
Characterization of Escherichia coli Strains for Novel Production of Plasmodium ovale Lactate Dehydrogenase
 , , , , , , , and
, , , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
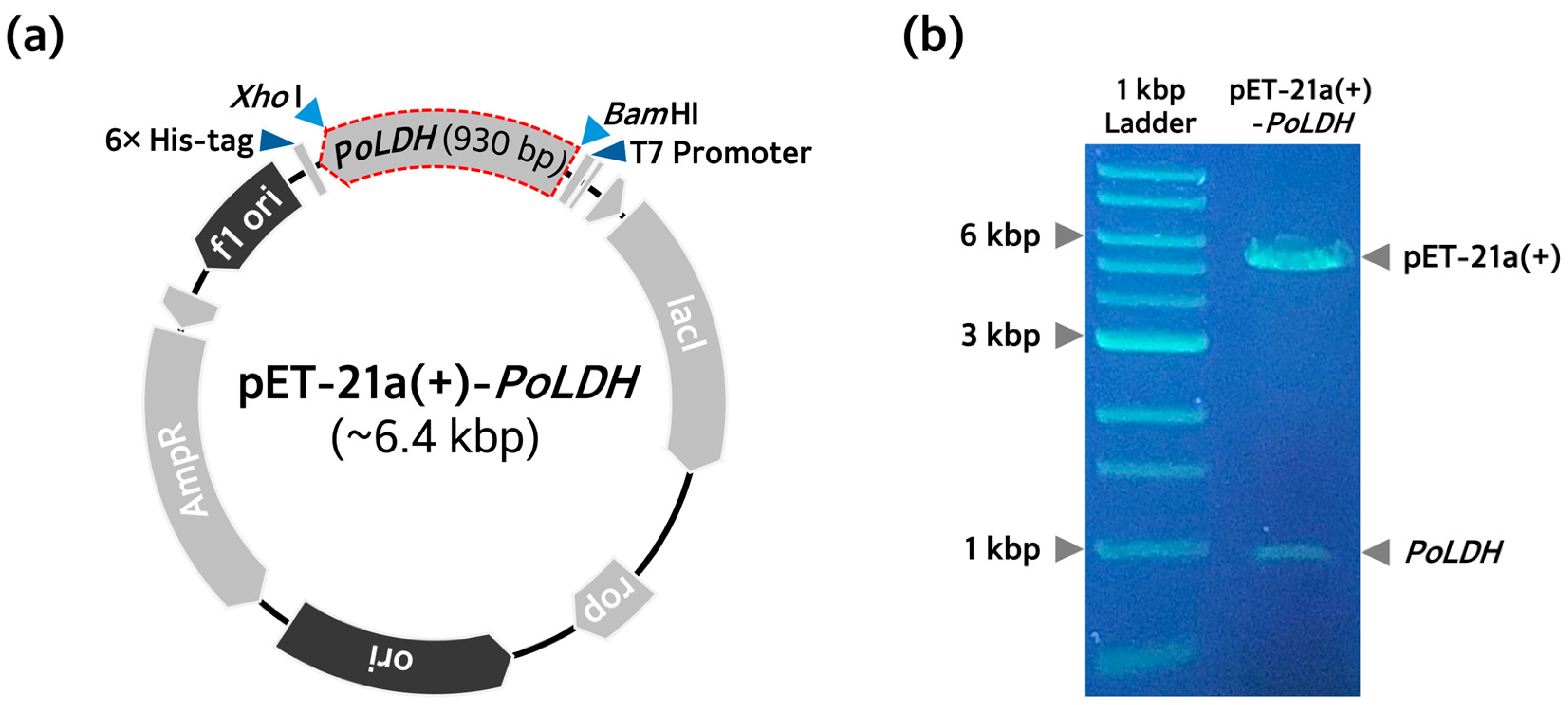
2.1. Cloning P. ovale LDH Gene
2.2. Selecting the Optimal Strain for PoLDH Expression
2.2.1. Transformation of E. coli Strains
2.2.2. Comparison of PoLDH Expression Patterns in the E. coli Strains
2.3. Establishing Optimized Operating Conditions for PoLDH Purification
2.3.1. Conditions for Purifying PoLDH Protein
2.3.2. Comparison of PoLDH Purification Yield for Each Strain
2.4. Analysis of Tetramer Formation and Enzyme Activity in the Purified PoLDH
3. Results and Discussion
3.1. PoLDH Recombinant Plasmid Construction and Transformation of E. coli Strains for Protein Expression
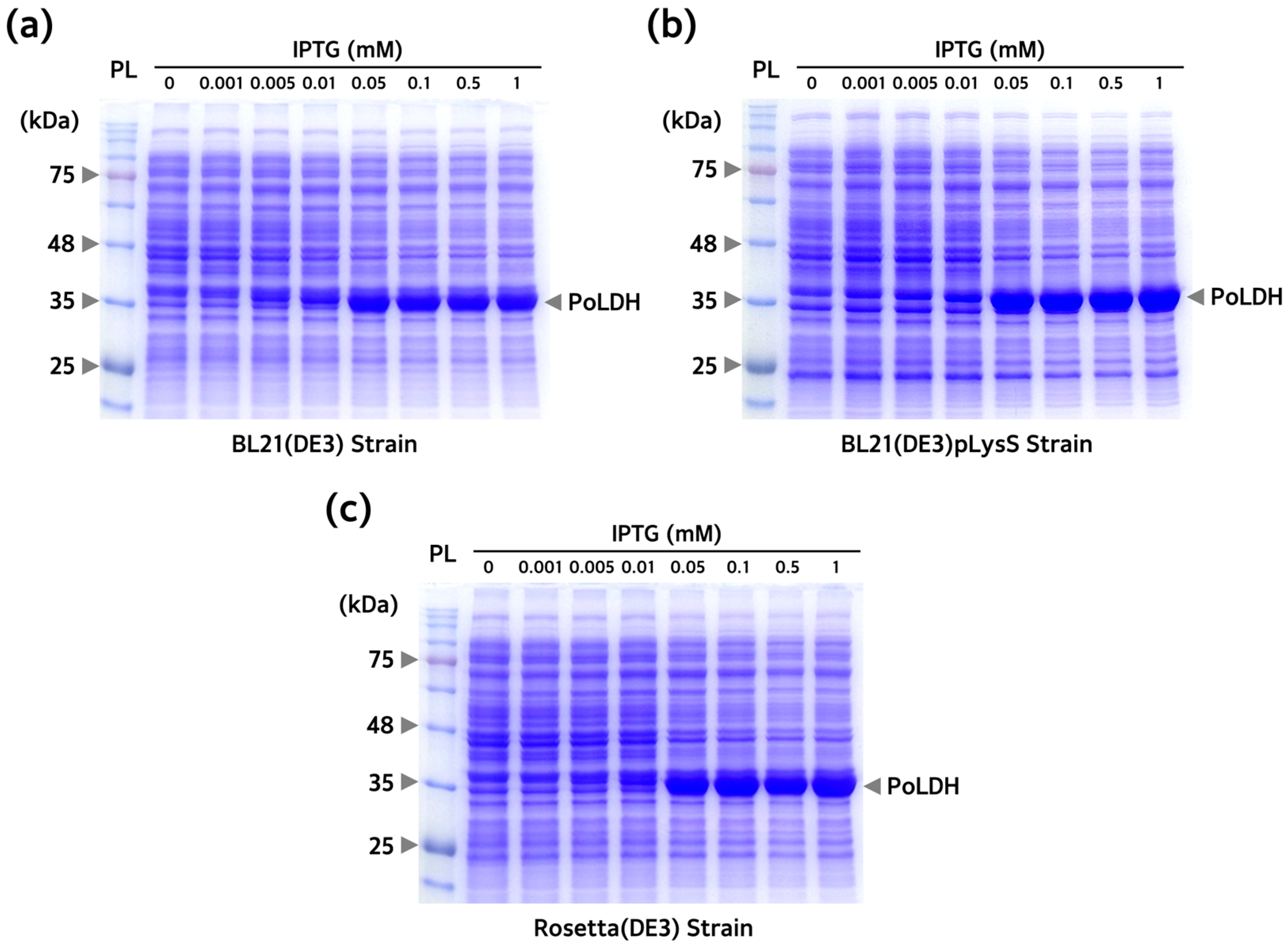
3.2. Comparison of PoLDH Expression Pattern and Overexpression Optimization for Different E. coli Strains
3.3. Optimization of Buffer Conditions for Isolating and Purifying PoLDH
3.4. Comparison of PoLDH Purification and Yields between Different E. coli Strains
3.5. Confirmation of Tetramer Formation in the Purified PoLDH
3.6. Analysis of Enzyme Activity of the Purified PoLDH
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fikadu, M.; Ashenafi, E. Malaria: An Overview. Infect. Drug Resist. 2023, 16, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023.
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Primers 2017, 3, 17050. [Google Scholar] [CrossRef] [PubMed]
- Cowman, A.F.; Healer, J.; Marapana, D.; Marsh, K. Malaria: Biology and Disease. Cell 2016, 167, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Cardenas, J.A.; González-Cerón, L.; García-Maroto, F.; Cebrián-Carmona, J.; Manzano-Agugliaro, F.; Mesa-Valle, C.M. Analysis of Fifty Years of Severe Malaria Worldwide Research. Pathogens 2023, 12, 373. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, K.; Hentzschel, F.; Valkiūnas, G.; Marti, M. Plasmodium Asexual Growth and Sexual Development in the Haematopoietic Niche of the Host. Nat. Rev. Microbiol. 2020, 18, 177–189. [Google Scholar] [CrossRef]
- Sato, S. Plasmodium—A Brief Introduction to the Parasites Causing Human Malaria and Their Basic Biology. J. Physiol. Anthropol. 2021, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Veletzky, L.; Groger, M.; Lagler, H.; Walochnik, J.; Auer, H.; Fuehrer, H.P.; Ramharter, M. Molecular Evidence for Relapse of an Imported Plasmodium ovale wallikeri Infection. Malar. J. 2018, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Kotepui, M.; Masangkay, F.R.; Kotepui, K.U.; Milanez, G.D.J. Misidentification of Plasmodium ovale as Plasmodium vivax Malaria by a Microscopic Method: A Meta-Analysis of Confirmed P. ovale Cases. Sci. Rep. 2020, 10, 21807. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Antimalarial Drug Resistance. J. Clin. Investig. 2004, 113, 1084–1092. [Google Scholar] [CrossRef]
- World Health Organization. Susceptibility of Plasmodium falciparum to Antimalarial Drugs: Report on Global Monitoring: 1996–2004; World Health Organization: Geneva, Switzerland, 2005.
- McKeage, K.; Scott, L.J. Atovaquone/Proguanil. Drugs 2003, 63, 597–623. [Google Scholar] [CrossRef]
- Wångdahl, A.; Sondén, K.; Wyss, K.; Stenström, C.; Björklund, D.; Zhang, J.; Askling, H.H.; Carlander, C.; Hellgren, U.; Färnert, A. Relapse of Plasmodium vivax and Plasmodium ovale Malaria with and without Primaquine Treatment in a Nonendemic Area. Clin. Infect. Dis. 2022, 74, 1199–1207. [Google Scholar] [CrossRef]
- Haston, J.C.; Hwang, J.; Tan, K.R. Guidance for Using Tafenoquine for Prevention and Antirelapse Therapy for Malaria—United States, 2019. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 1062–1068. [Google Scholar] [CrossRef]
- Kotepui, M.; Kotepui, K.U.; Milanez, G.D.; Masangkay, F.R. Severity and Mortality of Severe Plasmodium ovale Infection: A Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0235014. [Google Scholar] [CrossRef]
- Groger, M.; Fischer, H.S.; Veletzky, L.; Lalremruata, A.; Ramharter, M. A Systematic Review of the Clinical Presentation, Treatment and Relapse Characteristics of Human Plasmodium ovale Malaria. Malar. J. 2017, 16, 112. [Google Scholar] [CrossRef]
- Fuehrer, H.P.; Campino, S.; Sutherland, C.J. The Primate Malaria Parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: Genomic Insights into Distribution, Dispersal and Host Transitions. Malar. J. 2022, 21, 138. [Google Scholar] [CrossRef]
- Sitali, L.; Miller, J.M.; Mwenda, M.C.; Bridges, D.J.; Hawela, M.B.; Hamainza, B.; Chizema-Kawesha, E.; Eisele, T.P.; Chipeta, J.; Lindtjørn, B. Distribution of Plasmodium Species and Assessment of Performance of Diagnostic Tools Used During a Malaria Survey in Southern and Western Provinces of Zambia. Malar. J. 2019, 18, 130. [Google Scholar] [CrossRef]
- Gibson, L.E.; Markwalter, C.F.; Kimmel, D.W.; Mudenda, L.; Mbambara, S.; Thuma, P.E.; Wright, D.W. Plasmodium falciparum HRP2 ELISA for Analysis of Dried Blood Spot Samples in Rural Zambia. Malar. J. 2017, 16, 350. [Google Scholar] [CrossRef]
- Priest, J.W.; Plucinski, M.M.; Huber, C.S.; Rogier, E.; Mao, B.; Gregory, C.J.; Candrinho, B.; Colborn, J.; Barnwell, J.W. Specificity of the IgG Antibody Response to Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, and Plasmodium ovale MSP119 Subunit Proteins in Multiplexed Serologic Assays. Malar. J. 2018, 17, 417. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, J.W. Enzyme-Linked Aptamer-Based Sandwich Assay (ELASA) for Detecting Plasmodium falciparum Lactate Dehydrogenase, a Malarial Biomarker. RSC Adv. 2022, 12, 29535–29542. [Google Scholar] [CrossRef]
- Tang, J.; Tang, F.; Zhu, H.; Lu, F.; Xu, S.; Cao, Y.; Gu, Y.; He, X.; Zhou, H.; Zhu, G.; et al. Assessment of False Negative Rates of Lactate Dehydrogenase-Based Malaria Rapid Diagnostic Tests for Plasmodium ovale Detection. PLoS Negl. Trop. Dis. 2019, 13, e0007254. [Google Scholar] [CrossRef] [PubMed]
- Kori, L.D.; Valecha, N.; Anvikar, A.R. Glutamate Dehydrogenase: A Novel Candidate to Diagnose Plasmodium falciparum through Rapid Diagnostic Test in Blood Specimen from Fever Patients. Sci. Rep. 2020, 10, 6307. [Google Scholar] [CrossRef]
- Mathema, V.B.; Na-Bangchang, K. A Brief Review on Biomarkers and Proteomic Approach for Malaria Research. Asian Pac. J. Trop. Med. 2015, 8, 253–262. [Google Scholar] [CrossRef]
- Plucinski, M.M.; McElroy, P.D.; Dimbu, P.R.; Fortes, F.; Nace, D.; Halsey, E.S.; Rogier, E. Clearance Dynamics of Lactate Dehydrogenase and Aldolase Following Antimalarial Treatment for Plasmodium falciparum Infection. Parasit. Vectors 2019, 12, 293. [Google Scholar] [CrossRef]
- Zhou, Y.; Qi, M.; Yang, M. Current Status and Future Perspectives of Lactate Dehydrogenase Detection and Medical Implications: A Review. Biosensors 2022, 12, 1145. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New Tools for Recombinant Protein Production in Escherichia coli: A 5-Year Update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef]
- Hayat, S.M.G.; Farahani, N.; Golichenari, B.; Sahebkar, A. Recombinant Protein Expression in Escherichia coli (E.coli): What We Need to Know. Curr. Pharm. Des. 2018, 24, 718–725. [Google Scholar] [CrossRef]
- Turgut-Balik, D.; Akbulut, E.; Shoemark, D.K.; Celik, V.; Moreton, K.M.; Sessions, R.B.; Holbrook, J.J.; Brady, R.L. Cloning, Sequence and Expression of the Lactate Dehydrogenase Gene from the Human Malaria Parasite, Plasmodium vivax. Biotechnol. Lett. 2004, 26, 1051–1055. [Google Scholar] [CrossRef]
- Shin, H.I.; Kim, J.Y.; Lee, W.J.; Sohn, Y.; Lee, S.W.; Kang, Y.J.; Lee, H.W. Polymorphism of the Parasite Lactate Dehydrogenase Gene from Plasmodium vivax Korean Isolates. Malar. J. 2013, 12, 166. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.; Lee, S.; Manjunatha, D.H.; Ban, C. A Colorimetric Aptasensor for the Diagnosis of Malaria Based on Cationic Polymers and Gold Nanoparticles. Anal. Biochem. 2013, 439, 11–16. [Google Scholar] [CrossRef]
- Sousa, L.P.; Mariuba, L.A.M.; Holanda, R.J.; Pimentel, J.P.; Almeida, M.E.M.; Chaves, Y.O.; Borges, D.; Lima, E.; Crainey, J.L.; Orlandi, P.P.; et al. A Novel Polyclonal Antibody-Based Sandwich ELISA for Detection of Plasmodium vivax Developed from Two Lactate Dehydrogenase Protein Segments. BMC Infect. Dis. 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Tegel, H.; Tourle, S.; Ottosson, J.; Persson, A. Increased Levels of Recombinant Human Proteins with the Escherichia coli Strain Rosetta(DE3). Protein Expr. Purif. 2010, 69, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Son, M.S.; Hoang, T.T. One Step Engineering of T7-Expression Strains for Protein Production: Increasing the Host-Range of the T7-Expression System. Protein Expr. Purif. 2007, 55, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Shin, J.S.; Lee, K.W.; Eom, H.J.; Jo, B.G.; Lee, J.W.; Kim, J.H.; Kim, S.Y.; Kang, J.H.; Choi, J.W. Expression, Purification, and Characterization of Plasmodium vivax Lactate Dehydrogenase from Bacteria without Codon Optimization. Int. J. Mol. Sci. 2023, 24, 11083. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein-Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef]
- Oeller, M.; Kang, R.; Bell, R.; Ausserwöger, H.; Sormanni, P.; Vendruscolo, M. Sequence-based prediction of pH-dependent protein solubility using CamSol. Brief Bioinform. 2023, 24, bbad004. [Google Scholar] [CrossRef]
- Keluskar, P.; Ingle, S. Ethnopharmacology Guided Screening of Traditional Indian Herbs for Selective Inhibition of Plasmodium Specific Lactate Dehydrogenase. J. Ethnopharmacol. 2012, 144, 201–207. [Google Scholar] [CrossRef]
- Salim, N.O.; Fuad, F.A.A.; Khairuddin, F.; Seman, W.M.K.W.; Jonet, M.A. Purifying and Characterizing Bacterially Expressed Soluble Lactate Dehydrogenase from Plasmodium knowlesi for the Development of Anti-Malarial Drugs. Molecules 2021, 26, 6625. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-W.; Ha, S.-O.; Kim, Y.-J.; Shin, J.-S.; Choi, M.-J.; Yu, S.-E.; Han, J.; Park, E.-J.; Park, K.S.; Kang, J.H. Characterization of Escherichia coli Strains for Novel Production of Plasmodium ovale Lactate Dehydrogenase. Microorganisms 2024, 12, 876. https://doi.org/10.3390/microorganisms12050876
Choi J-W, Ha S-O, Kim Y-J, Shin J-S, Choi M-J, Yu S-E, Han J, Park E-J, Park KS, Kang JH. Characterization of Escherichia coli Strains for Novel Production of Plasmodium ovale Lactate Dehydrogenase. Microorganisms. 2024; 12(5):876. https://doi.org/10.3390/microorganisms12050876
Chicago/Turabian StyleChoi, Jae-Won, Sang-Oh Ha, Yeon-Jun Kim, Jun-Seop Shin, Min-Ji Choi, Si-Eun Yu, Junghun Han, Eun-Ji Park, Kyoung Sik Park, and Jung Hoon Kang. 2024. "Characterization of Escherichia coli Strains for Novel Production of Plasmodium ovale Lactate Dehydrogenase" Microorganisms 12, no. 5: 876. https://doi.org/10.3390/microorganisms12050876