1. Introduction
Artemia, more commonly recognized as brine shrimp, is a small aquatic crustacean inhabiting diverse saline environments across the world. Among its notable adaptations,
Artemia show a remarkable capability to undergo embryonic diapause—a phenomenon observed in numerous plants, insects, and mammals [
1,
2]. Diapause serves as a biological strategy enabling embryos to enter a state of suspended animation or dormancy until environmental conditions become conducive to hatching and subsequent survival [
3]. This mechanism assumes a crucial role in the endurance and perpetuation of
Artemia populations within unpredictable and frequently hostile aquatic habitats.
The onset of diapause is primarily triggered by adverse environmental conditions, such as fluctuations in temperature, salinity, and oxygen levels, or the presence of predators [
4]. Upon detecting these unfavorable conditions, embryos of
Artemia defer their developments to avoid hatching into potentially inhospitable surroundings. Throughout diapause,
Artemia embryos undergo various physiological transformations, notably a reduction in metabolic activity. Entering a state of metabolic arrest, they conserve energy to endure adverse conditions for prolonged periods [
5]. A high-energy substance called diguanosine (Gp4G) is preferentially utilized over conventional energy storage substances such as trehalose, glycogen, and glycerol during diapause [
6]. The duration of diapause varies depending on environmental conditions. In some instances, diapause may persist for months or even years until the external environment become more conducive. After diapause termination, metabolic processes within the embryo are reactivated, leading to resumption of protein synthesis, cell division, and overall growth.
The mechanisms governing EDT in
Artemia are complex and involve a combination of factors including environmental cues, hormonal fluctuations, and the engagement of specific genes and molecular pathways. The primary trigger for diapause termination is the recognition of improved environmental conditions, such as stable temperatures, optimal salinity levels, and the absence of stressors or predators [
7,
8]. Upon detection of these favorable cues, the embryo receives signals to resume development. Hormonal regulation likely constitutes another critical aspect of diapause termination. A diapause hormone receptor-like gene (Ar-DHR) has been identified in
Artemia. It is located on the cell membrane of the pre-diapause cyst but is then found in the cytoplasm of the diapause cyst [
9]. Changes in hormone levels within the embryo likely play a role in initiating the exit from diapause and promoting subsequent development. Additionally, genetic and molecular factors contribute to the termination of diapause in
Artemia. Specific genes are either activated or suppressed to coordinate the resumption of embryonic development. Jia et al. [
10] discovered a correlation between EDT and
Artemia DEK (Ar-DEK), a nuclear factor protein. This correlation was observed through an increase in euchromatin and a decrease in heterochromatin. Furthermore, their research identified a connection between the Wnt signaling pathway and the EDT process in
Artemia. Lin’s research team identified several genes crucial for diapause termination and cell cycle regulation during early embryonic development in
Artemia. These genes include apoptosis inhibitor 5 (API5) [
11], P53 and DNA damage-regulated gene 1 (pdrg1) [
12], retinoblastoma binding protein 4 (RBBP4) [
13], and glycerol kinase (GK) [
14]. The activation of embryos is a swift process, with alterations in transcription levels observable within 30 min after dormancy is disrupted, peaking after 5 h [
15,
16].
The mechanism of Artemia EDT is a finely tuned survival strategy, enabling these small crustaceans to optimize the likelihood of their offspring hatching and flourishing in the dynamic aquatic environments they inhabit. Comprehensive investigation into the diapause termination mechanism aids researchers in comprehending how Artemia’s physiological activities are regulated in response to environmental shifts. However, to date, only a handful of environmental factors and genes have been identified as influencing the EDT process. The molecular mechanism of EDT remains elusive, particularly for the signal transduction process.
To gain a deeper insight into the signal transduction process of EDT, we conducted an analysis of the gene expression profile of Artemia cysts at 30 min after EDT, which represents the initial phase of the EDT process, using ATAC-seq and RNA-seq techniques. The profile was compared with that of the diapause stage and 5 h after EDT to validate the differentially expressed genes (DEGs) related to the signal transduction pathways and GPCRs in different stages of EDT through GO enrichment analysis and structural examination. The identification of signal transduction pathways and GPCRs provides crucial insights for further exploring the EDT mechanism in Artemia.
2. Materials and Methods
2.1. Artemia Hatching and Culture
Artemia parthenogenetica cysts (provided by the Asian Regional Artemia Reference Center, ARARC, Tianjin, China) were collected at Ebinur Lake and reactivated after dehydration and refrigeration treatment to break diapause. At 30 min after diapause breaking, the dry cysts were thoroughly rehydrated in ice-cold 30‰ artificial seawater and reactivated in 30‰ artificial seawater at 28 °C under continuous illumination. The reactivated cysts were collected, placed in liquid nitrogen immediately, and then preserved in a −80 °C refrigerator. The cell activity of the samples was assessed with a Trypan blue assay and quantified.
2.2. Experiment Design for ATAC-Seq and RNA-Seq
The
A. parthenogenetica cysts collected 30 min after EDT were categorized as the ArR_30min group. A total of three biological replicates were collected, labeled as ArR_30min_1, ArR_30min_2, and ArR_30min_3. In preparation for subsequent sequencing, each of the samples was divided into two parts: one for ATAC-seq and the other for RNA-seq analysis. The sequencing results of ArR_30min were then compared with the ATAC-seq and RNA-seq data of
A. parthenogenetica cysts collected at the diapause stage (ArD_0h group) and 5 h after EDT (ArR_5h group), all obtained from the same batch of cyst samples. These data were sourced from the GEO database under the accession numbers GSE248452 (ATAC-seq data for ArD_0h and ArR_5h groups) and GSE249417 (RNA-seq data for ArD_0h and ArR_5h groups). Each group comprised three biological replicates, labeled as ArD_0h_1, ArD_0h_2, ArD_0h_3, ArR_5h_1, ArR_5h_2, and ArR_5h_3. The comparison of ATAC-seq datasets resulted in the ATAC-seq DEGs for ArR_30min vs. ArD_0h (referred to as the ‘0–30 min’ group) and ArR_5h vs. ArR_30min (referred to as the ‘30 min–5 h’ group). Similarly, the comparison of RNA-seq datasets resulted in the RNA-seq DEGs for the ‘0–30 min’ group and ‘30 min–5 h’ group. To identify the genes with consistent expression patterns and chromatin accessibility, the DEGs in ATAC-seq and RNA-seq of the ‘30 min–5 h’ group were compared to identify the integrated DEGs (IDEGs) of the ‘0–30 min’ group. Similarly, the DEGs in ATAC-seq and RNA-seq of the ‘30 min–5 h’ group were compared to identify the IDEGs of the ‘0–30 min’ group. An illustration of the experimental design is shown in
Figure 1.
2.3. ATAC Sequencing and Analysis
ATAC-seq was conducted as previously reported [
17,
18,
19]. In brief, nuclei were extracted from each sample, and the nuclei pellet was re-suspended in Tn5 transposase reaction mix. The transposition reaction was then incubated at 37 °C for 30 min. Following transposition, equimolar amounts of adapter 1 and adapter 2 were added. Subsequently, PCR was performed to amplify the libraries. After PCR amplification, the libraries were purified using AMPure beads, and their quality was assessed with a Qubit instrument. The index-coded samples were clustered on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Once cluster generation was completed, the library preparations were sequenced on an Illumina HiSeq platform (Illumina, San Diego, CA, USA), producing 150 bp paired-end reads.
After sequencing, Nextera adaptor sequences were trimmed from the reads using skewer (0.2.2). Subsequently, these reads were aligned to the
A. franciscana genome (NCBI assembly ID ASM3288406v1) using BWA (version 0.7.12-r1039, Cambridge, UK) [
20] with standard parameters. Following alignment, the reads were filtered for high quality based on criteria including a MAPQ ≥ 13 (i.e.,
p ≤ 0.05), exclusion of mitochondrial chromosomes, and retention of properly paired reads longer than 18 nucleotides. Data quality was assessed using FastQC (
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, accessed on 10 May 2023, Cambridge, UK), and peak calling was performed using Macs2 software (version 2.2.7.1, Boston, MA, USA) [
21]. Simulations of peaks called per input read utilized aligned and de-duplicated BAM files without any additional filtering.
The enrichments of peaks in the ArR_30min group were compared with those of the ArD_0h and ArR_5h groups, respectively. Differential peak analysis was carried out using the DESeq2 R package (version 1.20.0, Boston, MA, USA) [
22], where peaks with |log2.FoldEnrich| > 1 were identified as differential peaks. The ChIPseeker R package (version 1.38.0, Hong Kong, China) [
23] was employed to analyze the distribution of peaks in different functional regions, following a designated priority order: promoter, UTR, exon, intron, downstream TTS, and distal intergenic regions. In cases where a peak spanned both the promoter region of one gene and the UTR region of another gene, the priority order determined it as belonging to the promoter region rather than the UTR region. For Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation of genes associated with differentially expressed peaks, GOseq (version 4.10.2, Parkville, Australia) [
24] and KOBAS (version 3.0, Beijing, China) [
25] software were utilized, respectively.
2.4. RNA Sequencing and Analysis
The mRNA-Seq experiments were conducted by Novogene (Beijing, China) using Illumina high-throughput sequencing technology. Initially, total RNA was isolated from Artemia cysts using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), followed by treatment with RNase-free DNase I to eliminate any potential genomic DNA contamination. RNA integrity was assessed using the RNA Nano 6000 Assay Kit on the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). Sequencing libraries were generated with the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Beverly, MA, USA). Library quality was evaluated using the Agilent Bioanalyzer 2100 system. Index-coded samples were clustered on a cBot Cluster Generation System with a TruSeq PE Cluster Kit v3-cBot-HS (Illumina). Subsequently, the prepared libraries were sequenced on an Illumina NovaSeq platform, yielding 150 bp paired-end reads.
After sequencing, the original image data were processed into sequencing data via base calling to generate raw reads. Clean data, including clean reads, were obtained by filtering out raw reads containing adapters or poly-N sequences or exhibiting low quality. Subsequently, the quality of clean data was assessed in terms of Q20, Q30, and GC content. All subsequent analyses were conducted using high-quality clean data. The
A. franciscana genome (NCBI assembly ID ASM3288406v1) served as the reference genome for genome mapping. An index of the reference genome was constructed using Hisat2 (version 2.0.5, Baltimore, MD, USA) [
26], and paired-end clean reads were aligned to the reference genome using the same software. Reads mapped to each gene were quantified using FeatureCounts (version 1.5.0-p3, Parkville, Australia) [
27]. The FPKM (fragments per kilobase of transcript sequence per million base pairs sequenced) value for each gene was calculated based on its length and the number of reads mapped to it.
Gene expression levels in the samples from the ArR_30min group were compared with those from the ArD_0h and ArR_5h groups. To identify DEGs, a differential expression analysis was conducted using the DESeq2 R package (1.20.0). The resulting
p-values were adjusted using the Benjamini and Hochberg method to control the false discovery rate. Genes with an adjusted
p-value (Padj) ≤ 0.05 and |log2.Fold_change| > 1 were considered differentially expressed. Enrichment analysis of GO and KEGG for the DEGs was performed using GOseq (version 4.10.2, Parkville, Australia) [
24] and KOBAS software (version 3.0, Beijing, China) [
25], respectively. The ATAC-seq and RNA-seq data were deposited in the GEO database with the accession numbers GSE254934 and GSE254935.
2.5. Integration Analysis of ATAC-Seq and RNA-Seq
The expression profiles of the DEGs in the ATAC-seq and RNA-seq results were compared. Specifically, the up-regulated DEGs in RNA-seq were compared with the genes associated with up-regulated peaks in ATAC-seq, while the down-regulated DEGs in RNA-seq were compared with the genes associated with down-regulated peaks in ATAC-seq. In cases where a gene was associated with both up-regulated and down-regulated peaks in the ATAC-seq results, its expression profile consistent with the RNA-seq data was retained. Genes exhibiting consistent expression profiles in both methods were subjected to GO and KEGG enrichment analysis.
2.6. Structural Analysis of Candidate GPCR Proteins
To further identify candidate GPCR genes, the transmembrane helix (TMH) structure, secondary structure, and tertiary structure of the proteins encoded by these candidate genes were analyzed. The TMH structure of the proteins was predicted using TMHMM (
https://services.healthtech.dtu.dk/service.php?TMHMM-2.0, accessed on 2 February 2024) [
28]. The secondary and tertiary structures of the proteins were predicted using PredictProtein (
https://predictprotein.org/, accessed on 2 February 2024) [
29] and SWISS-MODEL (
https://swissmodel.expasy.org/, accessed on 2 February 2024) [
30], respectively.
4. Discussion
4.1. Regulation of EDT Process
Embryonic diapause is a fascinating and widely observed biological phenomenon observed in various animal species, particularly invertebrates and some mammals [
31]. It serves as a remarkable survival strategy, enabling developing organisms to endure adverse conditions by entering a state of suspended animation [
32]. The widespread distribution of embryonic diapause suggests its ancient evolutionary origin and potential shared molecular basis. During diapause, the embryo essentially enters a period of dormancy or quiescence, characterized by significantly reduced metabolic activity and halted development [
33].
The initiation and termination of diapause are closely related to environmental factors, but their molecular mechanisms remain unclear. In this study, ATAC-seq and RNA-seq sequencing were conducted on Artemia cyst samples at 30 min after EDT, and the results were compared with the sequencing results from cyst samples at the diapause stage and 5 h after EDT to explore the regulatory mechanisms of EDT. During comparative analysis of high-throughput sequencing data at different time points, we observed significant differences in gene expression levels during the first 30 min and at 30 min to 5 h after EDT. By integrating ATAC-seq and RNA-seq sequencing results, 786 and 850 IDEGs were identified in the ‘0–30 min’ and ‘30 min–5 h’ groups, respectively. However, there were only 131 common IDEGs between these two groups, with only 92 IDEGs showing consistent up-/down-regulation patterns. This suggests significant alterations in metabolism and regulation processes within Artemia cyst cells during the first 30 min and the subsequent 30 min to 5 h after EDT, with notable differences in the mechanisms underlying these changes. Among the DEGs exhibiting consistent patterns in both stages, there were more genes related to metabolic processes than regulation processes (34 vs. 17), indicating that changes in regulation processes are more pronounced than metabolic processes during the first 30 min and the subsequent 30 min to 5 h after EDT. Among the regulation-related genes, there were 60 and 66 signaling genes in the two stages, respectively, with only 8 of them being the same. Furthermore, when comparing results for the GPCR signaling pathway, which played a dominant role in both groups, it was found that the genes involved in this pathway in the two groups were entirely different. This suggests that the mechanisms of signal transduction processes may be completely different in these two periods. A more detailed stage division should be carried out in future research to deeply analyze the dynamic regulation process of EDT.
In both the ‘0–30 min’ and ‘30 min–5 h’ groups, signaling regulation was mainly mediated through the GPCR signaling pathway and cell surface receptor signaling pathway. In the ‘0–30 min’ group, hormone-mediated signaling pathways also played important roles, which was distinct from the ‘30 min–5 h’ group. In terms of other signaling pathways, both groups involved enzyme-linked receptor protein signaling pathways and Wnt and Notch signaling pathways. However, the specific genes involved in these pathways differed between the two groups. Additionally, the ‘0–30 min’ group included the tachykinin receptor signaling pathway and the neurotrophin signaling pathway, while the ‘30 min–5 h’ group included the neuropeptide signaling pathway. These results indicated that hormones play a crucial role in the first 30 min of EDT. In the 5 h following EDT, classical signaling pathways such as GPCR, Wnt, and Notch are then involved. However, the genes involved in these pathways vary between different time periods. The relationship between the Wnt signaling pathway and EDT is consistent with the findings of Jia et al. [
10]. Furthermore, Ouellet et al.’s study [
34] also suggests that the Notch signaling pathway is associated with the maintenance and termination of diapause in
C. elegans embryos. Additionally, we propose that the ‘0–30 min’ and ‘30 min–5 h’ stages contain their own unique signaling regulation pathways.
Based on the time-series analysis of the common IDEGs between the ‘0–30 min’ and ‘30 min–5 h’ groups, most metabolic and regulated related genes were found in the up–up group, indicating that metabolic and regulation processes were activated during the 5 h after EDT. As there were more IDGEs in the ‘30 min–5 h’ group compared to the ‘0–30 min’ group, we surmise that this activation is a gradual process and peaks at 5 h after EDT, which is consistent with the research of Chen et al. [
15] and Yu et al. [
16]. In the six signal-transduction-related genes, evm.TU.ctg33.11 is a GTPase-activating protein (GAP) for RhoA/Rho1, which plays an essential role in the regulation of the RhoA/Rho1-Drok-MRLC signaling pathway [
35]. evm.TU.ctg470.4 is an epidermal growth factor receptor gene that is involved in developmental decisions by transducing signals through the ras-raf-MAPK pathway. evm.TU.ctg275.1 participates in the proton transfer required for signaling transduction through proton-selective channels [
36]. evm.TU.ctg756.7 encodes a regulator of G-protein signaling, Loco, which is required for dorsal/ventral axis formation of the egg and embryo [
37].
4.2. GPCRs Participating in Artemia EDT Process
The GPCR, also known as a seven-transmembrane receptor or heptahelical receptor, is a type of cell membrane receptor protein that spans the cell membrane seven times, forming a distinctive helical structure. GPCRs constitute a diverse and extensive family of proteins present in the cell membranes of various organisms, including human [
38], mouse [
39], and
Takifugu rubripes [
40]. These receptors play a fundamental role in cellular signal transduction, serving as mediators for transmitting signals from the extracellular environment into the interior of the cell. They are crucial in facilitating the cellular response to a wide range of signals, including neurotransmitters, hormones, ions, and even light [
41,
42]. In insects, GPCRs have been found to interact with diapause hormones as ligands, suggesting their potential involvement in the signaling transduction of the diapause process [
43,
44].
In this study, we identified 5 and 10 genes with GPCR activity (GO:0004930) in the ‘0–30 min’ and ‘30 min–5 h’ groups, respectively. To further confirm whether these genes encode GPCR proteins, the secondary and tertiary structures of the proteins encoded by them were analyzed using three tools: TMHMM, PredictProtein, and SWISS-MODEL. Based on the distinctive 7-TMH structural characteristic of GPCRs, it was determined that three genes (evm.TU.ctg485.29, evm.TU.ctg71.25, evm.TU.ctg321.8) in the ‘0–30 min’ group and two genes (evm.TU.ctg441.12, evm.TU.ctg179.30) in the ‘30 min–5 h’ group encode proteins with structural characteristics consistent with GPCRs. evm.TU.ctg485.29, evm.TU.ctg71.25, and evm.TU.ctg321.8 were all found to be up-regulated in the ‘0–30 min’ group, suggesting that they are involved in processes occurring during the initial 30 min after EDT. evm.TU.ctg441.12 was found to be down-regulated exclusively in the ‘30 min–5 h’ group, indicating its potential significance as a GPCR during both the diapause stage and the 30 min period after EDT. Additionally, evm.TU.ctg179.30 exhibited up-regulation in the ‘30 min–5 h’ group, suggesting its primary function occurs in the 5 h after EDT.
In the genome annotation of
A. franciscana, evm.TU.ctg485.29 was identified as an Adhesion GPCR E5, a member of the LN-TM7 subfamily of GPCRs. This receptor is potentially involved in both adhesion and signaling processes shortly after leukocyte activation, playing a crucial role in leukocyte migration. evm.TU.ctg321.8 was annotated as Frizzled-10, a receptor for Wnt proteins. It has been shown to function in the canonical Wnt/beta-catenin signaling pathway and is activated by WNT7A to induce the expression of beta-catenin target genes, as observed in
Gallus gallus [
45]. It belongs to the Fz/Smo family of GPCRs. evm.TU.ctg441.12 is a RYamide receptor belonging to the GPCR1 family, serving as a receptor for the neuropeptides RYamide-1 and RYamide-2 in
Drosophila melanogaster [
46,
47]. evm.TU.ctg179.30 falls within the Opsin family of GPCRs, which are integral components of visual pigments. Visual pigments are molecules responsible for light absorption and mediating vision. They consist of an apoprotein, opsin, covalently linked to 11-cis-retinal [
48].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
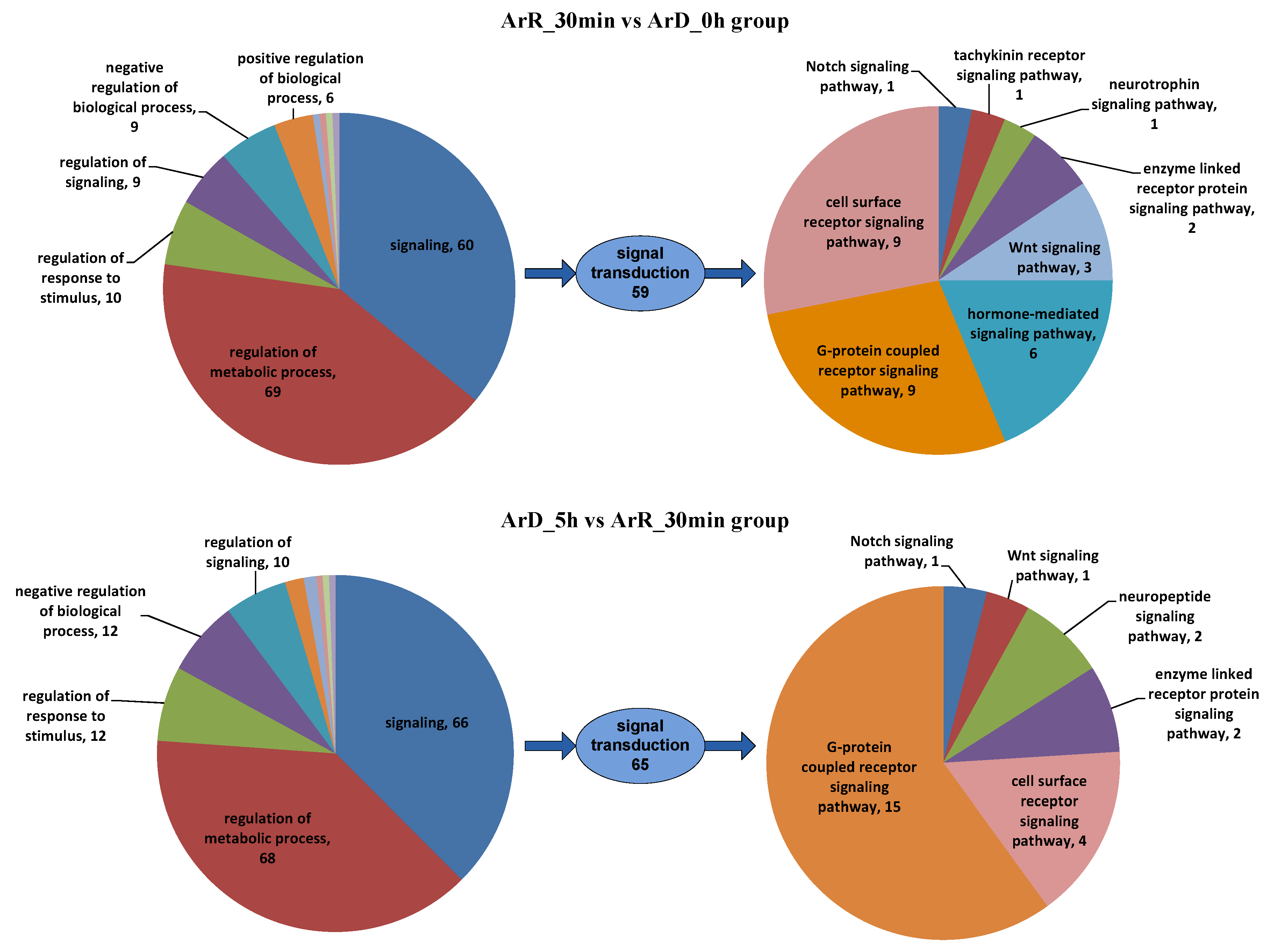{kind=link}
{kind=link}