17873. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.A.; Dos Santos, L.; Turri, J.A.; Nonogaki, S.; Buim, M.; Lima, J.F.; de Jesus Viana Pinheiro, J.; Bueno de Toledo Osório, C.A.; Soares, F.A.; Freitas, V.M. Prognostic value of ADAMTS proteases and their substrates in epithelial ovarian cancer. Pathobiology 2016, 83, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Anti-apoptotic genes are synergistically activated in OVSAYO cells cultured under conditions of serum starvation and hypoxia. Genom. Data 2015, 5, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Kyle, A.H. Drug penetration and therapeutic resistance. In Tumor Microenvironment; Wiley-Blackwell: West Sussex, UK, 2011; pp. 329–352. [Google Scholar]
- Asher, V.; Lee, J.; Bail, A. Preoperative serum albumin is an independent prognostic predictor of survival in ovarian cancer. Med. Oncol. 2012, 29, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Jin, C.; Zheng, H.-M.; Zhou, K.; Shi, B.-B.; Zhang, Q.; Zheng, F.-Y.; Lin, F. A novel prognostic inflammation score predicts outcomes in patients with ovarian cancer. Clin. Chim. Acta 2016, 456, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.J.; Howarth, D.J.; Attfield, J.C.; Cooke, C.J.; Nanjee, M.N.; Olszewski, W.L.; Morrissey, J.H.; Miller, N.E. Haemostatic factors in human peripheral afferent lymph. Thromb. Haemost. 2000, 83, 427–432. [Google Scholar] [PubMed]
- Haslene-hox, H.; Oveland, E.; Woie, K.; Salvesen, H.B.; Tenstad, O.; Wiig, H. Distribution volumes of macromolecules in human ovarian and endometrial cancers—effects of extracellular matrix structure. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H18–H28. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Takeda, J.; Fukase, M.; Motoyama, T. Hyalinized stroma in clear cell carcinoma of the ovary: How is it formed? Hum. Pathol. 2012, 43, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Krol, A.; Maresca, J.; Dewhirst, M.W.; Yuan, F. Available volume fraction of macromolecules in the extravascular space of a fibrosarcoma: Implications for drug delivery. Cancer Res. 1999, 59, 4136–4141. [Google Scholar] [PubMed]
- John, E.; Laskow, T.C.; Buchser, W.J.; Pitt, B.R.; Basse, P.H.; Butterfield, L.H.; Kalinski, P.; Lotze, M.T. Zinc in innate and adaptive tumor immunity. J. Transl. Med. 2010, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.U.; Kazi, T.G.; Afridi, H.I.; Jamali, M.K.; Arain, M.B.; Jalbani, N.; Syed, N. Evaluation of zinc status in whole blood and scalp hair of female cancer patients. Clin. Chim. Acta 2007, 379, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Siren, P.M.A.; Siren, M.J. Systemic zinc redistribution and dyshomeostasis in cancer cachexia. J. Cachexia Sarcopenia Muscle 2010, 1, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ezeoke, C.C.; Morley, J.E. Pathophysiology of anorexia in the cancer cachexia syndrome. J. Cachexia Sarcopenia Muscle 2015, 6, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the hallmarks of cancer. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Marrero, A.; Gomis-Rüth, F.X. Matrix metalloproteinases: Fold and function of the catalytic domains. Biochim. Biophys. Acta 2010, 1803, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Duan, H.; Liu, J.; Mo, Q.; Sun, C.; Ma, D.; Wang, J. Effect of LIV1 on the sensitivity of ovarian cancer cells to trichostatin A. Oncol. Rep. 2015, 33, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.; Smith, L.M.; Anderson, M.E.; Duniho, S.; Hunter, J.H.; Kostner, H.; Miyamoto, J.B.; Nesterova, A.; Westendorf, L.; Van Epps, H.A.; et al. SGN-LIV1A: A novel antibody-drug conjugate targeting LIV-1 for the treatment of metastatic breast cancer. Mol. Cancer Ther. 2014, 13, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Desouki, M.M.; Franklin, R.B.; Costello, L.C.; Fadare, O. Persistent low expression of hZip1 in mucinous carcinomas of the ovary, colon, stomach, and lung. J. Ovarian Res. 2015, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Kelleher, S.L. Cellular mechanisms of zinc dysregulation: A perspective on zinc homeostasis as an etiological factor in the development and progression of breast cancer. Nutrients 2012, 4, 875–903. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-C.; Jacobs, B.; Becker, E.B.E.; Glitsch, M.D. Reciprocal regulation of two G protein-coupled receptors sensing extracellular concentrations of Ca2+ and H+. Proc. Natl. Acad. Sci. USA 2015, 112, 10738–10743. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, N.; Benabbas, C.; Vidal, S.; Daryadel, A.; Bourgeois, S.; Velic, A.; Ludwig, M.-G.; Seuwen, K.; Wagner, C.A. The proton-activated G protein coupled receptor OGR1 acutely regulates the activity of epithelial proton transport proteins. Cell Physiol. Biochem. 2012, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Swietach, P.; Vaughan-Jones, R.D.; Ansorge, O.; Glitsch, M.D. Extracellular acidification elicits spatially and temporally distinct Ca2+ signals. Curr. Biol. 2008, 18, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Casey, G. Identification of human OGR1, a novel protein-coupled receptor tha maps to chromosome 14. Genomics 1996, 35, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, T.; Simmen, T. Endoplasmic reticulum chaperones and oxidoreductases: Critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 2014, 4, 291. [Google Scholar] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER stress-mediated signaling: Action potential and Ca2+ as key players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Avezov, E.; Konno, T.; Zyryanova, A.; Chen, W.; Laine, R.; Crespillo-Casado, A.; Melo, E.P.; Ushioda, R.; Nagata, K.; Kaminski, C.F.; et al. Retarded PDI diffusion and a reductive shift in poise of the calcium depleted endoplasmic reticulum. BMC Biol. 2015, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Peters, T. Ligand binding by albumin. In All about Albumin: Biochemistry, Genetics and Medical Applications; Peters, T., Ed.; Academic Press: Cambridge, MA, USA, 1995; pp. 79–132. [Google Scholar]
- Schauer, I.G.; Sood, A.K.; Mok, S.; Liu, J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 2011, 13, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; Howell, V.M.; Colvin, E.K. The extracellular matrix in epithelial ovarian cancer—A piece of a puzzle. Front. Oncol. 2015, 5, 245. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endotherial cells: Thromboxane A2 as a new player. Vasc. Pharmacol. 2014, 62, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Carneiro-Lobo, T.; Lima, M.T.; Mariano-Oliveira, A.; Dutra-Oliveira, A.; Oba-Shinjo, S.; Marie, S.K.N.; Sogayar, M.C.; Monteiro, R.Q. Expression of tissue factor signaling pathway elements correlates with the production of vascular endothelial growth factor and interleukin-8 in human astrocytoma patients. Oncol. Rep. 2014, 31, 679–686. [Google Scholar] [PubMed]
- Zacharski, L.R.; Memoli, V.A.; Ornstein, D.L.; Rousseau, S.M.; Kisiel, W.; Kudryk, B.J. Tumor cell procoagulant and urokinase expression in carcinoma of the ovary. J. Natl. Cancer Inst. 1993, 85, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Dunwiddie, C.; Nutt, E.M.; Hunt, J.; Memoli, V.A. Cellular localization of activated factor X by Xa-specific probes. Thromb. Haemost. 1991, 65, 545–548. [Google Scholar] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumor metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C.; Bennett-Cain, A.L.; DeMars, C.S.; Doellgast, G.J.; Grant, K.W.; Jones, N.L.; Gupta, M. Procoagulant activity after exposure of monocyte-derived macrophages to minimally oxidized low density lipoprotein. Co-localization of tissue factor antigen and Nascent fibrin fibers at the cell surface. Am. J. Pathol. 1995, 147, 1029–1040. [Google Scholar] [PubMed]
- Furlan-Freguia, C.; Marchese, P.; Gruber, A.; Ruggeri, Z.M.; Ruf, W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J. Clin. Investig. 2011, 121, 2932–2944. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumor-associated macrophages by tumor-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.O.; Simpson-Haidaris, P.J. Regulated de novo biosynthesis of fibrinogen in extrahepatic epithelial cells in response to inflammation. Thromb. Haemost. 2004, 92, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Simpson-Haidaris, P.J.; Sahni, S.-K.; Vadays, G.G.; Francis, C.W. Fibrinogen synthesized by cancer cells augments the proliferative effect of fibroblast growth factor-2 (FGF-2). J. Thromb. Haemost. 2007, 6, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Svenson, K.J.; Kucharzewska, P.; Christianson, H.C.; Skold, S.; Lofstedt, T.; Johansson, M.C.; Morgelin, M.; Benqzon, J.; Ruf, W.; Belting, M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13147–13152. [Google Scholar] [CrossRef] [PubMed]
- Haslene-Hox, H.; Madani, A.; Berg, K.C.G.; Woie, K.; Salvesen, H.B.; Wiig, H.; Tenstad, O. Quantification of the concentration gradient of biomarkers between ovarian carcinoma interstitial fluid and blood. BBA Clin. 2014, 2, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Alberio, L. The new direct oral anticoagulants in special indications: Rationale and preliminary data in cancer, mechanical heart valves, antiphospholipid syndrome, and heparin-induced thrombocytopenia and beyond. Semin. Hematol. 2014, 51, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Minton, A.R.; Peters, M.C.; van Ryn, J.; Gilmour, S.K. Thrombin inhibition and cisplatin block tumor progression in ovarian cancer by alleviating the immunosuppressive microenvironment. Oncotarget 2016, 7, 85291–85305. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Geddings, J.E.; Ay, C.; Mackman, N. Venous thrombosis and cancer: From mouse models to clinical trials. J. Thromb. Haemost. 2015, 13, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Price, J.M.; Robinson, S.P.; Koh, D.M. Imaging hypoxia in tumours with advanced MRI. Q. J. Nucl. Med. Mol. Imaging 2013, 57, 257–270. [Google Scholar] [PubMed]
- Metran-Nascente, C.; Yeung, I.; Vines, D.C.; Metser, U.; Dhani, N.C.; Green, D.; Milosevic, M.; Jaffray, D.; Hedley, D.W. Measurement of tumor hypoxia in patients with advanced pancreatic cancer based on 18F-fluoroazomycin. J. Nucl. Med. 2016, 57, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Nemieboka, B.; Sala, E.; Lewis, J.S.; Zeglis, B.M. Molecular imaging of ovarian cancer. J. Nucl. Med. 2016, 57, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Canese, R.; Mezzanzanica, D.; Bagnoli, M.; Indraccolo, S.; Canevari, S.; Podo, F.; Iorio, E. In vivo magnetic resonance metabolic and morphofunctional fingerprints in experimental models of human ovarian cancer. Front. Oncol. 2016, 6, 164. [Google Scholar] [CrossRef] [PubMed]
- Philips, R.M. Targeting the hypoxic fraction of tumor using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Boss, M.-K.; Dewhirst, M.W. Imaging tumor hypoxia to advance radiation oncology. Antioxid. Redox Signal. 2014, 21, 313–337. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, Y.; Ohno, T.; Kato, S.; Suzuki, M.; Morita, S.; Sato, S.; Oka, K.; Tsujii, H. Carbon beam therapy overcomes the radiation resistance of uterine cervical cancer originating from hypoxia. Clin. Cancer Res. 2006, 12, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Armaiz-Pena, G.N.; Lin, Y.G.; Davis, A.N.; Pradeep, S.; Dalton, H.J.; Hansen, J.M.; Merritt, W.M.; Nick, A.M.; Langley, R.R.; et al. Dual metronomic chemotherapy with nab-paclitaxel and topotecan has potent antiangiogenic activity in ovarian cancer. Mol. Cancer Ther. 2015, 14, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Duda, D.G.; Sahani, D.V.; Ancukiewicz, M.; Fidias, P.; Sequist, L.V.; Temel, J.S.; Shaw, A.T.; Pennel, N.A.; Neal, J.W.; et al. Improved tumor vascularization after anti-VEGF therapy with carboplatin and nab-paclitaxel associates with survival in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef] [PubMed]
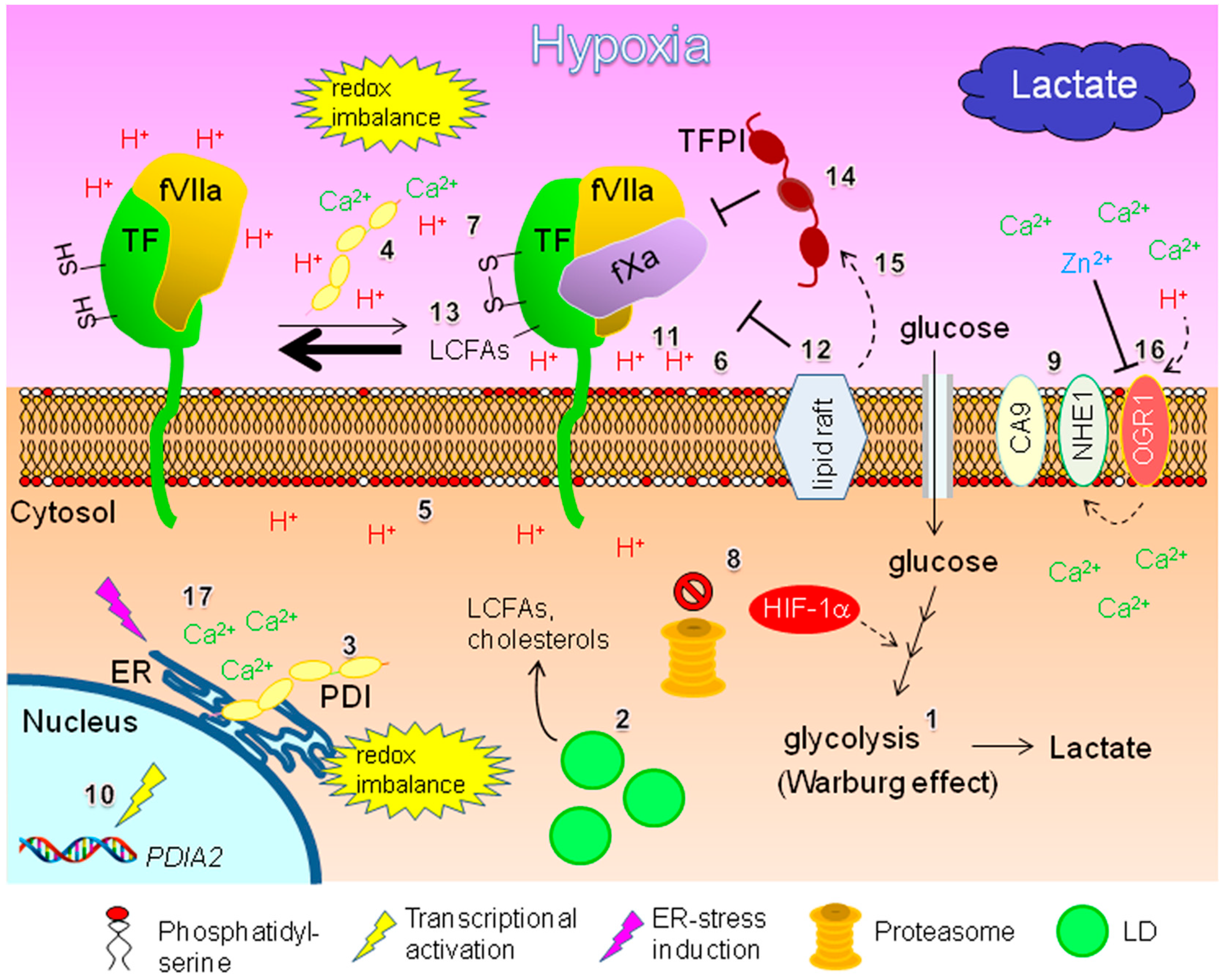
 : cleavage;
: cleavage;  : conversion.
: cleavage; : conversion.
: conversion.
: cleavage; : conversion.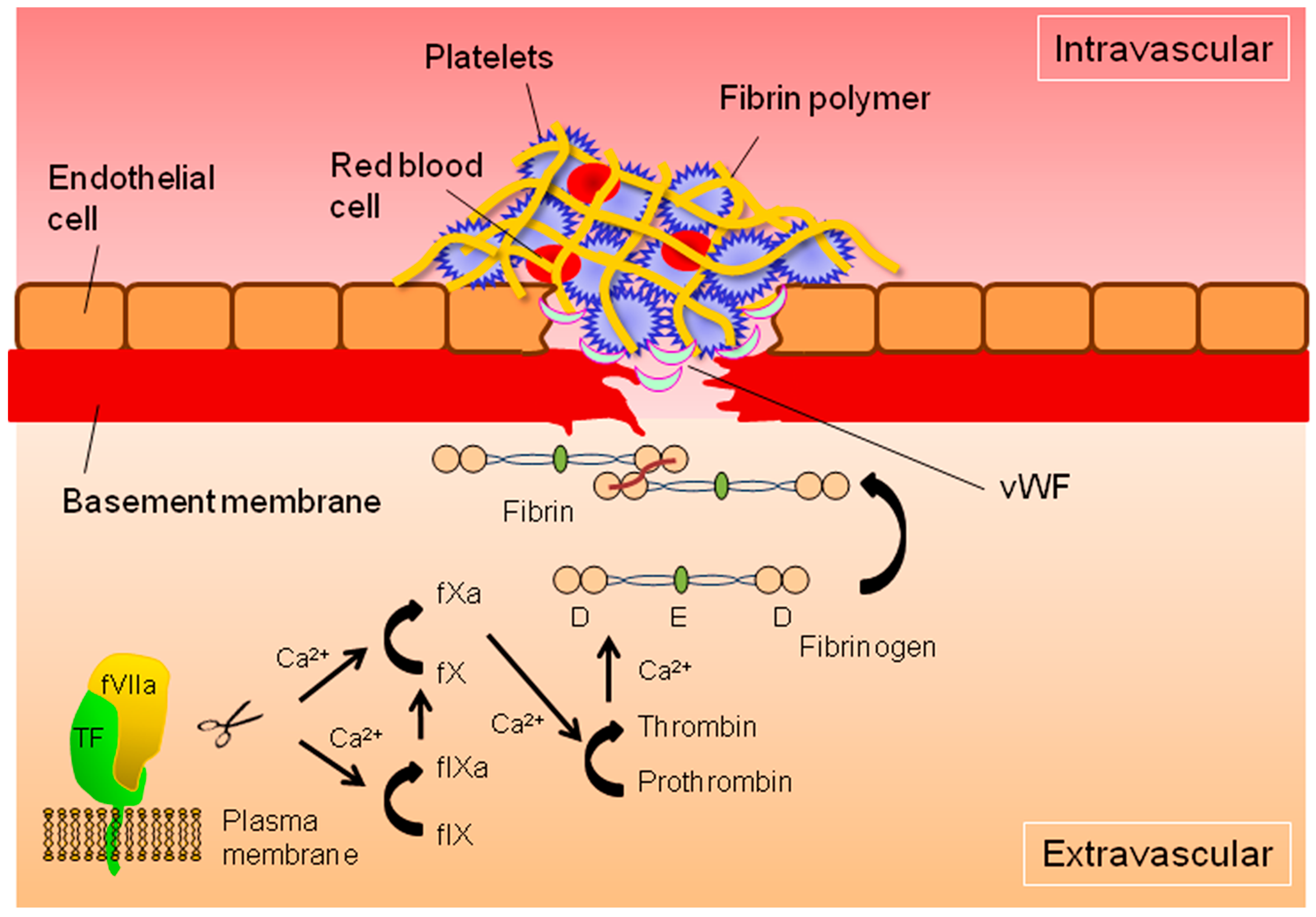 : cleavage; : conversion;
: cleavage; : conversion;  : binding;
: binding;  : intracellular signaling.
: cleavage; : conversion; : binding; : intracellular signaling.
: intracellular signaling.
: cleavage; : conversion; : binding; : intracellular signaling.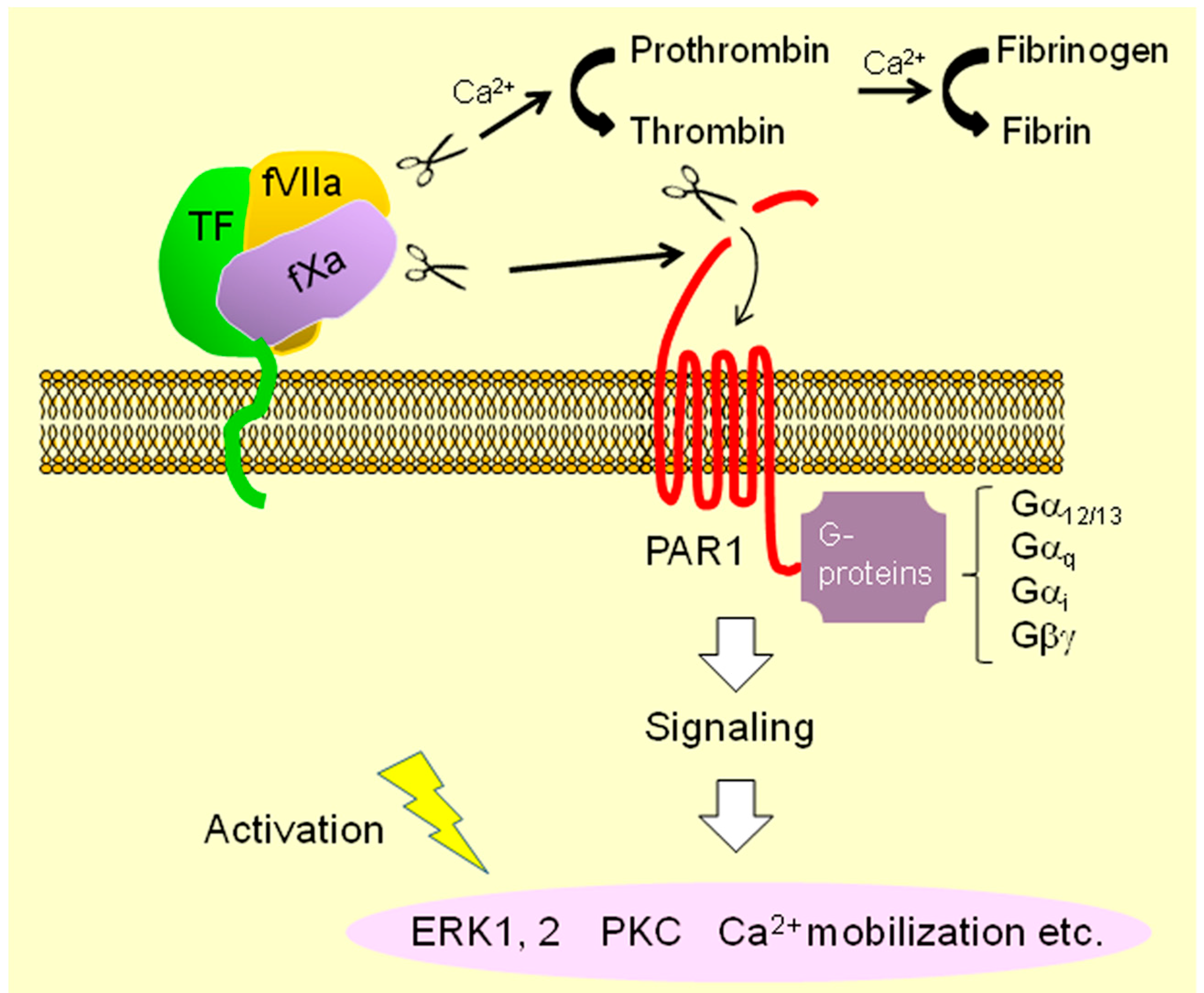
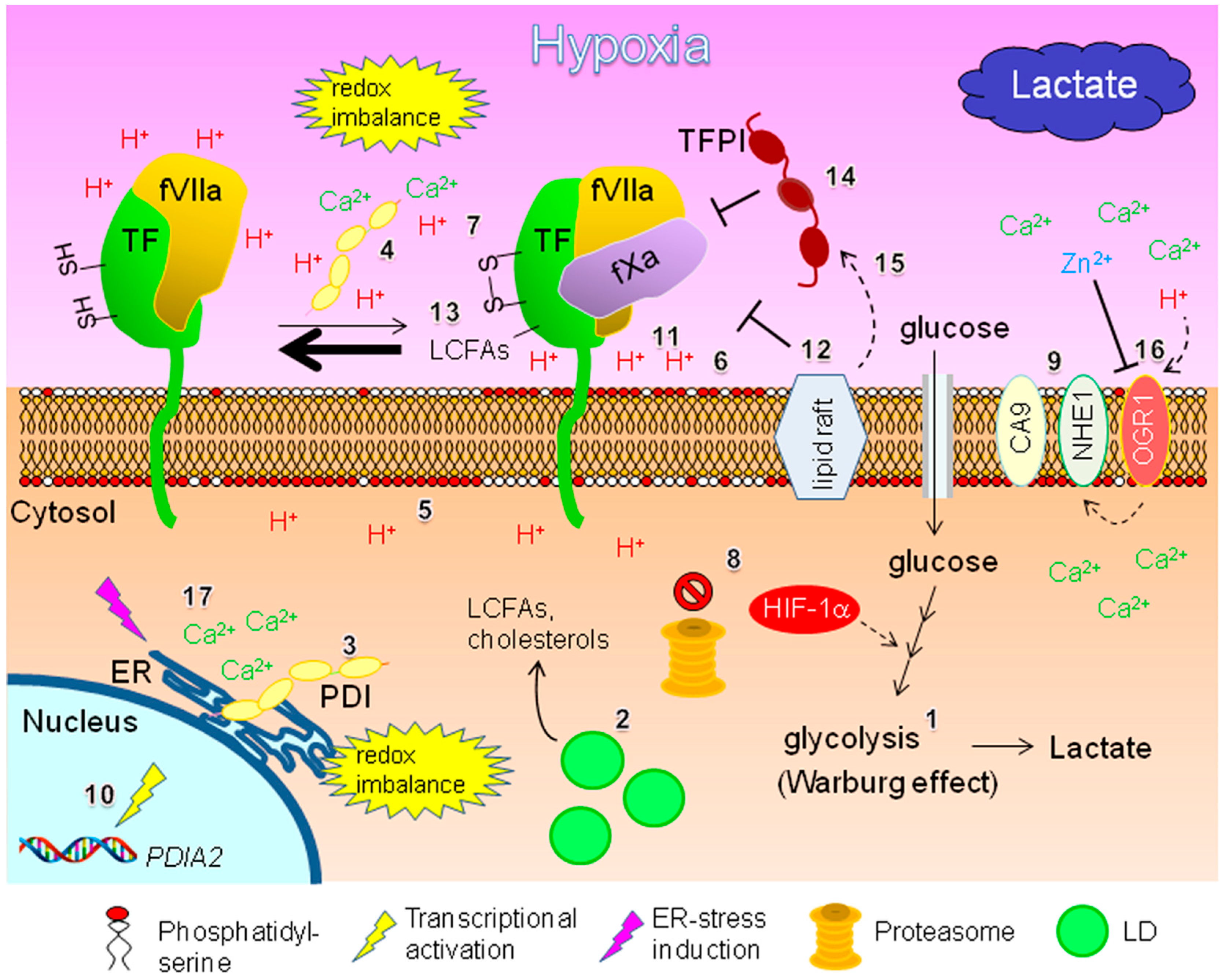
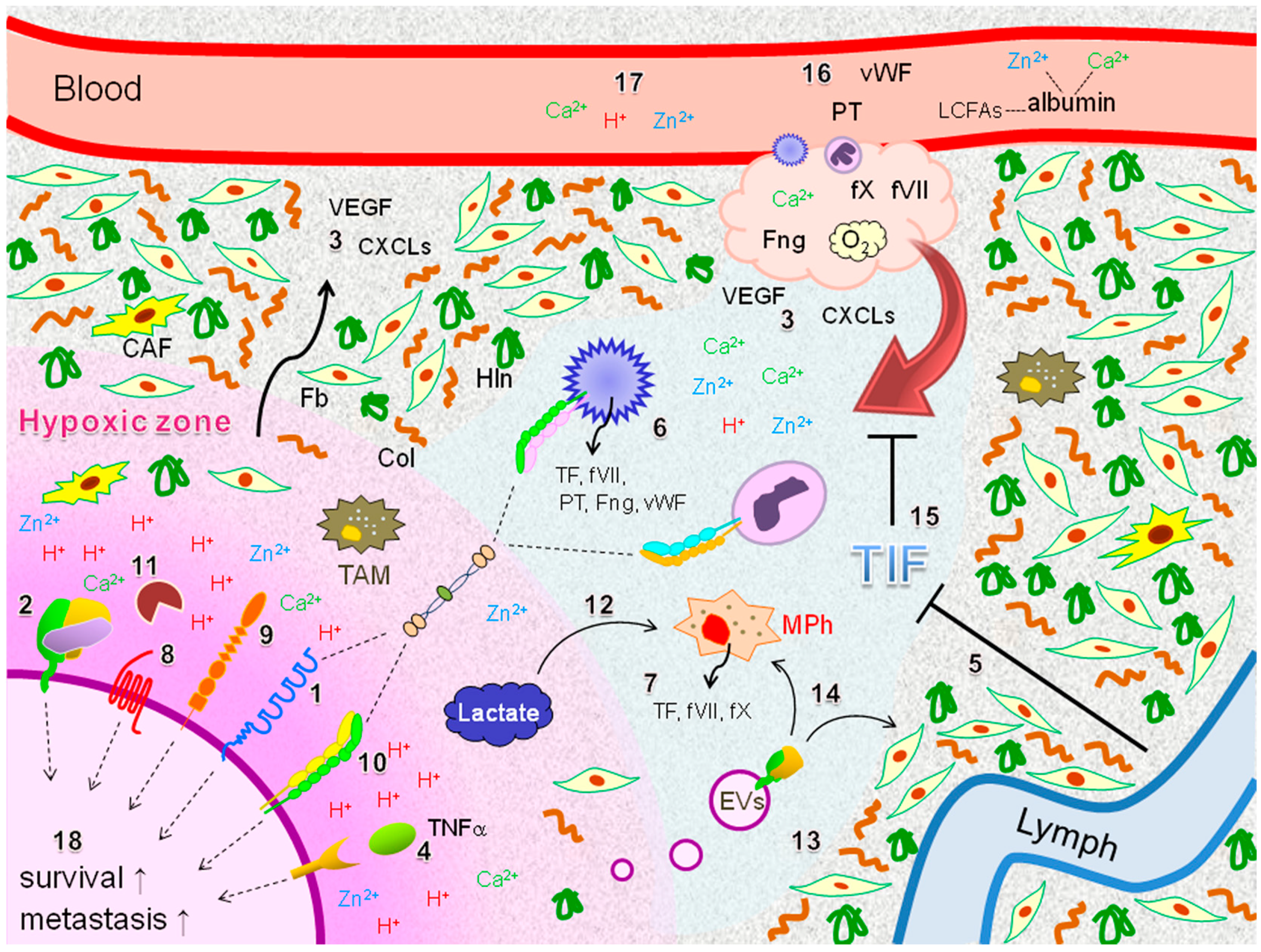
 : secretion;
: secretion;  : interaction;
: interaction;  : intracellular signaling.
: secretion; : interaction; : intracellular signaling.
: intracellular signaling.
: secretion; : interaction; : intracellular signaling.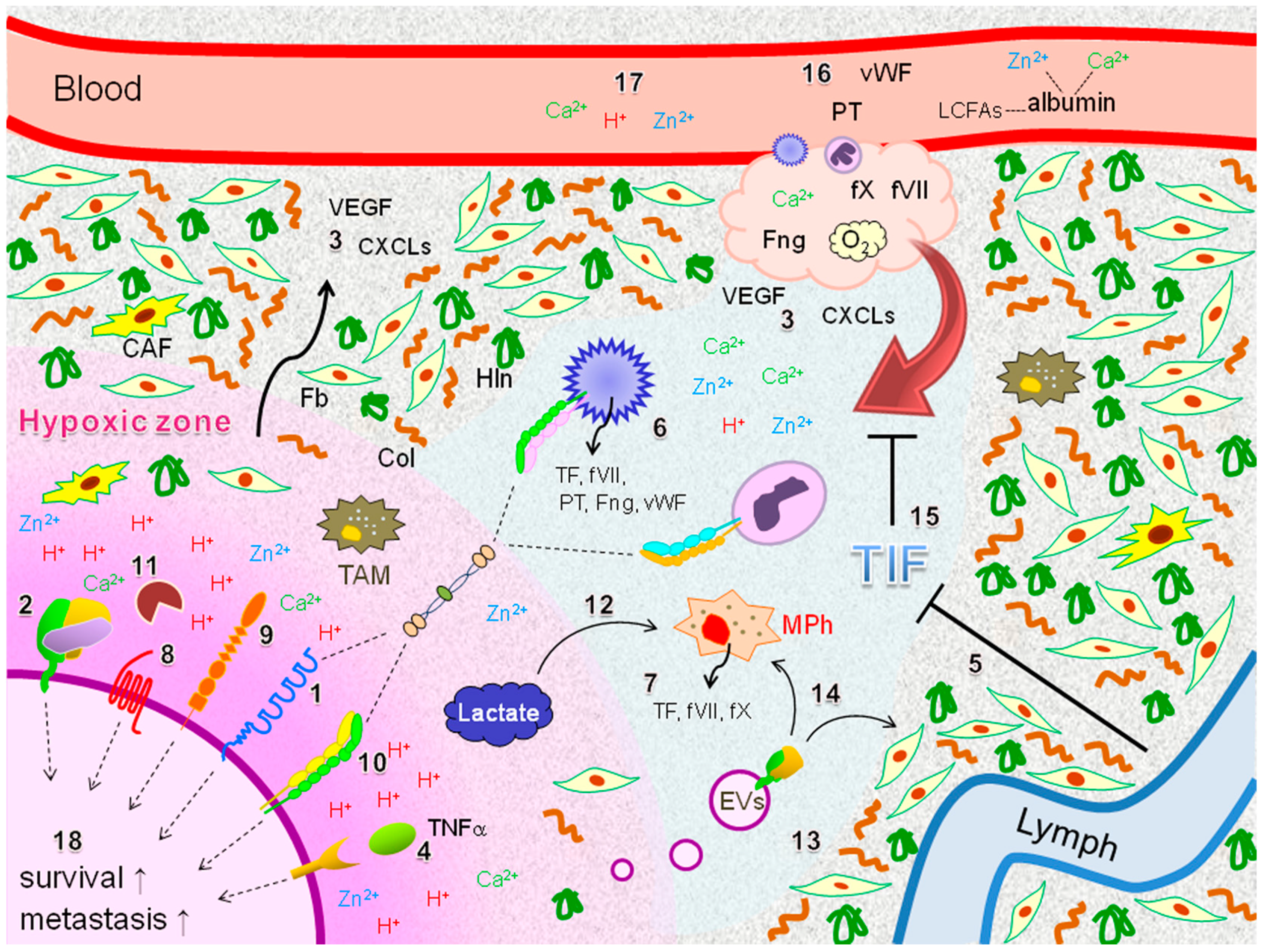
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koizume, S.; Miyagi, Y. Potential Coagulation Factor-Driven Pro-Inflammatory Responses in Ovarian Cancer Tissues Associated with Insufficient O2 and Plasma Supply. Int. J. Mol. Sci. 2017, 18, 809. https://doi.org/10.3390/ijms18040809
Koizume S, Miyagi Y. Potential Coagulation Factor-Driven Pro-Inflammatory Responses in Ovarian Cancer Tissues Associated with Insufficient O2 and Plasma Supply. International Journal of Molecular Sciences. 2017; 18(4):809. https://doi.org/10.3390/ijms18040809
Chicago/Turabian StyleKoizume, Shiro, and Yohei Miyagi. 2017. "Potential Coagulation Factor-Driven Pro-Inflammatory Responses in Ovarian Cancer Tissues Associated with Insufficient O2 and Plasma Supply" International Journal of Molecular Sciences 18, no. 4: 809. https://doi.org/10.3390/ijms18040809