
The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress

1
Clinical Pharmacology Research Center, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing 210009, China
2
Center of System Pharmacology and Pharmacometrics, College of Pharmacy, University of Florida, Gainesville, FL 32611, USA
*
Authors to whom correspondence should be addressed.
Antioxidants 2024, 13(1), 132; https://doi.org/10.3390/antiox13010132
Submission received: 29 November 2023
/
Revised: 6 January 2024
/
Accepted: 12 January 2024
/
Published: 22 January 2024
(This article belongs to the Special Issue Crosstalk Between Oxidative Stress and Inflammation in Cardiovascular Diseases, Cancer and Neurodegeneration)
Abstract
:Myeloperoxidase (MPO) is a heme-containing peroxidase, mainly expressed in neutrophils and, to a lesser extent, in monocytes. MPO is known to have a broad bactericidal ability via catalyzing the reaction of Cl− with H2O2 to produce a strong oxidant, hypochlorous acid (HOCl). However, the overproduction of MPO-derived oxidants has drawn attention to its detrimental role, especially in diseases characterized by acute or chronic inflammation. Broadly speaking, MPO and its derived oxidants are involved in the pathological processes of diseases mainly through the oxidation of biomolecules, which promotes inflammation and oxidative stress. Meanwhile, some researchers found that MPO deficiency or using MPO inhibitors could attenuate inflammation and tissue injuries. Taken together, MPO might be a promising target for both prognostic and therapeutic interventions. Therefore, understanding the role of MPO in the progress of various diseases is of great value. This review provides a comprehensive analysis of the diverse roles of MPO in the progression of several diseases, including cardiovascular diseases (CVDs), neurodegenerative diseases, cancers, renal diseases, and lung diseases (including COVID-19). This information serves as a valuable reference for subsequent mechanistic research and drug development.
1. Introduction
Myeloperoxidase (MPO), a member of the heme peroxidase enzyme family, is predominantly found in neutrophils, with only a small amount present in monocytes, which is lost during their maturation into macrophages [1,2]. The early recognition of MPO’s presence dates back to 1868 when Klebs observed that guaiac tincture, a substance reacting with peroxidase and used for peroxidase activity assays, could be oxidized by pus, suggesting the existence of MPO in leukocytes. In 1898, Linossier found that H2O2 is required for the peroxidase reaction in leukocytes [3]. Initially known as verdoperoxidase due to its green color [4], MPO was isolated from leucocytes in 1941 [5]. However, the extraction of peroxidase with a brown-green color from milk indicated its distinction from verdoperoxidase [6]. In 1943, Theorell coined the name MPO, elucidating its source, “myeloid”, and peroxidase activity, “peroxidase” [7].
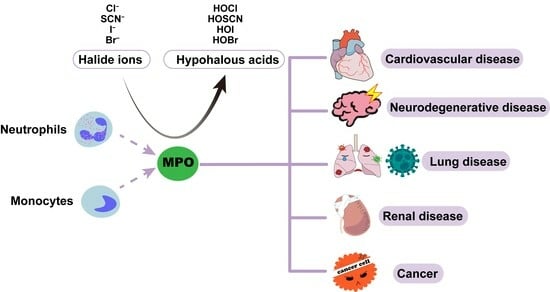
Due to the bactericidal effects of neutrophils, researchers hypothesized that MPO could also play an antimicrobial role. However, Klebanoff found that MPO, either alone or with semi-lethal amounts of H2O2, had little bactericidal effect on microorganisms in 1961–1962 [8]. Five years later, Klebanoff proposed and demonstrated that MPO exerted its antimicrobial effect via the MPO-H2O2-iodide system [9]. In a subsequent study, it was demonstrated that MPO and H2O2 form antibacterial systems with other halide and pseudo-halide ions. Among these, the MPO-H2O2-Cl− system exhibited the strongest antibacterial activity [10]. The formation of the MPO-H2O2-iodide system is as follows: Upon the invasion of pathogens, neutrophils are activated and secrete MPO into extracellular and phagocytic vesicles [11]. Then, MPO takes two electrons from halogen ions and oxidizes them to produce their corresponding hypohalous acids, using the H2O2 produced by respiratory burst as a co-substrate. MPO, with its powerful oxidation products, is an important factor in the role of neutrophils in innate immunity. Inhibiting MPO results in neutrophils maintaining normal phagocytic activity but with reduced antimicrobial activity [12].
If the antibacterial activity of MPO is not properly terminated, inflammation ceases to be salutary and becomes pathogenic. During oxidative stress or chronic inflammatory processes, MPO is secreted outside the cell, and the concentration of MPO-derived HOCl in the extracellular fluid increases, which leads to the oxidation of DNA [13], RNA, proteins, and lipids [14] due to their strong reactivity with biomolecules, resulting in tissue damage and impaired biological functions [15,16]. Furthermore, these injuries cause inflammation [8], which is thought to be a common mechanism in the pathological processes of many diseases, including but not limited to cardiovascular, respiratory, renal, and neurodegenerative diseases, in which MPO levels in patients’ plasma or other body fluids were significantly elevated and correlated with disease severity [17,18,19,20]. In this review, we focus on the basic information of MPO and integrate extensive information about the relationship between MPO and its active reaction products and multiple diseases, hoping to provide a reference for the development of MPO-related biological targets. In addition, MPO was found to be relevant to the COVID-19 pandemic in the past three years [21,22,23,24]; therefore, the most recent information on the role of MPO in COVID-19 has also been included and discussed in detail in this review.
2. Generation and Structure of MPO
MPO was mainly found in neutrophils and monocytes, with a dry weight of 5% and 1%, respectively [25]. The circulating neutrophil count in mice is 10–15%, much lower than that in humans, 60–70%, and the MPO level in murine neutrophils is approximately 10–20% of that in human neutrophils [26]. MPO is actively synthesized in promyelocytes and promonocytes during myelopoiesis in the bone marrow, while it remains inactive in fully differentiated myeloid cells [27]. The specific biosynthetic process of MPO is as follows (as shown in Figure 1) [28]: (a) The production of primary translation, pre-MPO, occurs in the endoplasmic reticulum (ER). Then, after the cotranslational cleavage of the signal peptide and incorporation of high-mannose oligosaccharide side chains, pre-MPO turns into apo-pro-MPO without enzymatic activity. (b) Apo-pro-MPO transiently associates with ER molecular chaperones (including calreticulin and calnexin [29]) in the ER, acquiring a heme group to generate enzymatically active pro-MPO [30]. (c) Subsequently, pro-MPO leaves the endoplasmic reticulum and mostly undergoes intramolecular protein hydrolysis cleavage to form heavy and light chains and form mature MPO dimers that are stored in neutrophils, while a small amount is secreted from the cells [31].
MPO is a dimer with a molecular weight ranging from 120 to 160 kDa [32]. It consists, symmetrically, of a light chain (14.5 kDa, 106 amino acids) and a heavy chain (58.5 kDa, 467 amino acids) [33]. Both of these chains have a biological action and are connected by a single disulfide bridge [34]. MPO is highly glycosylated, which is important for its enzymatic activity. Deglycosylated MPO, on the other hand, exhibits a significant decrease in chlorination activity, low-density lipoprotein (LDL) oxidation, and the ability to produce ROS [35].
The heme group in MPO is the derivative of protoporphyrin IX, with modified methyl groups on pyrrole rings A and C [34]. This modification helps form two ester bonds with the heavy-chain ester bond Glu (408) and the light-chain Asp (260). Heme is attached to MPO via three covalent bonds, in addition to the above two ester bonds, and there is a sulfonate bond between the pyrrole ring A and Met (243) [36], which causes the planar distortion and asymmetry of the heme group, resulting in the unique spectral properties and characteristic green color of MPO.
3. MPO-Derived Oxidants
The function of MPO mainly depends on its oxidative catalytic cycle (as shown in Figure 1). The native state of MPO is normally in the ferric form (MPO-Fe3+), which reacts with H2O2 to generate H2O and Compound I, which is a ferryl π-cation radical (FeIV = O) [34]. Compound I can be regenerated into ferric MPO via two different pathways. On the one hand, Compound I can accept two electrons from halide (Cl−, Br−, and I−) or pseudohalide thiocyanate (SCN−). In the meantime, it generates corresponding oxidants [37], which are commonly termed the halogenation cycle [38]. The concentrations in the plasma of Cl−, Br−, I−, and SCN− are 100–140 mM, 20–100 μM, less than 1 μM, and 20–120 μM, respectively [10]. On the other hand, Compound I can also be reduced via a two-step one-electron reduction [39] with radicals (nitric oxide, •NO, and O2•−) or organic compounds (including tyrosine, ascorbate, and steroidal hormones), generating an intermediate, Compound II, which then undergoes a second one-electron reduction to give the ferric species. This process is termed the peroxidase cycle. Native MPO can also directly react with O2•− to form Compound III [40], which can be reduced by certain reducing agents [41].
3.1. HOCl
H2O2 is catalyzed by MPO to react with halogen ions, of which 45% reacts with Cl− to form HOCl [10], partly due to the high concentration of Cl− in tissues [42] and its higher reactivity. Also, HOCl is considered the main oxidant product of MPO [43]. The antibacterial activity of HOCl is superior to that of other MPO-catalyzed products [44]. With its short-lived but highly reactive characteristic, HOCl can potentially oxidize most oxidizable groups in most substrates.
However, excessive or misplaced production of HOCl is associated with the onset and development of various pathological processes. HOCl can rapidly react with downstream macromolecules, such as proteins, DNA [13], and lipids [14]. Researchers determined the reactivity of HOCl with potential reactive sites of proteins under physiological pH conditions, showing the following order: Met > Cys Cystine His α-amino > Trp > Lys Tyr > Arg > Gln Asn [45].
3.2. HOSCN
As the most favorable substrate for MPO (with decreased protonation and a standard redox potential), HOSCN can regulate the final ratio of HOSCN and HOX (X = Cl−, Br−, and I−) produced by MPO [46], and in extreme cases, a high enough amount of SCN− can completely replace other halogen ions. HOSCN can be produced either via the MPO catalysis or direct reaction of SCN− and HOCl [47]. HOSCN can be produced in greater amounts in smokers, who have a higher level of SCN− compared with nonsmokers [48]. The antibacterial ability of HOSCN is inferior to that of HOCl because its oxidizing ability is much weaker than that of HOCl [49]. However, HOSCN can penetrate cells to oxidize intracellular sulfhydryl groups [50].
HOSCN reacts with thiols [51] and selenocysteine [52] in thiol/disulfide-like exchange reactions. As stated above, HOCl can produce irreversible oxidative damage to biomolecules, whereas HOSCN reacts reversibly with less impact on biological functions. The definite effects of HOSCN on diseases are unclear, and some investigators believe that HOSCN can induce cellular dysfunction in some specific cell types, such as macrophages and endothelial cells [53,54], which may be related to the pathological processes of some diseases. Others suggest that the formation of HOSCN is a protective mechanism that competitively inhibits the production of HOCl while consuming H2O2 [55].
3.3. ONOO−
When neutrophils are stimulated, respiratory burst occurs with increased oxygen consumption and the production of NADPH oxidase [56,57], and then superoxide (O2•−) reacts with •NO to form peroxynitrite (ONOO−), which contributes to the bactericidal activity of phagosomes [58]. ONOO− further generates a more active oxide, nitrogen dioxide (NO2) [59], which mainly exerts its bactericidal effect by oxidizing the sulfhydryl groups of biomolecules [60].
The half-life of ONOO−/ONOOH at physiological pH is approximately 1 s, while ONOO− is relatively stable and can react directly with proteins, lipids, DNA [61], and other biomolecules, promoting oxidation and nitration reactions [62], separately or collectively, in some pathophysiological processes [63]. Numerous studies have shown that ONOO− has a close association with some diseases, including but not limited to atherosclerosis [64], myocardial ischemia–reperfusion injury (MIR) [65], stroke [66], sepsis [67] and Alzheimer’s disease (AD) [68]. Currently, there are three primary categories of ONOO− inhibition: first, the use of antioxidants, such as NAC [69] and LA [70], to resist its oxidation; second, the inhibition of the raw materials and enzymes (such as MPO) for ONOO− synthesis; and third, the promotion of the degradation of ONOO−, such as via metalloporphyrins and organo-seleno derivatives [71].
4. Role of MPO in Innate Immunity
Neutrophils, constituting 60–70% of all human leukocytes [72], employ both oxidative and non-oxidative mechanisms to combat bacteria. Stored in the azurophilic granules and released upon neutrophil activation (triggered by contact with pathogens) [8], MPO plays a major role in the innate immune response [73]. Neutrophil activation by pathogen ingestion triggers the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme system [74], catalyzing the generation of O2•− with O2 as the substrate. O2•− produces hydrogen peroxide (H2O2) via a disproportionation reaction, which can react with halides to form the respective sub-halogenated acids catalyzed by MPO [75]. Among these, HOCl, synthesized via the reaction of H2O2 and Cl−, is a very effective component involved in antimicrobial defense and is capable of destroying biomolecules in the cells and tissues of host organisms [39]. HOCl is widely considered to be located in neutrophil phagosomes [76]. An in vitro experiment indicated that HOCI inhibited E. coli growth and division rapidly and selectively and impaired its protein synthesis [77]. Researchers found that MPO-knockout mice were more susceptible to bacterial infection, and further experiments indicated that MPO could protect the host against Klebsiella pneumoniae [78]. Neutrophils release both MPO and serine proteinases, and their relationships decide the final antimicrobial effect. Low levels of HOCl can inactivate α1-antitrypsin (the specific endogenous inhibitor of neutrophil elastase) [79], while high levels of HOCl can increase the susceptibility of neutrophil-derived proteins to proteolysis [80], implying that the MPO concentration can modulate the immune effect. Pseudomonas infections are more common in patients with MPO deficiency [81]. Population studies utilizing Bayer-Technicon hematological devices have found that a small number of patients with complete MPO deficiency experience severe complications related to infection or inflammation pathologies [82]. However, increased MPO levels are connected to some autoimmune diseases such as eosinophilic granulomatosis [83] and Kawasaki’s disease [84].
The antimicrobial activity of MPO is associated with three main antimicrobial modalities of neutrophils: phagocytosis [85], degranulation, and the formation of neutrophil extracellular traps (NETs). NETs are reticular structures composed of DNA and granular proteins [86], serving as extracellular anti-bacterial structures [87]. However, dysregulated NET formation can also lead to diseases, such as thrombosis [88] and respiratory failure [89]. Pathogens and inflammatory mediators activate the formation and release of NETs [90], which can be divided into three main steps: (a) The activation of NADPH oxidase results in the production of ROS, leading to chromatin denudation. (b) The translocation of neutrophil elastase (NE) and MPO to the nucleus [91] promotes further chromatin unfolding and nuclear membrane rupture [86], which results in the release of chromatin into the cytoplasm, where it is decorated with granules and cytoplasmic proteins. (c) After the disruption of the cytoplasmic membrane, NETs are released with the death of neutrophils. Previous articles have demonstrated that MPO is critical to the formation of NETs [92], as it is a major particle-resident protein that promotes the decondensation of chromatin [93]. In chronic granulomatous disease (CGD) patients who had damaged NADPH oxidase function affecting the production of hydrogen peroxide, it was difficult for the reaction substrate of MPO to form NETs [90]. Also, a study demonstrated that neutrophils from MPO-deficient patients failed to release NETs upon stimulation [94].
Even though MPO plays a pivotal role in the innate immune system, some researchers have differing views on the antimicrobial effect of MPO. It has been observed that MPO primarily targets specific microorganisms, such as Candida albicans and Staphylococcus aureus [11,27]. Moreover, individuals with MPO deficiency do not seem to experience significant health problems [95]. Some researchers have speculated on this phenomenon, suggesting that other antimicrobial systems may compensate for the long-term absence of MPO at the developmental stage. Additionally, the lack of production of active substances also prevents the inactivation of innate immune molecules within phagocytosed bodies [96].
5. Role of MPO in Diseases
5.1. Cardiovascular Diseases
Numerous studies suggest a robust association between MPO and CVDs, such as atherosclerosis, MIR [97], coronary artery disease [98], and stroke [99,100,101]. Elevated MPO levels are correlated with a poorer prognosis and increased severity of CVDs [102]. In a long-term clinical trial with 1302 asymptomatic adults, researchers found that adults with MPO at or above the median had greater body mass indices (p < 0.001), increased LDL-C levels (p = 0.001), decreased HDL-C levels (p = 0.001), and elevated systolic and diastolic blood pressure (p = 0.001 and p = 0.02, respectively) compared with those below the median. Moreover, the CVD event rate was twofold higher in adults with MPO levels at or above the median compared with adults with MPO levels below the median, at 2.3% and 4.6%, respectively [103]. Researchers built an unstable atherosclerotic plaque animal model, showing that the enzyme activity of MPO in unstable plaques was three times higher than that in stable phenotypic plaques. After inhibiting MPO or MPO gene deletion, the unstable phenotype was significantly attenuated, indicating that MPO is a potential therapeutic target for the identification and stabilization of unstable plaque [104]. Then, they collected carotid endarterectomy specimens from 31 patients and coronary plaques removed from 12 patients with cardiac transplantation, and the researchers found that MPO levels were higher in unstable carotid and coronary plaques than in stable plaques [105], providing significant new insights into the role of MPO in vulnerable plaques. MPO disrupts the stability of plaque by reducing the thickness of the fibrous cap, consequently leading to a rise in intraplaque bleeding and an increased likelihood of thrombotic incidents. MPO serum levels were considered as a powerful independent prognostic determinant of clinical outcomes in patients with ACS [18]. Combined with cardiac troponin T, the established prognostic markers of ACS, MPO identified 95% of all adverse events in the c7E3 Anti-Platelet Therapy in Unstable Refractory Angina trial.
Since MPO and its oxidative products are involved in all stages of atherosclerosis, atherosclerosis is highlighted in this review among CVDs (the main mechanisms are shown in Figure 2). A study that detected MPO levels in detergent extracts of atherosclerotic arteries demonstrated that MPO was expressed in human atherosclerotic lesions [106]. Moreover, after using the MPO inhibitor AZM198 in a tandem stenosis model of plaque instability, a study found that inhibiting MPO can stabilize existing vulnerable plaque [107]. Stroke, attributed to vascular disease, was induced by MPO by destroying the integrity of blood vessels, either directly or indirectly [108].
MPO is involved in CVD processes in many ways, contributing to the oxidation of LDL [109], the impairment of high-density lipoprotein (HDL) function, the reduction in nitric oxide (NO) bioavailability leading to endothelial dysfunction [110], and the activation of matrix metalloproteinases (MMPs) [111]. Additionally, MPO-derived-oxidant HOCl promotes endothelial cell apoptosis and shedding, leading to plaque instability [112]. The combination of these functions of MPO makes it an active mediator in the development of CVDs.
5.1.1. LDL
The oxidative conversion of LDL to atherogenic forms is a key event in cardiovascular disease development [113]. MPO is capable of generating a large number of reactive products, including HOCl, chloramines, tyrosine radicals, and nitrogen dioxide. These oxidative products oxidize the proteins, lipids, and antioxidant components in LDL, leading to the formation of oxidized LDL (oxLDL) [114]. In vitro studies showed that the MPO-H2O2-Cl− system oxidizes L-tyrosine, leading to the production of 3-chlorotyrosine, which serves as a specific marker of oxLDL. Furthermore, HOCl is an intermediate of this reaction. The detection of 3-chlorotyrosine in human atherosclerotic lesions and isolated LDL from atherosclerotic lesions strongly supports the hypothesis that MPO plays a crucial role in the oxidative modification of lipoproteins [115]. OxLDL can inhibit reverse cholesterol transport by promoting the formation of oxidized HDL (oxHDL), thereby impairing the protective effect of HDL on LDL [116].
The oxidative modification of LDL is an early event in the development of atherosclerosis [117]. OxLDL promotes atherogenesis by promoting cholesterol deposition, converting macrophages into foam cells. Retention in the subendothelial space makes LDL a major target of pro-oxidant oxidation produced by arterial wall cells [118]. OxLDL is more readily taken up by macrophages than natural LDL, potentially leading to the generation of foam cells as well as an enlarged lipid core that exacerbates the stress on the fibrous cap matrix, making atherosclerotic plaques more prone to rupture [119]. Furthermore, when LDL is oxidized to oxLDL, it is recognized by CD36, the scavenger receptor of macrophages, contributing to irregular uptake and the formation of foam cells [116].
5.1.2. HDL
HDL possesses several functions that help protect against CVDs. These functions include cholesterol reverse transport [120], anti-oxidation [121], anti-inflammation [122], protecting endothelial cells, and inhibiting the formation of oxLDL [123]. The major protein of HDL [124], ApoA-1, is the main location where HDL exerts its anti-oxidative effect [125], as circulating levels of ApoA-1 or HDL are negatively associated with CVDs [126]. It is worth noting that dysfunctional apoA1 can have a pro-inflammatory effect [127]. HDL, isolated from atherosclerotic lesions, contains a large number of MPO-modified peptides, such as chlorinated, nitrated, and sulfoxidated apoA-I [128], demonstrating the correlation between the modifying effect of MPO on HDL and the development of atherosclerosis.
Studies indicate that both HDL and apoA1 undergo extensive oxidation by MPO in human atherosclerotic lesions, resulting in decreased ABCA1-dependent cholesterol efflux [129] via the specific chlorination of the tyrosine residue site of apoA-1 [130]. Moreover, MPO-modified HDL forms a tighter connection with MPO when HDL is oxidized, creating a vicious cycle [131]. Modified HDL also competes with natural HDL as a ligand for the scavenger receptor BI, potentially interfering with cholesterol mobilization from peripheral tissues to the liver [132]. Besides the direct oxidation of apoA-I, MPO-derived oxidants modify lipids to produce highly reactive dicarbonates, such as malondialdehyde and isoprenoid adenosine, which can form a covalent bond with apoA-I and reduce cholesterol efflux [133,134]. In addition, MPO can induce the production of apoA-I/apoA-II heterodimers in HDL [135]. These heterodimers impaired wound-healing cell migration, and MPO-mediated HDL was shown to have an impaired endothelial healing function [136]. Taken together, MPO is considered to contribute to HDL dysfunction, which participates in the pathologic process of atherosclerosis.
5.1.3. Endothelial Dysfunction
Endothelial dysfunction, which is considered an early marker of atherosclerosis [137], has been linked to the potential role of MPO [138]. Researchers observed that the inhibition of MPO in a mice model of atherosclerosis reduced both the inflammatory response and endothelial dysfunction [139].
NO is produced by endothelial NO synthase (eNOS), which plays a key role in vascular homeostasis [140]. Insufficient NO can increase arterial oxidative stress and endothelial cell damage. MPO can be localized on the surface of endothelial cells and internalized [141], oxidizing NO and limiting its bioavailability [142], causing impaired endothelium-dependent diastole and resulting in endothelial dysfunction. MPO interferes with the eNOS/NO pathway to reduce eNOS activity [143], affecting endothelium-derived relaxation. In an in vitro experiment, MPO induced endothelial dysfunction by decreasing eNOS Ser1177 phosphorylation [144]. Meanwhile, HOCl-modified proteins were found in endothelial cells overlying atherosclerotic lesions in an immunostaining experiment [145]. When exposed to HOCl, the arterial rings of rabbits showed dose- and time-dependent endothelium impairment [146].
Endothelial glycocalyx (EG), playing a key role in maintaining vascular homeostasis [147], is highly susceptible to structural changes due to charge modifications. MPO has a high cationic charge at physiological pH [8] due to its arginine and lysine residues, while EG has a highly anionic nature [148]. Researchers demonstrated that MPO was bound to EG via the heparan sulfate side chain, leading to the collapse of the EG [149], and this reaction was not related to the catalytic activity of MPO.
While completely inhibiting MPO activity with non-selective inhibitors may affect its antimicrobial activity, selectively targeting extracellular MPO shows promise in eliminating MPO-induced endothelial dysfunction without compromising its bactericidal effect. However, currently, there are insufficient data to support whether specific MPO activity inhibition can still impact endothelial function in vivo.
5.2. Neurodegenerative Diseases
MPO plays an important role in neurodegenerative diseases, including AD, Parkinson’s disease (PD), cerebral ischemia, and multiple sclerosis (MS). MPO is a critical inflammatory enzyme and therapeutic target, triggering both oxidative stress and neuroinflammation in the pathological process of cerebral ischemia–reperfusion injury. It induces chloride stress and nitrosative stress. MPO catalyzes the reaction of H2O2 with Cl− to form HOCl, which causes chloride stress [150]. It also catalyzes the formation of NO2− from NO, leading to nitrosative stress [151], leading to protein nitrification and oxidation, lipid peroxidation, oxidative DNA damage, and the activation of MMP [152], which are all related to neurodegenerative diseases.
The microglia in the brain are usually in a “resting state” [153], with MPO mainly found in the microglia of diseased brains [154], whereas normal-brain microglia rarely express this enzyme [155]. Activated microglia release inducible nitric oxide synthase (iNOS), producing NO, which is converted into reactive oxidants such as NO2Cl and ONOO−, leading to neuronal damage [152].
HOCl triggers apoptosis at low doses and induces necrosis, including that of neurons and astrocytes, which are major components of the blood–brain barrier, at high doses [156]. It can also interact with ATP to interfere with energy metabolic processes [157]. HOCl-mediated activation of MMP induces tight junction degradation, leading to blood–brain barrier catabolism [158]. Elevated levels of 3-chlorotyrosine (a biomarker of HOCl) were found in damaged brain regions in [159].
5.2.1. Alzheimer’s Disease
The deposition of β-amyloid (Aβ) is a major pathological feature of AD [160]. MPO is co-localized with the Aβ protein in the senile plaques of cortical sections of AD patients [161]. The gene encoding MPO was involved in the pathway leading to Aβ deposition. AD patients had increased levels of neuronal expression of MPO [162]. In a meta-analysis, the concentration of MPO in peripheral blood was significantly higher in AD patients than that in healthy controls [163]. MPO polymorphisms have been identified as risk factors for AD [164].
MPO has been widely recognized to stimulate macrophages and cause the production of ROS and inflammatory factors. In a study about how MPO and microglia play a role in neurodegenerative Alzheimer’s disease, researchers found that MPO could cause the production of ROS and TNFα, the leading proinflammatory cytokines in this disease, in and around microglia, inducing neuronal apoptosis and necrosis [165]. In AD, microglia can interact with Aβ, thereby stimulating microglia inflammatory responses, which can lead to neuronal loss [166]. Therefore, MPO is a promising biomarker for AD that can help in the detection and risk stratification of AD patients.
5.2.2. Parkinson’s Disease
Neuronal expression of MPO is increased in the substantia nigra [167] in PD. MPO-deficient mice showed resistance to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity, a PD model [168]. As mentioned previously, MPO is found mainly in the microglia of the diseased brain [169], and microglia are mainly located in the substantia nigra, which is the brain region most susceptible to the influence of PD. HOCl chloritizes dopamine and neuromelanin to produce chloro-dopamine [170] and neuromelanin, which can enter dopaminergic cells via dopamine transporters to poison the mitochondria [171], contributing to selective dopaminergic neuron death in the substantia nigra [172].
5.2.3. Multiple Sclerosis
MS is an autoimmune disease that causes inflammatory damage to the central nervous system [173]. Immunohistochemical detection found that MPO is located in macrophages/microglia in nearby MS lesions, showing the role of MPO in the pathogenesis of MS [174]. Moreover, a study that collected the white matter of nine MS patients and seven healthy controls found that MPO levels were the highest in demyelinated white matter, followed by non-demyelinated white matter, and were lowest in control white matter [175]. This means elevated MPO levels were associated with MS, and MPO may contribute to axonal injury within plaques. However, in the EAE model, which is the animal model for MS, MPO−/− mice showed an increase in MS morbidity compared with wild-type (WT) mice, at 90% and 30%, respectively [176]. The study indicated that MPO may have a protective role in MS, which could be due to immunosuppressive effects. However, in consideration of the different MPO levels in humans and mice (which are six-fold higher in humans than in mice), the balance between its immunosuppressive and pathogenic effects might vary. The MPO GG genotype with the high-expression property was related to higher disability, a secondary progressive course of MS (p < 0.05), and the degree of brain atrophy (p < 0.05) in [177]. However, in a later study, the researchers did not find an association between MPO and susceptibility to the course and severity of MS [178]. This might be due to ethnic differences. As a result, the real association is not clear and should be investigated in different populations.
5.3. Cancers
MPO plays a dual role in tumor progression. On the one hand, MPO is involved in promoting tumor initiation, development, and migration; on the other hand, MPO enhances innate immune action during tumor elimination [179]. Increased levels of MPO have been found in biological samples of cancers, such as serum from lung cancer patients [180], plasma from subjects with gynecologic cancer, the neoplastic tissue of colon tumors [181], and so on. Previous research demonstrated that MPO had gene polymorphisms, the most common of which was MPO-463G, a polymorphism that affects MPO gene transcriptional levels [182]. This polymorphism, located 463 bp upstream of the transcription start site, binds to specificity protein 1 (SP1) [183]. The -463G site enhances the binding to SP1 compared with -463A so that the expression of the G allele is several times higher than that of the A allele [184,185]. Previous studies have shown that -463A is associated with a decreased risk of developing lung cancer, liver cancer, bladder cancer, and ovarian cancer [185], while -463G with a higher mRNA expression is believed to be connected to increased risks of many types of cancers [186].
MPO plays multiple roles in the progression of cancer. Caspase-3 plays a key role in the control of apoptosis, mediating cellular autophagy by participating in a cascade of reactions triggered in response to pro-apoptotic signals [187], while the non-enzymatic function of MPO acts by protecting cancer cells from caspase-3-mediated cell apoptosis [188]. The nitrosonium ion, an oxidative product of NO catalyzed by MPO, reduces caspase-3 activity via the nitroxylation of the caspase-3 thiol group, protecting tumor cells from apoptosis [189]. Meanwhile, MPO has been shown to catalyze the bioactivation of polycyclic aromatic hydrocarbons [190] and aromatic amines [191], leading to the production of carcinogenically active metabolites.
Due to increased exposure to oxidative stress, prolonged inflammation can lead to DNA damage [192] that induces malignant cell transformation [193]. Also, the concentration of HOCl is higher in tumor cells, especially in transformed cells, than in normal cells, which can also explain this viewpoint [194]. HOCl is the main oxidation product of MPO in vivo, and researchers have found that apoptosis could be induced by inhibiting the chlorination of MPO and thus reducing HOCl levels [195], implying a new anti-cancer strategy by targeting HOCl. HOCl has been shown to cause slow but effective DNA damage by breaking hydrogen bonds, which leads to DNA double-strand dissociation [196]. Also, HOCl can contribute to DNA structural changes and chemical modifications of the heterocyclic NH groups of guanosine and thymine [13]. Reactions with these groups result in the formation of chloramine, which can cause the double-strand dissociation of DNA [197].
On the plus side, MPO can protect against cancers associated with serious infections, such as cervical cancer. In a clinical study with 100 invasive cervical cancer patients and 122 healthy controls, the GG genotype was protective against cancer compared with the GA genotype, possibly due to the role of MPO oxidation products in killing HPV-transformed cells [198].
5.4. Renal Diseases
Elevated levels of MPO and its biological products are found in a variety of kidney diseases [199], including chronic kidney disease (CKD), pyelonephritis, and glomerulonephritis [200]. In a prospective cohort study of 3872 participants with CKD who were grouped by MPO levels, higher MPO levels were associated with a 10% higher risk for CKD progression (p = 0.03) compared with participants who held lower baseline MPO concentrations [201].
In a renal ablation model simulating CKD, researchers compared MPO−/− mice with WT mice for the incidence of nephropathy [202]. Compared with WT mice, MPO−/− mice showed significantly diminished glomerular injury, decreased gene expression of renal fibrosis markers, and reduced renal monocytes and macrophage infiltration. Also, they found that the level of plasma MPO tripled after renal ablation, suggesting a role of MPO in fibrotic remodeling [203]. In a clinical trial, the level of MPO was higher in the CKD group than in the control group and increased as CKD progressed [204]. As previously described, MPO, being a cationic protein, interacts with glomerular anionic sites [205], and MPO was also co-localized with the glomerulus after perfusion. With the infusion of MPO and H2O2 into the kidney, the urine protein content increased significantly, showing severe glomerular injury and swelling of endothelial cells [206]. MPO and non-toxic concentrations of H2O2 perfused alone did not show the above, suggesting that the injury was dependent on the action of the MPO-H2O2-halide system. MPO was considered to induce neutrophil recruitment [92], while neutrophils can produce a series of reactive oxygen intermediates when they are activated, mediating tissue injury with subsequent renal failure [207].
5.5. Lung Diseases and COVID-19
MPO and its derived oxidant HOCl may induce lung diseases by causing oxidative stress and inflammatory responses. 3-chlorotyrosine is considered a biomarker in respiratory diseases, including asthma, cystic fibrosis (CF), and chronic obstructive pulmonary disease, which are associated with the accumulation of inflammatory cells and oxidative stress [208]. Neutrophil-derived MPO is thought to be a major source of oxidative stress on the pulmonary airway surface in CF [209]. Researchers also administered MPO inhibitors at different stages of lung tumors in model mice. They found that MPO catalytic activity was present in the early stage of the tumors, the inflammation phase [188]. HOCl inactivates protease inhibitors at the inflammation site, leading to the dysregulation of elastase activity in the lungs, which may inadvertently destroy connective tissue fibers [210]. In the airways of children with CF, a great deal of HOCl was produced, which may oxidize reduced proteins like GSH in the airways to destroy the lung epithelium [211]. Also, HOCl was discovered to induce ROS and have potential roles in the pyroptosis of acute lung injury (ALI) death by using the fluorescent probe technique [212].
Researchers found that the use of SAAE, a syzygium aromaticum aqueous extract, effectively reduced MPO activity, LPS-induced lung inflammation, and MMP-2 and MMP-9 activity in mice [213]. Also, the inhibition of MPO reduced morbidity and oxidative stress in mice with cystic-fibrosis-like lung inflammation [214]. Researchers found that intranasally injecting MPO−/− mice with LPS showed reduced pro-inflammatory cytokines and chemokines compared with WT mice [215]. These results showed MPO is involved in the inflammatory process of lung diseases, which has also been observed in the worldwide COVID-19 pandemic in the past three years.
COVID-19 exploded at the end of 2019, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [216], and seriously disrupted healthcare systems, suppressed economic development, and threatened social stability [217], directly and indirectly. Recent studies have shown a correlation between COVID-19 and MPO, the major particle-resident protein of NETs, as mentioned above [218]. Clinical research on COVID-19 found that levels of MPO were significantly upregulated in the serum of COVID-19 patients compared with healthy individuals, and serum MPO levels in the recovered population were reduced to levels comparable to those in healthy people [21]. Also, another clinical trial focusing on COVID-19-positive patients found that plasma MPO levels in patients were significantly higher than those in healthy controls [219]. In an in-silico analysis to assess oxidative stress gene expression levels in COVID-19 patients, these gene expression levels were evaluated using COVID-19 transcriptomic datasets and single-cell datasets from specimens collected from the broncho-alveolar lavage fluid (BALF), whole blood, and lung autopsies of COVID-19 patients with varying disease severities [220]. The researchers found MPO levels were not only increased in the samples of COVID-19 patients in comparison with healthy individuals but also in those of severe COVID-19 patients compared with asymptomatic COVID-19 patients. These results showed a relationship between MPO and COVID-19 severity and the redox homeostasis dysfunction caused by this pandemic disease. The symptoms caused by COVID-19 are related to the cytokine storm that it triggers: the excessive immune response leads to the excessive activation of neutrophils and the subsequent production of large amounts of MPO, resulting in the series of clinical manifestations of COVID-19 [221]. It seems that maintaining a normal inflammatory response is as important as anti-viral treatment. COVID-19-induced dysfunctions, such as vascular inflammation and platelet aggregation, were associated with NO synthase, suppressed by over-generated HOCl [222]. Increased levels of MPO generated more HOCl, affecting NO synthase and contributing to COVID-19-induced dysfunctions, such as vascular inflammation and platelet aggregation.
In response to SARS-CoV-2 invasion, NADPH oxidase is activated, MPO is then released and enters into the nuclei of neutrophils [91], and, eventually, NETs are created and released extracellularly [223]. As stated above, NETs are at risk of triggering and spreading inflammation and thrombosis when not properly regulated [224]. The levels of DNA-MPO complexes in COVID-19 patients have been shown to be higher than in healthy individuals [22]. NETs are considered to bring about increased pulmonary morbidity [22,225], organ damage, and mortality rates, and the formation of microthrombosis, [226] during COVID-19 infection.
However, some researchers view MPO’s role in COVID-19 differently, emphasizing its antibacterial action [227]. Researchers produced recombinant MPO (establishing a human HEK293 cell line stably expressing recombinant MPO) and demonstrated its ability to kill a broad spectrum of pathogens, including bacteria and fungi with or without drug resistance, suggesting it is a promising antibacterial agent for COVID-19.
6. MPO Inhibitors in Clinical Trials
MPO has been proven to play many roles in the pathological processes of diseases, which are shown in Figure 3; thus, the inhibition of MPO has received much attention. According to the different mechanisms of MPO inhibitors, they are divided into irreversible inhibitors and reversible inhibitors (as shown in Table 1).
Irreversible inhibitors can be oxidized by MPO to generate a radical that binds to the active site, finally leading to irreversible inhibition [228]. p-aminobenzoic acid hydrazide (ABAH) was the first irreversible MPO inhibitor [229]. This inhibitor has been proven to be effective in various diseases. For example, treatment with ABAH can improve neurogenesis after stroke in the mice transient middle cerebral artery occlusion model [158]. An important series of irreversible inhibitors based on the xanthine structure were developed by AstraZeneca [230]. AZD3241 (verdiperstat), a first-class, oral, selective, and irreversible MPO inhibitor, might protect neurons by alleviating MPO-induced pathological oxidative stress and inflammatory effects [231]. Although no significant protective effects have been seen in phase III trials for the treatment of multiple-system atrophy, it has demonstrated efficacy in amyotrophic lateral sclerosis (NCT04436510). Several researchers have studied AZD3241 at the animal level for other disease applications, such as ALI [232], and some results have been achieved both in vitro (a reduced distribution of β-catenin in the nuclei of pulmonary microvascular endothelial cells after treatment with AZD3241) and in vivo (decreased lung coefficient and pathology scores in a rat ALI model injected with AZD3241). Also, this inhibitor could help improve the response in immune checkpoint therapy for patients with melanoma and immune checkpoint therapy resistance for melanoma [233]. However, the clinical application of AZD3241 still has a long way to go. AZD4831, another MPO inhibitor, has completed clinical trials in healthy volunteers and patients with CVDs, renal impairment, and HF. A double-blind phase 2a clinical study identified biomarker profiles associated with clinical outcomes in heart failure with preserved ejection fraction (HFpEF) and the levels of these biomarkers were downregulated after MPO inhibitor AZD4831 treatment, indicating that the effective inhibition of MPO is a promising strategy for HFpEF patients [234]. Meanwhile, in another study, there were no new safety or tolerability matters in HFpEF patients when extracellular MPO was inhibited by AZD4831 [235]. A sequential phase 2b–3 randomized clinical trial has been registered to evaluate the effects of AZD4831 on symptoms and exercise capacity in HFpEF patients [235]. PF-06282999, based on thiouracil, was developed by Pfizer [236]. There was a phase 1 study evaluating the safety and pharmacodynamic effects of PF-06282999 using LPS to induce inflammation in healthy subjects; however, the study was terminated early due to the safety of LPS. Depending on ligand-based pharmacophore modeling, several compounds via a virtual screening procedure were obtained in [237]. Among them, MPO-IN-28, containing a guanidinium-based structure, had the strongest inhibitory activity against MPO.
Reversible inhibitors bind to the MPO active site via non-covalent interactions with high affinity and low dissociation rates [228]. Researchers found MPO has a binding site for aromatic substrates, as the hydroxamic side chains of salicylhydroxamic (SHA) acid and benzohydroxamic acid (BHA) can bind to the hydrophobic pocket at the entrance of the heme [238]. Approximately two decades later, based on the effect of hydroxamic acid, three substituted aromatic hydroxamates were developed [239]. Among them, a trifluoromethyl-substituted aromatic hydroxamate, 2-(3,5-bistrifluoromethylbenzylamino)-6-oxo-1H-pyrimidine-5-carbohydroxamicv acid (HX1), was the most potent inhibitor with an IC50 of 5 nM. N-acetyl lysyltyrosylcysteine amide (KYC), a tripeptide inhibitor, has been widely used in different pharmacological studies and can suppress the production of HOCl and the oxidation of LDL [240].
To further the understanding of the relationship between MPO’s structure and functions, and with the help of new technologies such as virtual screening, a series of innovative MPO inhibitors are being investigated. The past decades have witnessed a surge in public interest in natural antioxidants globally, and some of them have been shown to be effective MPO inhibitors. Due to the high efficiency and low toxicity of natural compounds, the use of natural MPO inhibitors as adjunctive therapy for anti-inflammatory treatments holds great potential. Recently, natural polyphenols and flavonoids, such as quercetin, were considered to reversibly inhibit MPO activity [241].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative MPO inhibitors.
| Category | Pharmacophore | Inhibitor | Structure | IC50 | Pharmacological Effects | References |
|---|---|---|---|---|---|---|
| Irreversible inhibitors | Hydrazide | 4-ABAH |  | 0.3 μM | Improves neurogenesis after ischemic stroke (mice model); improves endothelial function and reduces atherosclerotic plaque development (mice model) | [158,229,242] |
| Xanthine | AZD4831 |  | 1.5 nM | Downregulates biomarkers associated with HFpEF (clinical trial) | [234,235,243,244] | |
| AZD3241 |  | 630 nM | Attenuates ALI (mice model); improves PD (clinical trial); and enhances immune checkpoint therapy for melanoma | [231,232,233] | ||
| AZD5904 |  | 140 nM | Alleviates the relaxation defect in hypertrophic human cardiomyocytes; enhances human sperm function in vitro | [245,246] | ||
| Thiouracil | PF-06282999 |  | 1.9 μM | Promotes atherosclerotic lesion stabilization and prevents atherosclerotic plaque rupture (mice model) | [236,247] | |
| Guanidine | MPO-IN-28 |  | 44 nM | Protects against endothelial glycocalyx degradation in primary human aortic endothelial cells cultured with plasma of COVID-19 patients | [237,248] | |
| Reversible inhibitors | Hydroxamic acid | SHA |  | 25 μM | No evidence of pharmacological effects; can be used to validate MPO inhibitors in silico | [238] |
| Tyrosine | KYC |  | 7 μM | Reduces bronchopulmonary dysplasia in hyperoxic neonatal rat pups; reduces oxidative injury and preserves neuronal function in MS (mice model); increases vasodilatation in sickle cell disease mice; promotes brain recovery from injury after stroke (mice model) | [240,249,250,251,252] | |
| Hydroxamate | HX1 |  | 5 nM | No evidence of pharmacological effects | [239,253] |
7. Conclusions
MPO, as the most abundant protein in neutrophils, plays a crucial role in immune responses via its catalytic cycle. MPO-derived HOCl is a strong oxidant and exerts antibacterial action, which is beneficial for the body in this aspect. However, both MPO and its product can react with biological molecules that cause tissue damage and cell dysfunction; hence, its roles in pathological processes have aroused vast attention. MPO has been implicated in many diseases, including CVDs, renal diseases, lung diseases (including COVID-19), neurodegenerative diseases, and cancers. The precise and detailed mechanisms of MPO’s effects on diseases require further exploration, and understanding these mechanisms will be crucial for the development of MPO-related drugs. Some MPO inhibitors have been found to be effective pharmacologically in in vitro and in vivo studies of these diseases. Clinical trials of some specific MPO inhibitors have been finished or continued based on their clinical outcomes. Despite the absence of MPO inhibitors on the market, the development of MPO inhibitors as investigational new drugs (INDs) is still significant and promising.
Author Contributions
Conceptualization, W.L. and C.G.; formal analysis, W.L.; writing—original draft preparation, W.L. and H.C.; writing—review and editing, W.L. and C.G.; visualization, W.L.; supervision, C.G. and X.C. funding acquisition, C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The National Natural Science Foundation of China (No. 82200510).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ortiz-Cerda, T.; Xie, K.; Mojadadi, A.; Witting, P.K. Myeloperoxidase in Health and Disease. Int. J. Mol. Sci. 2023, 24, 7725. [Google Scholar] [CrossRef] [PubMed]
- Bos, A.; Wever, R.; Roos, D. Characterization and quantification of the peroxidase in human monocytes. Biochim. Biophys. Acta 1978, 525, 37–44. [Google Scholar] [CrossRef]
- Siraki, A.G. The many roles of myeloperoxidase: From inflammation and immunity to biomarkers, drug metabolism and drug discovery. Redox Biol. 2021, 46, 102109. [Google Scholar] [CrossRef] [PubMed]
- Agner, K. Verdoperoxidase; a ferment isolated from leucocytes. Acta Physiol. Scand. 1941, 2, 1–64. [Google Scholar]
- Agner, K. Detoxicating effect of verdoperoxidase on toxins. Nature 1947, 159, 271. [Google Scholar] [CrossRef] [PubMed]
- Marzouki, S.M.; Limam, F.; Smaali, M.I.; Ulber, R.; Marzouki, M.N. A new thermostable peroxidase from garlic Allium sativum: Purification, biochemical properties, immobilization, and use in H2O2 detection in milk. Appl. Biochem. Biotechnol. 2005, 127, 201–214. [Google Scholar] [CrossRef]
- Maehly, A.C. [142] Myeloperoxidase. Methods Enzymol. 1955, 2, 794–801. [Google Scholar]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 1968, 95, 2131–2138. [Google Scholar] [CrossRef]
- Van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327 Pt 2, 487–492. [Google Scholar] [CrossRef]
- Prokopowicz, Z.; Marcinkiewicz, J.; Katz, D.R.; Chain, B.M. Neutrophil myeloperoxidase: Soldier and statesman. Arch. Immunol. Ther. Exp. 2012, 60, 43–54. [Google Scholar] [CrossRef]
- Odell, E.W.; Segal, A.W. The bactericidal effects of the respiratory burst and the myeloperoxidase system isolated in neutrophil cytoplasts. Biochim. Biophys. Acta 1988, 971, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Prütz, W.A. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch. Biochem. Biophys. 1996, 332, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; van den Berg, J.J.; Roitman, E.; Kuypers, F.A. Chlorohydrin formation from unsaturated fatty acids reacted with hypochlorous acid. Arch. Biochem. Biophys. 1992, 296, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Karakas, M.; Koenig, W. Myeloperoxidase production by macrophage and risk of atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J. Kinetic analysis of the reactions of hypobromous acid with protein components: Implications for cellular damage and use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 2004, 43, 4799–4809. [Google Scholar] [CrossRef]
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003, 349, 1595–1604. [Google Scholar] [CrossRef]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef]
- Kothari, N.; Keshari, R.S.; Bogra, J.; Kohli, M.; Abbas, H.; Malik, A.; Dikshit, M.; Barthwal, M.K. Increased myeloperoxidase enzyme activity in plasma is an indicator of inflammation and onset of sepsis. J. Crit. Care 2011, 26, 435.e1–435.e7. [Google Scholar] [CrossRef]
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum myeloperoxidase in chronic obstructive pulmonary disease. Eur. J. Med. Res. 2014, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Chelluboina, S.; Jedge, P.; Doke, P.; Palkar, S.; Mishra, A.C.; Arankalle, V.A. Elevated Levels of Neutrophil Activated Proteins, Alpha-Defensins (DEFA1), Calprotectin (S100A8/A9) and Myeloperoxidase (MPO) Are Associated with Disease Severity in COVID-19 Patients. Front. Cell. Infect. Microbiol. 2021, 11, 751232. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, E.P.; Jakobs, K.; Puccini, M.; Reinshagen, L.; Friebel, J.; Haghikia, A.; Kränkel, N.; Landmesser, U.; Rauch-Kröhnert, U. The Role of NETosis and Complement Activation in COVID-19-Associated Coagulopathies. Biomedicines 2023, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-Mediated Regulation of Innate and Adaptive Immunity: The Role of Myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef]
- Noguchi, N.; Nakano, K.; Aratani, Y.; Koyama, H.; Kodama, T.; Niki, E. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J. Biochem. 2000, 127, 971–976. [Google Scholar] [CrossRef]
- Van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Posttranslational processing of a human myeloid lysosomal protein, myeloperoxidase. Blood 1987, 70, 1143–1150. [Google Scholar] [CrossRef]
- Nauseef, W.M.; McCormick, S.J.; Clark, R.A. Calreticulin functions as a molecular chaperone in the biosynthesis of myeloperoxidase. J. Biol. Chem. 1995, 270, 4741–4747. [Google Scholar] [CrossRef]
- Olsson, I.; Egesten, A.; Gullberg, U.; Lantz, M.; Strömberg, K.; Winqvist, I. The biosynthesis of neutrophil and eosinophil granule proteins. Folia Histochem. Cytobiol. 1986, 24, 89–97. [Google Scholar]
- Nauseef, W.M. Biosynthesis of human myeloperoxidase. Arch. Biochem. Biophys. 2018, 642, 1–9. [Google Scholar] [CrossRef]
- Ikeda-Saito, M. On the analogy in the structure of the spleen green heme protein and granulocyte myeloperoxidase. FEBS Lett. 1986, 202, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.C.; Krinsky, N.I. The reductive cleavage of myeloperoxidase in half, producing enzymically active hemi-myeloperoxidase. J. Biol. Chem. 1981, 256, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- Furtmüller, P.G.; Zederbauer, M.; Jantschko, W.; Helm, J.; Bogner, M.; Jakopitsch, C.; Obinger, C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006, 445, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerpen, P.; Slomianny, M.C.; Boudjeltia, K.Z.; Delporte, C.; Faid, V.; Calay, D.; Rousseau, A.; Moguilevsky, N.; Raes, M.; Vanhamme, L.; et al. Glycosylation pattern of mature dimeric leukocyte and recombinant monomeric myeloperoxidase: Glycosylation is required for optimal enzymatic activity. J. Biol. Chem. 2010, 285, 16351–16359. [Google Scholar] [CrossRef] [PubMed]
- Kooter, I.M.; Koehler, B.P.; Moguilevsky, N.; Bollen, A.; Wever, R.; Johnson, M.K. The Met243 sulfonium ion linkage is responsible for the anomalous magnetic circular dichroism and optical spectral properties of myeloperoxidase. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 1999, 4, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. Commun. Free Radic. Res. 1997, 3, 3–15. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal 2008, 10, 1199–1234. [Google Scholar] [CrossRef]
- Metodiewa, D.; Dunford, H.B. The reactions of horseradish peroxidase, lactoperoxidase, and myeloperoxidase with enzymatically generated superoxide. Arch. Biochem. Biophys. 1989, 272, 245–253. [Google Scholar] [CrossRef]
- Marquez, L.A.; Dunford, H.B. Reaction of compound III of myeloperoxidase with ascorbic acid. J. Biol. Chem. 1990, 265, 6074–6078. [Google Scholar] [CrossRef] [PubMed]
- Delporte, C.; Van Antwerpen, P.; Vanhamme, L.; Roumeguère, T.; Zouaoui Boudjeltia, K. Low-density lipoprotein modified by myeloperoxidase in inflammatory pathways and clinical studies. Mediat. Inflamm. 2013, 2013, 971579. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.; Skaff, O.; Senthilmohan, R.; Kettle, A.J.; Davies, M.J. Hypobromous acid and bromamine production by neutrophils and modulation by superoxide. Biochem. J. 2009, 417, 773–781. [Google Scholar] [CrossRef]
- Lymar, S.V.; Hurst, J.K. Role of compartmentation in promoting toxicity of leukocyte-generated strong oxidants. Chem. Res. Toxicol. 1995, 8, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 1453–1464. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Reactions of myeloperoxidase-derived oxidants with biological substrates: Gaining chemical insight into human inflammatory diseases. Curr. Med. Chem. 2006, 13, 3271–3290. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.T.; Carlson, A.C.; Scott, M.J. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J. Am. Chem. Soc. 2004, 126, 15976–15977. [Google Scholar] [CrossRef]
- Jantschko, W.; Furtmüller, P.G.; Zederbauer, M.; Neugschwandtner, K.; Lehner, I.; Jakopitsch, C.; Arnhold, J.; Obinger, C. Exploitation of the unusual thermodynamic properties of human myeloperoxidase in inhibitor design. Biochem. Pharmacol. 2005, 69, 1149–1157. [Google Scholar] [CrossRef]
- Ashby, M.T. Inorganic chemistry of defensive peroxidases in the human oral cavity. J. Dent. Res. 2008, 87, 900–914. [Google Scholar] [CrossRef]
- Wang, J.; Slungaard, A. Role of eosinophil peroxidase in host defense and disease pathology. Arch. Biochem. Biophys. 2006, 445, 256–260. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 422, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.E.; Tan, J.T.; Hawkins, C.L.; Heather, A.K.; Davies, M.J. The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem. J. 2010, 430, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.M.; van Reyk, D.M.; Davies, M.J.; Hawkins, C.L. Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem. J. 2008, 414, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Szép, S.; Lu, Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 20515–20519. [Google Scholar] [CrossRef]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H.; Grimm, M.J.; Khan, A.N.; Han, W.; Blackwell, T.S. Regulation of innate immunity by NADPH oxidase. Free Radic. Biol. Med. 2012, 53, 72–80. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Hogg, N.; Darley-Usmar, V.M.; Wilson, M.T.; Moncada, S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem. J. 1992, 281 Pt 2, 419–424. [Google Scholar] [CrossRef]
- Denicola, A.; Rubbo, H.; Rodríguez, D.; Radi, R. Peroxynitrite-mediated cytotoxicity to Trypanosoma cruzi. Arch. Biochem. Biophys. 1993, 304, 279–286. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Denicola, A.; Radi, R. Red blood cells in the metabolism of nitric oxide-derived peroxynitrite. IUBMB Life 2006, 58, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Wattanapitayakul, S.K.; Piao, S.F.; Hoyt, D.G.; Bauer, J.A. Effects of angiotensin II on vascular endothelial cells: Formation of receptor-mediated reactive nitrogen species. Biochem. Pharmacol. 2003, 65, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Alvarez, S.; Boveris, A. Mitochondrial nitric oxide metabolism in rat muscle during endotoxemia. Free Radic. Biol. Med. 2004, 37, 1472–1478. [Google Scholar] [CrossRef]
- Oh-hashi, K.; Maruyama, W.; Yi, H.; Takahashi, T.; Naoi, M.; Isobe, K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem. Biophys. Res. Commun. 1999, 263, 504–509. [Google Scholar] [CrossRef]
- Cabassi, A.; Dumont, E.C.; Girouard, H.; Bouchard, J.F.; Le Jossec, M.; Lamontagne, D.; Besner, J.G.; de Champlain, J. Effects of chronic N-acetylcysteine treatment on the actions of peroxynitrite on aortic vascular reactivity in hypertensive rats. J. Hypertens. 2001, 19, 1233–1244. [Google Scholar] [CrossRef]
- Ziegler, D.; Nowak, H.; Kempler, P.; Vargha, P.; Low, P.A. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: A meta-analysis. Diabet. Med. J. Br. Diabet. Assoc. 2004, 21, 114–121. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: Kinetics, diffusion, and radicals. ACS Chem. Biol. 2009, 4, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rizo-Téllez, S.A.; Sekheri, M.; Filep, J.G. Myeloperoxidase: Regulation of Neutrophil Function and Target for Therapy. Antioxidants 2022, 11, 2302. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Iodination of bacteria: A bactericidal mechanism. J. Exp. Med. 1967, 126, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Takeuchi, C.; Ruedi, J.; Gutierrez, A.; Wentworth, P., Jr. Investigating antibody-catalyzed ozone generation by human neutrophils. Proc. Natl. Acad. Sci. USA 2003, 100, 3031–3034. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Maroz, A.; Woodroffe, G.; Winterbourn, C.C.; Anderson, R.F. Spectral and kinetic evidence for reaction of superoxide with compound I of myeloperoxidase. Free Radic. Biol. Med. 2011, 51, 2190–2194. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Walczewska, M. Neutrophils as Sentinel Cells of the Immune System: A Role of the MPO-halide-system in Innate and Adaptive Immunity. Curr. Med. Chem. 2020, 27, 2840–2851. [Google Scholar] [CrossRef]
- McKenna, S.M.; Davies, K.J. The inhibition of bacterial growth by hypochlorous acid. Possible role in the bactericidal activity of phagocytes. Biochem. J. 1988, 254, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hirche, T.O.; Gaut, J.P.; Heinecke, J.W.; Belaaouaj, A. Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: Effects on host defense. J. Immunol. 2005, 174, 1557–1565. [Google Scholar] [CrossRef]
- Montecucco, F.; Bertolotto, M.; Ottonello, L.; Pende, A.; Dapino, P.; Quercioli, A.; Mach, F.; Dallegri, F. Chlorhexidine prevents hypochlorous acid-induced inactivation of alpha1-antitrypsin. Clin. Exp. Pharmacol. Physiol. 2009, 36, e72–e77. [Google Scholar] [CrossRef]
- Olszowska, E.; Olszowski, S.; Zgliczyński, J.M.; Stelmaszyńska, T. Enhancement of proteinase-mediated degradation of proteins modified by chlorination. Int. J. Biochem. 1989, 21, 799–805. [Google Scholar] [CrossRef]
- Romano, M.; Dri, P.; Da Dalt, L.; Patriarca, P.; Baralle, F.E. Biochemical and molecular characterization of hereditary myeloperoxidase deficiency. Blood 1997, 90, 4126–4134. [Google Scholar] [CrossRef]
- Kutter, D. Prevalence of myeloperoxidase deficiency: Population studies using Bayer-Technicon automated hematology. J. Mol. Med. 1998, 76, 669–675. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Stegeman, C.A.; Tervaert, J.W. -463 G/A myeloperoxidase promoter polymorphism is associated with clinical manifestations and the course of disease in MPO-ANCA-associated vasculitis. Clin. Immunol. 2002, 103, 154–160. [Google Scholar] [CrossRef]
- Weng, K.P.; Hsieh, K.S.; Huang, S.H.; Wu, H.W.; Chien, J.H.; Lin, C.C.; Tang, C.W.; Ou, S.F.; Huang, S.J.; Ger, L.P. Myeloperoxidase genetic polymorphisms and susceptibility to Kawasaki disease in Taiwanese children. J. Microbiol. Immunol. Infect. 2016, 49, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respir. Int. Rev. Thorac. Dis. 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Klinke, A.; Nussbaum, C.; Kubala, L.; Friedrichs, K.; Rudolph, T.K.; Rudolph, V.; Paust, H.J.; Schröder, C.; Benten, D.; Lau, D.; et al. Myeloperoxidase attracts neutrophils by physical forces. Blood 2011, 117, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency: Prevalence and clinical significance. Ann. Intern. Med. 1981, 95, 293–301. [Google Scholar] [CrossRef]
- Vanhamme, L.; Zouaoui Boudjeltia, K.; Van Antwerpen, P.; Delporte, C. The other myeloperoxidase: Emerging functions. Arch. Biochem. Biophys. 2018, 649, 1–14. [Google Scholar] [CrossRef]
- Baldus, S.; Heitzer, T.; Eiserich, J.P.; Lau, D.; Mollnau, H.; Ortak, M.; Petri, S.; Goldmann, B.; Duchstein, H.J.; Berger, J.; et al. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic. Biol. Med. 2004, 37, 902–911. [Google Scholar] [CrossRef]
- Trentini, A.; Rosta, V.; Spadaro, S.; Bellini, T.; Rizzo, P.; Vieceli Dalla Sega, F.; Passaro, A.; Zuliani, G.; Gentili, V.; Campo, G.; et al. Development, optimization and validation of an absolute specific assay for active myeloperoxidase (MPO) and its application in a clinical context: Role of MPO specific activity in coronary artery disease. Clin. Chem. Lab. Med. 2020, 58, 1749–1758. [Google Scholar] [CrossRef]
- Matsuo, Y.; Onodera, H.; Shiga, Y.; Nakamura, M.; Ninomiya, M.; Kihara, T.; Kogure, K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke 1994, 25, 1469–1475. [Google Scholar] [CrossRef]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 35, 485–493. [Google Scholar] [CrossRef]
- Kim, H.J.; Wei, Y.; Wojtkiewicz, G.R.; Lee, J.Y.; Moskowitz, M.A.; Chen, J.W. Reducing myeloperoxidase activity decreases inflammation and increases cellular protection in ischemic stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1864–1877. [Google Scholar] [CrossRef]
- Li, J.; Cao, T.; Wei, Y.; Zhang, N.; Zhou, Z.; Wang, Z.; Li, J.; Zhang, Y.; Wang, S.; Wang, P.; et al. A Review of Novel Cardiac Biomarkers in Acute or Chronic Cardiovascular Diseases: The Role of Soluble ST2 (sST2), Lipoprotein-Associated Phospholipase A2 (Lp-PLA2), Myeloperoxidase (MPO), and Procalcitonin (PCT). Dis. Markers 2021, 2021, 6258865. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.D.; Gransar, H.; Narula, J.; Shaw, L.; Moon, J.H.; Miranda-Peats, R.; Rozanski, A.; Hayes, S.W.; Thomson, L.E.; Friedman, J.D.; et al. Myeloperoxidase, subclinical atherosclerosis, and cardiovascular disease events. JACC Cardiovasc. Imaging 2009, 2, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.; Tumanov, S.; Kong, S.M.Y.; Chen, W.; Giannotti, N.; Sivasubramaniam, V.; Rashid, I.; Ugander, M.; Jabbour, A.; Stocker, R. Intraplaque Myeloperoxidase Activity as Biomarker of Unstable Atheroma and Adverse Clinical Outcomes in Human Atherosclerosis. JACC Adv. 2023, 2, 100310. [Google Scholar] [CrossRef]
- Rashid, I.; Maghzal, G.J.; Chen, Y.C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.K.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur. Heart J. 2018, 39, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Chen, W.; Tumanov, S.; Kong, S.M.Y.; Cheng, D.; Michaëlsson, E.; Bongers, A.; Power, C.; Ayer, A.; Stocker, R. Therapeutic inhibition of MPO stabilizes pre-existing high risk atherosclerotic plaque. Redox Biol. 2022, 58, 102532. [Google Scholar] [CrossRef]
- Dhote, V.; Balaraman, R. Anti-oxidant activity mediated neuroprotective potential of trimetazidine on focal cerebral ischaemia-reperfusion injury in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 630–637. [Google Scholar] [CrossRef]
- Podrez, E.A.; Schmitt, D.; Hoff, H.F.; Hazen, S.L. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J. Clin. Investig. 1999, 103, 1547–1560. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Hazen, S.L. Nitric oxide is a physiological substrate for mammalian peroxidases. J. Biol. Chem. 2000, 275, 37524–37532. [Google Scholar] [CrossRef]
- Teng, N.; Maghzal, G.J.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. Commun. Free Radic. Res. 2017, 22, 51–73. [Google Scholar] [CrossRef]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef]
- Schindhelm, R.K.; van der Zwan, L.P.; Teerlink, T.; Scheffer, P.G. Myeloperoxidase: A useful biomarker for cardiovascular disease risk stratification? Clin. Chem. 2009, 55, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Prasad, A.; Tsimikas, S. Candidate biomarkers for the detection of coronary plaque destabilization and rupture. Curr. Atheroscler. Rep. 2008, 10, 309–317. [Google Scholar] [CrossRef]
- Zhang, Y.; Zanotti, I.; Reilly, M.P.; Glick, J.M.; Rothblat, G.H.; Rader, D.J. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 2003, 108, 661–663. [Google Scholar] [CrossRef]
- Pan, B.; Yu, B.; Ren, H.; Willard, B.; Pan, L.; Zu, L.; Shen, X.; Ma, Y.; Li, X.; Niu, C.; et al. High-density lipoprotein nitration and chlorination catalyzed by myeloperoxidase impair its effect of promoting endothelial repair. Free Radic. Biol. Med. 2013, 60, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Anantharamaiah, G.M.; Hassan, K.; Hough, G.P.; Watson, A.D.; Reddy, S.T.; Sevanian, A.; Fonarow, G.C.; Fogelman, A.M. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Steps 2 and 3. J. Lipid Res. 2000, 41, 1495–1508. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Davidson, W.S.; Thompson, T.B. The structure of apolipoprotein A-I in high density lipoproteins. J. Biol. Chem. 2007, 282, 22249–22253. [Google Scholar] [CrossRef]
- Ertek, S. High-density Lipoprotein (HDL) Dysfunction and the Future of HDL. Curr. Vasc. Pharmacol. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Johansen, O.; Abdelnoor, M.; Brekke, M.; Seljeflot, I.; Høstmark, A.T.; Arnesen, H. Predictors of restenosis after coronary angioplasty. A study on demographic and metabolic variables. Scand. Cardiovasc. J. SCJ 2001, 35, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef]
- Jin, Z.; Zhou, L.; Tian, R.; Lu, N. Myeloperoxidase Targets Apolipoprotein A-I for Site-Specific Tyrosine Chlorination in Atherosclerotic Lesions and Generates Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2021, 34, 1672–1680. [Google Scholar] [CrossRef]
- Shao, B.; Tang, C.; Sinha, A.; Mayer, P.S.; Davenport, G.D.; Brot, N.; Oda, M.N.; Zhao, X.Q.; Heinecke, J.W. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ. Res. 2014, 114, 1733–1742. [Google Scholar] [CrossRef]
- Marsche, G.; Furtmüller, P.G.; Obinger, C.; Sattler, W.; Malle, E. Hypochlorite-modified high-density lipoprotein acts as a sink for myeloperoxidase in vitro. Cardiovasc. Res. 2008, 79, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 2009, 50, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Pennathur, S.; Pagani, I.; Oda, M.N.; Witztum, J.L.; Oram, J.F.; Heinecke, J.W. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 2010, 285, 18473–18484. [Google Scholar] [CrossRef] [PubMed]
- May-Zhang, L.S.; Yermalitsky, V.; Huang, J.; Pleasent, T.; Borja, M.S.; Oda, M.N.; Jerome, W.G.; Yancey, P.G.; Linton, M.F.; Davies, S.S. Modification by isolevuglandins, highly reactive γ-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J. Biol. Chem. 2018, 293, 9176–9187. [Google Scholar] [CrossRef]
- Kameda, T.; Usami, Y.; Shimada, S.; Haraguchi, G.; Matsuda, K.; Sugano, M.; Kurihara, Y.; Isobe, M.; Tozuka, M. Determination of myeloperoxidase-induced apoAI-apoAII heterodimers in high-density lipoprotein. Ann. Clin. Lab. Sci. 2012, 42, 384–391. [Google Scholar] [PubMed]
- Kameda, T.; Horiuchi, Y.; Shimano, S.; Yano, K.; Lai, S.J.; Ichimura, N.; Tohda, S.; Kurihara, Y.; Tozuka, M.; Ohkawa, R. Effect of myeloperoxidase oxidation and N-homocysteinylation of high-density lipoprotein on endothelial repair function. Biol. Chem. 2022, 403, 265–277. [Google Scholar] [CrossRef]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Hartman, C.L.; Ford, D.A. MPO (Myeloperoxidase) Caused Endothelial Dysfunction. Arter. Thromb. Vasc. Biol. 2018, 38, 1676–1677. [Google Scholar] [CrossRef]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Naqvi, T.; Wu, Y.; Vogel, S.M.; Minshall, R.D.; Malik, A.B. Albumin mediates the transcytosis of myeloperoxidase by means of caveolae in endothelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7699–7704. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef]
- Harrison, D.G. Cellular and molecular mechanisms of endothelial cell dysfunction. J. Clin. Investig. 1997, 100, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Etwebi, Z.; Landesberg, G.; Preston, K.; Eguchi, S.; Scalia, R. Mechanistic Role of the Calcium-Dependent Protease Calpain in the Endothelial Dysfunction Induced by MPO (Myeloperoxidase). Hypertension 2018, 71, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Huang, A.; Jeranian, E.; Hou, J.Y.; Wu, T.T.; Thomas, S.R.; Keaney, J.F., Jr. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2028–2033. [Google Scholar] [CrossRef]
- Hazell, L.J.; Arnold, L.; Flowers, D.; Waeg, G.; Malle, E.; Stocker, R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Investig. 1996, 97, 1535–1544. [Google Scholar] [CrossRef]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1541–1547. [Google Scholar] [CrossRef]
- Manchanda, K.; Kolarova, H.; Kerkenpaß, C.; Mollenhauer, M.; Vitecek, J.; Rudolph, V.; Kubala, L.; Baldus, S.; Adam, M.; Klinke, A. MPO (Myeloperoxidase) Reduces Endothelial Glycocalyx Thickness Dependent on Its Cationic Charge. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1859–1867. [Google Scholar] [CrossRef]
- Wagner, B.A.; Buettner, G.R.; Oberley, L.W.; Darby, C.J.; Burns, C.P. Myeloperoxidase is involved in H2O2-induced apoptosis of HL-60 human leukemia cells. J. Biol. Chem. 2000, 275, 22461–22469. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Yoshikawa, N.; Kagota, S.; Nakamura, K.; Haginaka, J.; Kunitomo, M. Elevated circulating levels of markers of oxidative-nitrative stress and inflammation in a genetic rat model of metabolic syndrome. Nitric Oxide Biol. Chem. 2006, 15, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.S.; Katyal, A. Myeloperoxidase: Bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 2016, 68, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Rivest, S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol. Psychiatry 2006, 11, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef] [PubMed]
- Pope, S.K.; Kritchevsky, S.B.; Ambrosone, C.; Yaffe, K.; Tylavsky, F.; Simonsick, E.M.; Rosano, C.; Stewart, S.; Harris, T. Myeloperoxidase polymorphism and cognitive decline in older adults in the Health, Aging, and Body Composition Study. Am. J. Epidemiol. 2006, 163, 1084–1090. [Google Scholar] [CrossRef]
- Whiteman, M.; Rose, P.; Siau, J.L.; Cheung, N.S.; Tan, G.S.; Halliwell, B.; Armstrong, J.S. Hypochlorous acid-mediated mitochondrial dysfunction and apoptosis in human hepatoma HepG2 and human fetal liver cells: Role of mitochondrial permeability transition. Free Radic. Biol. Med. 2005, 38, 1571–1584. [Google Scholar] [CrossRef]
- Schraufstätter, I.U.; Browne, K.; Harris, A.; Hyslop, P.A.; Jackson, J.H.; Quehenberger, O.; Cochrane, C.G. Mechanisms of hypochlorite injury of target cells. J. Clin. Investig. 1990, 85, 554–562. [Google Scholar] [CrossRef]
- Kim, H.; Wei, Y.; Lee, J.Y.; Wu, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase Inhibition Increases Neurogenesis after Ischemic Stroke. J. Pharmacol. Exp. Ther. 2016, 359, 262–272. [Google Scholar] [CrossRef]
- Aratani, Y.; Koyama, H.; Nyui, S.; Suzuki, K.; Kura, F.; Maeda, N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect. Immun. 1999, 67, 1828–1836. [Google Scholar] [CrossRef]
- Serý, O.; Povová, J.; Míšek, I.; Pešák, L.; Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 2013, 51, 1–9. [Google Scholar] [CrossRef]
- Crawford, F.C.; Freeman, M.J.; Schinka, J.A.; Morris, M.D.; Abdullah, L.I.; Richards, D.; Sevush, S.; Duara, R.; Mullan, M.J. Association between Alzheimer’s disease and a functional polymorphism in the Myeloperoxidase gene. Exp. Neurol. 2001, 167, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Wu, C.Y.; Bawa, K.K.; Ouk, M.; Leung, N.; Yu, D.; Lanctôt, K.L.; Herrmann, N.; Pakosh, M.; Swardfager, W. Neutrophil activation in Alzheimer’s disease and mild cognitive impairment: A systematic review and meta-analysis of protein markers in blood and cerebrospinal fluid. Ageing Res. Rev. 2020, 62, 101130. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, D.L.; Lefkowitz, S.S. Microglia and myeloperoxidase: A deadly partnership in neurodegenerative disease. Free Radic. Biol. Med. 2008, 45, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-α-synuclein-A53T mouse model, correlating with increased nitration and aggregation of α-synuclein and exacerbation of motor impairment. Free Radic. Biol. Med. 2019, 141, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Omiatek, D.M.; Bressler, A.J.; Cans, A.S.; Andrews, A.M.; Heien, M.L.; Ewing, A.G. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci. Rep. 2013, 3, 1447. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Patrick, P.A.; Gomolin, I.; Palaia, T.; Ragolia, L.; Brand, D.; Delikatny, E.J. Inflaming the diseased brain: A role for tainted melanins. Biochim. Biophys. Acta 2015, 1852, 937–950. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomolin, I.; Peltier, M.R.; Moran, G.R. Linking Inflammation and Parkinson Disease: Hypochlorous Acid Generates Parkinsonian Poisons. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 153, 410. [Google Scholar] [CrossRef]
- Olek, M.J. Multiple Sclerosis. Ann. Intern. Med. 2021, 174, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci. Lett. 2008, 444, 195–198. [Google Scholar] [CrossRef]
- Brennan, M.; Gaur, A.; Pahuja, A.; Lusis, A.J.; Reynolds, W.F. Mice lacking myeloperoxidase are more susceptible to experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2001, 112, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska-Pniewska, B.; Styczynska, M.; Podlecka, A.; Samocka, R.; Peplonska, B.; Barcikowska, M.; Kwiecinski, H. Association of apolipoprotein E and myeloperoxidase genotypes to clinical course of familial and sporadic multiple sclerosis. Mult. Scler. 2004, 10, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, O.H.; Atkinson, E.J.; Hebrink, D.D.; McMurray, C.T.; Weinshenker, B.G. Association of a myeloperoxidase promoter polymorphism with multiple sclerosis. J. Neuroimmunol. 2000, 105, 189–194. [Google Scholar] [CrossRef]
- Dissemond, J.; Weimann, T.K.; Schneider, L.A.; Schneeberger, A.; Scharffetter-Kochanek, K.; Goos, M.; Wagner, S.N. Activated neutrophils exert antitumor activity against human melanoma cells: Reactive oxygen species-induced mechanisms and their modulation by granulocyte-macrophage-colony-stimulating factor. J. Investig. Dermatol. 2003, 121, 936–938. [Google Scholar] [CrossRef]
- Larsson, S.; Nordenson, A.; Glader, P.; Yoshihara, S.; Lindén, A.; Slinde, F. A gender difference in circulating neutrophils in malnourished patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 83–88. [Google Scholar] [CrossRef]
- Rainis, T.; Maor, I.; Lanir, A.; Shnizer, S.; Lavy, A. Enhanced oxidative stress and leucocyte activation in neoplastic tissues of the colon. Dig. Dis. Sci. 2007, 52, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.P.; Piedrafita, F.J.; Reynolds, W.F. Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the -463GA promoter polymorphism. J. Biol. Chem. 2004, 279, 8300–8315. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Casali, B.; Farnetti, E.; Pipitone, N.; Nicoli, D.; Macchioni, P.L.; Cimino, L.; Bajocchi, G.L.; Catanoso, M.G.; Pattacini, L.; et al. -463 G/A myeloperoxidase promoter polymorphism in giant cell arteritis. Ann. Rheum. Dis. 2008, 67, 485–488. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Kumar, A.P.; Piedrafita, F.J. The human myeloperoxidase gene is regulated by LXR and PPARalpha ligands. Biochem. Biophys. Res. Commun. 2006, 349, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Van Schooten, F.J.; Boots, A.W.; Knaapen, A.M.; Godschalk, R.W.; Maas, L.M.; Borm, P.J.; Drent, M.; Jacobs, J.A. Myeloperoxidase (MPO) -463G->A reduces MPO activity and DNA adduct levels in bronchoalveolar lavages of smokers. Cancer Epidemiol. Biomark. Prev. 2004, 13, 828–833. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Kutikhin, A.G. Common genetic variants in the myeloperoxidase and paraoxonase genes and the related cancer risk: A review. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2012, 30, 287–322. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Rymaszewski, A.L.; Tate, E.; Yimbesalu, J.P.; Gelman, A.E.; Jarzembowski, J.A.; Zhang, H.; Pritchard, K.A., Jr.; Vikis, H.G. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers 2014, 6, 1111–1127. [Google Scholar] [CrossRef]
- Saed, G.M.; Ali-Fehmi, R.; Jiang, Z.L.; Fletcher, N.M.; Diamond, M.P.; Abu-Soud, H.M.; Munkarah, A.R. Myeloperoxidase serves as a redox switch that regulates apoptosis in epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 276–281. [Google Scholar] [CrossRef]
- Mallet, W.G.; Mosebrook, D.R.; Trush, M.A. Activation of (+-)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to diolepoxides by human polymorphonuclear leukocytes or myeloperoxidase. Carcinogenesis 1991, 12, 521–524. [Google Scholar] [CrossRef]
- Kadlubar, F.F.; Butler, M.A.; Kaderlik, K.R.; Chou, H.C.; Lang, N.P. Polymorphisms for aromatic amine metabolism in humans: Relevance for human carcinogenesis. Environ. Health Perspect. 1992, 98, 69–74. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Bauer, G. HOCl and the control of oncogenesis. J. Inorg. Biochem. 2018, 179, 10–23. [Google Scholar] [CrossRef]
- An, Z.; Zhao, Z.; Zhao, L.; Yue, Q.; Li, K.; Zhao, B.; Miao, J.; Su, L. The novel HOCl fluorescent probe CAN induced A549 apoptosis by inhibiting chlorination activity of MPO. Bioorganic Med. Chem. Lett. 2020, 30, 127394. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to nucleosides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2001, 14, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: Formation of chloramines and nitrogen-centered radicals. Chem. Res. Toxicol. 2002, 15, 83–92. [Google Scholar] [CrossRef]
- Castelão, C.; da Silva, A.P.; Matos, A.; Inácio, Â.; Bicho, M.; Medeiros, R.; Bicho, M.C. Association of myeloperoxidase polymorphism (G463A) with cervix cancer. Mol. Cell. Biochem. 2015, 404, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Noor, S.; Alam, F.; Fatima, F.; Orakzai, S. Myeloperoxidase: Indicator of cardiovascular disease in chronic kidney disease patients of tertiary care hospital of Karachi. JPMA J. Pak. Med. Assoc. 2021, 71, 1128–1132. [Google Scholar] [CrossRef]
- Gröne, H.J.; Gröne, E.F.; Malle, E. Immunohistochemical detection of hypochlorite-modified proteins in glomeruli of human membranous glomerulonephritis. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 5–14. [Google Scholar] [CrossRef]
- Correa, S.; Pena-Esparragoza, J.K.; Scovner, K.M.; Waikar, S.S.; Mc Causland, F.R. Myeloperoxidase and the Risk of CKD Progression, Cardiovascular Disease, and Death in the Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2020, 76, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Klinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol. Ren. Physiol. 2014, 307, F407–F417. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Curtis, K.A.; Waikar, S.S.; Mc Causland, F.R. Serum Myeloperoxidase, Uric Acid, and the Risk of Atrial Fibrillation in Chronic Kidney Disease. Circ. Arrhythmia Electrophysiol. 2021, 14, e009483. [Google Scholar] [CrossRef]
- Tsai, M.S.; Shaw, H.M.; Li, Y.J.; Lin, M.T.; Lee, W.T.; Chan, K.S. Myeloperoxidase in chronic kidney disease: Role of visceral fat. Nephrology 2014, 19, 136–142. [Google Scholar] [CrossRef]
- Rennke, H.G.; Venkatachalam, M.A. Glomerular permeability: In vivo tracer studies with polyanionic and polycationic ferritins. Kidney Int. 1977, 11, 44–53. [Google Scholar] [CrossRef]
- Johnson, R.J.; Couser, W.G.; Chi, E.Y.; Adler, S.; Klebanoff, S.J. New mechanism for glomerular injury. Myeloperoxidase-hydrogen peroxide-halide system. J. Clin. Investig. 1987, 79, 1379–1387. [Google Scholar] [CrossRef]
- Heinzelmann, M.; Mercer-Jones, M.A.; Passmore, J.C. Neutrophils and renal failure. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1999, 34, 384–399. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relationship with airway inflammation. COPD 2010, 7, 411–417. [Google Scholar] [CrossRef]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef]
- He, J.; Turino, G.M.; Lin, Y.Y. Characterization of peptide fragments from lung elastin degradation in chronic obstructive pulmonary disease. Exp. Lung Res. 2010, 36, 548–557. [Google Scholar] [CrossRef]
- Magon, N.J.; Turner, R.; Gearry, R.B.; Hampton, M.B.; Sly, P.D.; Kettle, A.J. Oxidation of calprotectin by hypochlorous acid prevents chelation of essential metal ions and allows bacterial growth: Relevance to infections in cystic fibrosis. Free Radic. Biol. Med. 2015, 86, 133–144. [Google Scholar] [CrossRef]
- Zhan, Z.; Lei, Q.; Dai, Y.; Wang, D.; Yu, Q.; Lv, Y.; Li, W. Simultaneous Monitoring of HOCl and Viscosity with Drug-Induced Pyroptosis in Live Cells and Acute Lung Injury. Anal. Chem. 2022, 94, 12144–12151. [Google Scholar] [CrossRef]
- Chniguir, A.; Zioud, F.; Marzaioli, V.; El-Benna, J.; Bachoual, R. Syzygium aromaticum aqueous extract inhibits human neutrophils myeloperoxidase and protects mice from LPS-induced lung inflammation. Pharm. Biol. 2019, 57, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Huang, J.; Min, E.; Michaëlsson, E.; Lindstedt, E.L.; Pearson, J.F.; Kettle, A.J.; Day, B.J. Myeloperoxidase inhibition decreases morbidity and oxidative stress in mice with cystic fibrosis-like lung inflammation. Free Radic. Biol. Med. 2020, 152, 91–99. [Google Scholar] [CrossRef]
- Haegens, A.; Heeringa, P.; van Suylen, R.J.; Steele, C.; Aratani, Y.; O’Donoghue, R.J.; Mutsaers, S.E.; Mossman, B.T.; Wouters, E.F.; Vernooy, J.H. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 2009, 182, 7990–7996. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Bio-Medica Atenei Parm. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiao, B.; Qu, L.; Yang, D.; Liu, R. The development of COVID-19 treatment. Front. Immunol. 2023, 14, 1125246. [Google Scholar] [CrossRef]
- Jamali, E.; Abbasi, M.; Tayer, A.H.; Monfared, A.A.; Tandel, P.; Tamaddon, G.; Kazerooni, E.S.; Rakhshandehroo, S.; Ranjbaran, R. The significance of surface neutrophilic MPO expression level in NETosis and NETosis-associated coagulopathies in COVID-19 infected patients. Blood Cells Mol. Dis. 2022, 96, 102676. [Google Scholar] [CrossRef]
- Huckriede, J.; de Vries, F.; Hultström, M.; Wichapong, K.; Reutelingsperger, C.; Lipcsey, M.; Garcia de Frutos, P.; Frithiof, R.; Nicolaes, G.A.F. Histone H3 Cleavage in Severe COVID-19 ICU Patients. Front. Cell. Infect. Microbiol. 2021, 11, 694186. [Google Scholar] [CrossRef]
- Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Mdkhana, B.; Hussain Alsayed, H.A.; Alsafar, H.; Alrais, Z.F.; Hamid, Q.; Halwani, R. Upregulation of oxidative stress gene markers during SARS-CoV-2 viral infection. Free Radic. Biol. Med. 2021, 172, 688–698. [Google Scholar] [CrossRef]
- Bonini, M.G.; Siraki, A.G.; Atanassov, B.S.; Mason, R.P. Immunolocalization of hypochlorite-induced, catalase-bound free radical formation in mouse hepatocytes. Free Radic. Biol. Med. 2007, 42, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.K.; Na, D.H.; Kim, T.H.; Choy, Y.B.; et al. Bioinspired DNase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated NETosis Dysregulation. Adv. Sci. 2020, 7, 2001940. [Google Scholar] [CrossRef]
- Twaddell, S.H.; Baines, K.J.; Grainge, C.; Gibson, P.G. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 2019, 156, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Holliday, Z.M.; Earhart, A.P.; Alnijoumi, M.M.; Krvavac, A.; Allen, L.H.; Schrum, A.G. Non-Randomized Trial of Dornase Alfa for Acute Respiratory Distress Syndrome Secondary to COVID-19. Front. Immunol. 2021, 12, 714833. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Cao, Z.; Cheng, G. Recombinant Myeloperoxidase as a New Class of Antimicrobial Agents. Microbiol. Spectr. 2022, 10, e0052221. [Google Scholar] [CrossRef]
- Soubhye, J.; Van Antwerpen, P.; Dufrasne, F. A patent review of myeloperoxidase inhibitors for treating chronic inflammatory syndromes (focus on cardiovascular diseases, 2013-2019). Expert Opin. Ther. Pat. 2020, 30, 595–608. [Google Scholar] [CrossRef]
- Kettle, A.J.; Gedye, C.A.; Hampton, M.B.; Winterbourn, C.C. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem. J. 1995, 308 Pt 2, 559–563. [Google Scholar] [CrossRef]
- Stamp, L.K.; Turner, R.; Khalilova, I.S.; Zhang, M.; Drake, J.; Forbes, L.V.; Kettle, A.J. Myeloperoxidase and oxidation of uric acid in gout: Implications for the clinical consequences of hyperuricaemia. Rheumatology 2014, 53, 1958–1965. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Deng, M. New developments and opportunities in drugs being trialed for amyotrophic lateral sclerosis from 2020 to 2022. Front. Pharmacol. 2022, 13, 1054006. [Google Scholar] [CrossRef]
- Ren, R.; Xu, Z.; Wang, X.; Jiang, W.; Yu, P. Verdiperstat attenuates acute lung injury by modulating MPO/μ-calpain/β-catenin signaling. Eur. J. Pharmacol. 2022, 924, 174940. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Gammon, S.T.; Yang, P.; Ma, W.; Wang, J.; Piwnica-Worms, D. Inhibition of myeloperoxidase enhances immune checkpoint therapy for melanoma. J. Immunother. Cancer 2023, 11. [Google Scholar] [CrossRef]
- Michaëlsson, E.; Lund, L.H.; Hage, C.; Shah, S.J.; Voors, A.A.; Saraste, A.; Redfors, B.; Grove, E.L.; Barasa, A.; Richards, A.M.; et al. Myeloperoxidase Inhibition Reverses Biomarker Profiles Associated With Clinical Outcomes in HFpEF. JACC Heart Fail. 2023, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Lam, C.S.P.; Pizzato, P.E.; Gabrielsen, A.; Michaëlsson, E.; Nelander, K.; Ericsson, H.; Holden, J.; Folkvaljon, F.; Mattsson, A.; et al. Rationale and design of ENDEAVOR: A sequential phase 2b-3 randomized clinical trial to evaluate the effect of myeloperoxidase inhibition on symptoms and exercise capacity in heart failure with preserved or mildly reduced ejection fraction. Eur. J. Heart Fail. 2023, 25, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, R.B.; Buckbinder, L.; Bagley, S.W.; Carpino, P.A.; Conn, E.L.; Dowling, M.S.; Fernando, D.P.; Jiao, W.; Kung, D.W.; Orr, S.T.; et al. Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases. J. Med. Chem. 2015, 58, 8513–8528. [Google Scholar] [CrossRef]
- Soubhye, J.; Chikh Alard, I.; Aldib, I.; Prévost, M.; Gelbcke, M.; De Carvalho, A.; Furtmüller, P.G.; Obinger, C.; Flemmig, J.; Tadrent, S.; et al. Discovery of Novel Potent Reversible and Irreversible Myeloperoxidase Inhibitors Using Virtual Screening Procedure. J. Med. Chem. 2017, 60, 6563–6586. [Google Scholar] [CrossRef]
- Hori, H.; Fenna, R.E.; Kimura, S.; Ikeda-Saito, M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J. Biol. Chem. 1994, 269, 8388–8392. [Google Scholar] [CrossRef]
- Forbes, L.V.; Sjögren, T.; Auchère, F.; Jenkins, D.W.; Thong, B.; Laughton, D.; Hemsley, P.; Pairaudeau, G.; Turner, R.; Eriksson, H.; et al. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J. Biol. Chem. 2013, 288, 36636–36647. [Google Scholar] [CrossRef]
- Zhang, H.; Jing, X.; Shi, Y.; Xu, H.; Du, J.; Guan, T.; Weihrauch, D.; Jones, D.W.; Wang, W.; Gourlay, D.; et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor. J. Lipid Res. 2013, 54, 3016–3029. [Google Scholar] [CrossRef]
- Boufadi, Y.M.; Soubhye, J.; Riazi, A.; Rousseau, A.; Vanhaeverbeek, M.; Nève, J.; Boudjeltia, K.Z.; Van Antwerpen, P. Characterization and antioxidant properties of six Algerian propolis extracts: Ethyl acetate extracts inhibit myeloperoxidase activity. Int. J. Mol. Sci. 2014, 15, 2327–2345. [Google Scholar] [CrossRef] [PubMed]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef]
- Jurva, U.; Weidolf, L.; Sandinge, A.S.; Leandersson, C.; Ekdahl, A.; Li, X.Q.; Antonsson, T.; Sundell, J.; Westerlund, K.; Amilon, C.; et al. Biotransformation of the Novel Myeloperoxidase Inhibitor AZD4831 in Preclinical Species and Humans. Drug Metab. Dispos. 2023, 51, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.M.; Lagerström-Fermér, M.; Ericsson, H.; Nelander, K.; Lindstedt, E.L.; Michaëlsson, E.; Kjaer, M.; Heijer, M.; Whatling, C.; Fuhr, R. Safety, tolerability, pharmacokinetics and effect on serum uric acid of the myeloperoxidase inhibitor AZD4831 in a randomized, placebo-controlled, phase I study in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Sucquart, I.E.; Whittaker, A.; Sanganee, H.J.; Barratt, C.L.R.; Martins da Silva, S.J. Myeloperoxidase inhibitor AZD5904 enhances human sperm function in vitro. Hum. Reprod. 2021, 36, 560–570. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Kp, M.M.J.; Chua, J.; Hernandez-Resendiz, S.; Liehn, E.A.; Knöll, R.; Gan, L.M.; Michaëlsson, E.; Jonsson, M.K.B.; Ryden-Markinhuhta, K.; et al. Inhibiting cardiac myeloperoxidase alleviates the relaxation defect in hypertrophic cardiomyocytes. Cardiovasc. Res. 2022, 118, 517–530. [Google Scholar] [CrossRef]
- Roth Flach, R.J.; Su, C.; Bollinger, E.; Cortes, C.; Robertson, A.W.; Opsahl, A.C.; Coskran, T.M.; Maresca, K.P.; Keliher, E.J.; Yates, P.D.; et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLoS ONE 2019, 14, e0214150. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.; Chan, L.L.Y.; Cheung, C.; Chia, P.Y.; Ong, S.W.X.; Fong, S.W.; Ng, L.F.P.; Renia, L.; Lye, D.C.; Young, B.E.; et al. Myeloperoxidase inhibition may protect against endothelial glycocalyx shedding induced by COVID-19 plasma. Commun. Med. 2023, 3, 62. [Google Scholar] [CrossRef]
- Teng, R.J.; Jing, X.; Martin, D.P.; Hogg, N.; Haefke, A.; Konduri, G.G.; Day, B.W.; Naylor, S.; Pritchard, K.A., Jr. N-acetyl-lysyltyrosylcysteine amide, a novel systems pharmacology agent, reduces bronchopulmonary dysplasia in hyperoxic neonatal rat pups. Free Radic. Biol. Med. 2021, 166, 73–89. [Google Scholar] [CrossRef]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. Neuroreport 2018, 29, 208–213. [Google Scholar] [CrossRef]
- Yu, G.; Liang, Y.; Zheng, S.; Zhang, H. Inhibition of Myeloperoxidase by N-Acetyl Lysyltyrosylcysteine Amide Reduces Oxidative Stress-Mediated Inflammation, Neuronal Damage, and Neural Stem Cell Injury in a Murine Model of Stroke. J. Pharmacol. Exp. Ther. 2018, 364, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Weihrauch, D.; Jones, D.W.; Jing, X.; Shi, Y.; Gourlay, D.; Oldham, K.T.; Hillery, C.A.; Pritchard, K.A., Jr. Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice. J. Lipid Res. 2013, 54, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Forbes, L.V.; Kettle, A.J. A multi-substrate assay for finding physiologically effective inhibitors of myeloperoxidase. Anal. Biochem. 2018, 544, 13–21. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The generation, structure, and catalytic cycle of MPO. The upper part of Figure 1 shows the MPO synthetic process. MPO primarily exists in the azurophil granules of the myeloid series of hematopoietic cells. MPO synthesis occurs at the promyelocyte differentiation stages, and MPO synthesis activity gradually decreases during this process, disappearing in fully differentiated myeloid cells. The first step occurs in the ER, where primary translation production apo-pre-pro-MPO becomes apo-pro-MPO via the cotranslational glycosylation process. The next step, also in the ER, involves apo-pro-MPO combining with ER molecular chaperones and a heme group to turn into enzymatically active pro-MPO. The final step is pro-MPO leaving the ER and entering the azurophilic granule, where the two monomers combine to generate mature MPO. The left side of the picture demonstrates the structure of MPO, constructed in the Protein Data Bank (accession code 7Z53, https://www.rcsb.org/structure/7Z53, accessed on 12 September 2023). MPO (120–160 kDa) is a dimeric enzyme containing a light chain and a heavy chain in each monomer. There is a disulfide bridge connecting the two monomers, and each of them has a heme group that is attached to MPO with two ester bonds (connecting the light chain and the heavy chain) and a sulfonate bond. The right side of the picture illustrates the catalytic cycle of MPO, representing the main ways in which MPO exerts its innate immune function. Ferric MPO reacts with H2O2 to generate Compound I. Compound I can be backward-reacted into ferric MPO via two pathways, which are marked in blue and red in the figure. Among them, the blue path is called the halogenation cycle: Compound I is reduced by halide ions (or pseudohalide ions) to form ferric MPO and respective hypohalous acids. Also in the red path, Compound I can transform into native MPO via an intermediate, Compound II. Ferric MPO and Compound III can mutually transform by consuming or generating O2•−.
Figure 1.
The generation, structure, and catalytic cycle of MPO. The upper part of Figure 1 shows the MPO synthetic process. MPO primarily exists in the azurophil granules of the myeloid series of hematopoietic cells. MPO synthesis occurs at the promyelocyte differentiation stages, and MPO synthesis activity gradually decreases during this process, disappearing in fully differentiated myeloid cells. The first step occurs in the ER, where primary translation production apo-pre-pro-MPO becomes apo-pro-MPO via the cotranslational glycosylation process. The next step, also in the ER, involves apo-pro-MPO combining with ER molecular chaperones and a heme group to turn into enzymatically active pro-MPO. The final step is pro-MPO leaving the ER and entering the azurophilic granule, where the two monomers combine to generate mature MPO. The left side of the picture demonstrates the structure of MPO, constructed in the Protein Data Bank (accession code 7Z53, https://www.rcsb.org/structure/7Z53, accessed on 12 September 2023). MPO (120–160 kDa) is a dimeric enzyme containing a light chain and a heavy chain in each monomer. There is a disulfide bridge connecting the two monomers, and each of them has a heme group that is attached to MPO with two ester bonds (connecting the light chain and the heavy chain) and a sulfonate bond. The right side of the picture illustrates the catalytic cycle of MPO, representing the main ways in which MPO exerts its innate immune function. Ferric MPO reacts with H2O2 to generate Compound I. Compound I can be backward-reacted into ferric MPO via two pathways, which are marked in blue and red in the figure. Among them, the blue path is called the halogenation cycle: Compound I is reduced by halide ions (or pseudohalide ions) to form ferric MPO and respective hypohalous acids. Also in the red path, Compound I can transform into native MPO via an intermediate, Compound II. Ferric MPO and Compound III can mutually transform by consuming or generating O2•−.

Figure 2.
The role of MPO in the pathogenesis of cardiovascular disease. The red arrows demonstrate the process promoted by MPO: the migration of monocytes, the oxidation of LDL, the dysfunction of HDL, the activation of MMP, the migration of SMC, the death of EC, the formation of foam cells, and the rupture of atherosclerotic plaque. The blue arrows show the inhibition process caused by MPO: the reduction in cholesterol efflux and NO bioavailability.
Figure 2.
The role of MPO in the pathogenesis of cardiovascular disease. The red arrows demonstrate the process promoted by MPO: the migration of monocytes, the oxidation of LDL, the dysfunction of HDL, the activation of MMP, the migration of SMC, the death of EC, the formation of foam cells, and the rupture of atherosclerotic plaque. The blue arrows show the inhibition process caused by MPO: the reduction in cholesterol efflux and NO bioavailability.

Figure 3.
MPO’s various roles in cardiovascular diseases, lung diseases, neurodegenerative diseases, renal diseases, and cancer. (1) In cardiovascular diseases, MPO can oxidize LDL, impair HDL, activate MMPs, as well as increase endothelial dysfunction and plaque instability. (2) In lung diseases, MPO can increase inflammatory responses, morbidity, oxidative stress, and MMPs, while decreasing GSH and elastase activity in the lung. Additionally, MPO is involved in the COVID-19 inflammatory process and is associated with disease severity. (3) In neurodegenerative diseases, MPO can increase BBB catabolism, nitrosative and chlorination stress, and lipid peroxidation as well as activate MMPs and induce oxidative DNA damage. (4) In renal diseases, MPO can increase the urine protein content and adhesion molecule expression and induce glomerular injury and the swelling of endothelial cells. (5) Unlike in other diseases, MPO acts as a two-edged sword in the cancer pathological process. On the one hand, MPO can increase carcinogenically active metabolites, DNA damage, and malignant cell transformation, as well as reduce caspase-3 activity to protect cancer cells from apoptosis. On the other hand, MPO has pharmacologic effects on cervical cancer via its innate immune action.
Figure 3.
MPO’s various roles in cardiovascular diseases, lung diseases, neurodegenerative diseases, renal diseases, and cancer. (1) In cardiovascular diseases, MPO can oxidize LDL, impair HDL, activate MMPs, as well as increase endothelial dysfunction and plaque instability. (2) In lung diseases, MPO can increase inflammatory responses, morbidity, oxidative stress, and MMPs, while decreasing GSH and elastase activity in the lung. Additionally, MPO is involved in the COVID-19 inflammatory process and is associated with disease severity. (3) In neurodegenerative diseases, MPO can increase BBB catabolism, nitrosative and chlorination stress, and lipid peroxidation as well as activate MMPs and induce oxidative DNA damage. (4) In renal diseases, MPO can increase the urine protein content and adhesion molecule expression and induce glomerular injury and the swelling of endothelial cells. (5) Unlike in other diseases, MPO acts as a two-edged sword in the cancer pathological process. On the one hand, MPO can increase carcinogenically active metabolites, DNA damage, and malignant cell transformation, as well as reduce caspase-3 activity to protect cancer cells from apoptosis. On the other hand, MPO has pharmacologic effects on cervical cancer via its innate immune action.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, W.; Chen, H.; Chen, X.; Guo, C. The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress. Antioxidants 2024, 13, 132. https://doi.org/10.3390/antiox13010132
AMA Style
Lin W, Chen H, Chen X, Guo C. The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress. Antioxidants. 2024; 13(1):132. https://doi.org/10.3390/antiox13010132
Chicago/Turabian StyleLin, Wei, Huili Chen, Xijing Chen, and Chaorui Guo. 2024. "The Roles of Neutrophil-Derived Myeloperoxidase (MPO) in Diseases: The New Progress" Antioxidants 13, no. 1: 132. https://doi.org/10.3390/antiox13010132
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.