The Quiescent Cellular State is Arf/p53-Dependent and Associated with H2AX Downregulation and Genome Stability

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
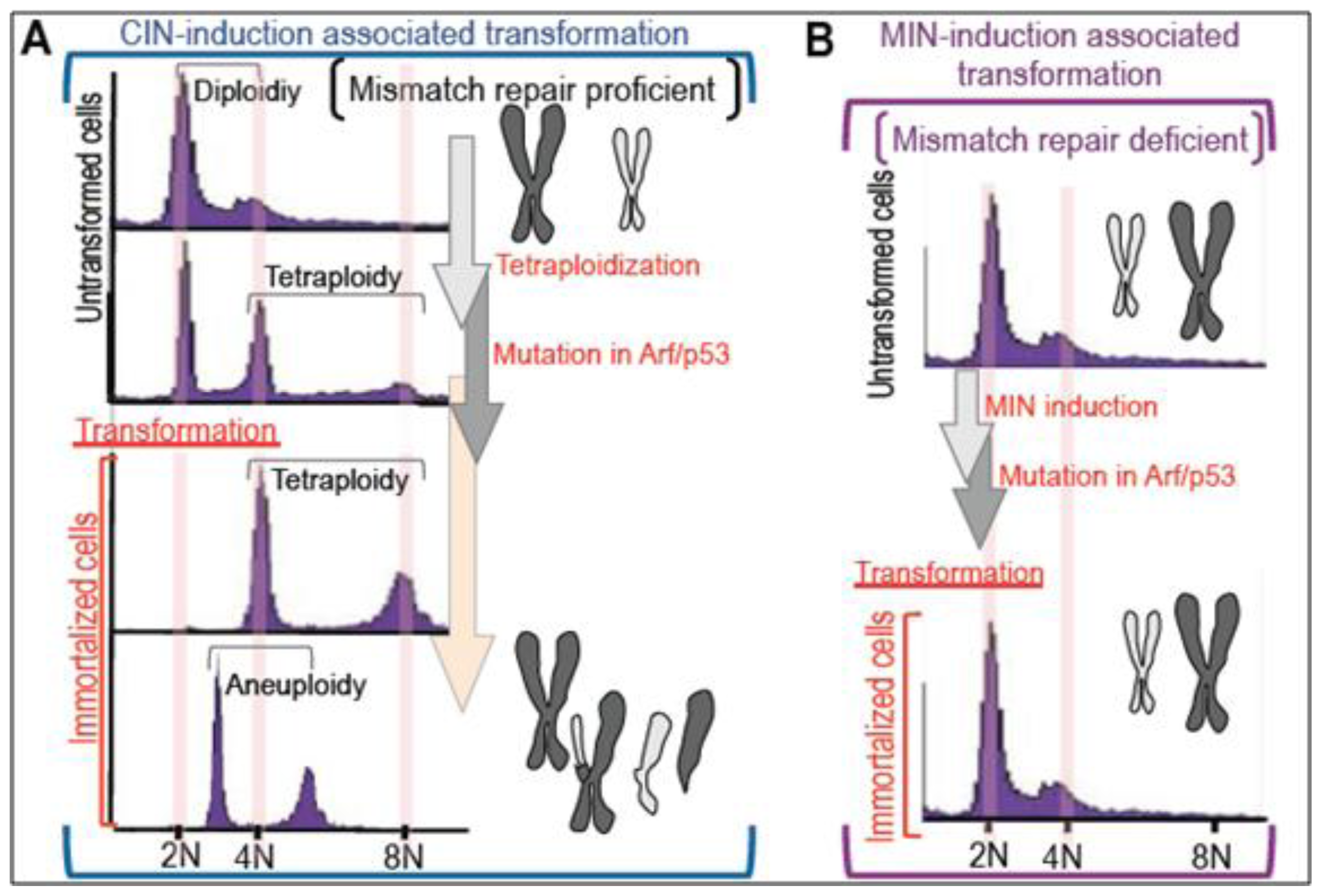
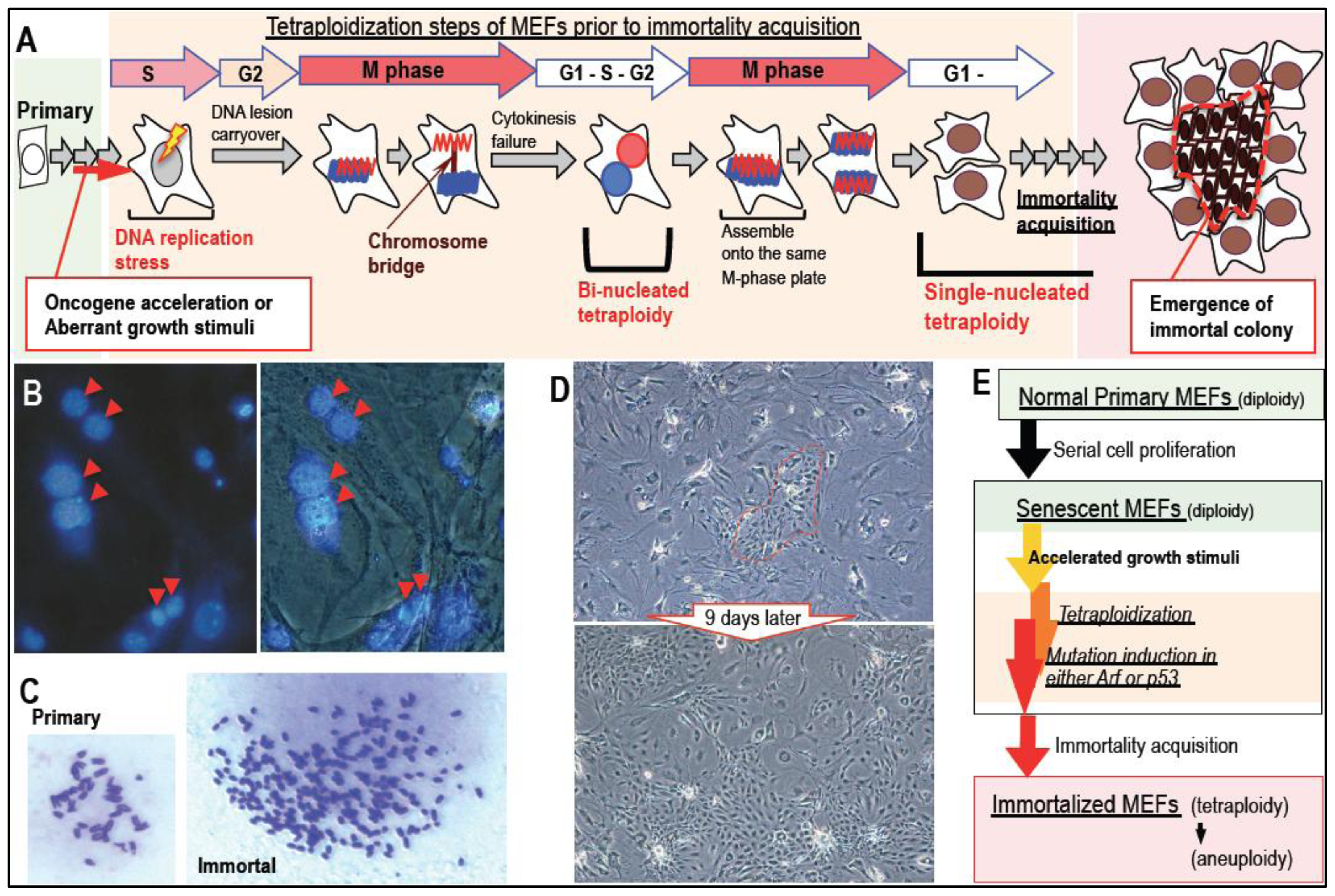
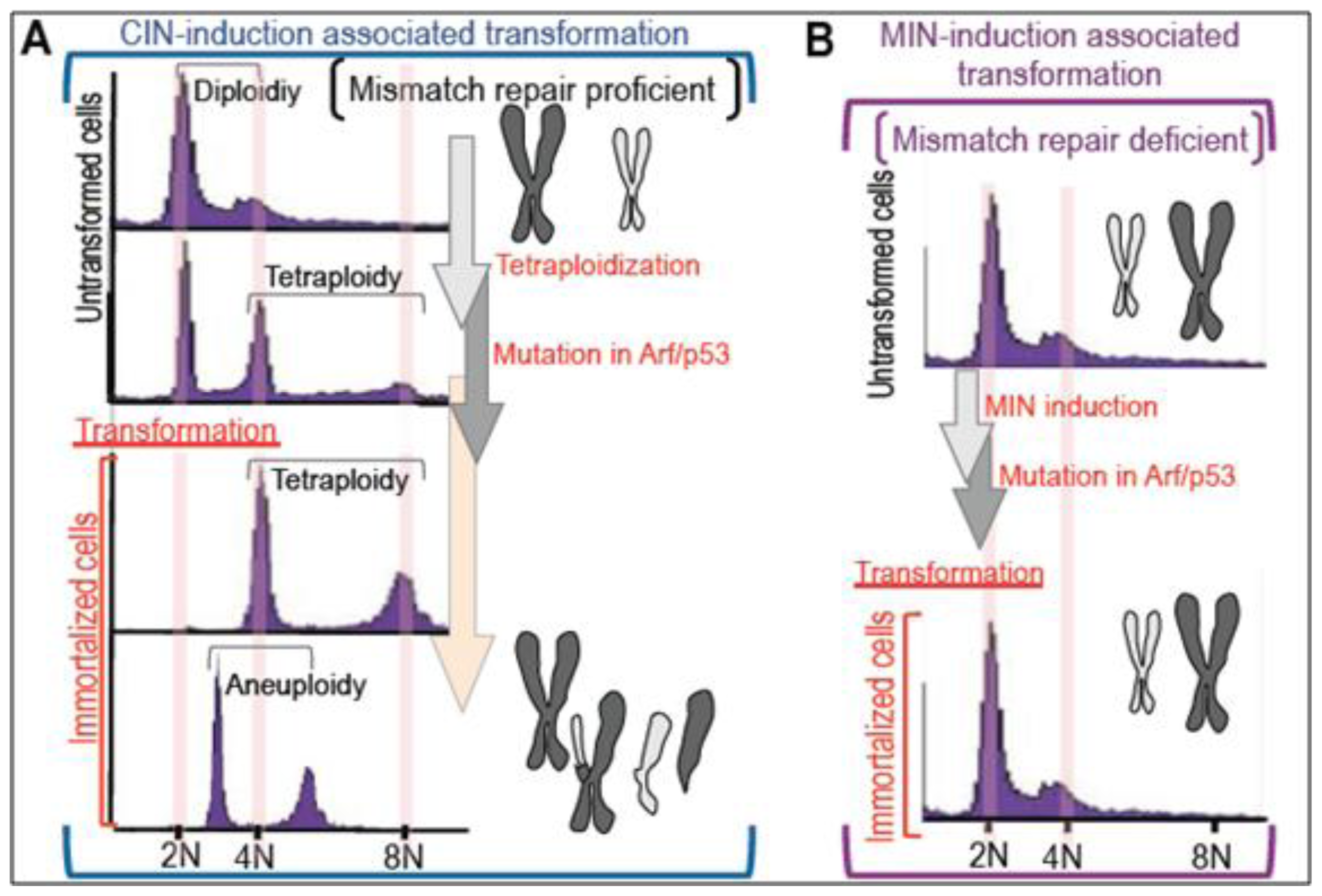
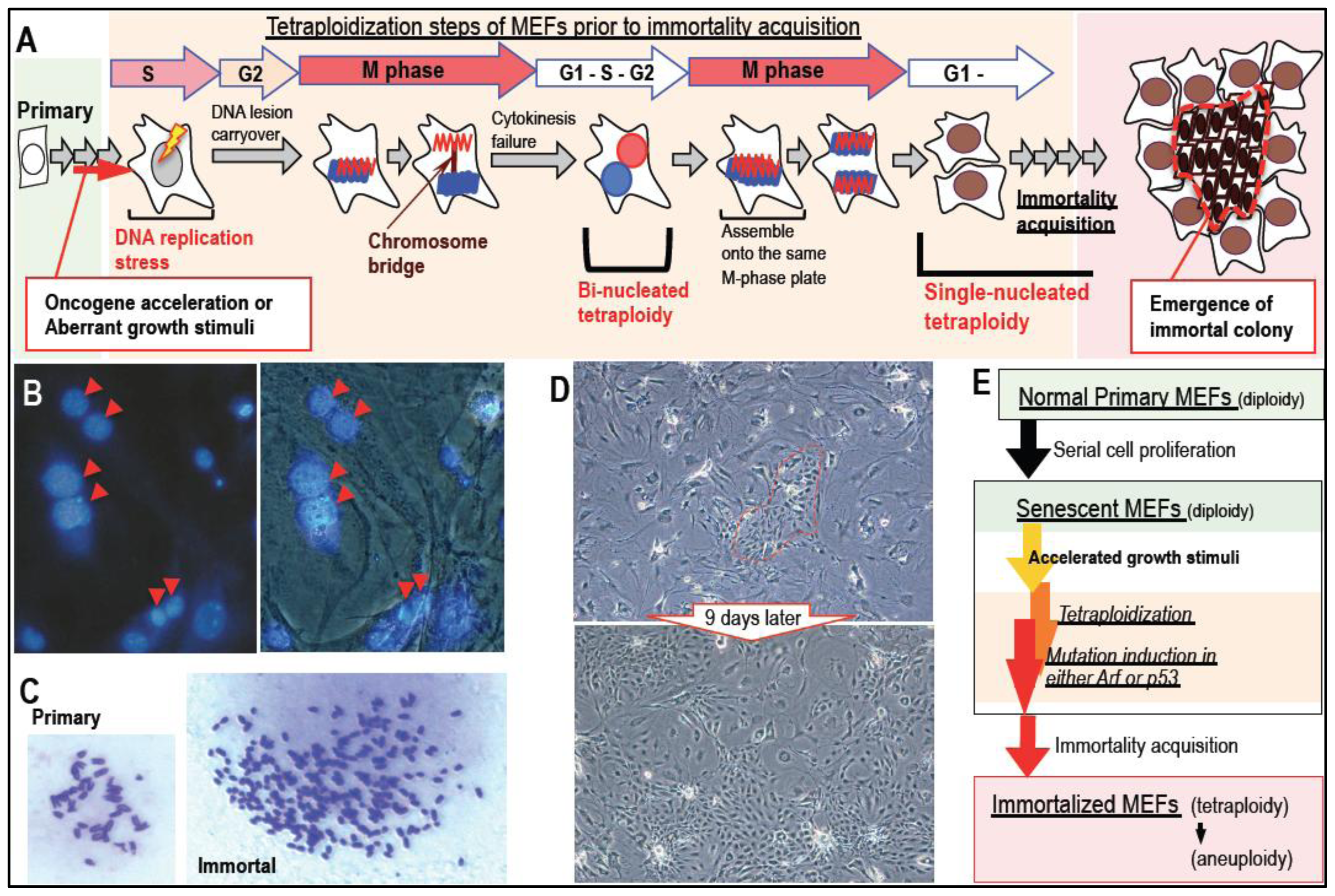
2. Massive Genomic Rearrangements during Cellular Transformation are Associated with Tetraploidization
3. Mutation Induction during the Development of Tetraploidy
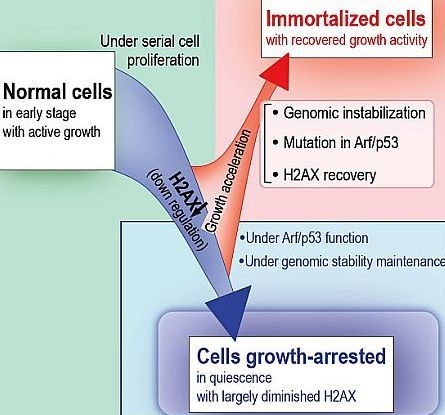
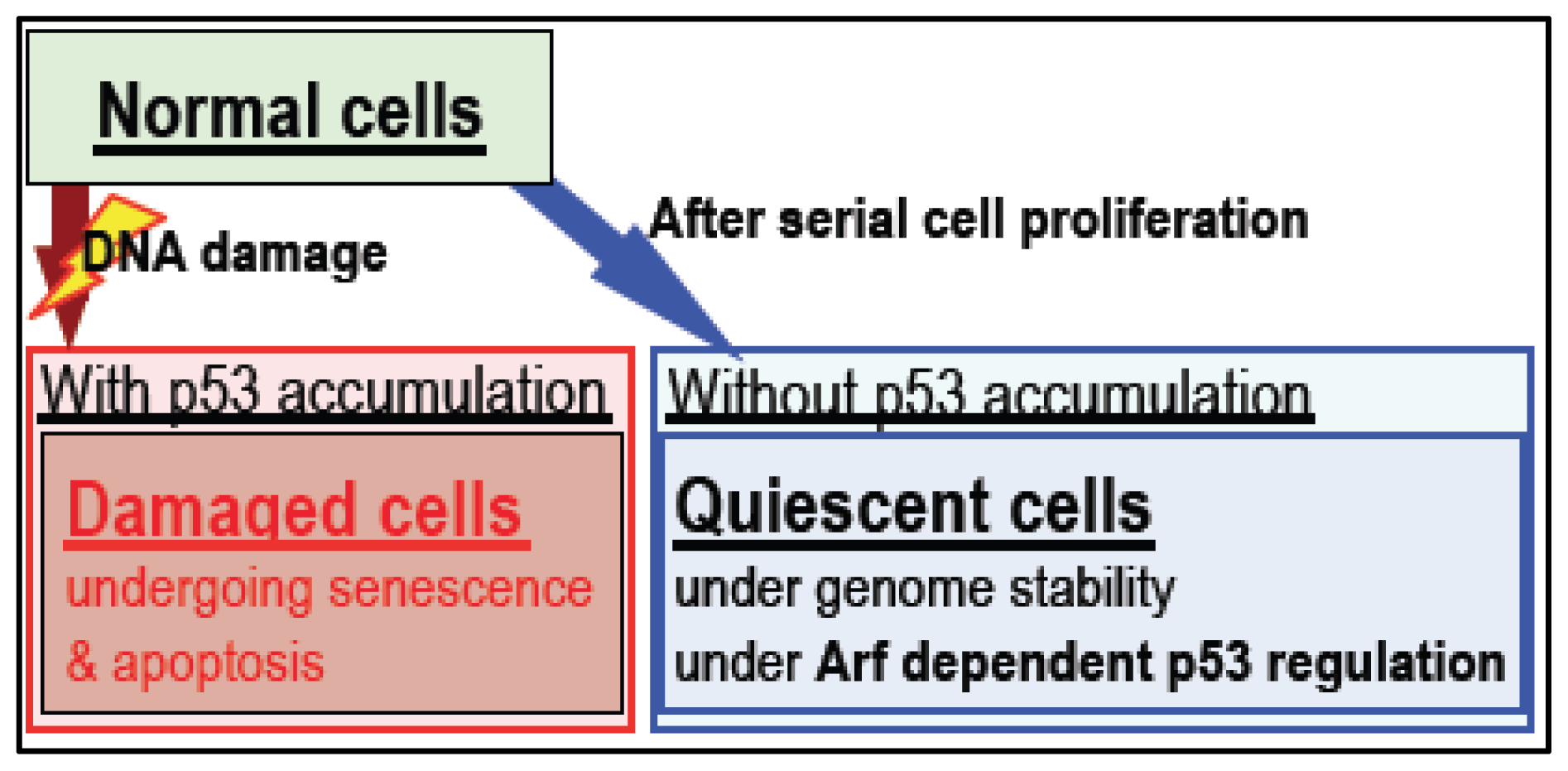
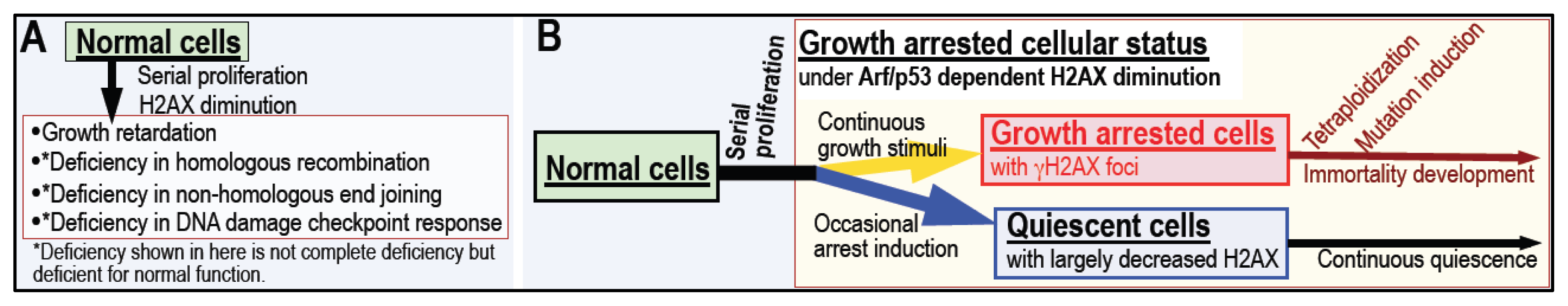
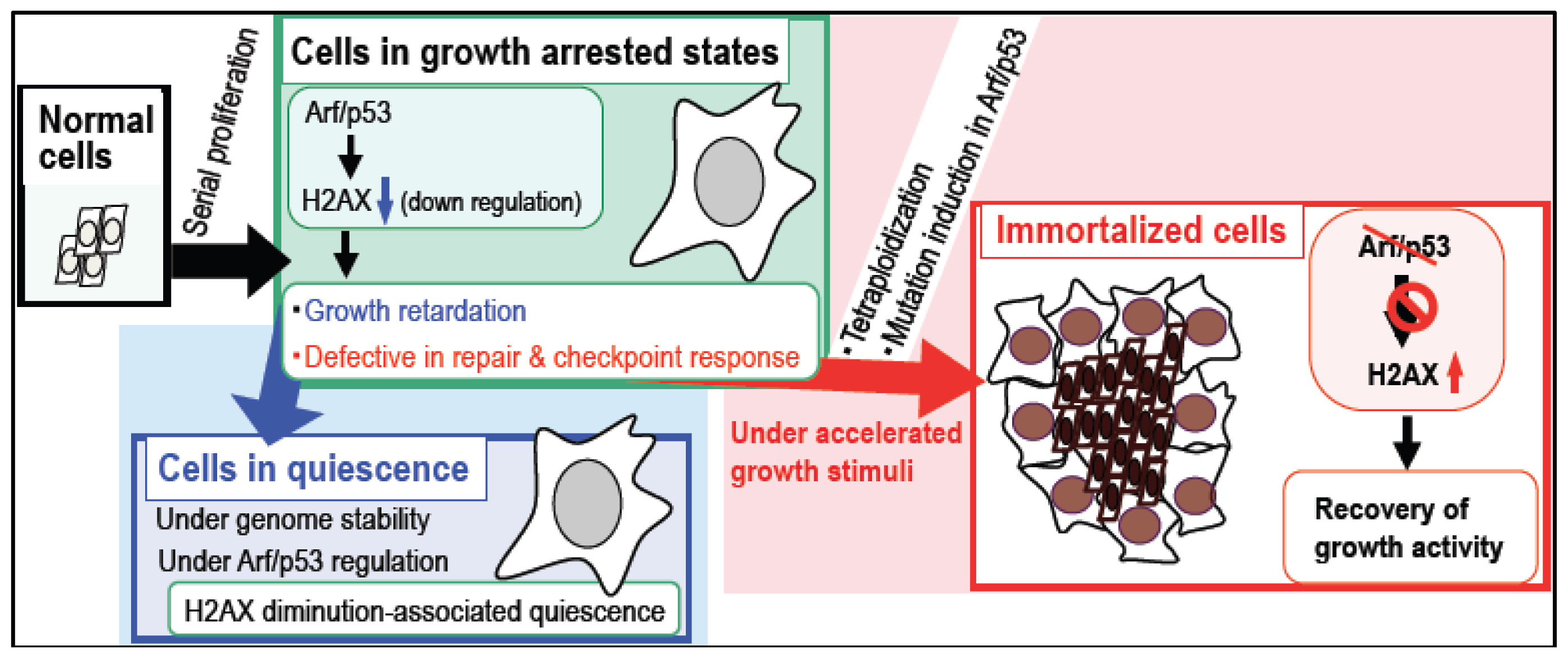
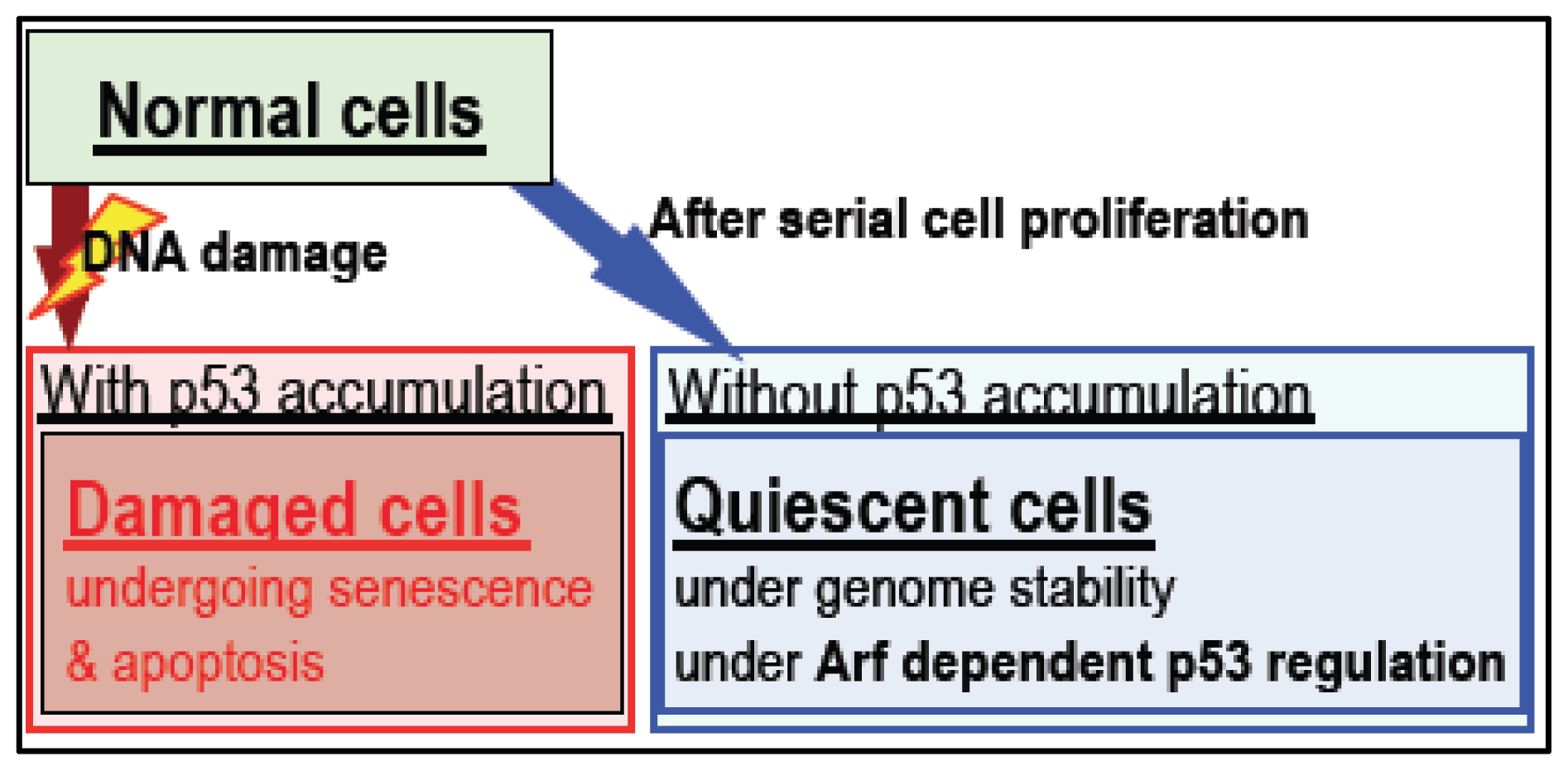
4. Arf/p53 Module-Dependent Quiescent Cellular Status
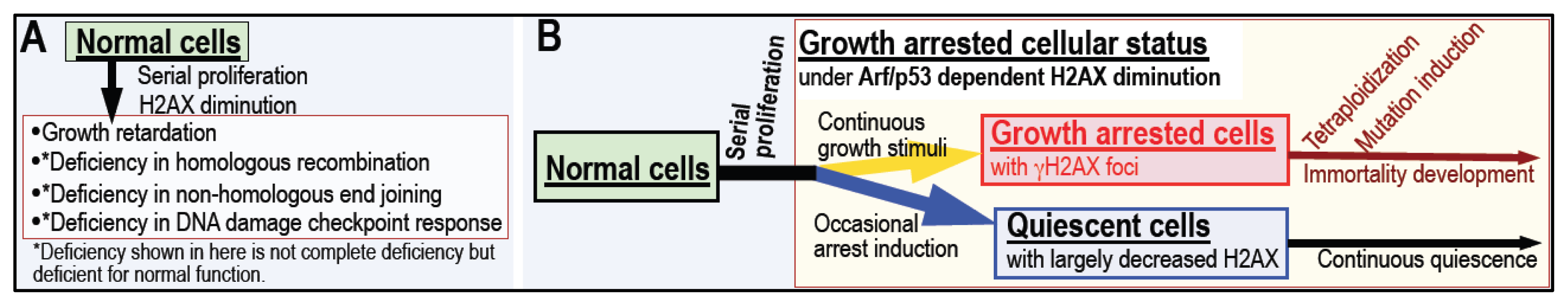
5. Cellular Quiescence Is Produced with Arf/p53-Dependent H2AX Diminution
6. Conclusions
Acknowledgments
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 632–637. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol 2010, 11, 220–228. [Google Scholar]
- Matheu, A.; Maraver, A.; Serrano, M. The Arf/p53 pathway in cancer and aging. Cancer Res 2008, 68, 6031–6034. [Google Scholar]
- Debies, M.T.; Gestl, S.A.; Mathers, J.L.; Mikse, O.R.; Leonard, T.L.; Moody, S.E.; Chodosh, L.A.; Cardiff, R.D.; Gunther, E.J. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16 Ink4a loss. J. Clin. Invest 2008, 118, 51–63. [Google Scholar]
- Mallakin, A.; Sugiyama, T.; Taneja, P.; Matise, L.A.; Frazier, D.P.; Choudhary, M.; Hawkins, G.A.; D’Agostino, R.B., Jr; Willingham, M.C.; Inoue, K. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 2007, 12, 381–394. [Google Scholar]
- Peng, C.Y.; Chen, T.C.; Hung, S.P.; Chen, M.F.; Yeh, C.T.; Tsai, S.L.; Chu, C.M.; Liaw, Y.F. Genetic alterations of INK4alpha/ARF locus and p53 in human hepatocellular carcinoma. Anticancer Res 2002, 22, 1265–1271. [Google Scholar]
- Gale, K.B.; Ford, A.M.; Repp, R.; Borkhardt, A.; Keller, C.; Eden, O.B.; Greaves, M.F. Backtracking leukemia to birth: Identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl. Acad. Sci. USA 1997, 94, 13950–13954. [Google Scholar]
- von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.H.; Lawlor, E.R. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One 2011, 6, e19305. [Google Scholar]
- Hu-Lieskovan, S.; Zhang, J.; Wu, L.; Shimada, H.; Schofield, D.E.; Triche, T.J. EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Res 2005, 65, 4633–4644. [Google Scholar]
- Gmidene, A.; Frikha, R.; Sennana, H.; Elghezal, H.; Elloumi, M.; Saad, A. T(1;21;8)(p34;q22;q22): A novel variant of t(8;21) in acute myeloblastic leukemia with maturation. Med. Oncol 2011, 28, S509–S512. [Google Scholar]
- Nanri, T.; Matsuno, N.; Kawakita, T.; Suzushima, H.; Kawano, F.; Mitsuya, H.; Asou, N. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21)(q22;q22). Leukemia 2005, 19, 1361–1366. [Google Scholar]
- Kozu, T.; Fukuyama, T.; Yamami, T.; Akagi, K.; Kaneko, Y. MYND-less splice variants of AML1-MTG8 (RUNX1-CBFA2T1) are expressed in leukemia with t(8;21). Genes Chrom. Cancer 2005, 43, 45–53. [Google Scholar]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol 2011, 192, 547–556. [Google Scholar]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 2010, 29, 273–283. [Google Scholar]
- Maslov, A.Y.; Vijg, J. Genome instability, cancer and aging. Biochim. Biophys. Acta 2009, 1790, 963–969. [Google Scholar]
- Vijg, J.; Dolle, M.E. Genome instability: Cancer or aging? Mech. Ageing Dev 2007, 128, 466–468. [Google Scholar]
- Hoeijmakers, J.H. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech. Ageing Dev 2007, 128, 460–462. [Google Scholar]
- Chaturvedi, S.; Hass, R. Extracellular signals in young and aging breast epithelial cells and possible connections to age-associated breast cancer development. Mech. Ageing Dev 2011, 132, 213–219. [Google Scholar]
- Keyes, M.K.; Jang, H.; Mason, J.B.; Liu, Z.; Crott, J.W.; Smith, D.E.; Friso, S.; Choi, S.W. Older age and dietary folate are determinants of genomic and p16-specific DNA methylation in mouse colon. J. Nutr 2007, 137, 1713–1717. [Google Scholar]
- Pal, S.K.; Hurria, A. Impact of age, sex, and comorbidity on cancer therapy and disease progression. J. Clin. Oncol 2010, 28, 4086–4093. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar]
- Laurent-Puig, P.; Blons, H.; Cugnenc, P.H. Sequence of molecular genetic events in colorectal tumorigenesis. Eur. J. Cancer Prev 1999, 1, S39–S47. [Google Scholar]
- Strauss, B.S. Frameshift mutation, microsatellites and mismatch repair. Mutat. Res 1999, 437, 195–203. [Google Scholar]
- Shibata, D. When does MMR loss occur during HNPCC progression? Cancer Biomark 2006, 2, 29–35. [Google Scholar]
- Pal, T.; Permuth-Wey, J.; Kumar, A.; Sellers, T.A. Systematic review and meta-analysis of ovarian cancers: Estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin. Cancer Res 2008, 14, 6847–6854. [Google Scholar]
- Shah, S.N.; Hile, S.E.; Eckert, K.A. Defective mismatch repair, microsatellite mutation bias, and variability in clinical cancer phenotypes. Cancer Res 2010, 70, 431–435. [Google Scholar]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar]
- Weigman, V.J.; Chao, H.H.; Shabalin, A.A.; He, X.; Parker, J.S.; Nordgard, S.H.; Grushko, T.; Huo, D.; Nwachukwu, C.; Nobel, A.; et al. Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res.Treat 2011. [Google Scholar] [CrossRef]
- Matheu, A.; Maraver, A.; Klatt, P.; Flores, I.; Garcia-Cao, I.; Borras, C.; Flores, J.M.; Vina, J.; Blasco, M.A.; Serrano, M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007, 448, 375–379. [Google Scholar]
- Ichijima, Y.; Yoshioka, K.; Yoshioka, Y.; Shinohe, K.; Fujimori, H.; Unno, J.; Takagi, M.; Goto, H.; Inagaki, M.; Mizutani, S.; et al. DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [Green Version]
- Ding, L.; Ellis, M.J.; Li, S.Q.; Larson, D.E.; Chen, K.; Wallis, J.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar]
- Kan, Z.Y.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.B.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar]
- Stephens, P.J.; Greenman, C.D.; Fu, B.Y.; Yang, F.T.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet 2011, 20, 1916–1924. [Google Scholar]
- Kloosterman, W.P.; Hoogstraat, M.; Paling, O.; Tavakoli-Yaraki, M.; Renkens, I.; Vermaat, J.S.; van Roosmalen, M.J.; van Lieshout, S.; Nijman, I.J.; Roessingh, W.; et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 2011, 12, R103:1–R103-11. [Google Scholar]
- Colnaghi, R.; Carpenter, G.; Volker, M.; O’Driscoll, M. The consequences of structural genomic alterations in humans: Genomic disorders, genomic instability and cancer. Semin. Cell Dev. Biol 2011, 22, 875–885. [Google Scholar]
- Gisselsson, D.; Jin, Y.; Lindgren, D.; Persson, J.; Gisselsson, L.; Hanks, S.; Sehic, D.; Mengelbier, L.H.; Ora, I.; Rahman, N.; et al. Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc. Natl. Acad. Sci. USA 2010, 107, 20489–20493. [Google Scholar]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci 2008, 121, 3859–3866. [Google Scholar]
- Maley, C.C.; Galipeau, P.C.; Li, X.H.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Reid, B.J. The combination of genetic instability and clonal expansion predicts progression to esophageal adenocarcinoma. Cancer Res 2004, 64, 7629–7633. [Google Scholar]
- Vitale, I.; Galluzzi, L.; Senovilla, L.; Criollo, A.; Jemaa, M.; Castedo, M.; Kroemer, G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ 2011, 18, 1403–1413. [Google Scholar]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol 2003, 5, 741–747. [Google Scholar]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol 1963, 17, 299–313. [Google Scholar]
- Atsumi, Y.; Fujimori, H.; Fukuda, H.; Inase, A.; Shinohe, K.; Yoshioka, Y.; Shikanai, M.; Ichijima, Y.; Unno, J.; Mizutani, S.; et al. Onset of quiescence following p53 mediated down-regulation of H2AX in normal cells. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar]
- Shi, Q.; King, R.W. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 2005, 437, 1038–1042. [Google Scholar]
- Heselmeyer, K.; Schrock, E.; duManoir, S.; Blegen, H.; Shah, K.; Steinbeck, R.; Auer, G.; Ried, T. Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc. Natl. Acad. Sci. USA 1996, 93, 479–484. [Google Scholar]
- Mullins, J.M.; Biesele, J.J. Terminal phase of cytokinesis in D-98s cells. J. Cell Biol 1977, 73, 672–684. [Google Scholar]
- Stewenius, Y.; Gorunova, L.; Jonson, T.; Larsson, N.; Hoglund, M.; Mandahl, N.; Mertens, F.; Mitelman, F.; Gisselsson, D. Structural and numerical chromosome changes in colon cancer develop through telomere-mediated anaphase bridges, not through mitotic multipolarity. Proc. Natl. Acad. Sci. USA 2005, 102, 5541–5546. [Google Scholar]
- Weaver, B.A.; Silk, A.D.; Cleveland, D.W. Cell biology: Nondisjunction, aneuploidy and tetraploidy. Nature 2006, 442, E9–E10, discussion E10. [Google Scholar]
- Steigemann, P.; Wurzenberger, C.; Schmitz, M.H.A.; Held, M.; Guizetti, J.; Maar, S.; Gerlich, D.W. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009, 136, 473–484. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; Kletsas, D.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar]
- White, J.M.; Blobel, C.P. Cell-to-cell fusion. Curr. Opin. Cell Biol 1989, 1, 934–939. [Google Scholar]
- Iida, S.; Hirota, T.; Morisaki, T.; Marumoto, T.; Hara, T.; Kuninaka, S.; Honda, S.; Kosai, K.; Kawasuji, M.; Pallas, D.C.; et al. Tumor suppressor WARTS ensures genomic integrity by regulating both mitotic progression and G(1) tetraploidy checkpoint function. Oncogene 2004, 23, 5266–5274. [Google Scholar]
- Elhajouji, A.; Cunha, M.; Kirsch-Volders, M. Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis-block assay. Mutagenesis 1998, 13, 193–198. [Google Scholar]
- Dai, W.; Wang, Q.; Liu, T.Y.; Swamy, M.; Fang, Y.Q.; Xie, S.Q.; Mahmood, R.; Yang, Y.M.; Xu, M.; Ra, C.V. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 2004, 64, 440–445. [Google Scholar]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol 2004, 6, 168–170. [Google Scholar]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 2008, 1. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145, 571–583. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar]
- Kamijo, T.; Zindym, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF). Cell 1997, 91, 649–659. [Google Scholar]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev 2004, 18, 306–319. [Google Scholar]
- Hinkal, G.W.; Gatza, C.E.; Parikh, N.; Donehower, L.A. Altered senescence, apoptosis, and DNA damage response in a mutant p53 model of accelerated aging. Mech. Ageing Dev 2009, 130, 262–271. [Google Scholar]
- Polyak, K.; Waldman, T.; He, T.C.; Kinzler, K.W.; Vogelstein, B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 1996, 10, 1945–1952. [Google Scholar]
- Sherr, C.J. Parsing Ink4a/Arf: “pure” p16-null mice. Cell 2001, 106, 531–534. [Google Scholar]
- Bassing, C.H.; Alt, F.W. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 2004, 3, 149–153. [Google Scholar]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2ax and Cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar]
- Sedelnikova, O.A.; Pilch, D.R.; Redon, C.; Bonner, W.M. Histone H2AX in DNA damage and repair. Cancer Biol. Ther 2003, 2, 233–235. [Google Scholar]
- Sokolov, M.V.; Dickey, J.S.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX in bystander cells: Not just a radiation-triggered event, a cellular response to stress mediated by intercellular communication. Cell Cycle 2007, 6, 2210–2212. [Google Scholar]
- Pilch, D.R.; Sedelnikova, O.A.; Redon, C.; Celeste, A.; Nussenzweig, A.; Bonner, W.M. Characteristics of γ-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol 2003, 81, 123–129. [Google Scholar]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair (Amst. ) 2004, 3, 959–967. [Google Scholar]
- Dickey, J.S.; Redon, C.E.; Nakamura, A.J.; Baird, B.J.; Sedelnikova, O.A.; Bonner, W.M. H2AX: Functional roles and potential applications. Chromosoma 2009, 118, 683–692. [Google Scholar]
- Lal, A.; Pan, Y.; Navarro, F.; Dykxhoorn, D.M.; Moreau, L.; Meire, E.; Bentwich, Z.; Lieberman, J.; Chowdhury, D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat. Struct. Mol. Biol 2009, 16, 492–498. [Google Scholar]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshioka, K.-i.; Atsumi, Y.; Fukuda, H.; Masutani, M.; Teraoka, H. The Quiescent Cellular State is Arf/p53-Dependent and Associated with H2AX Downregulation and Genome Stability. Int. J. Mol. Sci. 2012, 13, 6492-6506. https://doi.org/10.3390/ijms13056492
Yoshioka K-i, Atsumi Y, Fukuda H, Masutani M, Teraoka H. The Quiescent Cellular State is Arf/p53-Dependent and Associated with H2AX Downregulation and Genome Stability. International Journal of Molecular Sciences. 2012; 13(5):6492-6506. https://doi.org/10.3390/ijms13056492
Chicago/Turabian StyleYoshioka, Ken-ichi, Yuko Atsumi, Hirokazu Fukuda, Mitsuko Masutani, and Hirobumi Teraoka. 2012. "The Quiescent Cellular State is Arf/p53-Dependent and Associated with H2AX Downregulation and Genome Stability" International Journal of Molecular Sciences 13, no. 5: 6492-6506. https://doi.org/10.3390/ijms13056492