
Syntheses of New Multisubstituted 1-Acyloxyindole Compounds

College of Pharmacy and Innovative Drug Center, Duksung Women’s University, 33 Samyangro 144-gil, Dobong-gu, Seoul 01369, Korea
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(19), 6769; https://doi.org/10.3390/molecules27196769
Submission received: 22 September 2022
/
Revised: 7 October 2022
/
Accepted: 7 October 2022
/
Published: 10 October 2022
(This article belongs to the Special Issue Recent Advances in Indole Derivatives in Medicinal and Synthetic Organic Chemistry)
Abstract
:The syntheses of novel 1-acyloxyindole compounds 1 and the investigations on reaction pathways are presented. Nitro ketoester substrate 2, obtained in a two-step synthetic process, underwent reduction, intramolecular addition, nucleophilic 1,5-addition, and acylation to afford 1-acyloxyindoles 1 in one pot. Based on the systematic studies, we established the optimized reaction conditions for 1 focusing on the final acylation step of the intermediate 1-hydroxyindole 8. With the optimized conditions, we succeeded in synthesizing 21 examples of new 1-acyloxyindole derivatives 1 in modest yields (Y = 24 − 35%). Among the 1-acyloxyindole compounds, 1-acetoxyindole compounds 1x were generally unstable, and their yields were relatively lower than the other 1-acyloxyindoles. We expect that a bulkier alkyl or aromatic group on R2 could stabilize the 1-acyloxyindole compounds. Significantly, one-pot reactions of a four-step sequence successfully generated compounds 1 that are all new and might be difficult to be synthesized otherwise.
1. Introduction
The indole structure is an important component of many biologically active natural and synthetic compounds. Indeed, 1-acyloxyindole compounds are one of the N(1)-substituted indole derivatives containing an acyloxy group (-OCOR) instead of hydrogen on N(1) position (Figure 1). Among 1-acyloxyindole compounds, 1-benzoyloxyindole was first reported by Acheson et al. in 1970 [1], and 1-acetoxyindole was then reported in 1974 [2]. Further, 1-hydroxyindole, 1-alkoxyindole, and 1-acyloxyindole derivatives have attracted attention by the emergence of several potent 1-hydroxyindole derivatives [3,4,5] and 1-methoxyindole derivatives [6,7] in natural products. Furthermore, these unique structures demonstrated potential as indole substitutes and indole precursors associated with metabolic transformations [8,9,10].
Several investigations reported that 1-hydroxyindole and 1-alkoxyindole compounds have various biological properties, including antitumor [11,12,13], antibiotic [14], and platelet aggregation inhibiting effects [15]. In particular, Granchi et al. discovered that 1-hydroxyindole derivatives inhibited lactate dehydrogenase isoform A (LDH-A) at low micromolar concentration [12] and thus were considered as potential compounds for cancer treatment [16]. Furthermore, these derivatives were employed as intermediates for synthesis of potent anticonvulsant and antiarrhythmic agents [17]. Despite their various biological activities, few studies on 1-hydroxy, 1-alkoxy, and 1-acyloxyindole derivatives have been conducted. Probably due to the instability of 1-hydroxyindole compounds [10,18], studies on synthetic methods and their derivatization have not been extensively explored. Somei et al. developed a synthetic method for structurally concise 1-hydroxyindoles by oxidation of 2,3-dihydroindoles with a catalytic amount of sodium tungstate (Na2WO4) [19,20]. Other groups employed Zn/NH4Cl [2], tin chloride (SnCl2) [21,22,23], or Pd/triethylammonium formate (TEAF) [24] for reductive cyclization to produce 1-hydroxyindole moiety, and 1-Alkoxyindole compounds were also synthesized by alkylation of 1-hydroxyindoles [25] or intramolecular cyclization of methoxime structure [26]. Although a few simple 1-acyloxyindole derivatives were synthesized by acylation of 1-hydroxyindoles [17,27,28,29,30], the generated 1-acyloxyindoles were unstable and easily hydrolyzed to afford 1-hydroxyindoles [31]. Consequently, these compounds suffer from chemical instabilities and difficult manipulation.
In this study, we aimed to create novel derivatives of multisubstituted 1-acyloxyindoles (Figure 2) with improved chemical stability and meaningful biological activity. With the nitro ketoester substrate obtained in a two-step sequence, we devised a convenient one-pot synthetic method of consecutive four-step sequence to afford the desired 1-acyloxyindole compounds 1. In addition, we expect that the compounds could serve as useful prodrugs for valuable medicinal agents or pharmacokinetic structural components for drug delivery.
2. Results and Discussion
2.1. Synthesis of Conjugate Nitro Ketoester 2
At first, we prepared the substrate 2 [25,32,33,34] in two-step reactions using 2-chloro-6-nitrotoluene as a starting material. For this purpose, we applied our previous procedures [32] with minor modifications. As shown in Scheme 1, 2-chloro-6-nitrotoluene 3 was reacted with dimethyloxalate in the presence of excess sodium hydride to afford 4 [32] in an excellent yield (Y = 96%). Subsequently, 4 was reacted with dimethylmethyleneimminium chloride to add a methylene group at α-carbon in 4, affording conjugate ketoester 2 [32] in a good yield (Y = 85%). The result of synthesis of 2 was slightly improved compared to the previous results [32].
2.2. Optimization for Formation of 1-Acyloxyindoles 1
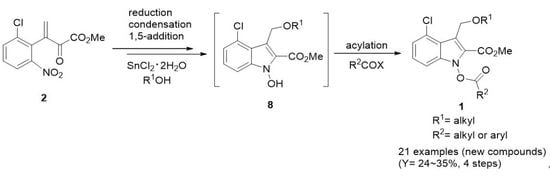
The reactions to generate 1-acyloxyindoles 1 consist of two main parts: formation of 1-hydroxyindole intermediates 8 and formation of 1-acyloxyindoles 1 by acylation of 8. Although we previously established the reaction conditions for 1-hydroxyindoles 8 [32], re-optimization for synthesis of 1-acyloxyindoles 1 is required because the whole process, including the acylation step, needs to be carried out in one pot. With substrate 2, we first attempted to perform systematic studies on the reaction conditions suitable for formation of 1-acyloxyindole 1. As indicated in Scheme 2, the substrate 2 was reduced to generate hydroxylamine 5, cyclized to provide hydroxyindoline 6, and dehydrated to produce conjugate nitrone 7. Then, nucleophilic 1,5-addition of alcohol to 7 produced the intermediate 1-hydroxyindole 8, and, finally, acylation of 8 afforded 1-acyloxyindole 1.
In particular, as a base for the acylation reaction, we tested several reagents, such as K2CO3, triethylamine (TEA), N,N-diisopropylethylamine (DIEA), 4-dimethylaminopyridine (DMAP), and 1,8-diazabicyclo [5.4.0]undece-7-ene (DBU). Among them, DBU provided the best results (data not shown), which are consistent with our previous reports [25]. Thus, we chose DBU for our purpose. Optimization of the reaction conditions was performed by varying the amount of SnCl2∙2H2O, DBU, alcohol (R1OH), and acylating agent (R2COX). SnCl2∙2H2O, an appropriate reducing agent for aromatic nitro group [35], was applied to convert 2 to 7 (2 → 5 → 6 → 7). We used benzyl alcohol (BnOH) as a template nucleophile and pivaloyl chloride as a template acylating agent in dimethoxyethane (DME) to produce 1dy (Table 1). Considering our previous procedure for synthesis of 1-alkoxyindoles [25], we applied the range of reagents as such: SnCl2∙2H2O 2.5–3.7 eq and DBU 10.6–15.7 eq for synthesis of 1. Here, we used an increased amount of DBU due to expected extra consumption by carboxylic acids that could be generated by partial hydrolysis of the acylating agents. At lower or higher amounts than 3.3 eq for SnCl2∙2H2O and 14.0 eq for DBU, the product 1dy was obtained in relatively poor yields (entries 1, 2, and 7, Table 1). We also compared the yields of 1-hydroxyindole intermediate 8d by varying the amount of SnCl2∙2H2O and found that the isolated yield of 1-hydroxyindole 8d with 3.3 eq of SnCl2∙2H2O was better (Y = 48%) than 3.7 eq (Y = 33%) and 2.5 eq (Y = 32%) in the case of 2.0 eq of BnOH. The orders of yields for intermediate 1-hydroxyindole 8d and 1-pivaloyloxyindole 1dy were generally correlated. The amount of SnCl2∙2H2O might be an important factor for construction of 1-acyloxyindole as well as 1-hydroxyindole intermediate by triggering the reduction of nitro group in 2. We further tested the amount of BnOH (1.5–3.0 eq) and pivaloyl chloride (1.5–3.0 eq). When using BnOH less than 1.5 eq, the product 1dy was obtained in poor yields (entries 3 and 4). More than 2.0 eq of BnOH and pivaloyl chloride did not seem to improve the yield (entry 6). Taken together, we chose the optimized condition for 1dy (entry 5): 1.0 eq of 2, 3.3 eq of SnCl2∙2H2O, 2.0 eq of BnOH at 40 °C, and then 14.0 eq of DBU and 2.0 eq of pivaloyl chloride at room temperature, which was applied to all other reactions unless otherwise noted.
2.3. Synthesis of New Derivatives of 1-Acyloxyindole 1
Under the optimized condition (entry 5, Table 1), we synthesized new 1-acyloxyindole derivatives by employing various nucleophiles and several acylating reagents (acetic anhydride and acyl chlorides) (Scheme 3). First, SnCl2·2H2O and 4Å molecular sieves were stirred in DME for 30 min at room temperature. We added alcohol and substrate 2, and then the reaction mixture was stirred at 40 °C for 1.5–3 h. After confirming that the starting material 2 was converted to 1-hydroxyindole 8 by checking TLC, we slowly added 14.0 eq of DBU with vigorous stirring. The reaction mixture was stirred for 30 min at room temperature and then acetic anhydride or acyl chloride was added in an ice bath. We kept stirring the reaction mixture at room temperature for 1.5–4 h, leading to formation of targeted 1-acyloxyindoles 1.
As acylating agents of 1-hydroxyindole intermediate 8, acetic anhydride, pivaloyl chloride, benzoyl chloride, butanoyl chloride, hexanoyl chloride, and hydrocinnamoyl chloride were employed (Table 2). For acetylation reactions, we used acetic anhydride instead of acetyl chloride due to the high reactivity and instability of acetyl chloride. For example, both acetic anhydride and acetyl chloride provided 1dx in similar yields (Y = ~30%), so we chose acetic anhydride. The yields for acetylation were generally lower than those for pivaloylation and benzoylation. For example, among 1dx, 1dy, and 1dz (entries 13–15), the yield of 1-acetoxyindole 1dx was lower than those for 1dy and 1dz with bulkier alkyl and aromatic group, respectively. Moreover, the yield of 1dw with phenethyl group (entry 12) was higher than that of 1dx. We expected that low yields of 1-acetoxyindoles might be due to the instability of the compounds and that a bulkier alkyl or aromatic group on R2 could stabilize the 1-acyloxyindole compounds. Interestingly, when we analyzed the spectroscopic features of these compounds, we found some consistence. For example, we found that the δ values (13C NMR) of carbonyl carbons of N-OC(O)CH3 in 1-acetoxyindoles 1x were ~168.5, which means an upfield shift (~2) compared with those of carbonyl carbons in corresponding esters (R-OC(O)CH3). In addition, the λmax values in UV–Vis were in the range of 229–236 nm. We also performed some of the reactions for 1du, 1dx, and 1dz in a larger scale (1.1 mmol of 2) and confirmed robust reproducibility of the established optimized conditions. Consequently, we successfully synthesized 21 new 1-acyloxyindole compounds 1 in modest yields (Y = 24–35%).
Furthermore, some degree of decomposition of 1-acyloxyindoles (1du, 1dv, and 1dx) with linear alkyl groups (R2 = n − Pr, n − Pen, and Me) on a TLC plate was observed. Partial degradation was observed for 1-acetoxyindole 1dx within 30 min, and, for 1-butanoyloxyindole 1du and 1-hexanoyloxyindole 1dv, within 2 h. However, 1-pivaloyloxyindole 1dy and 1-benzoyloxyindole 1dz were not easily decomposed on TLC. Consequently, we found that these 1-acyloxyindole compounds seem to exhibit significantly different stabilities depending on the R2 in the acyl group (R2CO). Furthermore, these observations prompted us to test the stability of these compounds under hydrolysis conditions. We found that 1-butanoyloxyindole 1du and 1-acetoxyindole 1dx were easily hydrolyzed to provide 1-hydroxyindole under mildly basic conditions (data not shown). It is expected that this instability is due to the labile ester bond of NO-C(O)R2. This bond seems easily cleavable in even weakly acidic or basic conditions, resulting in 1-hydroxyindole and carboxylic acids (Scheme 4). We believed that this labile ester bond might provide us with an interesting possibility of its application in a prodrug strategy, which aims to explore drug delivery by lowering the polarity of the compounds by acylation of 1-hydroxyindole. Thus, further application studies on stability are in progress.
2.4. Mechanistic Investigations on Reaction Pathways
We investigated the reaction mechanisms and pathways based on the observed products, as shown in Scheme 5. We suggest that three pathways, A1, A2, and B, are involved in the reaction mechanism, which derives some support from our previous work [34]. The nitro group of conjugate ketoester derivative 2 was reduced to afford hydroxylamine compound 5 (or conformer 5′). The pathways A1 and A2 proceeded through conformer 5, and pathway B through conformer 5′. The intramolecular addition of N-H of two conformers, 5 and 5′, provided two different indoline derivatives, 6 and 10, respectively. Following dehydration of 6, it was possible to generate conjugate nitrone 7. Nucleophilic 1,5-addition of alcohol (R1OH) produced 1-hydroxyindole 8 (Path A1); subsequent acylation of the hydroxy group with R2COX provided 1-acyloxyindole 1. However, instead of alcohol, H2O as a nucleophile could be added to conjugate nitrone 7 to produce dihydroxy species 9 (Path A2). Dihydroxy compound 9 could be acylated with R2COX to provide diacylated compound 12. In the process of synthesizing 1dy, dipivaloylated compound 12 (R2 = t-Bu) was obtained and identified by mass analysis (446 [M + Na]+). On the other hand, conformer 5′ produced enolic compound 10 through intramolecular conjugate addition (aza-Michael addition) (Path B). Then, subsequent oxidative aromatization provided 1-hydroxyindole 11 [34]. Although we expected that acylation of 11 could produce 13, the acylated product 13 was difficult to be isolated and even identified. In most of the reactions in Table 2, we believed that substrate 2 proceeded through not only Path A1 but also Path A2 and Path B, which might explain the low yields of the products 1.
3. Experimental
3.1. General
Reagents were obtained from Sigma-Aldrich (Darmstad, Germany), Thermo Fisher (Waltham, MA, USA), and TCI (Tokyo, Japan). They were of commercial quality and used without further purification unless otherwise stated. Reactions were periodically monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Merck silica gel plates (20 × 20 cm; Merck F254) (Darmstad, Germany) and visualized by UV light. Purifications were performed by preparative TLC (PTLC) and column chromatography. PTLC separations were carried out on the same silica gel plates. Column chromatography was performed using Merck silica gels (230–400 mesh) (Zvornik, Bosnia and Herzegovina). Melting points (uncorrected) were determined in Deckgläser microscope cover glasses (Lauda-Königshofen, Germany) using a Thermo Scientific 00590Q apparatus (Dubuque, Iowa, USA). 1H (300 MHz) and 13C (75 MHz) NMR spectra were obtained by a Bruker DRX 300 spectrometer (Zürich, Switzerland), and chemical shifts (δ) are expressed with respect to tetramethylsilane (TMS). NMR spectra are presented in the Supplementary Materials. Mass spectra were obtained in EI or ESI ionization modes (Agilent, Santa Clara, CA, USA). High resolution mass spectra were obtained using JEOL apparatus (Tokyo, Japan) at the Korea Basic Science Institute, Republic of Korea. HPLC analyses were performed using the following Waters Associate Units: 515 A pump, 515 B pump, dual λ absorbance 2487 detector, and COSMOSIL 5C18-AR-Ⅱ Packed Column (4.6 × 250 mm) (Worcester, MA, USA). The products were analyzed using a linear gradient: from 70% A (aqueous) and 30% B (acetonitrile) for 3 min (isocratic) to 10% A and 90% B over 30 min at a flow rate of 1 mL/min with eluent monitoring at 254 nm. HPLC solvents were filtered (aqueous solution with PALL FP-450, 0.45 μm, 47 mm; acetonitrile with PALL TF-450, 0.45 μm, 47 mm) and degassed before use.
3.2. Substrate Synthesis
- Methyl 3-(2′-Chloro-6′-Nitrophenyl)-2-Oxopropanoate (4) [32]
- 2-Chloro-6-nitrotoluene (3, 1.17 g, 6.8 mmol, 1.0 eq) and dimethyl oxalate (4.02 g, 34.0 mmol, 5.0 eq) were dissolved in anhydrous DMF (8.2 mL). To a stirred mixture of NaH (60% in mineral oil, 1.09 g, 27.2 mmol, 4.0 eq) in anhydrous DMF (4.1 mL) at 0 °C was added dropwise a solution of dimethyl oxalate and 2-chloro-6-nitrotoluene. The reaction mixture was stirred at 0 °C for 1 h and at room temperature for 4.5 h. The reaction mixture was quenched with saturated NH4Cl (15 mL) at 0 °C, extracted with methylene chloride (2 × 50 mL), and washed with H2O (2 × 50 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to obtain compound 4 (1.68 g, 96%) as a pale-yellow solid. Spectral data are in accordance with literature information [32].
- Methyl 3-(2′-chloro-6′-nitrophenyl)-2-oxobut-3-enoate (2) [32]
- Ketoester (4, 1.53 g, 5.95 mmol, 1.0 eq) was dissolved in anhydrous THF (50 mL). To a stirred mixture of NaH (60% in mineral oil, 262 mg, 6.54 mmol, 1.1 eq) in anhydrous THF (100 mL) at 0 °C was added dropwise a solution of ketoester. After stirring for 1 h at 0 °C, N,N-dimethylmethyleneiminium chloride (1.85 g, 17.84 mmol, 3.0 eq) was added and the reaction mixture was stirred for 1 h at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for additional 5 h. The reaction mixture was quenched with saturated NH4Cl (10 mL) at 0 °C, extracted with EtOAc (2 × 250 mL), and washed with H2O (2 × 250 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to obtain compound 2 (1.36 g, 85%) as a pale-yellow solid. Spectral data are in accordance with literature information [32].
3.3. General Procedure for Synthesis of 1-Acyloxyindoles 1
SnCl2·2H2O and 4Å molecular sieves stirred in DME for 30 min at room temperature. To a stirred mixture was added alcohol and conjugate ketoester 2. The resulting mixture was stirred for 1.5–3 h at 40 °C. After confirming that the starting material was disappeared by using TLC, DME and DBU were added and stirred strongly for 30 min at room temperature. The acetic anhydride or acyl chloride was added in ice bath and kept stirring at room temperature for 1.5–4 h until reaction ends. The reaction mixture was diluted with CH2Cl2 and washed with diluted water, saturated aqueous ammonium chloride, sodium bicarbonate, and brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified by preparative TLC (PTLC) and column chromatography to provide 1-acyloxyindoles 1.
- Methyl 4-chloro-1-acetoxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1ax)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 4 h in general procedure afforded the title compound 1ax (9.6 mg, 28%) as a yellow solid. Mp 104–106 °C; Rf 0.35 (1:2 EtOAc/hexanes); HPLC tR 12.1 min; UV–Vis (CH3CN-H2O) λmax 213, 232, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.30–7.14 (m, 3H, Ar), 5.12 (s, 2H, C(3)CH2O), 3.95 (s, 3H, CO2CH3), 3.46 (s, 3H, CH2OCH3), 2.43 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 136.9, 128.9, 127.1, 125.0, 123.7, 120.1, 118.2, 108.0 (Ar), 63.5 (CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3), 18.2 (OC(O)CH3); MS m/z 311 [M]+; HRMS (+ESI) calcd for C14H14ClNO5 [M]+ 311.0561, found 311.0559.
- Methyl 4-chloro-1-pivaloyloxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1ay)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ay (14.1 mg, 35%) as a pale-yellow solid. Mp 88–89 °C; Rf 0.47 (1:2 EtOAc/hexanes); HPLC tR 37.9 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.28–7.18 (m, 2H, Ar), 7.06 (dd, J = 7.8, 0.9 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.92 (s, 3H, CO2CH3), 3.44 (s, 3H, CH2OCH3), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.2 (CO2CH3), 137.0, 128.9, 127.0, 125.7, 123.6, 120.3, 118.3, 107.8 (Ar), 63.5 (CH2OCH3), 58.0 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (C(CH3)3), 27.4 (C(CH3)3); MS m/z 353 [M]+; HRMS (+ESI) calcd for C17H20ClNO5 [M]+ 353.1030, found 353.1028.
- Methyl 4-chloro-1-benzoyloxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1az)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1az (13.5 mg, 33%) as a pale-yellow solid. Mp 112–114 °C; Rf 0.39 (1:2 EtOAc/hexanes); HPLC tR 37.1 min; UV–Vis (CH3CN-H2O) λmax 236, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.6 Hz, 2H, Ar), 7.72 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.7 Hz, 2H, Ar), 7.29–7.19 (m, 3H, Ar), 5.18 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.48 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 137.4, 134.9, 130.6, 129.2, 128.9, 127.2, 126.4, 125.5, 123.8, 120.4, 118.8, 108.3 (Ar), 63.5 (CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 373 [M]+; HRMS (+ESI) calcd for C19H16ClNO5 [M]+ 373.0717, found 373.0718.
- Methyl 4-chloro-1-acetoxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1bx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1bx (9.7 mg, 25%) as a pale-yellow solid. Mp 48–50 °C; Rf 0.26 (1:4 EtOAc/hexanes); HPLC tR 27.9 min; UV–Vis (CH3CN-H2O) λmax 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.32–7.13 (m, 3H, Ar), 5.11 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.59 (t, J = 6.4 Hz, 2H, OCH2CH2), 2.43 (s, 3H, OC(O)CH3), 1.60 (quintet, J = 7.0 Hz, 2H, OCH2CH2), 1.39 (sextet, J = 7.4 Hz, 2H, O(CH2)2CH2), 0.89 (t, J = 7.2 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.3, 118.6, 108.0 (Ar), 70.4 (OCH2CH2), 62.0 (C(3)CH2O), 52.4 (CO2CH3), 32.1(OCH2CH2), 19.6 (O(CH2)2CH2), 18.2 (OC(O)CH3) 14.1 (O(CH2)3CH3); MS m/z 353 [M]+; HRMS (+ESI) calcd for C17H20ClNO5 [M]+ 353.1030, found 353.1031.
- Methyl 4-chloro-1-pivaloyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1by)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1by (12.0 mg, 28%) as a pale-yellow solid. Mp 66–68 °C; Rf 0.53 (1:4 EtOAc/hexanes); HPLC tR 33.3 min; UV–Vis (CH3CN-H2O) λmax 211, 230, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.27–7.17 (m, 2H, Ar), 7.04 (d, J = 7.8 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.91 (s, 3H, CO2CH3), 3.58 (t, J = 6.5 Hz, 2H, OCH2CH2), 1.68–1.32 (m, 4H, OCH2(CH2)2), 1.47 (s, 9H, C(CH3)3), 0.89 (t, J = 7.3 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.4, 118.8, 107.8 (Ar), 70.3 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (C(CH3)3), 32.1 (OCH2CH2), 27.4 (C(CH3)3), 19.6 (O(CH2)2CH2), 14.1 (O(CH2)3CH3); MS m/z 395 [M]+; HRMS (+ESI) calcd for C20H26ClNO5 [M]+ 395.1500, found 395.1500.
- Methyl 4-chloro-1-benzoyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1bz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1bz (14.4 mg, 32%) as a pale-yellow solid. Mp 56–58 °C; Rf 0.38 (1:4 EtOAc/hexanes); HPLC tR 32.6 min; UV–Vis (CH3CN-H2O) λmax 229, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.3 Hz, 2H, Ar), 7.72 (t, J = 7.5 Hz, 1H, Ar), 7.57 (t, J = 7.8 Hz, 2H, Ar), 7.29–7.18 (m, 3H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.62 (t, J = 6.5 Hz, 2H, OCH2CH2), 1.63 (quintet, J = 6.5 Hz, 2H, OCH2CH2), 1.41 (sextet, J = 7.2 Hz, 2H, O(CH2)2CH2), 0.91 (t, J = 7.3 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 137.5, 134.9, 130.6, 129.2, 129.0, 127.1, 126.3, 125.5, 123.7, 120.5, 119.2, 108.2 (Ar), 70.4 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 32.1 (OCH2CH2), 19.6 (O(CH2)2CH2), 14.1 (O(CH2)3CH3); MS m/z 415 [M]+; HRMS (+ESI) calcd for C22H22ClNO5 [M]+ 415.1187, found 415.1185.
- Methyl 4-chloro-1-acetoxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1cx (11.3 mg, 27%) as a pale-yellow solid. Mp 51–53 °C; Rf 0.26 (1:4 EtOAc/hexanes); HPLC tR 31.4 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.19 (m, 2H, Ar), 7.14 (dd, J = 7.1, 0.9 Hz, 1H, Ar), 5.12 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.58 (t, J = 6.6 Hz, 2H, OCH2CH2), 2.42 (s, 3H, OC(O)CH3), 1.62 (quintet, J = 6.8 Hz, 2H, OCH2CH2), 1.40–1.25 (m, 6H, O(CH2)2(CH2)3), 0.86 (t, J = 6.5 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.3, 118.6, 108.0 (Ar), 70.7 (OCH2CH2), 62.0 (C(3)CH2O), 52.4 (CO2CH3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 26.1 (O(CH2)3CH2), 22.8 (O(CH2)4CH2), 18.2 (OC(O)CH3), 14.3 (O(CH2)5CH3); MS m/z 381 [M]+; HRMS (+ESI) calcd for C19H24ClNO5 [M]+ 381.1343, found 381.1339.
- Methyl 4-chloro-1-pivaloyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1cy (13.7 mg, 29%) as a pale-yellow solid. Mp 57–58 °C; Rf 0.55 (1:4 EtOAc/hexanes); HPLC tR 36.5 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.27–7.18 (m, 2H, Ar), 7.05 (dd, J = 7.9, 0.9 Hz, 1H, Ar), 5.14 (s, 2H, C(3)CH2O), 3.92 (s, 3H, CO2CH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2CH2), 1.68–1.25 (m, 8H, OCH2(CH2)4), 1.47 (s, 9H, C(CH3)3), 0.86 (t, J = 6.6 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 175.6 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.5, 118.8, 107.8 (Ar), 70.5 (OCH2CH2), 61.9 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (C(CH3)3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 27.4 (C(CH3)3), 26.1 (O(CH2)3CH2), 22.8 (O(CH2)4CH2), 14.2 (O(CH2)5CH3); MS m/z 423 [M]+; HRMS (+ESI) calcd for C22H30ClNO5 [M]+ 423.1813, found 423.1815.
- Methyl 4-chloro-1-benzoyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 1 h in general procedure afforded the title compound 1cz (14.2 mg, 30%) as a pale-yellow solid. Mp 62–64 °C; Rf 0.44 (1:4 EtOAc/hexanes); HPLC tR 35.4 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 7.4 Hz, 2H, Ar), 7.72 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.9 Hz, 2H, Ar), 7.28–7.17 (m, 3H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.82 (s, 3H, CO2CH3), 3.60 (t, J = 6.6 Hz, 2H, OCH2CH2), 1.63 (quintet, J = 7.0 Hz, 2H, OCH2CH2), 1.41–1.22 (m, 6H, O(CH2)2(CH2)3), 0.86 (t, J = 6.6 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 164.7 (NOC(O)), 160.4 (CO2CH3), 137.6, 134.9, 130.6, 129.2, 129.0, 127.1, 126.4, 126.0, 123.7, 120.5, 119.3, 108.3 (Ar), 70.7 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 26.1 (O(CH2)3CH2), 22.9 (O(CH2)4CH2), 14.3 (O(CH2)5CH3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1500.
- Methyl 4-chloro-1-butanoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1du)
- Use of SnCl2·2H2O (138 mg, 0.62 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and butanoyl chloride (38 μL, 0.37 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1du (20.4 mg, 26%) as a white solid. Mp 81–83 °C; Rf 0.56 (1:2 EtOAc/hexanes); HPLC tR 30.2 min; UV–Vis (CH3CN-H2O) λmax 214, 234, 297 nm; 1H NMR (300 MHz, CDCl3) δ 7.42–7.20 (m, 7H, Ar), 7.12 (d, J = 7.9 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.67 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.68 (t, J = 7.4 Hz, 2H, OC(O)CH2), 1.86 (sextet, J = 7.4 Hz, 2H, OC(O)CH2CH2), 1.10 (t, J = 7.4 Hz, 3H, OC(O)(CH2)2CH3); 13C NMR (75 MHz, CDCl3) δ 171.2 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.0, 128.9, 128.5, 128.2, 127.7, 127.0, 125.2, 123.6, 120.2, 117.9, 108.0 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3), 33.4 (OC(O)CH2), 18.3 (OC(O)CH2CH2), 13.9 (OC(O)(CH2)2CH3); MS m/z 415 [M]+; HRMS (+ESI) calcd for C22H22ClNO5 [M]+ 415.1187, found 415.1184.
- Methyl 4-chloro-1-hexanoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dv)
- Use of SnCl2·2H2O (138 mg, 0.61 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and hexanoyl chloride (52 μL, 0.37 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dv (23.9 mg, 30%) as a pale-yellow solid. Mp 64–66 °C; Rf 0.62 (1:2 EtOAc/hexanes); HPLC tR 34.0 min; UV–Vis (CH3CN-H2O) λmax 211, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.20 (m, 7H, Ar), 7.11 (dd, J = 7.9, 1.2 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.68 (t, J = 7.5 Hz, 2H, OC(O)CH2), 1.83 (quintet, J = 7.4 Hz, 2H, OC(O)CH2CH2), 1.49–1.34 (m, 4H, OC(O)(CH2)2(CH2)2), 0.94 (t, J = 6.9 Hz, 3H, OC(O)(CH2)4CH3); 13C NMR (75 MHz, CDCl3) δ 171.4 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.1, 129.0, 128.5, 128.2, 127.7, 127.0, 125.9, 123.6, 120.3, 118.0, 108.0 (Ar) 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.2 (CO2CH3), 31.5 (OC(O)CH2), 31.4 (OC(O)CH2CH2), 24.4 (OC(O)(CH2)2CH2), 22.4 (OC(O)(CH2)3CH2), 14.1 (OC(O)(CH2)4CH3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1496.
- Methyl 4-chloro-1-hydrocinnamoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dw)
- Use of SnCl2·2H2O (138 mg, 0.61 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 3 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and hydrocinnamoyl chloride (55 μL, 0.37 mmol, 2.0 eq) for 3 h in general procedure afforded the title compound 1dw (28.6 mg, 32%) as a pale-yellow solid. Mp 103–104 °C; Rf 0.48 (1:2 EtOAc/hexanes); HPLC tR 32.7 min; UV–Vis (CH3CN-H2O) λmax 211, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.14 (m, 13H, Ar), 5.19 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.77 (s, 3H, CO2CH3), 3.14 (t, J = 7.1 Hz, 2H, (OC(O)CH2), 3.03 (t, J = 7.1 Hz, 2H, (OC(O)CH2CH2); 13C NMR (75 MHz, CDCl3) δ 170.6 (NOC(O)), 160.3 (CO2CH3), 139.7, 138.7, 137.0, 129.0, 128.8, 128.7, 128.5, 128.2, 127.7, 127.0, 126.9, 125.2, 123.6, 120.2, 118.1, 108.1 (Ar), 72.6 (OCH2Ph), 61.6 (C(3)CH2O), 52.2 (CO2CH3), 33.3 (OC(O)CH2), 30.7 (OC(O)CH2CH2); MS m/z 477 [M]+; HRMS (+ESI) calcd for C27H24ClNO5 [M]+ 477.1343, found 477.1345.
- Methyl 4-chloro-1-acetoxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dx (13.0 mg, 30%) as a pale-yellow solid. Mp 68–69 °C; Rf 0.31 (1:2 EtOAc/hexanes); HPLC tR 26.6 min; UV–Vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3) δ 7.42–7.14 (m, 8H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.67 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.42 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 138.6, 137.0, 129.0, 128.5, 128.2, 127.7, 127.1, 125.0, 123.7, 120.2, 118.0, 108.0 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3), 18.2 (OC(O)CH3); MS m/z 387 [M]+; HRMS (+ESI) calcd for C20H18ClNO5 [M]+ 387.0874, found 387.0875.
- Methyl 4-chloro-1-pivaloyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dy (15.7 mg, 33%) as a pale-yellow solid. Mp 80–82 °C; Rf 0.65 (1:2 EtOAc/hexanes); HPLC tR 31.6 min; UV–Vis (CH3CN-H2O) λmax 213, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.19 (m, 7H, Ar), 7.06 (d, J = 7.7 Hz, 1H, Ar), 5.22 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.2 (CO2CH3), 138.8, 137.1, 129.0, 128.4, 128.2, 127.7, 126.9, 125.8, 123.6, 120.4, 118.2, 107.8 (Ar), 72.5 (OCH2Ph), 61.7 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (OC(CH3)3), 27.4 (OC(CH3)3); MS m/z 429 [M]+; HRMS (+ESI) calcd for C23H24ClNO5 [M]+ 429.1343, found 429.1342.
- Methyl 4-chloro-1-benzoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dz (16.5 mg, 33%) as a pale-yellow solid. Mp 112–114 °C; Rf 0.50 (1:2 EtOAc/hexanes); HPLC tR 31.0 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 7.2 Hz, 2H, Ar), 7.70 (t, J = 7.5 Hz, 1H, Ar), 7.55 (t, J = 7.9 Hz, 2H, Ar), 7.42–7.17 (m, 8H, Ar), 5.25 (s, 2H, C(3)CH2O), 4.68 (s, 2H, OCH2Ph), 3.72 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.5, 134.9, 130.6, 129.2, 129.0, 128.2, 128.1, 127.7, 127.1, 126.7, 125.6, 123.7, 120.5, 118.6, 108.3 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3); MS m/z 449 [M]+; HRMS (+ESI) calcd for C25H20ClNO5 [M]+ 449.1030, found 449.1028.
- Methyl 4-chloro-1-acetoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ex)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ex (11.5 mg, 26%) as a pale-yellow solid. Mp 70–71 °C; Rf 0.21 (1:4 EtOAc/hexanes); HPLC tR 27.6 min; UV–Vis (CH3CN-H2O) λmax 211, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.13 (m, 8H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.89 (s, 3H, CO2CH3), 3.80 (t, J = 7.2 Hz, 2H, OCH2CH2), 2.94 (t, J = 7.4 Hz, 2H, OCH2CH2), 2.42 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 139.2, 137.0, 129.1, 129.0, 128.4, 127.0, 126.2, 125.0, 123.7, 120.2, 118.3, 108.0 (Ar), 71.4 (OCH2CH2), 62.1 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2), 18.2 (OC(O)CH3); MS m/z 401 [M]+; HRMS (+ESI) calcd for C21H20ClNO5 [M]+ 401.1030, found 401.1029.
- Methyl 4-chloro-1-pivaloyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ey)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1ey (13.1 mg, 27%) as a pale-yellow solid. Mp 83–84 °C; Rf 0.59 (1:4 EtOAc/hexanes); HPLC tR 33.1 min; UV–Vis (CH3CN-H2O) λmax 212, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.32–7.18 (m, 7H, Ar), 7.05 (d, J = 8.0 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 3.88 (s, 3H, CO2CH3), 3.80 (t, J = 7.3 Hz, 2H, OCH2CH2), 2.94 (t, J = 7.4 Hz, 2H, OCH2CH2), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 139.3, 137.2, 129.2, 129.0, 128.5, 127.0, 126.4, 125.9, 123.6, 120.4, 118.6, 108.1 (Ar), 71.3 (OCH2CH2), 62.1 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (OC(CH3)3), 36.6 (OCH2CH2), 27.4 (OC(CH3)3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1503.
- Methyl 4-chloro-1-benzoyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ez)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ez (16.3 mg, 32%) as a pale-yellow solid. Mp 112–113 °C; Rf 0.44 (1:4 EtOAc/hexanes); HPLC tR 32.0 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.2 Hz, 2H, Ar), 7.73 (t, J = 7.4 Hz, 1H, Ar), 7.58 (t, J = 7.7 Hz, 2H, Ar), 7.31–7.18 (m, 8H, Ar), 5.24 (s, 2H, C(3)CH2O), 3.84 (t, J = 7.4Hz, 2H, OCH2CH2), 3.79 (s, 3H, CO2CH3), 2.97 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 139.3, 137.5, 134.9, 130.6, 129.2, 129.1, 129.0, 128.4, 127.1, 126.4, 126.2, 125.5, 123.7, 120.4, 119.0, 108.3 (Ar), 71.4 (OCH2CH2), 62.0 (C(3)CH2O), 52.3 (CO2CH3), 36.5 (OCH2CH2); MS m/z 463 [M]+; HRMS (+ESI) calcd for C26H22ClNO5 [M]+ 463.1187, found 463.1188.
- Methyl 4-chloro-1-acetoxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1fx (10.2 mg, 24%) as a pale-yellow solid. Mp 80–82 °C; Rf 0.42 (1:2 EtOAc/hexanes); HPLC tR 24.9 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.13 (m, 3H, Ar), 5.14 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.51–3.44 (m, 1H, OCH), 2.43 (s, 3H, OC(O)CH3), 2.06–1.25 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 168.6 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.2, 119.0, 108.0 (Ar), 77.9 (OCH), 59.4 (C(3)CH2O), 52.3 (CO2CH3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2), 18.2 (N(1)OC(O)CH3); MS m/z 379 [M]+; HRMS (+ESI) calcd for C19H22ClNO5 [M]+ 379.1187, found 379.1183.
- Methyl 4-chloro-1-pivaloyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1fy (13.9 mg, 30%) as a pale-yellow solid. Mp 98–100 °C; Rf 0.63 (1:2 EtOAc/hexanes); HPLC tR 34.7 min; UV–Vis (CH3CN-H2O) λmax 212, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.24–7.15 (m, 2H, Ar), 7.01 (d, J = 7.9 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.89 (s, 3H, CO2CH3), 3.48–3.41 (m, 1H, OCH), 1.44 (s, 9H, (CH3)3), 2.10–1.23 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.5, 119.3, 107.8 (Ar), 77.7 (OCH), 59.4 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (OC(CH3)3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2), 27.4 (OC(CH3)3); MS m/z 421 [M]+; HRMS (+ESI) calcd for C22H28ClNO5 [M]+ 421.1656, found 421.1655.
- Methyl 4-chloro-1-benzoyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1fz (13.1 mg, 27%) as a pale-yellow solid. Mp 96–97 °C; Rf 0.52 (1:2 EtOAc/hexanes); HPLC tR 33.8 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.2 Hz, 2H, Ar), 7.73 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.9 Hz, 2H, Ar), 7.29–7.17 (m, 8H, Ar), 5.20 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.58–3.47 (m, 1H, OCH), 2.08–1.22 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 164.7 (NOC(O)), 160.4 (CO2CH3), 137.6, 134.9, 130.6, 129.2, 129.0, 127.1, 126.4, 125.5, 123.7, 120.5, 119.7, 108.3 (Ar), 77.9 (OCH), 59.4 (C(3)CH2O), 52.3 (CO2CH3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2); MS m/z 441 [M]+; HRMS (+ESI) calcd for C24H24ClNO5 [M]+ 441.1343, found 441.1340.
4. Conclusions
We reported the studies on one-pot synthesis of new 1-acyloxyindoles 1 through four-step reactions. With substrate 2 obtained by a two-step synthetic sequence, we performed the reactions using SnCl2·2H2O as a reducing agent and alcohol (R1OH) as a nucleophile through reduction, intramolecular addition, and nucleophilic 1,5-addition, affording intermediate 1-hydroxyindole 8. Subsequent acylation of 8 using acetic anhydride or acyl chlorides (R2COX) in a basic condition provided target compound 1-acyloxyindoles 1. Optimization of the reaction conditions was established as follows: 1) conjugate ketoester 2 (1.0 eq), SnCl2·2H2O (3.3 eq), and ROH (2.0 eq) in DME at 40 °C; and 2) DBU (14.0 eq) and acetic anhydride or acyl chloride (2.0 eq) at room temperature. Consequently, using the optimized conditions, 21 examples of new 1-acyloxyindole derivatives were successfully synthesized in modest yields (Y = 24–35%) through one-pot reaction of a four-step sequence.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27196769/s1, The charts for 1H- and 13C-NMR spectroscopies are available online.
Author Contributions
Conceptualization, S.H.L. and H.C.; methodology, Y.E.K., Y.J.L., C.K., Y.R.J. and H.C.; writing—original draft preparation, Y.E.K.; writing—review and editing, S.H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Research Foundation of Korea (NRF) Grants (2018R1D1A1B07048631 and 2019M3E5D5066543), by Priority Research Centers Program through NRF (2016R1A6A1A03007648) funded by the Ministry of Education, Science and Technology (MEST), and by Korea Environment Industry & Technology Institute (KEITI) through Technology Development Project for Safety Management of Household Chemical Products, funded by Korea Ministry of Environment (MOE) (2022002980001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in insert article or Supplementary Materials here. Samples of compounds 1ax–1fz are available from the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Acheson, R.M.; Bolton, R.G.; Hunter, I. 1-Hydroxyindoles, and Products from the Reaction of 2-Nitrophenylsuccinic Anhydride with Fluorosulphonic Acid. J. Chem. Soc. C 1970, 1067–1070. [Google Scholar] [CrossRef]
- Acheson, R.M.; Littlewood, D.M.; Rosenberg, H.E. Synthesis of 1-Methoxyindoles. J. Chem. Soc. Chem. Commun. 1974, 671a. [Google Scholar] [CrossRef]
- Bartsch, A.; Bross, M.; Spiteller, P.; Spiteller, M.; Steglich, W. Birnbaumin A and B: Two Unusual 1-Hydroxyindole Pigments from the “Flower Pot Parasol” Leucocoprinus Birnbaumii. Angew. Chem. Int. Ed. 2005, 44, 2957–2959. [Google Scholar] [CrossRef]
- Wang, J.; Pearce, A.N.; Chan, S.T.S.; Taylor, R.B.; Page, M.J.; Valentin, A.; Bourguet-Kondracki, M.; Dalton, J.P.; Wiles, S.; Copp, B.R. Biologically Active Acetylenic Amino Alcohol and N-Hydroxylated 1,2,3,4-Tetrahydro-β-carboline Constituents of the New Zealand Ascidian Pseudodistoma opacum. J. Nat. Prod. 2016, 79, 607–610. [Google Scholar] [CrossRef]
- Escolano CStephacidin, B. The Avrainvillamide Dimer: A Formidable Synthetic Challenge. Angew. Chem. Int. Ed. 2005, 44, 7670–7673. [Google Scholar] [CrossRef] [PubMed]
- Gmelin, R.; Virtanen, A.I. Neoglucobrassicin, ein zweiter SCN--Precursor vom Indoltyp in Brassica-Arten. Acta. Chem. Scand. 1962, 16, 1378–1384. [Google Scholar] [CrossRef]
- Johns, S.R.; Lamberton, J.A.; Occolowitz, J.L. 1,5-Dimethoxy-3-(dimethylaminomethyl)indole, the major alkaloid from Gymnacranthera paniculata (A. DC.) Warb. var. zippeliana (Miq.) J. Sinclair (family Myristicaceae). Aust. J. Chem. 1967, 20, 1737–1742. [Google Scholar] [CrossRef]
- Acheson, R.M.; Nwankwo, J.O. The metabolism of some 1-hydroxylated Indoles in the rat. Xenobiotica 1984, 14, 877–883. [Google Scholar] [CrossRef]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
- Somei, M. Recent Advances in the Chemistry of 1-Hydroxyindoles, 1-Hydroxytryptophans, and 1-Hydroxytryptamines. Adv. Heterocycl. Chem. 2002, 82, 101–155. [Google Scholar]
- Jump, S.M.; Kung, J.; Staub, R.; Kinseth, M.A.; Cram, E.J.; Yudina, L.N.; Preobrazhenskaya, M.N.; Bjeldanes, L.F.; Firestone, G.L. N-Alkoxy derivatization of indole-3-carbinol Increases the efficacy of the G1 cell cycle arrest and of I3C-specific regulation of cell cycle gene transcription and activity in human breast cancer cells. Biochem. Pharmacol. 2008, 75, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-Hydroxyindole-Based Inhibitors of Human Lactate Dehydrogenase Isoform A (LDH-A) as Starvation Agents against Cancer Cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Calvaresi, E.C.; Tuccinardi, T.; Paterni, I.; Macchia, M.; Martinelli, A.; Hergenrother, P.J.; Minutolo, F. Assessing the differential action on cancer cells of LDH-A inhibitors based on the N-hydroxyindole-2-carboxylate (NHI) and malonic (Mal) scaffolds. Org. Biomol. Chem. 2013, 11, 6588–6596. [Google Scholar] [CrossRef] [Green Version]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide Antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Somei, M.; Yamada, K.; Hasegawa, M.; Tabata, M.; Nagahama, Y.; Morikawa, H.; Yamada, F. Preparations of 1-Hydroxyindole Derivatives and Their Potent Inhibitory Activities on Platelet Aggregation. Heterocycles 1996, 43, 1855–1858. [Google Scholar] [CrossRef]
- Fiume, L. Inhibition of Aerobic Glycolysis in Yoshida Ascites Hepatoma by Tartronic Acid. Nature 1960, 187, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Munshi, K.L.; Kohl, H.; de Souza, N.J. Synthesis of 2-Amino-3-carbethoxy-1-hydroxyindoles. J. Heterocycl. Chem. 1977, 14, 1145–1146. [Google Scholar] [CrossRef]
- Belley, M.; Beaudoin, D.; Duspara, P.; Sauer, E.; St-Pierre, G.; Trimble, L.A. Synthesis and Reactivity of N-Hydroxy-2-Amino-3-Arylindoles. Synlett 2007, 19, 2991–2994. [Google Scholar] [CrossRef]
- Somei, M.; Kawasaki, T. A New and Simple Synthesis of 1-Hydroxyindole Derivatives. Heterocycles 1989, 29, 1251–1254. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kodama, A.; Nishida, T.; Shimizu, K.; Somei, M. Preparation of 1-Hydroxyindole Derivatives and a New Route to 2-Substituted Indoles. Heterocycles 1991, 32, 221–227. [Google Scholar]
- Chirkova, Z.V.; Kabanova, M.V.; Sergeev, S.S.; Filimonov, S.I.; Abramov, I.G.; Samet, A.V.; Suponitsky, K.Y. Synthesis of 3-acyl-1-hydroxy-1H-indole-5,6-dicarbonitriles. Mendeleev Commun. 2015, 25, 315–317. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Lee, S.H.; Estrada, A.A.; Zak, M. Construction of Substituted N-Hydroxyindoles: Synthesis of a Nocathiacin I Model System. Angew. Chem. 2005, 117, 3802–3806. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Estrada, A.A.; Freestone, G.C.; Lee, S.H.; Alvarez-Mico, X. New synthetic technology for the construction of N-hydroxyindoles and synthesis of nocathiacin I Model Systems. Tetrahedron 2007, 63, 6088–6114. [Google Scholar] [CrossRef]
- Wong, A.; Kuethe, J.T.; Davies, I.W. A General Synthesis of N-Hydroxyindoles. J. Org. Chem. 2003, 68, 9865–9866. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Cho, H.; Lim, Y.J.; Kim, C.; Lee, S.H. One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles. Molecules 2021, 26, 1466. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Cheng, R.; Sun, J.; Zhang-Negrerie, D.; Du, Y. Iodobenzene Dichloride/Zinc Chloride-Mediated Synthesis of N-Alkoxyindole-3-carbonitriles from 3-Alkoxyimino-2-arylalkylnitriles via Intramolecular Heterocyclization. Adv. Synth. Catal. 2018, 360, 250–254. [Google Scholar] [CrossRef]
- Acheson, R.M.; Hunt, P.G.; Littlewood, D.M.; Murrer, B.A.; Rosenberg, H.E. The Synthesis, Reactions, and Spectra of 1-Acetoxy-, 1-Hydroxy-, and 1-Methoxy-Indoles. J. Chem. Soc. Perkin Trans. 1 1978, 1, 1117–1125. [Google Scholar] [CrossRef]
- Somei, M.; Shoda, T. A Facile Route to 1-Acetoxy- and 1-Methoxyindols. Heterocycles 1981, 16, 1523–1525. [Google Scholar] [CrossRef]
- Nagayoshi, T.; Saeki, S.; Hamana, M. Studies on Tertiary Amine Oxides. LXXIX. Reactions of 2-Ethoxycarbonyl-1-hydroxyindole in the Presence of Acylating Agents. Chem. Pharm. Bull. 1984, 32, 3678–3682. [Google Scholar] [CrossRef] [Green Version]
- Chirkova, Z.V.; Kabanova, M.V.; Filimonov, S.I.; Abramov, I.G.; Samet, A.V.; Stashina, G.A.; Suponitsky, K.Y. The C-3 acylation of 1-hydroxyindoles. Tetrahedron Lett. 2017, 58, 755–757. [Google Scholar] [CrossRef]
- Belley, M.; Sauer, E.; Beaudoin, D.; Duspara, P.; Trimble, L.A.; Dubé, P. Synthesis and reactivity of N-hydroxy-2-aminoindoles. Tetrahedron Lett. 2006, 47, 159–162. [Google Scholar] [CrossRef]
- Park, Y.K.; Kim, H.; Kim, D.S.; Cho, H.; Moon, A.; Jeong, C.; Yoon, H.; Lee, S.H. Synthesis of New 2,3-Disubstituted 4-Chloro-1-Hydroxyindoles. Bull. Korean Chem. Soc. 2015, 36, 2095–2100. [Google Scholar] [CrossRef]
- Park, Y.K.; Kim, H.; Lee, S.H. Synthesis of New Highly Substituted and Hindered 1-Hydroxyindole-2-Carboxylates. Bull. Korean Chem. Soc. 2016, 37, 82–90. [Google Scholar] [CrossRef]
- Park, Y.K.; Lee, S.H. Synthesis of New 1-Hydroxyindole-2-Carboxylates and Mechanistic Studies on Reaction Pathways. J. Heterocycl. Chem. 2017, 54, 1995–2002. [Google Scholar] [CrossRef]
- Bellamy, F.D.; Ou, K. Selective reduction of aromatic nitro compounds with stannous chloride in non acidic and non aqueous medium. Tetrahedron Lett. 1984, 25, 839–842. [Google Scholar] [CrossRef]
Figure 1.
Indole, 1-hydroxyindole, 1-alkoxyindole, and 1-acyloxyindole.

Figure 2.
Structure of multisubstituted 1-acyloxyindoles 1.

Scheme 1.
Synthesis of conjugate ketoester 2.

Scheme 2.
Proposed scheme for synthesis of multisubstituted 1-acyloxyindoles 1.

Scheme 3.
Synthesis of various 1-acyloxyindoles 1.

Scheme 4.
Decomposition of 1-acyloxyindole under weakly acidic and basic conditions.

Scheme 5.
Proposed pathways for 1, 12, and 13.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of the reaction conditions for 1dy a.
 | |||||
|---|---|---|---|---|---|
| Entry | 8d Formation | Acylation | Yield (%) | ||
| SnCl2∙2H2O (eq) | BnOH (eq) | DBU (eq) | Pivaloyl Chloride (eq) | ||
| 1 | 2.5 | 2.0 | 10.6 | 2.0 | 21 |
| 2 | 2.9 | 2.0 | 12.3 | 2.0 | 27 |
| 3 | 3.3 | 1.5 | 14.0 | 1.5 | 25 |
| 4 | 3.3 | 1.5 | 14.0 | 3.0 | 26 |
| 5 | 3.3 | 2.0 | 14.0 | 2.0 | 35 |
| 6 | 3.3 | 3.0 | 14.0 | 3.0 | 35 |
| 7 | 3.7 | 2.0 | 15.7 | 2.0 | 22 |
a All reactions were run in 0.056 mmol scale of conjugate ketoester 2 (1.0 eq, [c] = 0.3 M) for formation of 8d in DME at 40 °C; [c] = 0.1 M at 25 °C for formation of 1dy.
Table 2.
Synthesis of derivatives of 1-acyloxyindole 1 a.
 | ||||
|---|---|---|---|---|
| Entry | ROH | RCOX | Product | Yield (%) |
| 1 | MeOH | acetic anhydride | 1ax | 28 |
| 2 | MeOH | pivaloyl chloride | 1ay | 35 |
| 3 | MeOH | benzoyl chloride | 1az | 33 |
| 4 | n-BuOH | acetic anhydride | 1bx | 25 |
| 5 | n-BuOH | pivaloyl chloride | 1by | 28 |
| 6 | n-BuOH | benzoyl chloride | 1bz | 32 |
| 7 | n-HexOH | acetic anhydride | 1cx | 27 |
| 8 | n-HexOH | pivaloyl chloride | 1cy | 29 |
| 9 | n-HexOH | benzoyl chloride | 1cz | 30 |
| 10 | BnOH | butanoyl chloride | 1du | 26 |
| 11 | BnOH | hexanoyl chloride | 1dv | 30 |
| 12 | BnOH | hydrocinnamoyl chloride | 1dw | 32 |
| 13 | BnOH | acetic anhydride | 1dx | 30 |
| 14 | BnOH | pivaloyl chloride | 1dy | 33 |
| 15 | BnOH | benzoyl chloride | 1dz | 33 |
| 16 | PhCH2CH2OH | acetic anhydride | 1ex | 26 |
| 17 | PhCH2CH2OH | pivaloyl chloride | 1ey | 27 |
| 18 | PhCH2CH2OH | benzoyl chloride | 1ez | 32 |
| 19 | c-HxOH | acetic anhydride | 1fx | 24 |
| 20 | c-HxOH | pivaloyl chloride | 1fy | 30 |
| 21 | c-HxOH | benzoyl chloride | 1fz | 27 |
a Reactions were run in 0.056–0.18 mmol scale of conjugate ketoester 2 (1.0 eq, [c] = 0.3 M) for formation of 8 in DME at 40 °C; [c] = 0.1 M at 25 °C for formation of 1.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, Y.E.; Lim, Y.J.; Kim, C.; Jeong, Y.R.; Cho, H.; Lee, S.H. Syntheses of New Multisubstituted 1-Acyloxyindole Compounds. Molecules 2022, 27, 6769. https://doi.org/10.3390/molecules27196769
AMA Style
Kim YE, Lim YJ, Kim C, Jeong YR, Cho H, Lee SH. Syntheses of New Multisubstituted 1-Acyloxyindole Compounds. Molecules. 2022; 27(19):6769. https://doi.org/10.3390/molecules27196769
Chicago/Turabian StyleKim, Ye Eun, Yoo Jin Lim, Chorong Kim, Yu Ra Jeong, Hyunsung Cho, and Sang Hyup Lee. 2022. "Syntheses of New Multisubstituted 1-Acyloxyindole Compounds" Molecules 27, no. 19: 6769. https://doi.org/10.3390/molecules27196769