
Computational Ensemble Gene Co-Expression Networks for the Analysis of Cancer Biomarkers
 , ,
, ,  , and
, and 
Abstract
:1. Introduction
- We propose a case study based on three different widely GCN-inferred algorithms.
- The use of the ensemble strategy derives from a more reliable inferred GCN.
- A case study of breast and prostate cancer is presented.
- We propose several genes as potential biomarkers for both breast and prostate cancers.
Related Works
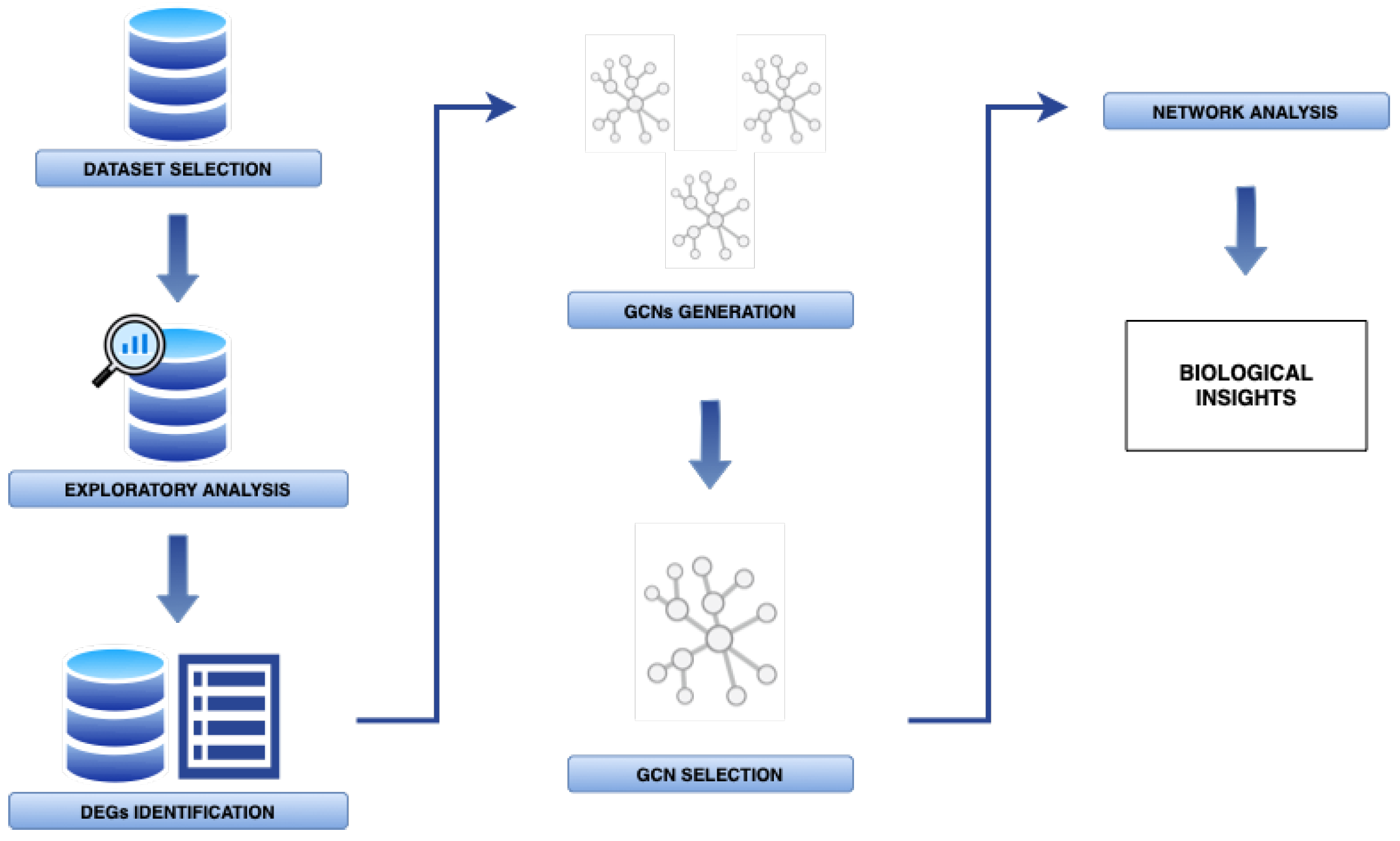
2. Materials and Methods
2.1. Sample Dataset
2.2. Data Preprocessing and Exploratory Analysis
2.3. Differential Gene Expression Analysis
2.4. Gene Co-Expression Network Reconstruction
- Pearson’s correlation coefficient: The values of Pearson’s correlation coefficient range from −1 to 1, with a score of −1 indicating a perfectly negative correlation, a score of 1 indicating a perfectly positive correlation, and a score of 0 indicating no correlation between the expression levels of the two genes under consideration.where refers to Pearson’s correlation value between gene x and gene y.
- Spearman’s correlation coefficient: The values of Spearman’s correlation coefficient range from −1 to 1, as in the case of Pearson’s.where refers to Spearman’s correlation value, to the differences between the ranks of x and y and n to the number of observations.
- Kendall’s correlation coefficient: this coefficient ranges from −1 to 1 and represents a valuable parameter for detecting non-linear relationships among genes.where refers to Kendall’s correlation value, to the number of concordant pairs of observations and to the number of discordant pairs of observations.
2.5. Candidate Gene Co-Expression Network Selection
2.6. Gene Co-Expression Network Analysis
3. Results
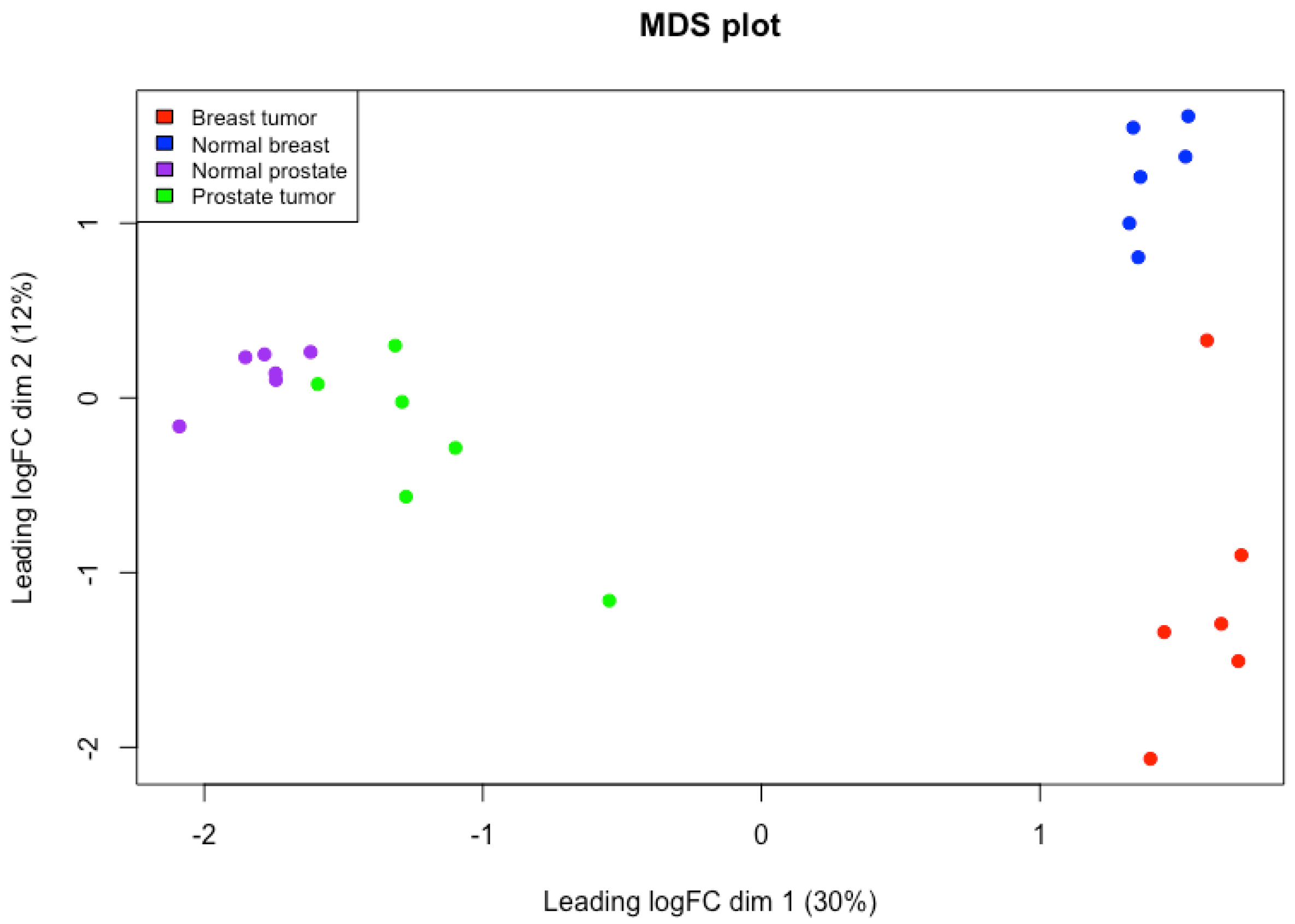
3.1. Exploratory Analysis
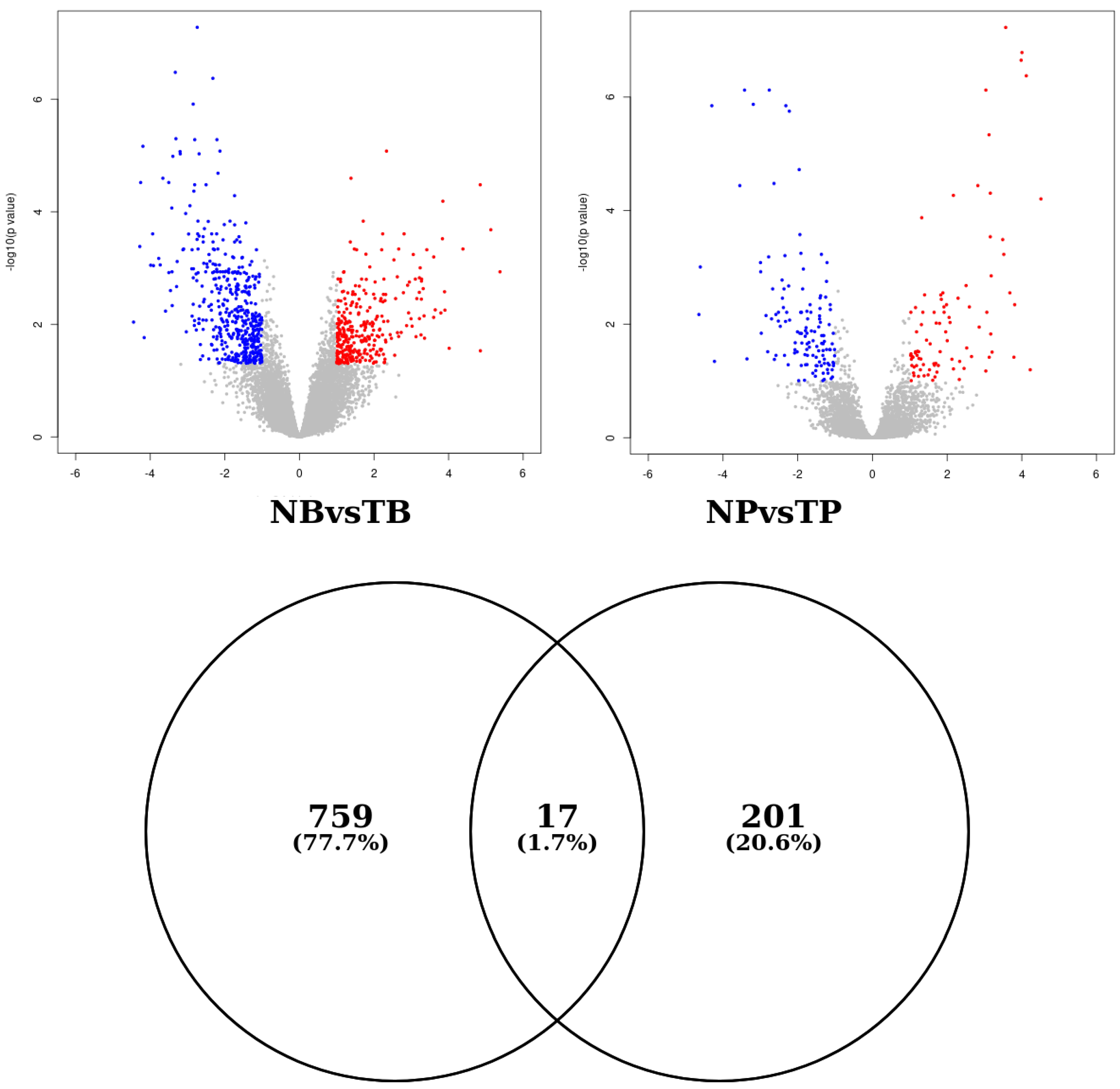
3.2. Differential Gene Expression Analysis
3.3. Performance Comparison between Different GCN Methods
3.4. Gene Co-Expression Network Reconstruction and Candidate Co-Expression Network Selection
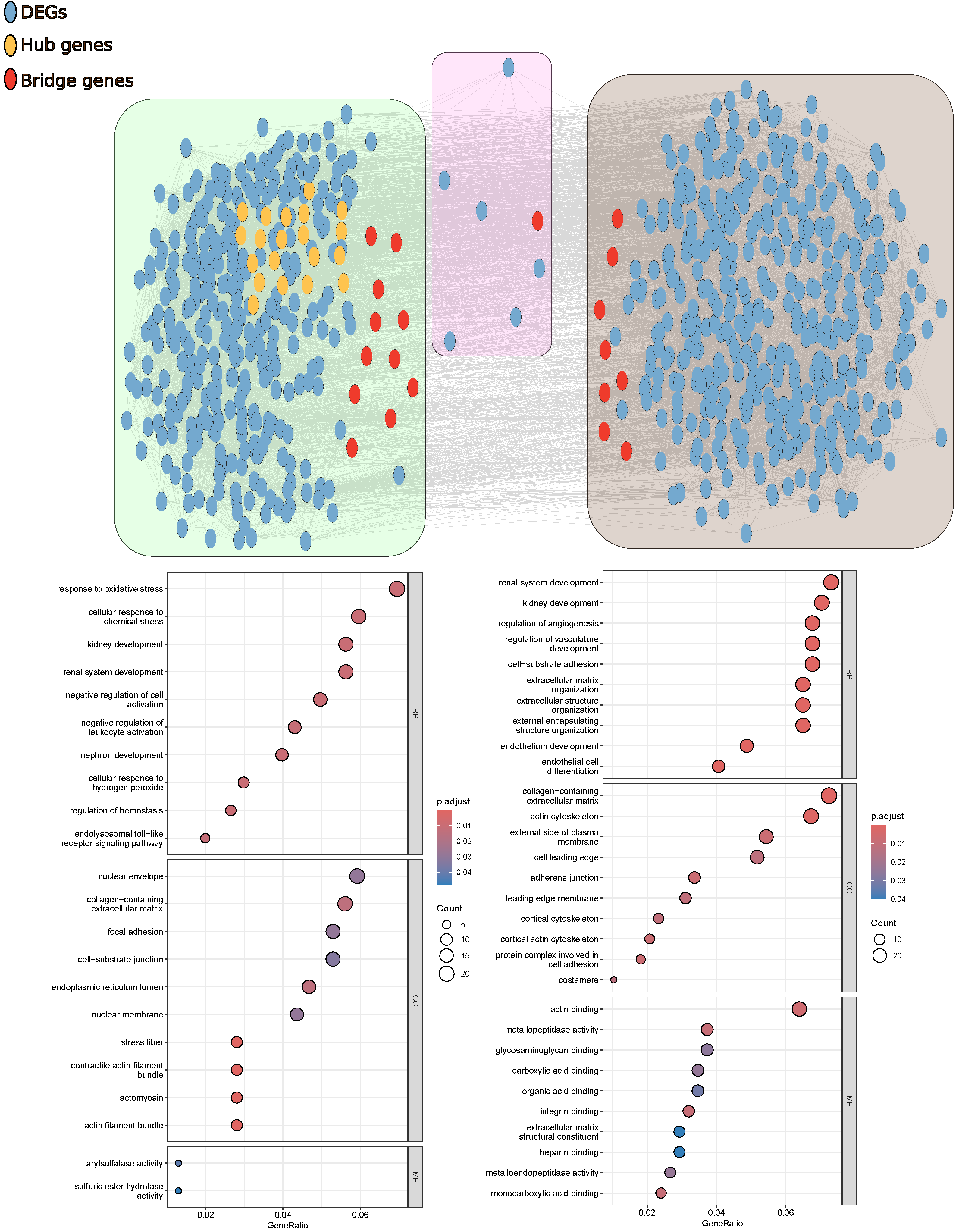
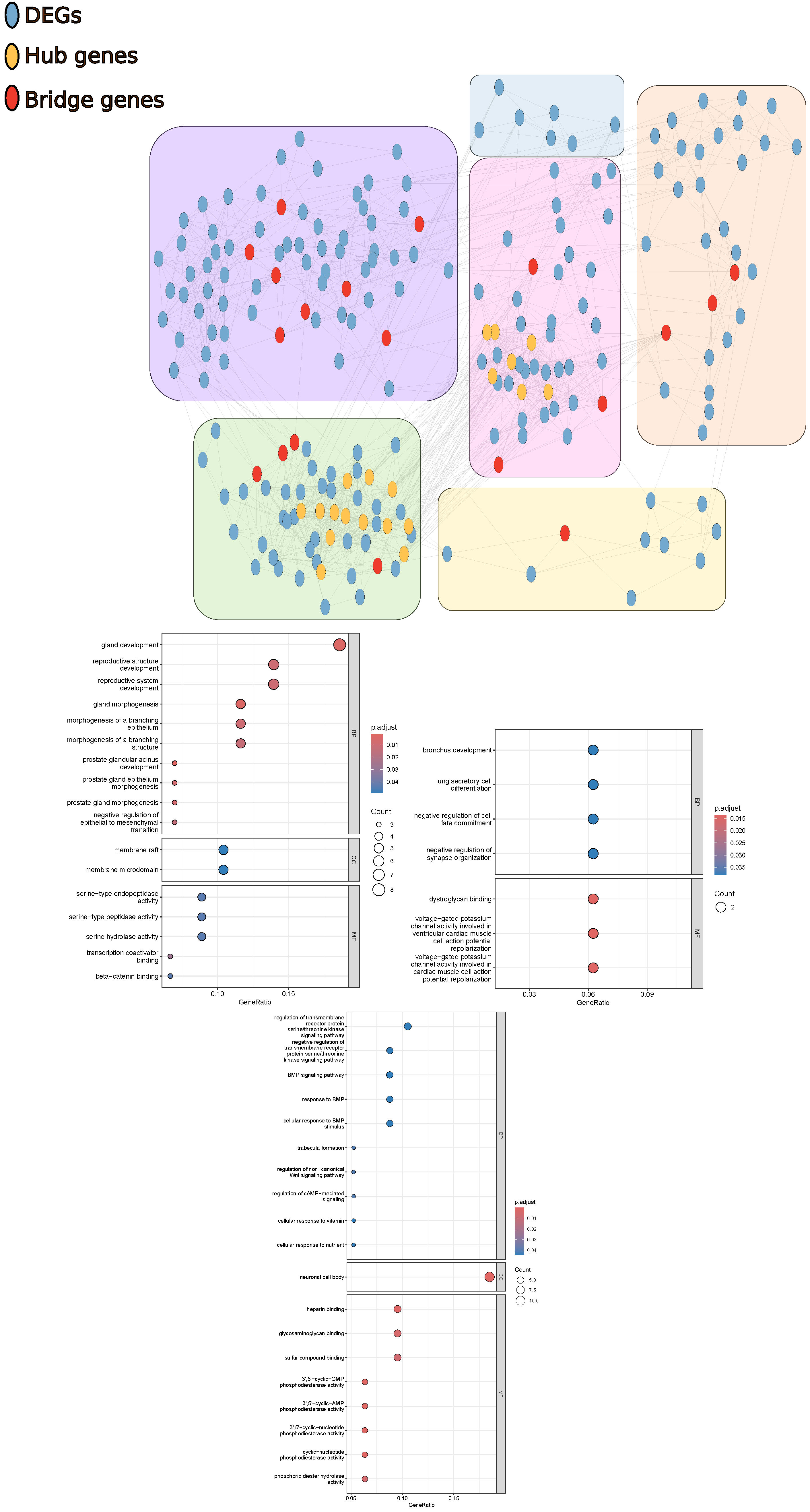
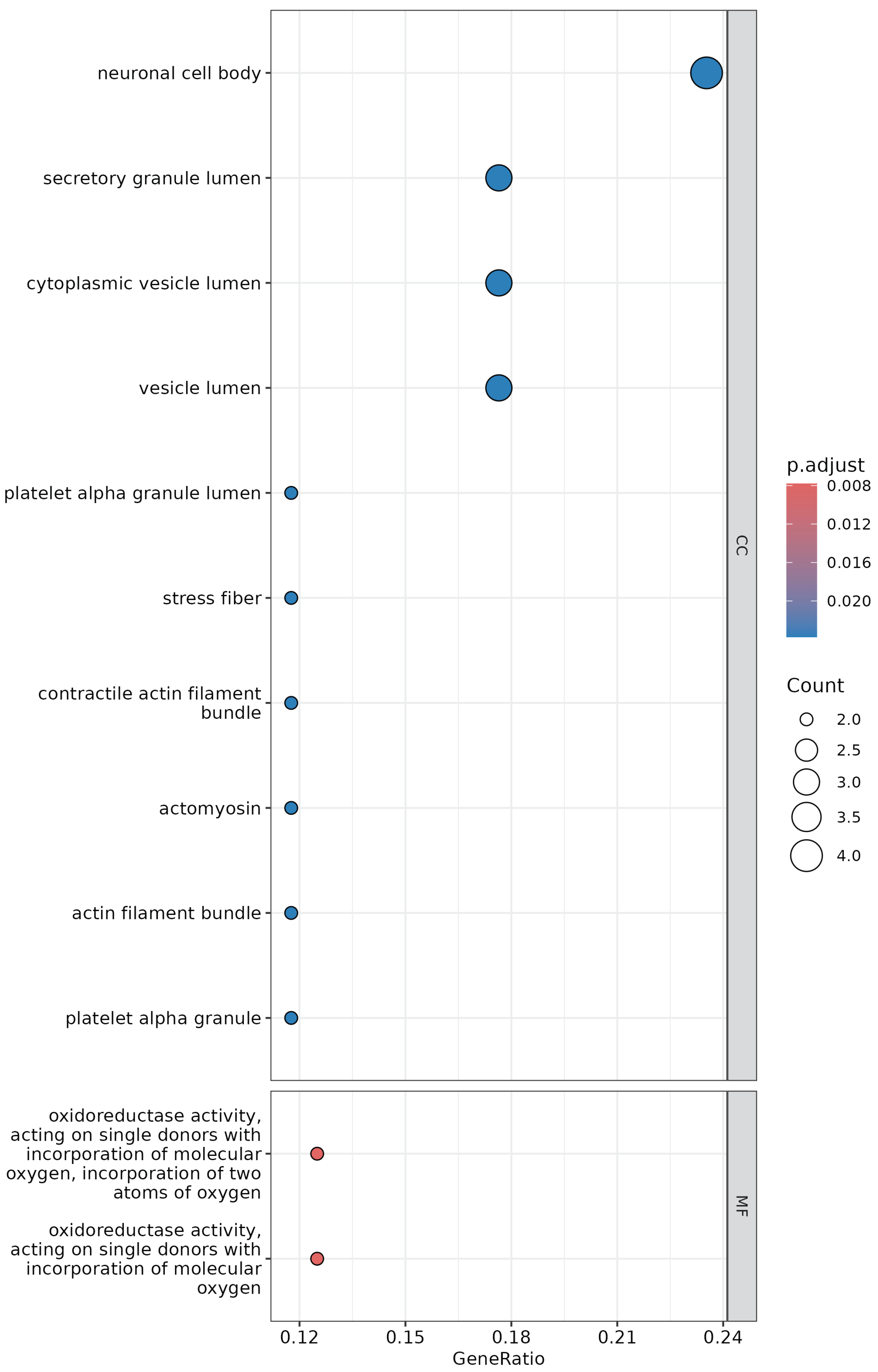
3.5. Gene Co-Expression Network Analysis
4. Discussion
4.1. Differential Expression Analysis Revealed Significant Differences between Stromal Breast Tumor and Stromal Prostate Tumor
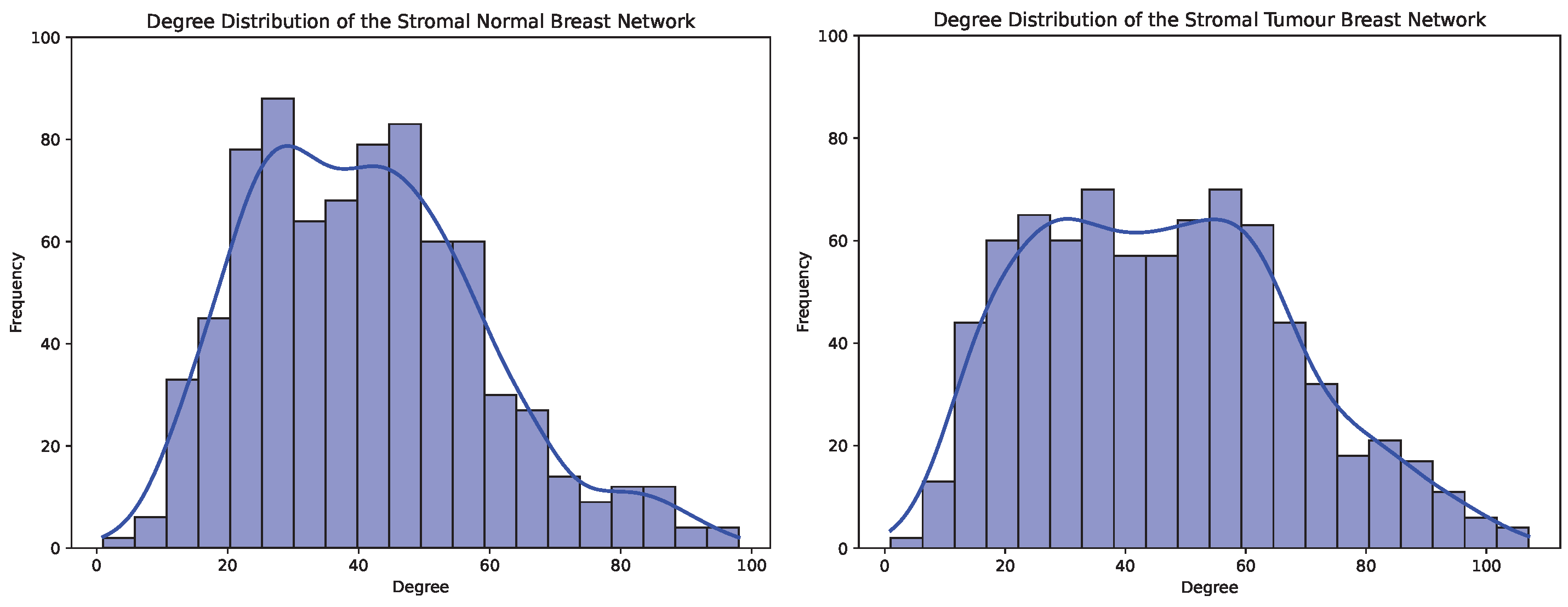
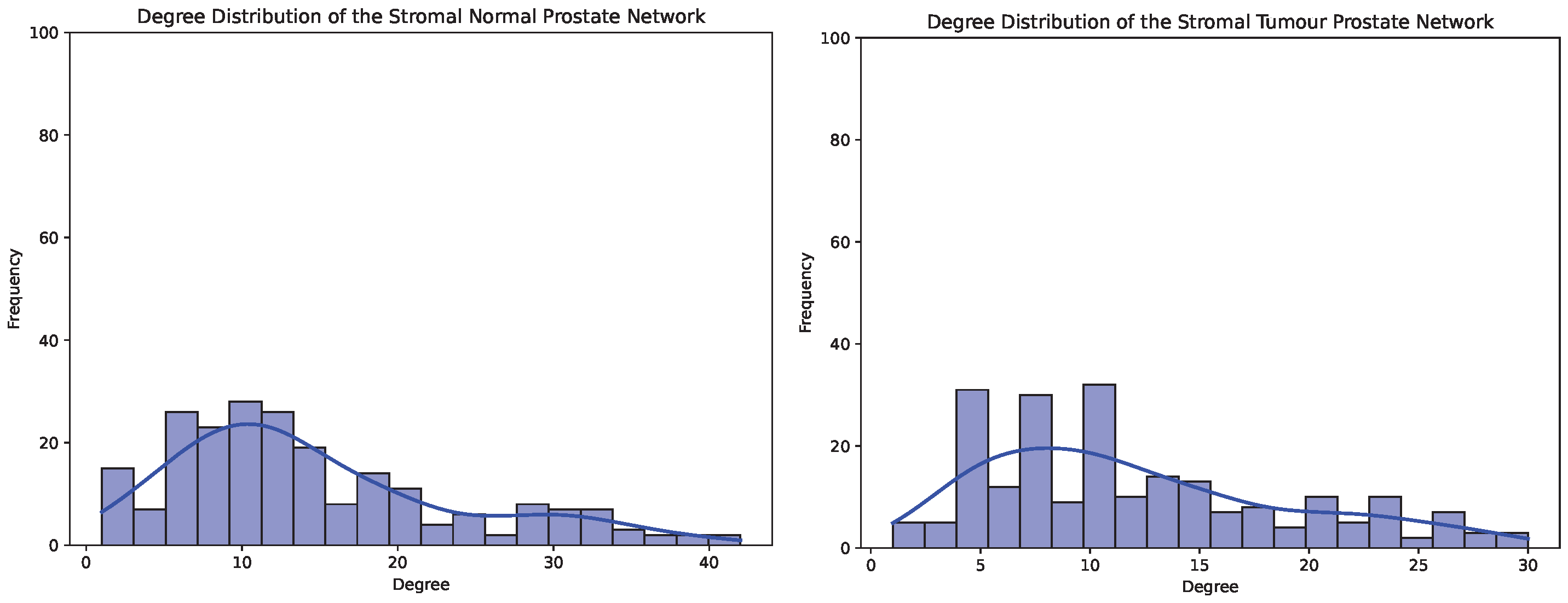
4.2. Exploratory Analysis in Stromal Breast and Prostate Co-Expression Networks
4.3. Gene Expression of Stromal Cells in Breast Tumors Is Related to Focal Adhesion Modifications, While That of Stromal Cells in Prostate Tumors Is Related to Organ Formation and Cell Differentiation
4.4. ST6GAL2, RIPOR3, COL5A1, and DEPDC7 Are Potential Biomarkers in the Breast Tumor Microenvironment
4.5. GATA6-AS1, ARFGEF3, PRR15L, and APBA2 Are Potential Biomarkers in the Prostate Tumor Microenvironment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Detailed Results from Experiments
Appendix A.1. Figures



Appendix A.2. Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | NB vs. TB | NP vs. TP |
|---|---|---|
| Downregulated | 451 | 129 |
| Not significant | 20,048 | 20,606 |
| Upregulated | 325 | 89 |
| Gene ID | Regulation NB vs. TB | Regulation NP vs. TP |
|---|---|---|
| KLHL14 | downregulated | downregulated |
| TGFB3 | upregulated | downregulated |
| PDLIM5 | upregulated | upregulated |
| PPL | downregulated | downregulated |
| PTGS1 | upregulated | downregulated |
| CFD | downregulated | downregulated |
| ASPA | downregulated | downregulated |
| FAM107A | downregulated | downregulated |
| DUSP6 | downregulated | upregulated |
| GPM6A | downregulated | downregulated |
| BMPR1B | upregulated | upregulated |
| SASH1 | downregulated | upregulated |
| PENK | downregulated | downregulated |
| FIGN | downregulated | upregulated |
| MAL2 | upregulated | upregulated |
| C16orf89 | downregulated | downregulated |
| BCO2 | downregulated | downregulated |
| Group | Threshold | GNC Value-BioGrid | Num. Nodes | Num. Edges | Thrscore |
|---|---|---|---|---|---|
| 0.7 | 0.503094 | 218 | 2693 | 0.285671 | |
| 0.75 | 0.419838 | 218 | 1677 | 0.082137 | |
| Prostate Normal | 0.8 | 0.342942 | 217 | 1303 | 0.218407 |
| 0.85 | 0.182962 | 205 | 659 | 0.082843 | |
| 0.9 | 0.079346 | 186 | 428 | 0.187738 | |
| 0.7 | 0.520096 | 218 | 2325 | 0.333839 | |
| 0.75 | 0.41008 | 218 | 1391 | 0.084419 | |
| Prostate tumor | 0.8 | 0.328616 | 218 | 1120 | 0.2957 |
| 0.85 | 0.157933 | 211 | 571 | 0.087541 | |
| 0.9 | 0.058878 | 203 | 385 | 0.200432 | |
| 0.7 | 0.508661 | 776 | 29320 | 0.026487 | |
| 0.75 | 0.516137 | 776 | 16,645 | 0.008006 | |
| Breast Normal | 0.8 | 0.468251 | 776 | 12,818 | 0.023706 |
| 0.85 | 0.305834 | 776 | 5799 | 0.009305 | |
| 0.9 | 0.159673 | 772 | 3640 | 0.024182 | |
| 0.7 | 0.50731 | 776 | 31,345 | 0.024690 | |
| 0.75 | 0.516461 | 776 | 18,594 | 0.007984 | |
| Breast tumor | 0.8 | 0.469527 | 776 | 14,098 | 0.020221 |
| 0.85 | 0.317097 | 775 | 6811 | 0.009268 | |
| 0.9 | 0.162074 | 770 | 4047 | 0.018492 |
| Nodes | Edges | Density | Avg Cluster Coef. | Community | Degree Assortativity | |
|---|---|---|---|---|---|---|
| Breast normal | 776 | 15,975 | 0.053 | 0.43 | 0.21 | 0.63 |
| Breast tumor | 776 | 17,924 | 0.059 | 0.45 | 0.47 | 0.61 |
| Prostate normal | 218 | 1619 | 0.067 | 0.47 | 0.37 | 0.64 |
| Prostate tumor | 218 | 1333 | 0.055 | 0.48 | 0.55 | 0.55 |
| Cluster | Avg Nodes | Avg Edges | |
|---|---|---|---|
| Breast tumor | 3 | 257.67 | 5418.33 |
| Prostate tumor | 6 | 36.33 | 188.83 |
| Stromal Breast Normal | |||||||
|---|---|---|---|---|---|---|---|
| Degree Hubs | Score | Betweenness | Score | Closeness | Score | Eigenvector | Score |
| DOC2B | 98.0 | AEBP1 | 5352 | ARSA | 352.5 | DOC2B | 0.13 |
| SH2D3C | 95.0 | GSN | 4042.1 | SH2D3C | 352.3 | SH2D3C | 0.12 |
| ARSA | 94.0 | LMO2 | 3853.0 | ADCK2 | 351.6 | ANKRD2-0A11P | 0.12 |
| MYH9 | 94.0 | MMP11 | 3844.5 | ANKRD2-0A11P | 350.9 | MYH9 | 0.12 |
| ANKRD2-0A11P | 91.0 | CRTAM | 3702.3 | RGMA | 350.6 | ARSA | 0.12 |
| RGMA | 91.0 | SH3BGRL2 | 3672 | SHISA2 | 350.1 | C1orf122 | 0.12 |
| FAM110D | 91.0 | ENTREP1 | 3489.3 | C1orf122 | 350.1 | DACT3 | 0.11 |
| GRP | 89.0 | NLRC3 | 3411.1 | DOC2B | 349.9 | SPP1 | 0.11 |
| ADCK2 | 88.0 | CAVIN2 | 3403.9 | GRP | 349.8 | RGMA | 0.11 |
| C1orf122 | 88.0 | USP36 | 3377.1 | CCDC178 | 347.9 | FAM110D | 0.11 |
| LY86 | 88.0 | RGS10 | 3303.2 | SPP1 | 347.9 | GRP | 0.11 |
| FDX2 | 87.0 | RSAD2 | 3251.7 | MFAP2 | 346.8 | FDX2 | 0.11 |
| DACT3 | 87.0 | LEPR | 3237.0 | EVC | 346.7 | EVC | 0.11 |
| SPP1 | 87.0 | ANLN | 3199 | LY86 | 346.5 | RAPGEF3 | 0.11 |
| SHISA2 | 85.0 | JPT1 | 3168.7 | OTULINL | 346.4 | ITGA7 | 0.11 |
| RAPGEF3 | 85.0 | TLL2 | 3159.5 | MYH9 | 346.4 | LY86 | 0.11 |
| EVC | 85.0 | EXOC6 | 3146.4 | RAPGEF3 | 346.2 | CCN4 | 0.11 |
| CCN4 | 85.0 | C19orf53 | 3131.7 | DACT3 | 345.7 | OTULINL | 0.11 |
| FAM110C | 85.0 | FICD | 3116.9 | FAM110C | 345.6 | ADCK2 | 0.10 |
| CCM2L | 84.0 | THBD | 3065.2 | LOXL2 | 345.1 | FAM241A | 0.10 |
| Stromal Breast Cancer | |||||||
|---|---|---|---|---|---|---|---|
| Degree Hubs | Score | Betweenness | Score | Closeness | Score | Eigenvector | Score |
| LONP2 | 107.0 | HMGN3 | 7460.8 | LONP2 | 361.1 | CRNDE | 0.20 |
| ST6GAL2 | 105.0 | LOXL2 | 6723.3 | ST6GAL2 | 360.4 | PRR15L | 0.19 |
| SUN1 | 104.0 | CMTM3 | 4674.1 | SUN1 | 359.9 | PDLIM5 | 0.18 |
| GPR137B | 102.0 | GLMP | 4422.6 | GPR137B | 358.9 | SPON2 | 0.18 |
| RIPOR3 | 99.0 | MYO5B | 4365.5 | MAP7D1 | 357.6 | GATA6-AS1 | 0.18 |
| DYRK2 | 99.0 | PTGS1 | 4295.2 | MCTS1 | 355.8 | LEF1-AS1 | 0.17 |
| MAP7D1 | 99.0 | NKTR | 4222.7 | COL5A1 | 355.0 | CDH1 | 0.17 |
| SNRK | 98.0 | SEMA3C | 4044.4 | LGALS1 | 353.6 | APBA2 | 0.16 |
| SYNDIG1 | 97.0 | CCR5 | 3963.5 | DYRK2 | 353.5 | GPR160 | 0.16 |
| MATN3 | 97.0 | KIF26B | 3924.3 | SNRK | 352.6 | ARFGEF3 | 0.16 |
| MCTS1 | 96.0 | VOPP1 | 3832.7 | CLK1 | 352.6 | RAB25 | 0.16 |
| SH3BGRL2 | 96.0 | JPT1 | 3769.8 | INPP1 | 351.8 | HOXB13 | 0.16 |
| CLK1 | 95.0 | FNDC1 | 3662.7 | SYNDIG1 | 351.5 | HOXC6 | 0.16 |
| COL5A1 | 95.0 | PCNA | 3603.4 | MMP13 | 351.3 | EHF | 0.16 |
| LGALS1 | 95.0 | SLIT3 | 3500.1 | MATN3 | 350.9 | IER5L | 0.15 |
| ALDH1L2 | 94.0 | LINC01614 | 3395.4 | LRRC59 | 350.8 | SPDEF | 0.15 |
| DEPDC7 | 93.0 | PIGP | 3349.7 | ALDH1L2 | 350.4 | FOLH1B | 0.15 |
| PROS1 | 92.0 | SPP1 | 3322.5 | PGM1 | 349.3 | DUSP6 | 0.15 |
| NAP1L5 | 92.0 | NR2F2 | 3260.2 | DEPDC7 | 349.2 | PCDH10-DT | 0.14 |
| LRRC59 | 92.0 | LOC1053-77134 | 3246 | HECW2-AS1 | 348.9 | SORD | 0.14 |
| Stromal Prostate Normal | |||||||
|---|---|---|---|---|---|---|---|
| Degree Hubs | Score | Betweenness | Score | Closeness | Score | Eigenvector | Score |
| TIMP4 | 42.0 | PENK | 1862.6 | TIMP4 | 101.5 | RBP4 | 0.20 |
| SMTNL2 | 40.0 | STAC | 1796.2 | SMTNL2 | 99.9 | NAT2 | 0.20 |
| RBP4 | 39.0 | AGR2 | 1771.1 | RBP4 | 99.1 | TIMP4 | 0.20 |
| NAT2 | 38.0 | MB | 1691.4 | CCDC85A | 99.1 | SMTNL2 | 0.20 |
| CCDC85A | 37.0 | PCDH10-DT | 1613.0 | NAT2 | 98.2 | ARHGA-P28 | 0.19 |
| KLHL14 | 36.0 | TBX5-AS1 | 1601.9 | ARHGA-P28 | 97.3 | CCDC85A | 0.19 |
| IGSF1 | 35.0 | ERG | 1517.0 | PDE3B | 96.8 | CFD | 0.18 |
| PDE3B | 35.0 | PEX14 | 1364.4 | MLC1 | 96.5 | IGSF1 | 0.18 |
| ARHGA-P28 | 35.0 | SPDEF | 1349 | TRHDE | 96.2 | LOC10192-7668 | 0.18 |
| RSPH9 | 33.0 | LINC03026 | 1334.4 | GRTP1-AS1 | 96.1 | PDE3B | 0.18 |
| TRHDE | 33.0 | CLGN | 1311.5 | IGSF1 | 96.0 | KLHL14 | 0.18 |
| ODAD3 | 33.0 | FXYD6 | 1300.3 | RNF112 | 95.7 | RSPH9 | 0.17 |
| CFD | 33.0 | PPL | 1287.9 | LY6G6D | 94.9 | FBXO2 | 0.17 |
| LOC10192-7668 | 33.0 | PRSS35 | 1270.1 | KLHL14 | 94.9 | ODAD3 | 0.17 |
| LINC01082 | 32.0 | FAM107A | 1257.9 | RSPH9 | 94.8 | CHGB | 0.17 |
| FBXO2 | 32.0 | ADGRD1 | 1246.4 | FBXL21P | 94.6 | LINC01082 | 0.17 |
| GRTP1-AS1 | 31.0 | C9orf24 | 1239.1 | LOC10192-7668 | 94.3 | GATA6-AS1 | 0.16 |
| GATA6-AS1 | 31.0 | TRHDE | 1203.1 | CFD | 94.2 | GRTP1-AS1 | 0.15 |
| CLGN | 31.0 | TMSB15A | 1150.5 | ODAD3 | 94.1 | TRHDE | 0.15 |
| MLC1 | 30.0 | LRP1B | 1150.4 | LINC01082 | 94.0 | CLGN | 0.15 |
| Stromal Prostate Cancer | |||||||
|---|---|---|---|---|---|---|---|
| Degree Hubs | Score | Betweenness | Score | Closeness | Score | Eigenvector | Score |
| CDH1 | 30.0 | SMIM31 | 2940.4 | EHF | 90.5 | CRNDE | 0.20 |
| ARFGEF3 | 29.0 | NELL2 | 2244.7 | CDH1 | 90 | PRR15L | 0.19 |
| PRR15L | 29.0 | SMOC1 | 2212.8 | ARFGEF3 | 89.7 | PDLIM5 | 0.18 |
| EHF | 28.0 | PRKAR2B | 2178 | HOXB13 | 88.5 | SPON2 | 0.18 |
| SPON2 | 28.0 | LOC10192-7668 | 2165.4 | PRR15L | 87.9 | GATA6-AS1 | 0.18 |
| CRNDE | 28.0 | PTGS1 | 2016.1 | CRNDE | 87.8 | LEF1-AS1 | 0.17 |
| HOXB13 | 27.0 | ST8SIA1 | 1826.6 | LEF1-AS1 | 87.8 | CDH1 | 0.17 |
| PDLIM5 | 27.0 | ADGRD1 | 1823.9 | SPON2 | 87.2 | APBA2 | 0.16 |
| CXADR | 26.0 | LRP1B | 1771.2 | PDLIM5 | 87.1 | GPR160 | 0.16 |
| FOXA1 | 26.0 | PRAC1 | 1676.0 | HOXC6 | 87 | ARFGEF3 | 0.16 |
| GATA6-AS1 | 26.0 | MCF2 | 1549.7 | DUSP6 | 87 | RAB25 | 0.16 |
| LEF1-AS1 | 26.0 | EHF | 1505.7 | CXADR | 86.8 | HOXB13 | 0.16 |
| HOXC6 | 26.0 | TSLP | 1492.5 | APBA2 | 86.6 | HOXC6 | 0.16 |
| GPR160 | 25.0 | CSGALN-ACT1 | 1487.7 | GATA6-AS1 | 86.6 | EHF | 0.16 |
| APBA2 | 25.0 | PENK | 1467.1 | SORD | 86.4 | IER5L | 0.15 |
| SORD | 24.0 | FRRS1L | 1429.8 | RBM47 | 86.1 | SPDEF | 0.15 |
| RAB25 | 24.0 | RBP4 | 1419.3 | MAP2 | 85.8 | FOLH1B | 0.15 |
| DUSP6 | 24.0 | RAB37 | 1304.7 | TRPM8 | 85.4 | DUSP6 | 0.15 |
| LINC03026 | 23.0 | PLA1A | 1300.7 | GPR160 | 85.3 | PCDH10-DT | 0.14 |
| TRPM8 | 23.0 | FOXF2 | 1299.4 | DKK1 | 84.9 | SORD | 0.14 |
References
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Withrow, D.; Pilleron, S.; Nikita, N.; Ferlay, J.; Sharma, S.; Nicholson, B.; Rebbeck, T.R.; Lu-Yao, G. Current and projected number of years of life lost due to prostate cancer: A global study. Prostate 2022, 82, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Alekseenko, I.V.; Chernov, I.P.; Kostrov, S.V.; Sverdlov, E.D. Are synapse-like structures a possible way for crosstalk of cancer with its microenvironment? Cancers 2020, 12, 806. [Google Scholar] [CrossRef] [PubMed]
- Sund, M.; Kalluri, R. Tumor stroma derived biomarkers in cancer. Cancer Metastasis Rev. 2009, 28, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef]
- Kawada, M.; Inoue, H.; Usami, I.; Ikeda, D. Phthoxazolin A inhibits prostate cancer growth by modulating tumor–stromal cell interactions. Cancer Sci. 2009, 100, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Tao, S.; Zhang, L.; Diao, L.T.; Huang, X.; Huang, S.; Xie, S.J.; Xiao, Z.D.; Zhang, H. RNA sequencing: New technologies and applications in cancer research. J. Hematol. Oncol. 2020, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Riquelme Medina, I.; Lubovac-Pilav, Z. Gene co-expression network analysis for identifying modules and functionally enriched pathways in type 1 diabetes. PLoS ONE 2016, 11, e0156006. [Google Scholar] [CrossRef]
- Redekar, S.S.; Varma, S.L.; Bhattacharjee, A. Gene co-expression network construction and analysis for identification of genetic biomarkers associated with glioblastoma multiforme using topological findings. J. Egypt. Natl. Cancer Inst. 2023, 35, 22. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Long, J.; Zhao, Z.B.; Li, L.; Lian, Z.X.; Liang, Z.; Wu, J.R. Gene co-expression network based on part mutual information for gene-to-gene relationship and gene-cancer correlation analysis. BMC Bioinform. 2022, 23, 194. [Google Scholar] [CrossRef]
- Huang, R.; He, Y.; Sun, B.; Liu, B. Bioinformatic Analysis Identifies Three Potentially Key Differentially Expressed Genes in Peripheral Blood Mononuclear Cells of Patients with Takayasu’s Arteritis. Cell J. 2018, 19, 647–653. [Google Scholar] [CrossRef]
- Brugere, I.; Gallagher, B.; Berger-Wolf, T.Y. Network structure inference, a survey: Motivations, methods, and applications. ACM Comput. Surv. (CSUR) 2018, 51, 24. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.M.; Chu, C.M.; Chang, Y.J.; Yu, J.C.; Chen, C.T.; Jian, C.E.; Lee, C.Y.; Chiang, Y.T.; Chang, C.W.; Chang, Y.T. Six novel immunoglobulin genes as biomarkers for better prognosis in triple-negative breast cancer by gene co-expression network analysis. Sci. Rep. 2019, 9, 4484. [Google Scholar] [CrossRef]
- Wang, L.; Liao, Z. Mixture prior for sparse signals with dependent covariance structure. PLoS ONE 2023, 18, e0284284. [Google Scholar] [CrossRef]
- van den Heuvel, E.; Zhan, Z. Myths about linear and monotonic associations: Pearson’sr, Spearman’s ρ, and Kendall’s τ. Am. Stat. 2022, 76, 44–52. [Google Scholar] [CrossRef]
- Delgado-Chaves, F.M.; Gómez-Vela, F.; García-Torres, M.; Divina, F.; Vazquez Noguera, J.L. Computational inference of gene co-expression networks for the identification of lung carcinoma biomarkers: An ensemble approach. Genes 2019, 10, 962. [Google Scholar] [CrossRef] [PubMed]
- Ovens, K.; Eames, B.F.; McQuillan, I. Comparative analyses of gene co-expression networks: Implementations and applications in the study of evolution. Front. Genet. 2021, 12, 695399. [Google Scholar] [CrossRef]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic genes of breast cancer identified by gene co-expression network analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef]
- Zhou, Q.; Ren, J.; Hou, J.; Wang, G.; Ju, L.; Xiao, Y.; Gong, Y. Co-expression network analysis identified candidate biomarkers in association with progression and prognosis of breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2383–2396. [Google Scholar] [CrossRef]
- Adhami, M.; MotieGhader, H.; Haghdoost, A.A.; Afshar, R.M.; Sadeghi, B. Gene co-expression network approach for predicting prognostic microRNA biomarkers in different subtypes of breast cancer. Genomics 2020, 112, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z. Identification of T cell-related biomarkers for breast cancer based on weighted gene co-expression network analysis. J. Chemother. 2023, 35, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.-Y.; Chao, F.; Zhuo, Z.; Ma, Z.; Li, W.; Chen, G. Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis. Aging 2019, 11, 4736. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Dong, R.N.; Lin, T.T.; Lin, T.; Lin, Y.Z.; Chen, S.H.; Zhu, J.M.; Ke, Z.B.; Huang, F.; Chen, Y.H.; et al. Development and validation of novel biomarkers related to M2 macrophages infiltration by weighted gene co-expression network analysis in prostate cancer. Front. Oncol. 2021, 11, 634075. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, M.Y.; Huang, J.M.; Liu, Q.; Wang, L.; Liu, R.; Yang, N.; Huang, W.H.; Zhang, W. LncRNA weighted gene co-expression network analysis reveals novel biomarkers related to prostate cancer metastasis. BMC Med. Genom. 2022, 15, 256. [Google Scholar] [CrossRef]
- Deng, J.L.; Xu, Y.-H.; Wang, G. Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front. Genet. 2019, 10, 695. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, Y.; Chen, C.Y.; Wang, L.; Huo, Z.; Kim, S.; Sotiriou, C.; Oesterreich, S.; Tseng, G.C. MetaDCN: Meta-analysis framework for differential co-expression network detection with an application in breast cancer. Bioinformatics 2017, 33, 1121–1129. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.E.; Obradovic, A.Z.; Dallos, M.C.; Lim, E.A.; Runcie, K.; Ager, C.R.; McKiernan, J.; Anderson, C.B.; Decastro, J.; Weintraub, J.; et al. Single-Cell RNAseq Analysis Reveals Robust, Anti-PD-1-Mediated Increase in Immune Infiltrate in Metastatic Castration-Sensitive Prostate Cancer. bioRxiv 2022, 490968. [Google Scholar] [CrossRef]
- Decker, A.M.; Decker, J.T.; Jung, Y.; Cackowski, F.C.; Daignault-Newton, S.; Morgan, T.M.; Shea, L.D.; Taichman, R.S. Adrenergic blockade promotes maintenance of dormancy in prostate cancer through upregulation of GAS6. Transl. Oncol. 2020, 13, 100781. [Google Scholar] [CrossRef]
- Mevik, B.H.; Wehrens, R. The pls Package: Principal Component and Partial Least Squares Regression in R. J. Stat. Softw. 2007, 18, 1–23. [Google Scholar] [CrossRef]
- Siletz, A.; Schnabel, M.; Kniazeva, E.; Schumacher, A.J.; Shin, S.; Jeruss, J.S.; Shea, L.D. Dynamic transcription factor networks in epithelial-mesenchymal transition in breast cancer models. PLoS ONE 2013, 8, e57180. [Google Scholar] [CrossRef]
- Haury, A.C.; Mordelet, F.; Vera-Licona, P.; Vert, J.P. TIGRESS: Trustful inference of gene regulation using stability selection. BMC Syst. Biol. 2012, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Cutler, A.; Cutler, D.R.; Stevens, J.R. Random forests. In Ensemble Machine Learning: Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2012; pp. 157–175. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Meyer, P.E.; Kontos, K.; Lafitte, F.; Bontempi, G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007, 2007, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, R.S.; Cavalcante, J.V.F.; Magalhães, L.; Ribeiro-dos Santos, Â.; Dalmolin, R.J.S. Revealing metastatic castration-resistant prostate cancer master regulator through lncRNAs-centered regulatory network. Cancer Med. 2023, 12, 19279–19290. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Delgado-Chaves, F.M.; Rodríguez-Baena, D.S.; García-Torres, M.; Divina, F. Ensemble and Greedy Approach for the Reconstruction of Large Gene Co-Expression Networks. Entropy 2019, 21, 1139. [Google Scholar] [CrossRef]
- Planche, A.; Bacac, M.; Provero, P.; Fusco, C.; Delorenzi, M.; Stehle, J.C.; Stamenkovic, I. Identification of Prognostic Molecular Features in the Reactive Stroma of Human Breast and Prostate Cancer. PLoS ONE 2011, 6, e18640. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5, 1408. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Nie, J.; Chen, H.; Ma, H.; Stewart, R.; Li, X.; Lu, M.; Taylor, W.; Wei, H. Evaluation of Gene Association Methods for Coexpression Network Construction and Biological Knowledge Discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef] [PubMed]
- de Siqueira Santos, S.; Takahashi, D.Y.; Nakata, A.; Fujita, A. A Comparative Study of Statistical Methods Used to Identify Dependencies Between Gene Expression Signals. Brief. Bioinform. 2014, 15, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Díaz-Montaña, J.J.; Gómez-Vela, F.; Díaz-Díaz, N. GNC–app: A new Cytoscape app to rate gene networks biological coherence using gene–gene indirect relationships. Biosystems 2018, 166, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Lagares, J.A.; Díaz-Díaz, N. Gene network coherence based on prior knowledge using direct and indirect relationships. Comput. Biol. Chem. 2015, 56, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.; Sibille, E. Beyond modules and hubs: The potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 2014, 13, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community structure analysis of biological networks. Bioinformatics 2010, 26, 3135–3137. [Google Scholar] [CrossRef]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tipney, H.; Hunter, L. An introduction to effective use of enrichment analysis software. Hum. Genom. 2010, 4, 202. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Chen, L.; Zhang, W. WGCNA application to proteomic and metabolomic data analysis. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 585, pp. 135–158. [Google Scholar]
- Liu, W.; Li, L.; Ye, H.; Tu, W. Weighted gene co-expression network analysis in biomedicine research. Chin. J. Biotechnol. 2017, 33, 1791–1801. [Google Scholar]
- Delgado-Chaves, F.M.; Gómez-Vela, F.; Divina, F.; García-Torres, M.; Rodriguez-Baena, D.S. Computational analysis of the global effects of Ly6E in the immune response to coronavirus infection using gene networks. Genes 2020, 11, 831. [Google Scholar] [CrossRef] [PubMed]
- Saz-Navarro, D.M.; López-Fernández, A.; Gómez-Vela, F.A.; Rodriguez-Baena, D.S. CyEnGNet—App: A new Cytoscape app for the reconstruction of large co-expression networks using an ensemble approach. SoftwareX 2024, 25, 101634. [Google Scholar] [CrossRef]
- Yu, Y.R.; Ho, P.C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef]
- Mukherjee, S.; Diaz Valencia, J.D.; Stewman, S.; Metz, J.; Monnier, S.; Rath, U.; Asenjo, A.B.; Charafeddine, R.A.; Sosa, H.J.; Ross, J.L.; et al. Human Fidgetin is a microtubule severing enzyme and minus-end depolymerase that regulates mitosis. Cell Cycle 2012, 11, 2359–2366. [Google Scholar] [CrossRef]
- Dauphinee, S.M.; Clayton, A.; Hussainkhel, A.; Yang, C.; Park, Y.J.; Fuller, M.E.; Blonder, J.; Veenstra, T.D.; Karsan, A. SASH1 is a scaffold molecule in endothelial TLR4 signaling. J. Immunol. 2013, 191, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Sostres, C.; Gargallo, C.J.; Lanas, A. Aspirin, cyclooxygenase inhibition and colorectal cancer. World J. Gastrointest. Pharmacol. Ther. 2014, 5, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Oulès, B.; Pisco, A.O.; Ly, T.; Liakath-Ali, K.; Walko, G.; Viswanathan, P.; Tihy, M.; Nijjher, J.; Dunn, S.J.; et al. A protein phosphatase network controls the temporal and spatial dynamics of differentiation commitment in human epidermis. eLife 2017, 6, e27356. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xu, S.; Dong, X.; Lu, C.; Springer, T.A. Prodomain-growth factor swapping in the structure of pro-TGF-β1. J. Biol. Chem. 2018, 293, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Hieda, M.; Yokoyama, Y.; Nishioka, Y.; Yoshidome, K.; Tsujimoto, M.; Matsuura, N. Pérdida global de un componente de lámina nuclear, lamina A/C y componentes complejos LINC SUN1, SUN2 y nesprin-2 en el cáncer de mama. Cancer Med. 2015, 4, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Moon, H.G.; Cho, B.I.; Jeong, C.Y.; Joo, Y.T.; Lee, Y.J.; Hong, S.C.; Choi, S.K.; Ha, W.S.; Kim, J.W.; et al. Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. Int. J. Cancer 2007, 120, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.L.; Zhang, X.; Chang, M.; Huang, C.; Qian, J.; Li, Q.; Yuan, F.; Sun, L.; Yu, X.; Cui, X.; et al. Genome-wide 5-Hydroxymethylcytosine Profiling Analysis Identifies MAP7D1 as A Novel Regulator of Lymph Node Metastasis in Breast Cancer. Genom. Proteom. Bioinform. 2021, 19, 64–79. [Google Scholar] [CrossRef]
- Borziak, K.; Finkelstein, J. Gene Expression Markers of Prognostic Importance for Prostate Cancer Risk in Patients with Benign Prostate Hyperplasia. In Proceedings of the 2022 44th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Glasgow, UK, 11–15 July 2022; pp. 73–76. [Google Scholar] [CrossRef]
- Zhou, C.; Dai, X.; Chen, Y.; Shen, Y.; Lei, S.; Xiao, T.; Bartfai, T.; Ding, J.; Wang, M. G protein-coupled receptor GPR160 is associated with apoptosis and cell cycle arrest of prostate cancer cells. Oncotarget 2016, 7, 12823–12839. [Google Scholar] [CrossRef]
- Joyce, J.; Fearon, D. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Gingis-Velitski, S.; Zetser, A.; Flugelman, M.; Vlodavsky, I.; Ilan, N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J. Biol. Chem. 2004, 279, 23536–23541. [Google Scholar] [CrossRef]
- Goldshmidt, O.; Zcharia, E.; Cohen, M.; Aingorn, H.; Cohen, I.; Nadav, L.; Katz, B.; Geiger, B.; Vlodavsky, I. Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 2003, 17, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Herrera-Abreu, M.; Rohn, J.; Racine, V.; Tajadura, V.; Suryavanshi, N.; Bechtel, S.; Wiemann, S.; Baum, B.; Ridley, A. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011, 9, 54. [Google Scholar] [CrossRef]
- Li, Z.; Bresnick, A.R. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res. 2006, 66, 5173–5180. [Google Scholar] [CrossRef]
- Torres-Rosado, A.; O’Shea, K.S.; Tsuji, A.; Chou, S.H.; Kurachi, K. Hepsin, a putative cell-surface serine protease, is required for mammalian cell growth. Proc. Natl. Acad. Sci. USA 1993, 90, 7181–7185. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Egevad, L. Prostatic stromal proliferations: A review. Pathology 2021, 53, 12–25. [Google Scholar] [CrossRef]
- Hansel, D.E.; Herawi, M.; Montgomery, E.; Epstein, J.I. Spindle cell lesions of the adult prostate. Mod. Pathol. 2007, 20, 148–158. [Google Scholar] [CrossRef]
- Liang, L.; Xu, J.; Wang, M.; Xu, G.; Zhang, N.; Wang, G.; Zhao, Y. LncRNA HCP5 promotes the progression of follicular thyroid carcinoma through miRNA sponging. Cell Death Dis. 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Da, J.; Liu, X.; Liu, X.; Wang, J.; Jin, H.; Li, Y.; Zhang, B. Downregulated Expression of RIPOR3 Correlated with Immune Infiltrates Predicts Poor Prognosis in Oral Tongue Cancer. Med. Sci. Monit. 2022, 28, e935055. [Google Scholar] [CrossRef]
- Yang, M.; Lu, Z.; Yu, B.; Zhao, J.; Li, L.; Zhu, K.; Ma, M.; Long, F.; Wu, R.; Hu, G.; et al. COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression. Cancers 2022, 14, 3244. [Google Scholar] [CrossRef]
- Liao, Z.; Wang, X.; Wang, X.; Li, L.; Lin, D. DEPDC7 inhibits cell proliferation, migration and invasion in hepatoma cells. Oncol. Lett. 2017, 14, 7332–7338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Chen, J.; Huang, M.; Mei, J.; Lin, M. The Functions of Long Noncoding RNAs on Regulation of F-Box Proteins in Tumorigenesis and Progression. Front. Oncol. 2022, 12, 963617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, L.; Yang, L.; Lv, P.; Mai, S.; Wang, Y. Long Non-Coding RNA GATA6-AS1 Sponges miR-324-5p to Inhibit Lung Cancer Cell Proliferation and Invasion. OncoTargets Ther. 2020, 13, 9741–9751. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, X.; Zhang, Y.; Zheng, W.; Zhang, H. GATA6-AS1 inhibits ovarian cancer cell proliferation and migratory and invasive abilities by sponging miR-19a-5p and upregulating TET2. Oncol. Lett. 2021, 22, 718. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Akiyama, M.; Park, J.; Lin, M.; Shimo, A.; Ueki, T.; Daigo, Y.; Tsunoda, T.; Nishidate, T.; Nakamura, Y.; et al. Activation of an estrogen/estrogen receptor signaling by BIG3 through its inhibitory effect on nuclear transport of PHB2/REA in breast cancer. Cancer Sci. 2009, 100, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Sakamoto, T.; Hashimoto, T.; Tsukamoto, S.; Iwasa, S.; Saito, Y.; Sekine, S. Identification of a novel PRR15L-RSPO2 fusion transcript in a sigmoid colon cancer derived from superficially serrated adenoma. Virchows Arch. 2019, 475, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, Y.; Zhu, R.; Li, Y.; Jiang, H.; Li, R.; Chen, Q.; Wang, Q.; Tang, L.; Ren, Z. Identification of epigenetic modifications mediating the antagonistic effect of selenium against cadmium-induced breast carcinogenesis. Environ. Sci. Pollut. Res. Int. 2022, 29, 22056–22068. [Google Scholar] [CrossRef]
- Dong, Q.; Yan, L.; Xu, Q.; Hu, X.; Yang, Y.; Zhu, R.; Xu, Q.; Yang, Y.; Wang, B. Pan-Cancer Analysis of Forkhead Box Q1 as a Potential Prognostic and Immunological Biomarker. Front. Genet. 2022, 13, 944970. [Google Scholar] [CrossRef]





| Group | Metric | Proposed Method | WGCNA | EnGNet |
|---|---|---|---|---|
| Prostate Normal | Mean | 0.26044 | 0.26575 | 0.10877 |
| Maximum value | 0.50309 | 0.47667 | 0.22808 | |
| Prostate tumor | Mean | 0.24832 | 0.26014 | 0.09970 |
| Maximum value | 0.52010 | 0.49204 | 0.22293 | |
| Breast Normal | Mean | 0.32993 | 0.13133 | 0.11581 |
| Maximum value | 0.51614 | 0.30467 | 0.28765 | |
| Breast tumor | Mean | 0.33407 | 0.12499 | 0.13853 |
| Maximum value | 0.51646 | 0.26190 | 0.35152 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa-Martínez, J.; Saz-Navarro, D.M.; López-Fernández, A.; Rodríguez-Baena, D.S.; Gómez-Vela, F.A. Computational Ensemble Gene Co-Expression Networks for the Analysis of Cancer Biomarkers. Informatics 2024, 11, 14. https://doi.org/10.3390/informatics11020014
Figueroa-Martínez J, Saz-Navarro DM, López-Fernández A, Rodríguez-Baena DS, Gómez-Vela FA. Computational Ensemble Gene Co-Expression Networks for the Analysis of Cancer Biomarkers. Informatics. 2024; 11(2):14. https://doi.org/10.3390/informatics11020014
Chicago/Turabian StyleFigueroa-Martínez, Julia, Dulcenombre M. Saz-Navarro, Aurelio López-Fernández, Domingo S. Rodríguez-Baena, and Francisco A. Gómez-Vela. 2024. "Computational Ensemble Gene Co-Expression Networks for the Analysis of Cancer Biomarkers" Informatics 11, no. 2: 14. https://doi.org/10.3390/informatics11020014