
(2,6-Dimethylphenyl)arsonic Acid Induces Apoptosis through the Mitochondrial Pathway, Downregulates XIAP, and Overcomes Multidrug Resistance to Cytostatic Drugs in Leukemia and Lymphoma Cells In Vitro
Abstract
:1. Introduction
2. Results
2.1. As2 as a Unique and Biologically Active Candidate for Cancer Treatment
2.2. As2 Inhibits Proliferation and Induces Apoptosis in Cancer Cells In Vitro
2.3. As2 Acts Selectively on Malignant Cells
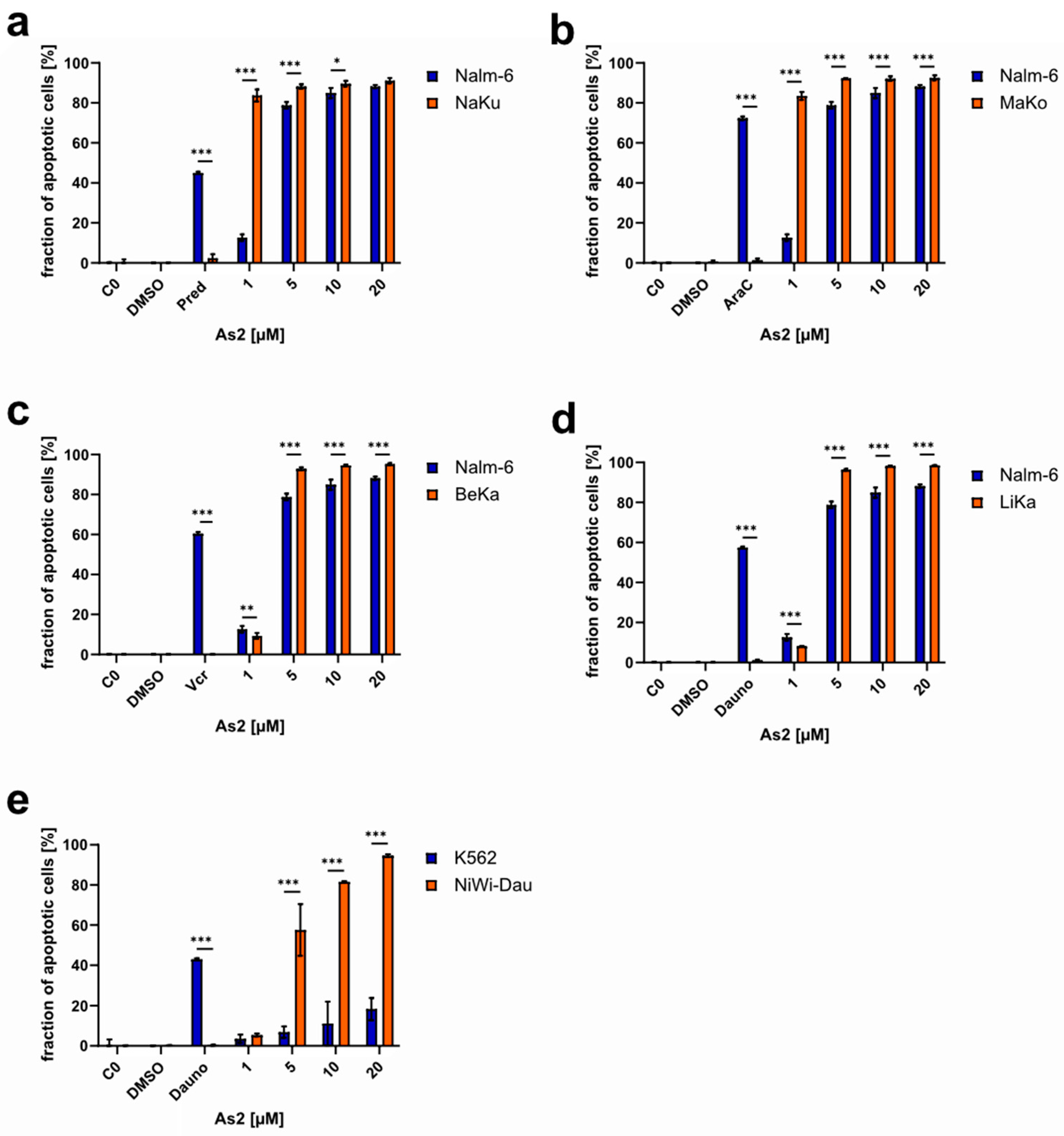
2.4. As2 Overcomes Multidrug Resistance In Vitro
2.5. As2 Increases Cancer Cell Sensitivity to Cytostatic Drugs
2.6. Disentangling As2-Induced Apoptosis Mechanisms
2.6.1. As2 Acts Independently of Bcl-2 and Caspase-3 Expression Levels
2.6.2. XIAP Downregulation by As2
2.6.3. As2 Induces Apoptosis through the Mitochondrial Pathway
3. Discussion
4. Materials and Methods
4.1. Chemicals and Drugs
4.2. Cell Lines and Cell Culture
4.3. Determination of Cell Viability
4.4. LDH Release Assay
4.5. Isolation of Human Leukocytes
4.6. Western Blot Analysis
4.7. Measurement of DNA Fragmentation
4.8. Annexin-V/Propidium Iodide Apoptosis Assay
4.9. Measurement of Mitochondrial Membrane Permeabilization
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Cheff, D.M.; Hall, M.D. Correction to A Drug of Such Damned Nature. Challenges and Opportunities in Translational Platinum Drug Research. J. Med. Chem. 2018, 61, 8944. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against novel anticancer metal compounds: Differences and similarities. Drug Resist. Updat. 2008, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Zheng, Y.; Xia, W.; Mao, Z.-W. Organometallic anti-tumor agents: Targeting from biomolecules to dynamic bioprocesses. Chem. Soc. Rev. 2023, 52, 2790–2832. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-based antitumor compounds: Beyond cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.; Podolski-Renić, A.; Poetsch, I.; Filipović, N.; López, Ó.; Turel, I.; Heffeter, P. Metal- and metalloid-based compounds to target and reverse cancer multidrug resistance. Drug Resist. Updat. 2021, 58, 100778. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef]
- Waxman, S.; Anderson, K.C. History of the development of arsenic derivatives in cancer therapy. Oncologist 2001, 6 (Suppl. S2), 3–10. [Google Scholar] [CrossRef]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Thang, N.D.; Yajima, I.; Kumasaka, M.Y.; Kato, M. Bidirectional functions of arsenic as a carcinogen and an anti-cancer agent in human squamous cell carcinoma. PLoS ONE 2014, 9, e96945. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-B.; Zhang, J.; Wang, Z.-Y.; Chen, S.-J.; Chen, Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Soignet, S.L.; Maslak, P.; Wang, Z.G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.X.; Chen, G.Q.; Ni, J.H.; Li, X.S.; Xiong, S.M.; Qiu, Q.Y.; Zhu, J.; Tang, W.; Sun, G.L.; Yang, K.Q.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997, 89, 3354–3360. [Google Scholar] [CrossRef]
- Hoonjan, M.; Jadhav, V.; Bhatt, P. Arsenic trioxide: Insights into its evolution to an anticancer agent. J. Biol. Inorg. Chem. 2018, 23, 313–329. [Google Scholar] [CrossRef]
- Ghiaur, A.; Doran, C.; Gaman, M.-A.; Ionescu, B.; Tatic, A.; Cirstea, M.; Stancioaica, M.C.; Hirjan, R.; Coriu, D. Acute Promyelocytic Leukemia: Review of Complications Related to All-Trans Retinoic Acid and Arsenic Trioxide Therapy. Cancers 2024, 16, 1160. [Google Scholar] [CrossRef]
- Carrall, J.A.; Lie, W.; Lambert, J.M.; Harris, H.H.; Lai, B.; Dillon, C.T. Optimizing Arsenic Therapy by Selectively Targeting Leukemia Cells. J. Med. Chem. 2023, 66, 12101–12114. [Google Scholar] [CrossRef] [PubMed]
- He, H.; An, R.; Hou, J.; Fu, W. Arsenic trioxide induced rhabdomyolysis, a rare but severe side effect, in an APL patient: A case report. Front. Med. 2017, 11, 284–286. [Google Scholar] [CrossRef]
- Mohan, D.; Prabhu, R.; Reghu, R. Arsenic trioxide-induced QT interval prolongation: A case report. Natl. J. Physiol. Pharm. Pharmacol. 2017, 1, 878. [Google Scholar] [CrossRef]
- Ding, W.; Zhang, L.; Kim, S.; Tian, W.; Tong, Y.; Liu, J.; Ma, Y.; Chen, S. Arsenic sulfide as a potential anti-cancer drug. Mol. Med. Rep. 2015, 11, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.H.; Ho, P.C. Arsenic compounds induce cytotoxicity and apoptosis in cisplatin-sensitive and -resistant gynecological cancer cell lines. Cancer Chemother. Pharmacol. 2001, 47, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [PubMed]
- Horsley, L.; Cummings, J.; Middleton, M.; Ward, T.; Backen, A.; Clamp, A.; Dawson, M.; Farmer, H.; Fisher, N.; Halbert, G.; et al. A phase 1 trial of intravenous 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO) in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Kim, W.-S.; Uchida, T.; Uike, N.; Suehiro, Y.; Ishizawa, K.; Nagai, H.; Nagahama, F.; Sonehara, Y.; Tobinai, K. Phase I studies of darinaparsin in patients with relapsed or refractory peripheral T-cell lymphoma: A pooled analysis of two phase I studies conducted in Japan and Korea. Jpn. J. Clin. Oncol. 2021, 51, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Skoczynska, A.; Skoczynska, M. Breast Cancer and Arsenic Anticancer Effects: Systematic Review of the Experimental Data from In Vitro Studies. Biomed Res. Int. 2022, 2022, 8030931. [Google Scholar] [CrossRef] [PubMed]
- She, W.; Shi, X.; Liu, T.; Liu, Y.; Liu, Y. Discovery of novel organoarsenicals as robust thioredoxin reductase inhibitors for oxidative stress mediated cancer therapy. Biochem. Pharmacol. 2023, 218, 115908. [Google Scholar] [CrossRef] [PubMed]
- Lucibello, M.; Gambacurta, A.; Zonfrillo, M.; Pierimarchi, P.; Serafino, A.; Rasi, G.; Rubartelli, A.; Garaci, E. TCTP is a critical survival factor that protects cancer cells from oxidative stress-induced cell-death. Exp. Cell Res. 2011, 317, 2479–2489. [Google Scholar] [CrossRef]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef]
- Hrgovic, I.; Zöller, E.; Doll, M.; Hailemariam-Jahn, T.; Jakob, T.; Kaufmann, R.; Meissner, M.; Kleemann, J. Arsenic Trioxide Decreases Lymphangiogenesis by Inducing Apoptotic Pathways and Inhibition of Important Endothelial Cell Receptors. Curr. Issues Mol. Biol. 2023, 46, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Lee, Y.-C.; Chiou, J.-T.; Shi, Y.-J.; Wang, L.-J.; Chang, L.-S. Arsenic trioxide-induced p38 MAPK and Akt mediated MCL1 downregulation causes apoptosis of BCR-ABL1-positive leukemia cells. Toxicol. Appl. Pharmacol. 2020, 397, 115013. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, C.; Wang, L.; Dai, Z.; Yang, K. Arsenic trioxide induces apoptosis and the formation of reactive oxygen species in rat glioma cells. Cell. Mol. Biol. Lett. 2018, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Meister, M.T.; Boedicker, C.; Graab, U.; Hugle, M.; Hahn, H.; Klingebiel, T.; Fulda, S. Arsenic trioxide induces Noxa-dependent apoptosis in rhabdomyosarcoma cells and synergizes with antimicrotubule drugs. Cancer Lett. 2016, 381, 287–295. [Google Scholar] [CrossRef]
- Basova, S.; Wilke, N.; Koch, J.C.; Prokop, A.; Berkessel, A.; Pradel, G.; Ngwa, C.J. Organoarsenic Compounds with In Vitro Activity against the Malaria Parasite Plasmodium falciparum. Biomedicines 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- van Genderen, H.; Kenis, H.; Lux, P.; Ungeth, L.; Maassen, C.; Deckers, N.; Narula, J.; Hofstra, L.; Reutelingsperger, C. In vitro measurement of cell death with the annexin A5 affinity assay. Nat. Protoc. 2006, 1, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Lehne, G. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr. Drug Targets 2000, 1, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Kater, L.; Claffey, J.; Hogan, M.; Jesse, P.; Kater, B.; Strauss, S.; Tacke, M.; Prokop, A. The role of the intrinsic FAS pathway in Titanocene Y apoptosis: The mechanism of overcoming multiple drug resistance in malignant leukemia cells. Toxicol. In Vitro 2012, 26, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L.A.; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W.S.; Wolber, G.; et al. Benzimidazol-2-ylidene gold(I) complexes are thioredoxin reductase inhibitors with multiple antitumor properties. J. Med. Chem. 2010, 53, 8608–8618. [Google Scholar] [CrossRef]
- Kater, B.; Hunold, A.; Schmalz, H.-G.; Kater, L.; Bonitzki, B.; Jesse, P.; Prokop, A. Iron containing anti-tumoral agents: Unexpected apoptosis-inducing activity of a ferrocene amino acid derivative. J. Cancer Res. Clin. Oncol. 2011, 137, 639–649. [Google Scholar] [CrossRef]
- Dragoun, M.; Günther, T.; Frias, C.; Berkessel, A.; Prokop, A. Metal-free salan-type compound induces apoptosis and overcomes multidrug resistance in leukemic and lymphoma cells in vitro. J. Cancer Res. Clin. Oncol. 2018, 144, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Schlagintweit, J.F.; Jakob, C.H.G.; Wilke, N.L.; Ahrweiler, M.; Frias, C.; Frias, J.; König, M.; Esslinger, E.-M.H.J.; Marques, F.; Machado, J.F.; et al. Gold(I) Bis(1,2,3-triazol-5-ylidene) Complexes as Promising Selective Anticancer Compounds. J. Med. Chem. 2021, 64, 15747–15757. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of Apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Juo, P.; Woo, M.S.; Kuo, C.J.; Signorelli, P.; Biemann, H.P.; Hannun, Y.A.; Blenis, J. FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ. 1999, 10, 797–804. [Google Scholar] [PubMed]
- Hopff, S.M.; Onambele, L.A.; Brandenburg, M.; Berkessel, A.; Prokop, A. Discovery of a cobalt (III) salen complex that induces apoptosis in Burkitt like lymphoma and leukemia cells, overcoming multidrug resistance in vitro. Bioorg. Chem. 2020, 104, 104193. [Google Scholar] [CrossRef]
- Wilke, N.L.; Burmeister, H.; Frias, C.; Ott, I.; Prokop, A. Ruthenium Complex HB324 Induces Apoptosis via Mitochondrial Pathway with an Upregulation of Harakiri and Overcomes Cisplatin Resistance in Neuroblastoma Cells In Vitro. Int. J. Mol. Sci. 2023, 24, 952. [Google Scholar] [CrossRef] [PubMed]
- Ahrweiler-Sawaryn, M.-C.; Biswas, A.; Frias, C.; Frias, J.; Wilke, N.L.; Wilke, N.; Berkessel, A.; Prokop, A. Novel gold(I) complexes induce apoptosis in leukemia cells via the ROS-induced mitochondrial pathway with an upregulation of Harakiri and overcome multi drug resistances in leukemia and lymphoma cells and sensitize drug resistant tumor cells to apoptosis in vitro. Biomed. Pharmacother. 2023, 161, 114507. [Google Scholar] [CrossRef]
- Forkner, C.E.; Scott, T.F.M. Arsenic as a therapeutic agent in chronic myleogenous leukemia. JAMA 1931, 97, 3. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in medicine: Past, present and future. Biometals 2022. [Google Scholar] [CrossRef]
- Alimoghaddam, K. A Review of Arsenic Trioxide and Acute Promyelocytic Leukemia. Int. J. Hematol. Oncol. Stem Cell Res. 2014, 8, 44–54. [Google Scholar] [PubMed]
- Woo, S.H.; Park, I.-C.; Park, M.-J.; Lee, H.-C.; Lee, S.-J.; Chun, Y.-J.; Lee, S.-H.; Hong, S.-I.; Rhee, C.H. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int. J. Oncol. 2002, 21, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Wen, T.; Gong, L.-B.; Deng, M.-M.; Hou, K.-Z.; Xu, L.; Shi, S.; Qu, X.-J.; Liu, Y.-P.; Che, X.-F.; et al. Inhibition of FEN1 Increases Arsenic Trioxide-Induced ROS Accumulation and Cell Death: Novel Therapeutic Potential for Triple Negative Breast Cancer. Front. Oncol. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, Y.; Fang, M.; Jehan, S.; Zhou, W. Current Advances of Nanomedicines Delivering Arsenic Trioxide for Enhanced Tumor Therapy. Pharmaceutics 2022, 14, 743. [Google Scholar] [CrossRef] [PubMed]
- Dilda, P.J.; Hogg, P.J. Arsenical-based cancer drugs. Cancer Treat. Rev. 2007, 33, 542–564. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.T.; Menale, C.; Crispi, S. Combined anticancer therapies: An overview of the latest applications. Anticancer. Agents Med. Chem. 2015, 15, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef]
- Ralph, S.J. Arsenic-based antineoplastic drugs and their mechanisms of action. Met. Based Drugs 2008, 2008, 260146. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Dai, J.; Zhao, J.; Guo, Y. The Mechanism of Apoptosis Regulation by IAP Antagonist Smac/DIABLO. In Molecular Mechanisms of Programmed Cell Death; Shi, Y., Cidlowski, J.A., Scott, D., Wu, J.-R., Shi, Y.-B., Eds.; Springer: Boston, MA, USA, 2003; pp. 195–211. ISBN 978-1-4419-3404-8. [Google Scholar]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Carlet, M.; Schmelz, K.; Vergalli, J.; Herold, T.; Senft, D.; Jurinovic, V.; Hoffmann, T.; Proba, J.; Weichert, N.; Junghanß, C.; et al. X-linked inhibitor of apoptosis protein represents a promising therapeutic target for relapsed/refractory ALL. EMBO Mol. Med. 2023, 15, e14557. [Google Scholar] [CrossRef] [PubMed]
- Fakler, M.; Loeder, S.; Vogler, M.; Schneider, K.; Jeremias, I.; Debatin, K.-M.; Fulda, S. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009, 113, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Loeder, S.; Drensek, A.; Jeremias, I.; Debatin, K.-M.; Fulda, S. Small molecule XIAP inhibitors sensitize childhood acute leukemia cells for CD95-induced apoptosis. Int. J. Cancer 2010, 126, 2216–2228. [Google Scholar] [CrossRef]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | AC50 [µM] |
|---|---|---|
| As1 |  | no effect |
| As2 |  | ~6.3 |
| As3 |  | no effect |
| As4 |  | no effect |
| As5 |  | >100 |
| As6 |  | no effect |
| As7 |  | no effect |
| As8 |  | no effect |
| As9 |  | no effect |
| As10 |  | no effect |
| As11 |  | no effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilke, N.; Frias, C.; Berkessel, A.; Prokop, A. (2,6-Dimethylphenyl)arsonic Acid Induces Apoptosis through the Mitochondrial Pathway, Downregulates XIAP, and Overcomes Multidrug Resistance to Cytostatic Drugs in Leukemia and Lymphoma Cells In Vitro. Int. J. Mol. Sci. 2024, 25, 4713. https://doi.org/10.3390/ijms25094713
Wilke N, Frias C, Berkessel A, Prokop A. (2,6-Dimethylphenyl)arsonic Acid Induces Apoptosis through the Mitochondrial Pathway, Downregulates XIAP, and Overcomes Multidrug Resistance to Cytostatic Drugs in Leukemia and Lymphoma Cells In Vitro. International Journal of Molecular Sciences. 2024; 25(9):4713. https://doi.org/10.3390/ijms25094713
Chicago/Turabian StyleWilke, Nathalie, Corazon Frias, Albrecht Berkessel, and Aram Prokop. 2024. "(2,6-Dimethylphenyl)arsonic Acid Induces Apoptosis through the Mitochondrial Pathway, Downregulates XIAP, and Overcomes Multidrug Resistance to Cytostatic Drugs in Leukemia and Lymphoma Cells In Vitro" International Journal of Molecular Sciences 25, no. 9: 4713. https://doi.org/10.3390/ijms25094713