
ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation
 , , and
, , and 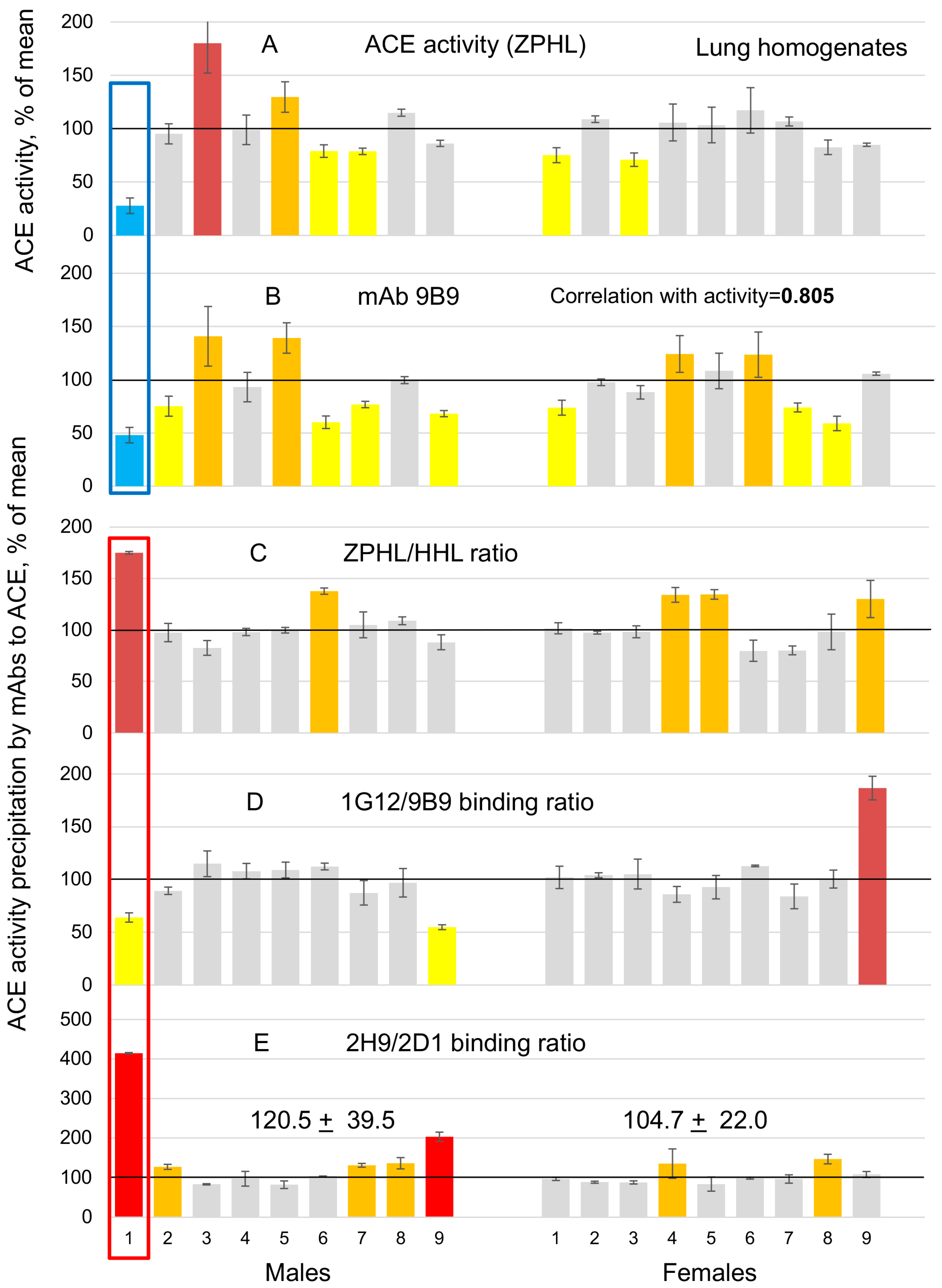{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Chemicals
2.2. Antibodies
2.3. Study Participants
2.4. ACE Activity Assay
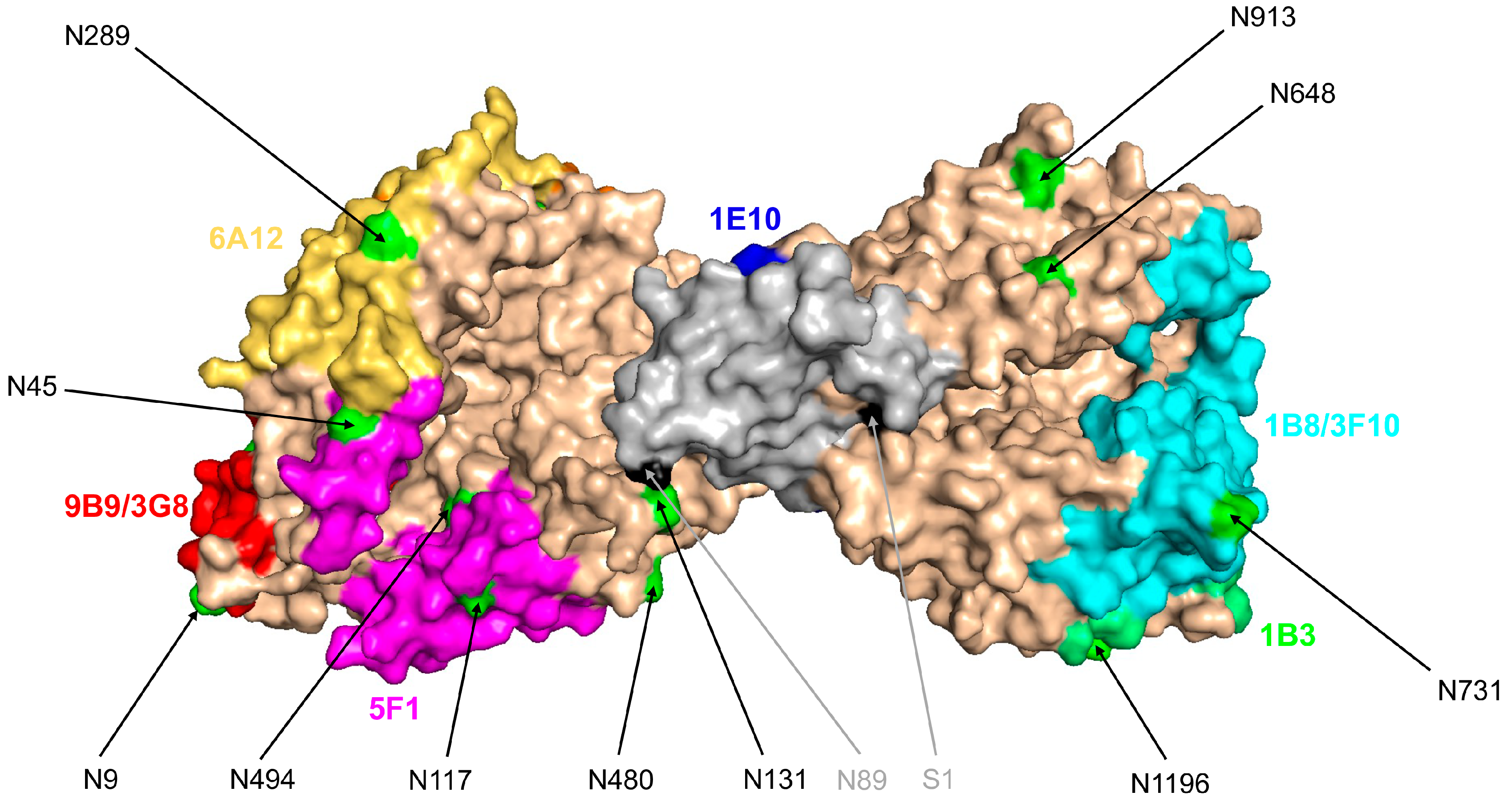
2.5. Immunological Characterization of the Blood ACE
2.6. Whole Exome Sequencing
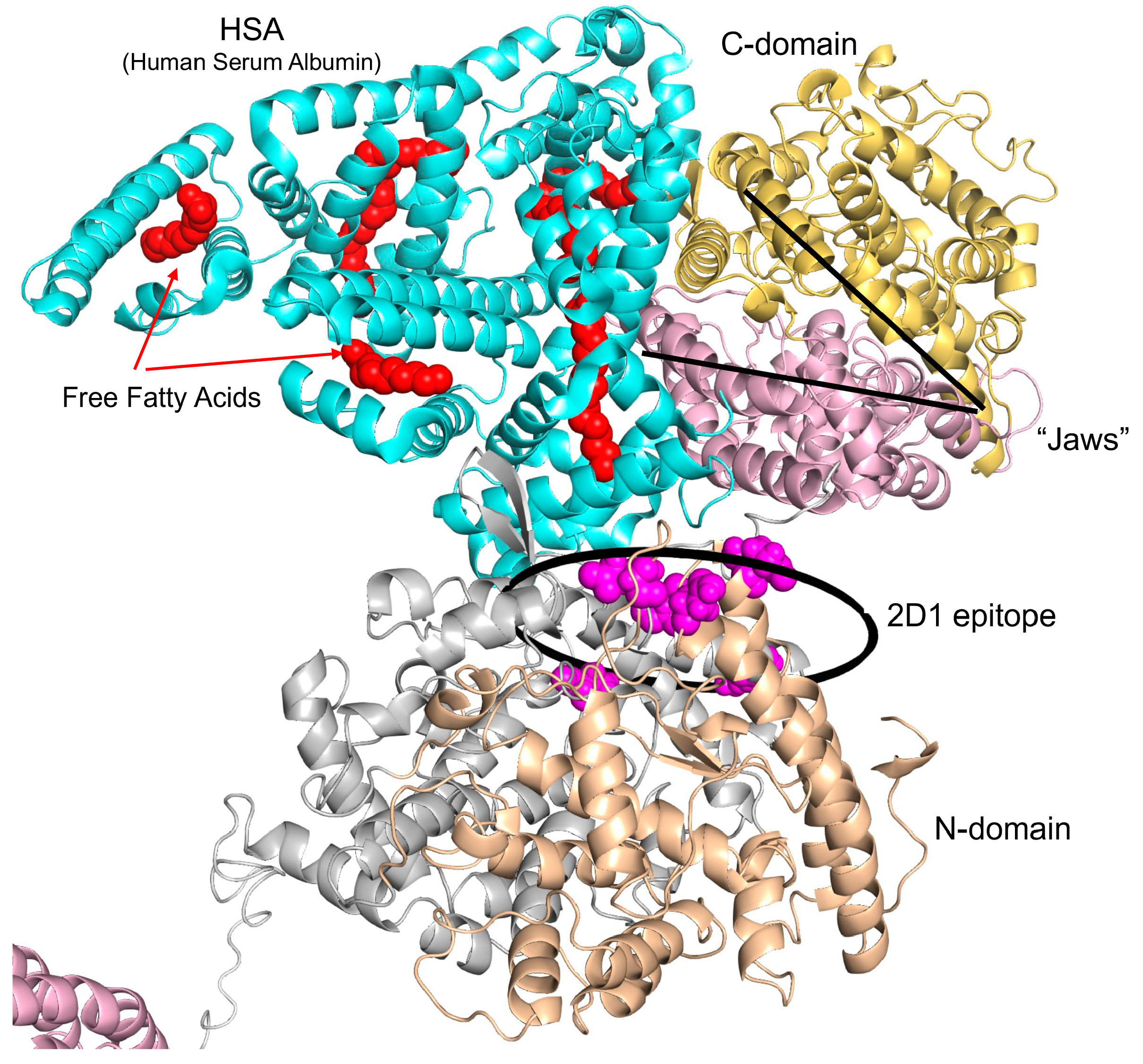
2.7. Computational Analysis of the Models of ACE Interaction with Human Serum Albumin (HSA) and CCL18
2.8. Statistical Analysis
3. Results and Discussion
3.1. ACE Phenotyping in Human Lung Homogenates and Corresponding Sera Samples
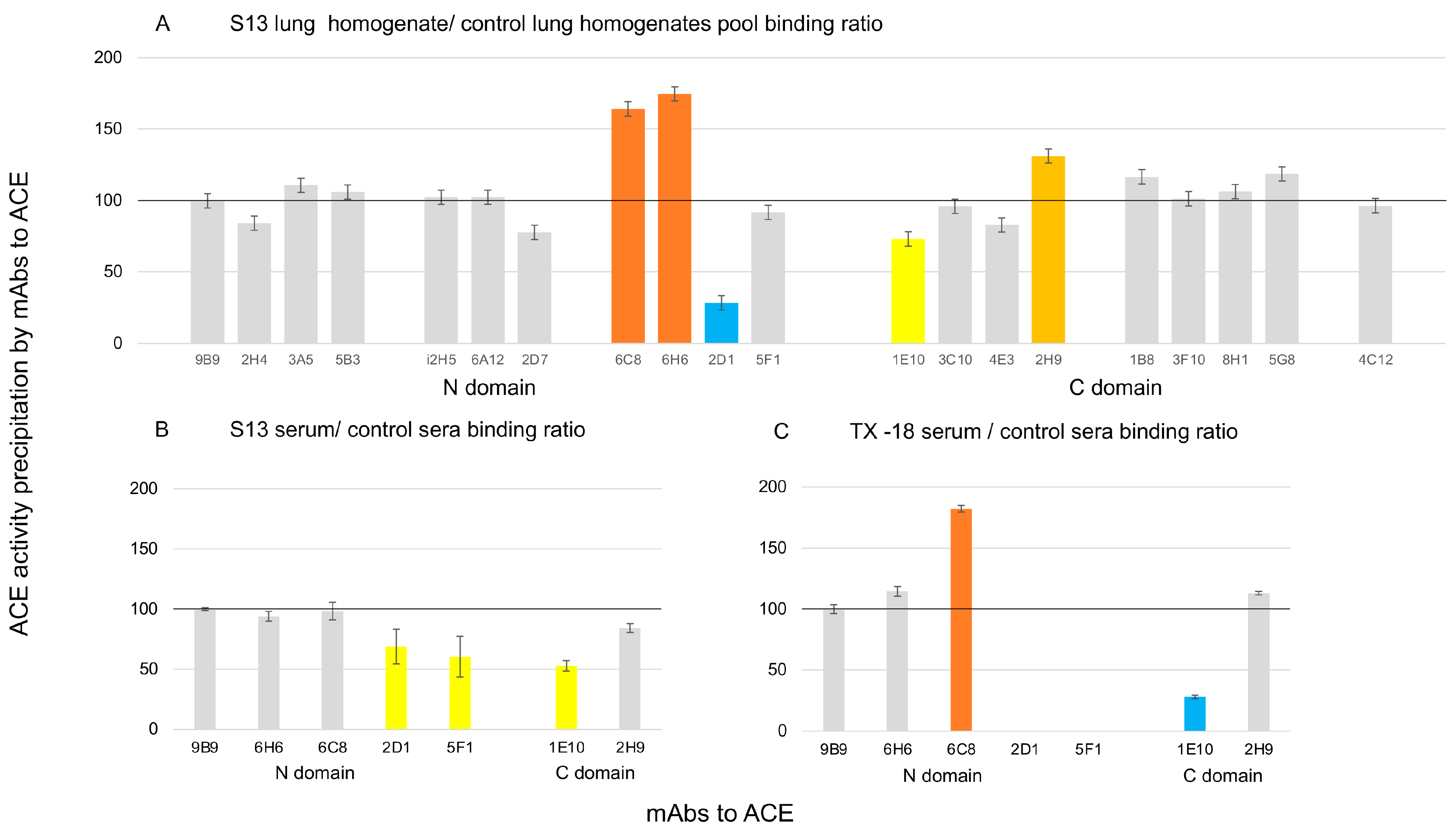
3.2. Further Analysis of ACE Outlier (from Patient LM1)
- SAA1—rs1136747 (V75A);
- ADAMTSL4—rs199599791 (R87P);
- NOD2—rs2066844 (R702W);
- SMPD1—rs550365194 (V36-V39 del).
3.3. Sex Differences in ACE Sialylation in Different Tissues
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sturrock, E.D.; Anthony, C.S.; Danilov, S.M. Peptidyl-dipeptidase a/angiotensin I-converting enzyme. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2012; pp. 480–494. [Google Scholar]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.-L.B.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev. 2012, 65, 1–46. [Google Scholar] [CrossRef]
- Ching, S.F.; Hayes, L.W.; Slakey, L.L. Angiotensin-converting enzyme in cultured endothelial cells. Synthesis, degradation, and transfer to culture medium. Arteriosclerosis 1983, 3, 581–588. [Google Scholar] [CrossRef]
- Metzger, R.; Franke, F.F.; Bohle, R.M.; Alhenc-Gelas, F.; Danilov, S.M. Heterogeneous distribution of Angiotensin I-converting enzyme (CD143) in the human and rat vascular systems: Vessels, organs, and species specificity. Microvasc. Res. 2011, 82, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Turner, A.J.; Hooper, N.M. Secretase-mediated cell surface shedding of the angiotensin-converting enzyme. Protein Pept. Lett. 2004, 11, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Alhenc-Gelas, F.; Richard, J.; Courbon, D.; Warnet, J.M.; Corvol, P. Distribution of plasma angiotensin I-converting enzyme levels in healthy men: Relationship to environmental and hormonal parameters. J. Lab. Clin. Med. 1991, 117, 33–39. [Google Scholar] [PubMed]
- Rømer, F.K. Clinical and biochemical aspects of sarcoidosis. With special reference to angiotensin-converting enzyme (ACE). Acta Med. Scand. Suppl. 1984, 690, 3–96. [Google Scholar] [PubMed]
- Xu, C.; Garcia, D.; Lu, Y.; Ozuna, K.; Adjeroh, D.A.; Wang, K. Levels of angiotensin-converting enzyme and apolipoproteins are associated with Alzheimer’s disease and cardiovascular diseases. Cells 2022, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, P.; Russ, C.; McIlory, S.; Williams, H.; Holmans, P.; Holmes, C.; Liolitsa, D.; Vahidassr, D.; Powell, J.; McGleenon, B.; et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer’s disease. Nat. Genet. 1999, 21, 71–72. [Google Scholar] [CrossRef]
- Liu, S.; Ando, F.; Fujita, Y.; Liu, J.; Maeda, T.; Shen, X.; Kikuchi, K.; Matsumoto, A.; Yokomori, M.; Tanabe-Fujimura, C.; et al. A clinical dose of angiotensin-converting enzyme (ACE) inhibitor and heterozygous ace deletion exacerbate Alzheimer’s disease pathology in mice. J. Biol. Chem. 2019, 294, 9760–9770. [Google Scholar] [CrossRef]
- Xie, X.-Y.; Zhao, Q.-H.; Huang, Q.; Dammer, E.; Chen, S.-D.; Ren, R.-J.; Wang, G. Genetic profiles of familial late-onset Alzheimer’s disease in China: The Shanghai FLOAD Study. Genes Dis. 2022, 9, 1639–1649. [Google Scholar] [CrossRef]
- Danilov, S.M.; Adzhubey, I.A.; Kozuch, A.J.; Petukhov, P.A.; Popova, I.A.; Choudhury, A.; Sengupta, D.; Dudek, S.M. Carriers of heterozygous loss-of-function ACE mutations are at risk for Alzheimer disease. Biomedicines 2024, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Jain, M.S.; Petukhov, P.A.; Goldman, C.; Goldman, M.; Vancavage, R.; Francuzevitch, L.Y.; Samokhodskaya, L.M.; Kamalov, A.A.; Arbieva, Z.H.; et al. Novel ACE mutations mimicking sarcoidosis by increasing blood ACE Levels. Transl. Res. 2021, 230, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Samokhodskaya, L.M.; Jain, M.S.; Kurilova, O.V.; Bobkov, A.P.; Kamalov, A.A.; Dudek, S.M.; Danilov, S.M. Phenotyping angiotensin-converting enzyme in blood: A necessary approach for precision medicine. J. Appl. Lab. Med. 2021, 6, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Fagyas, M.; Úri, K.; Siket, I.M.; Fülöp, G.Á.; Csató, V.; Daragó, A.; Boczán, J.; Bányai, E.; Szentkirályi, I.E.; Maros, T.M.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) II: Albumin suppresses angiotensin converting enzyme (ACE) activity in human. PLoS ONE 2014, 9, e87844. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, A.J.; Petukhov, P.A.; Fagyas, M.; Popova, I.A.; Lindeblad, M.O.; Bobkov, A.P.; Kamalov, A.A.; Toth, A.; Dudek, S.M.; Danilov, S.M. Urinary ACE phenotyping as a research and diagnostic tool: Identification of sex-dependent ACE immunoreativity. Biomedicines 2023, 11, 953. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Balyasnikova, I.V.; Danilova, A.S.; Naperova, I.A.; Arablinskaya, N.E.; Borisov, S.E.; Metzger, R.; Franke, F.E.; Schwartz, D.E.; Gachok, I.V.; et al. Conformational fingerprinting of the angiotensin I-converting enzyme (ACE). 1. application in sarcoidosis. J. Proteome Res. 2010, 9, 5782–5793. [Google Scholar] [CrossRef] [PubMed]
- Popova, I.A.; Lubbe, L.; Petukhov, P.A.; Kalantarov, G.F.; Trakht, I.N.; Chernykh, E.R.; Leplina, O.Y.; Lyubimov, A.V.; Garcia, J.G.N.; Dudek, S.M.; et al. Epitope mapping of novel monoclonal antibodies to human angiotensin I-converting enzyme. Protein Sci. 2021, 30, 1577–1593. [Google Scholar] [CrossRef] [PubMed]
- Bánhegyi, V.; Enyedi, A.; Fülöp, G.Á.; Oláh, A.; Siket, I.M.; Váradi, C.; Bottyán, K.; Lódi, M.; Csongrádi, A.; Umar, A.J.; et al. Human Tissue Angiotensin Converting Enzyme (ACE) Activity Is Regulated by Genetic Polymorphisms, Posttranslational Modifications, Endogenous Inhibitors and Secretion in the Serum, Lungs and Heart. Cells 2021, 10, 1708. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Savoie, F.; Lenoir, B.; Jeunemaitre, X.; Azizi, M.; Tarnow, L.; Alhenc-Gelas, F. Development of enzyme-linked imunoassays for human angiotensin-I converting enzyme suitable for large-scale studies. J. Hypertens. 1996, 14, 719–727. [Google Scholar] [CrossRef]
- Danilov, S.M.; Balyasnikova, I.V.; Albrecht, R.F.; Kost, O.A. Simultaneous determination of ACE activity with 2 substrates provides information on the status of somatic ace and allows detection of inhibitors in human blood. J. Cardiovasc. Pharmacol. 2008, 52, 90–103. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grune, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.A.; Curry, S.; Franks, N.P. Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. J. Biol. Chem. 2000, 275, 38731–38738. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 2.5.2; Schrodinger, LLC: New York, NY, USA, 2022.
- Danilov, S.M.; Gordon, K.; Nesterovitch, A.B.; Luensdorf, H.; Chen, Z.; Castellon, M.; Popova, I.A.; Kalinin, S.; Mendonca, E.; Petukhov, P.A.; et al. Angiotensin I-converting enzyme mutation (Y465D) causes dramatic increase in blood ACE via accelerated ACE shedding due to changes of ACE dimerization. PLoS ONE 2011, 6, e25952. [Google Scholar] [CrossRef] [PubMed]
- Cozier, G.E.; Lubbe, L.; Sturrock, E.D.; Acharya, K.R. Angiotensin-converting enzyme open for business: Structural insights into the subdomain dynamics. FEBS J. 2020, 288, 2238–2256. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Tikhomirova, V.E.; Kryukova, O.V.; Balatsky, A.V.; Bulaeva, N.I.; Golukhova, E.Z.; Bokeria, L.A.; Samokhodskaya, L.M.; Kost, O.A. Conformational fingerprint of blood and tissue ACEs: Personalized approach. PLoS ONE 2018, 13, e0209861. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Lünsdorf, H.; Akinbi, H.T.; Nesterovitch, A.B.; Epshtein, Y.; Letsiou, E.; Kryukova, O.V.; Piegeler, T.; Golukhova, E.Z.; Schwartz, D.E.; et al. Lysozyme and bilirubin bind to ACE and regulate its conformation and shedding. Sci. Rep. 2016, 6, 34913. [Google Scholar] [CrossRef] [PubMed]
- Cídl, K.; Strelcová, L.; Znojil, V.; Váchal, J. Angiotensin I-converting enzyme (ACE) polymorphism and ABO blood groups as factors codetermining plasma ACE activity. Exp. Hematol. 1996, 24, 790–794. [Google Scholar] [PubMed]
- Zhang, Y.F.; Cheng, Q.; Tang, N.L.; Chu, T.T.; Tomlinson, B.; Liu, F.; Kwok, T.C.Y. Gender difference of serum angiotensin-converting enzyme (ACE) activity in DD genotype of ACE insertion/deletion polymorphism in elderly Chinese. J. Renin Angiotensin Aldosterone Syst. 2013, 15, 547–552. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.H.; Danahey, K.; Ratain, M.J. The outlier in all of us: Why implementing pharmacogenomics could matter for everyone. Clin. Pharmacol. Ther. 2016, 99, 401–404. [Google Scholar] [CrossRef]
- Zhu, Z.; Ihle, N.T.; Rejto, P.A.; Zarrinkar, P.P. Outlier analysis of functional genomic profiles enriches for oncology targets and enables precision medicine. BMC Genom. 2016, 17, 455. [Google Scholar] [CrossRef]
- Kaiser, J. The Hunt for Missing Genes. Science 2014, 344, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Wade, M.S.; Schwager, S.L.; Douglas, R.G.; Nesterovitch, A.B.; Popova, I.A.; Hogarth, K.D.; Bhardwaj, N.; Schwartz, D.E.; Sturrock, E.D.; et al. A novel angiotensin I-converting enzyme mutation (S333W) impairs N-domain enzymatic cleavage of the anti-fibrotic peptide, AcSDKP. PLoS ONE 2014, 9, e88001. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirova, V.E.; Kost, O.A.; Kryukova, O.V.; Golukhova, E.A.; Bulaeva, N.I.; Zholbaeva, A.Z.; Bokeria, L.A.; Garcia, J.G.N.; Danilov, S.M. ACE phenotyping in human heart. PLoS ONE 2017, 12, e0181976. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, K.; Fujimura, A.; Ebisu, H.; Sakai, T.; Sadakane, Y.; Fujii, N.; Tanimura, T. Acein-1, a novel angiotensin-I-converting enzyme inhibitory peptide isolated from tryptic hydrolysate of human plasma. FEBS Lett. 1998, 438, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, K.; Yamada, R.; Ebisu, H.; Sadakane, Y.; Akizawa, T.; Tanimura, T. Isolation of acein-2, a novel angiotensin-I-converting enzyme inhibitory peptide derived from a tryptic hydrolysate of human plasma. FEBS Lett. 2000, 467, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Skirgello, O.E.; Balyasnikova, I.V.; Binevski, P.V.; Sun, Z.-L.; Baskin, I.I.; Palyulin, V.A.; Nesterovitch, A.B.; Albrecht, R.F.; Kost, O.A.; Danilov, S.M. Inhibitory antibodies to human angiotensin-converting enzyme: Fine epitope mapping and mechanism of action. Biochemistry 2006, 45, 4831–4847. [Google Scholar] [CrossRef] [PubMed]
- Watermeyer, J.M.; Sewell, B.T.; Schwager, S.L.; Natesh, R.; Corradi, H.R.; Acharya, K.R.; Sturrock, E.D. Structure of testis ACE glycosylation mutants and evidence for conserved domain movement. Biochemistry 2006, 45, 12654–12663. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, L.; Sewell, B.T.; Sturrock, E.D. The influence of angiotensin converting enzyme mutations on the kinetics and dynamics of N-domain selective inhibition. FEBS J. 2016, 283, 3941–3961. [Google Scholar] [CrossRef]
- Vy, T.T.; Heo, S.Y.; Jung, W.K.; Yi, M. Spontaneous Hinge-Bending Motions of Angiotensin I Converting Enzyme: Role in Activation and Inhibition. Molecules 2020, 25, 1288. [Google Scholar] [CrossRef]
- Lubbe, L.; Sewell, B.T.; Woodward, J.D.; Sturrock, E.D. Cryo-EM reveals mechanisms of angiotensin I-converting enzyme allostery and dimerization. EMBO J. 2022, 41, e110550. [Google Scholar] [CrossRef]
- Santhamma, K.R.; Sen, I. Specific cellular proteins associated with angiotensin-converting enzyme and regulate its intracellular transport and cleavage-secretion. J. Biol. Chem. 2000, 275, 23253–23258. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.M.; Tikhomirova, V.E.; Metzger, R.; Naperova, I.A.; Bukina, T.M.; Goker-Alpan, O.; Tayebi, N.; Gayfullin, N.M.; Schwartz, D.E.; Samokhodskaya, L.M.; et al. ACE phenotyping in Gaucher disease. Mol. Genet. Metab. 2018, 123, 501–510. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Wei, L.; Alhenc-Gelas, F.; Corvol, P.; Clauser, E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 1991, 266, 9002–9008. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Trindale, B.C.; Chen, G.Y. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol. Rev. 2020, 297, 136–161. [Google Scholar]
- Málaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; Souza, C.F.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42 (Suppl. 1), 197–206. [Google Scholar] [CrossRef]
- Moshkovskii, S.S. Why do cancer cells produce Serum Amyloid A acute-phase protein? Biochemistry 2012, 77, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Sack, G.H. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Griffiths, K.; Maxwell, A.P.; McCarter, R.V.; Nicol, P.; Hogg, R.E.; Harbinson, M.; McKay, G.J. Serum amyloid A levels are associated with polymorphic variants in the serum amyloid A1 and A2 genes. Irish J. Med. Sci. 2019, 188, 1175–1183. [Google Scholar] [CrossRef]
- Cho, W.G.S.; Yip, T.T.; Cheng, W.W.; Au, J.S.K. Serum amyloid A is elevated in the serum of lung cancer patients with poor prognosis. Br. J. Cancer 2010, 102, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Hollak, C.E.; Verhoek, M.; Alberts, C.; Jonkers, R.E.; Aerts, J.M. Plasma chitotriosidase and CCL18 as surrogate markers for granulomatous macrophages in sarcoidosis. Clin. Chim. Acta 2010, 411, 31–36. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Orós, I.; Irún, P.; Cebolla, J.J.; Rodriguez-Sureda, V.; Mallén, M.; Pueyo, M.J.; Mozas, P.; Dominguez, C.; Pocoví, M. Assessment of plasma chitotriosidase activity, CCL18/PARC concentration and NP-C suspicion index in the diagnosis of Niemann-Pick disease type C: A prospective observational study. J. Transl. Med. 2017, 15, 43. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enyedi, E.E.; Petukhov, P.A.; Kozuch, A.J.; Dudek, S.M.; Toth, A.; Fagyas, M.; Danilov, S.M. ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation. Biomedicines 2024, 12, 940. https://doi.org/10.3390/biomedicines12050940
Enyedi EE, Petukhov PA, Kozuch AJ, Dudek SM, Toth A, Fagyas M, Danilov SM. ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation. Biomedicines. 2024; 12(5):940. https://doi.org/10.3390/biomedicines12050940
Chicago/Turabian StyleEnyedi, Enikő E., Pavel A. Petukhov, Alexander J. Kozuch, Steven M. Dudek, Attila Toth, Miklós Fagyas, and Sergei M. Danilov. 2024. "ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation" Biomedicines 12, no. 5: 940. https://doi.org/10.3390/biomedicines12050940