- Case Report

A Heterozygous ABCC6 Variant as a Potential Contributor to Choroidal Neovascularization in a β-Thalassemia Patient
- Debashis Pal,
- Dipankar Saha and
- Anupam Basu
- + 2 authors
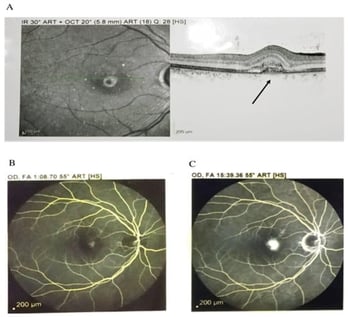
β-thalassemia patients often experience ocular abnormalities such as angioid streaks (ASs), retinal pigmented epithelium degradation, visual field defects, and in rare instances choroidal neovascularization (CNV). Although ASs are common in individuals with hemoglobinopathies, the occurrence of choroidal neovascularization without preceding ASs is exceptionally rare. In this report, we describe a β-thalassemia patient who had developed CNV at the age of 27 years and also had experience of renal stones at the age of 19 years. He had undergone splenectomy and was under conservative therapy of iron supplementation. We conducted whole-exome sequencing (WES) in search of CNV-associated variants. Through variant filtering and Phenolyzer analysis, we have identified a rare heterozygous missense variant in the ABCC6 gene, ABCC6:NM_001171:exon25:c.3524T>C (rs376062004). In silico analysis revealed that this variant is present in the highly conserved region and is likely to decrease the stability of the protein. Mutation in the ABCC6 gene leads to pseudoxanthoma elasticum (PXE). Previously, it was believed that ASs and subsequent CNV-like ocular complication may develop due to the pathophysiological condition of thalassemia. However, our study provides compelling evidence that rare mutations in the ABCC6 gene, in combination with oxygen insufficiency, may contribute to the development of CNV in β-thalassemia patients. This finding highlights the potential genetic basis of PXE-mediated CNV development in β-thalassemia.
29 January 2026