
First Pangenome of Corynebacterium rouxii, a Potentially Toxigenic Species of Corynebacterium diphtheriae Complex
 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Data Retrieval from Public Database
2.2. Taxonomy, Typing, and Phylogeny
2.3. Prediction of Mobile Genetic Elements, CRISPR-Cas Systems, and Genomic Islands
2.4. Gene Synteny
2.5. Virulence Factors and Antimicrobial Resistance Genes
2.6. Pangenome Development and Functional Annotation
3. Results
3.1. Taxonomy, Typing, and Phylogeny
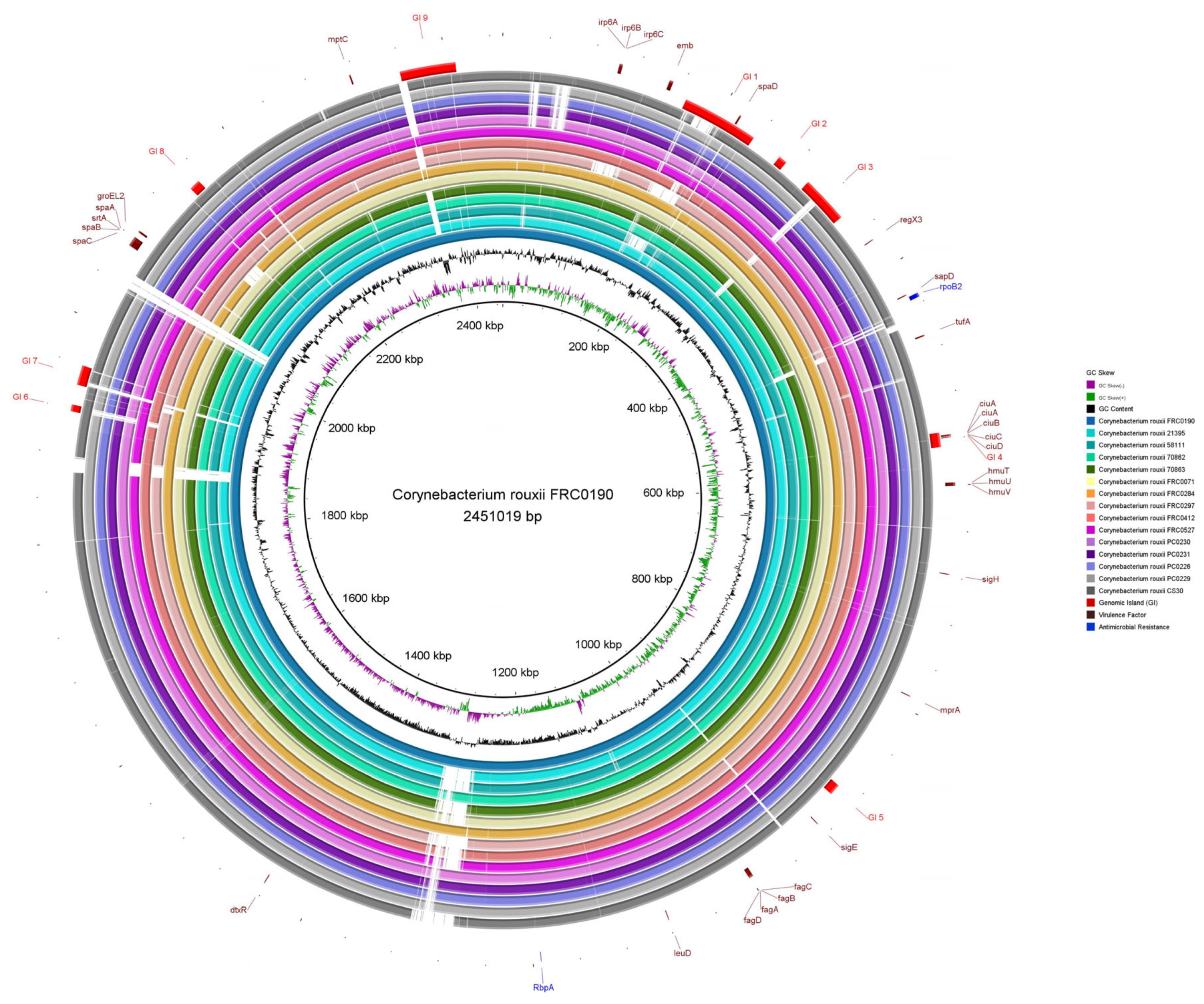
3.2. Prediction of Mobile Genetic Elements, CRISPR-Cas Systems, and Genomic Islands
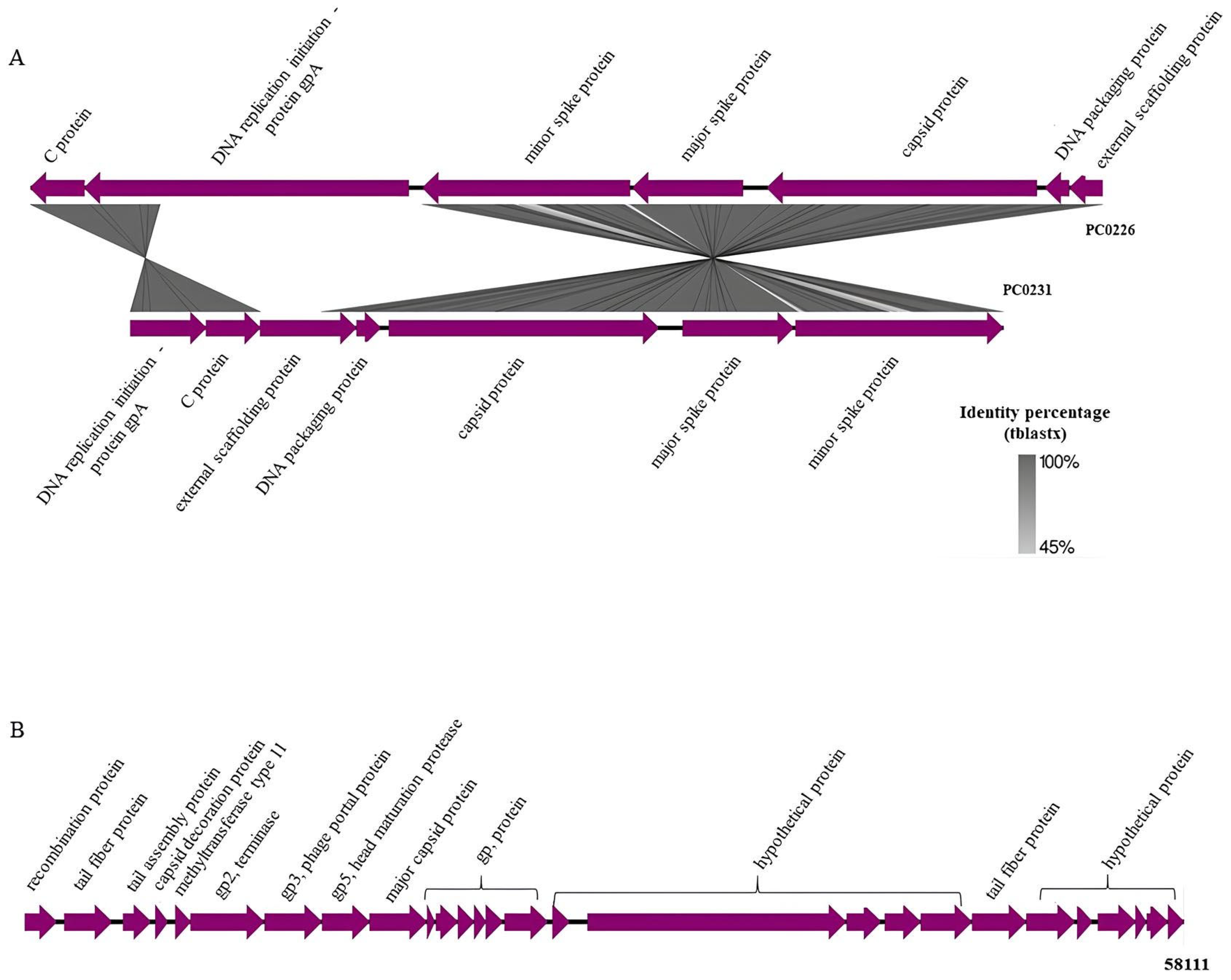
3.3. Gene Synteny
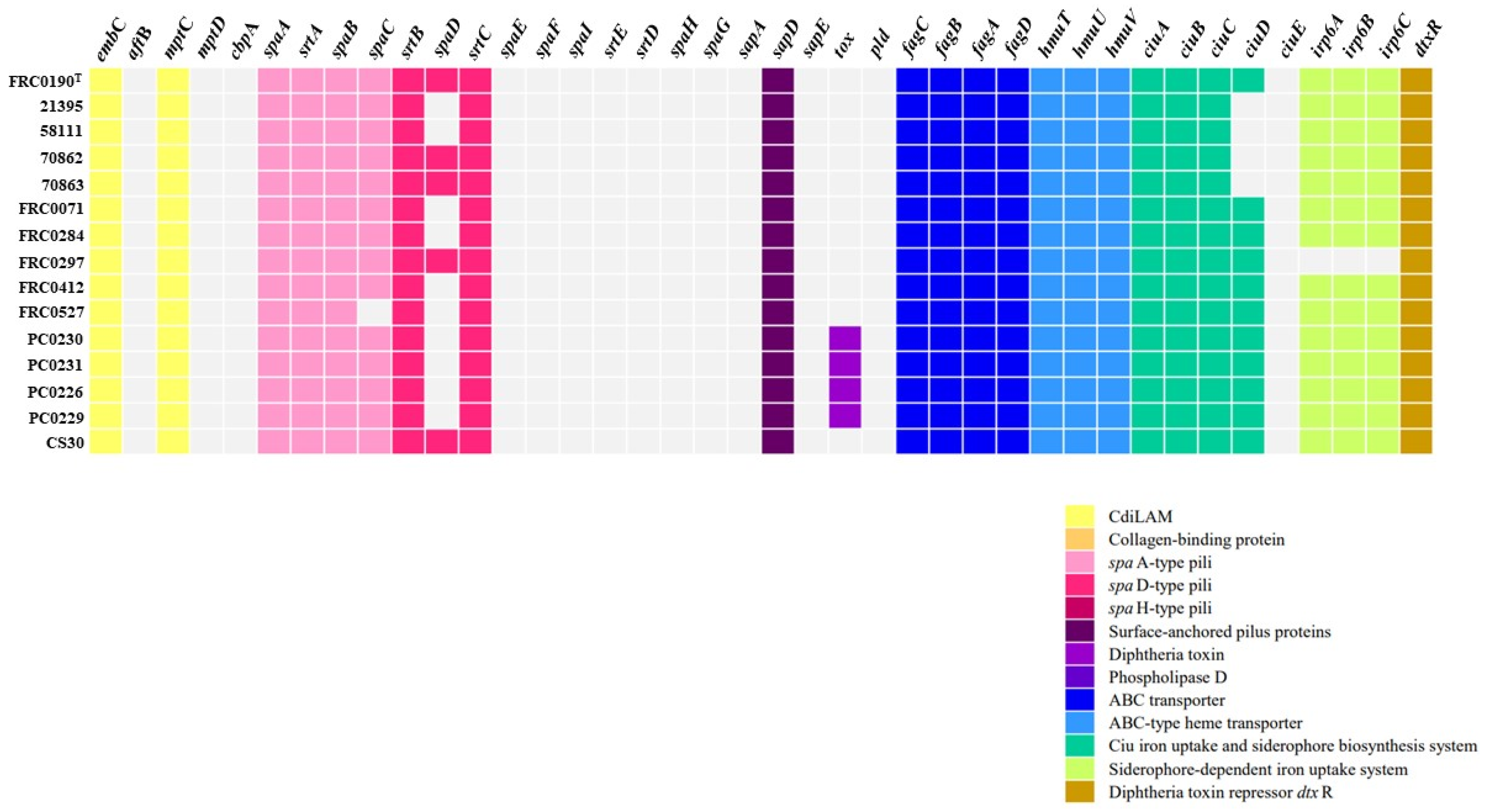
3.4. Virulence Factors and Antimicrobial Resistance Genes
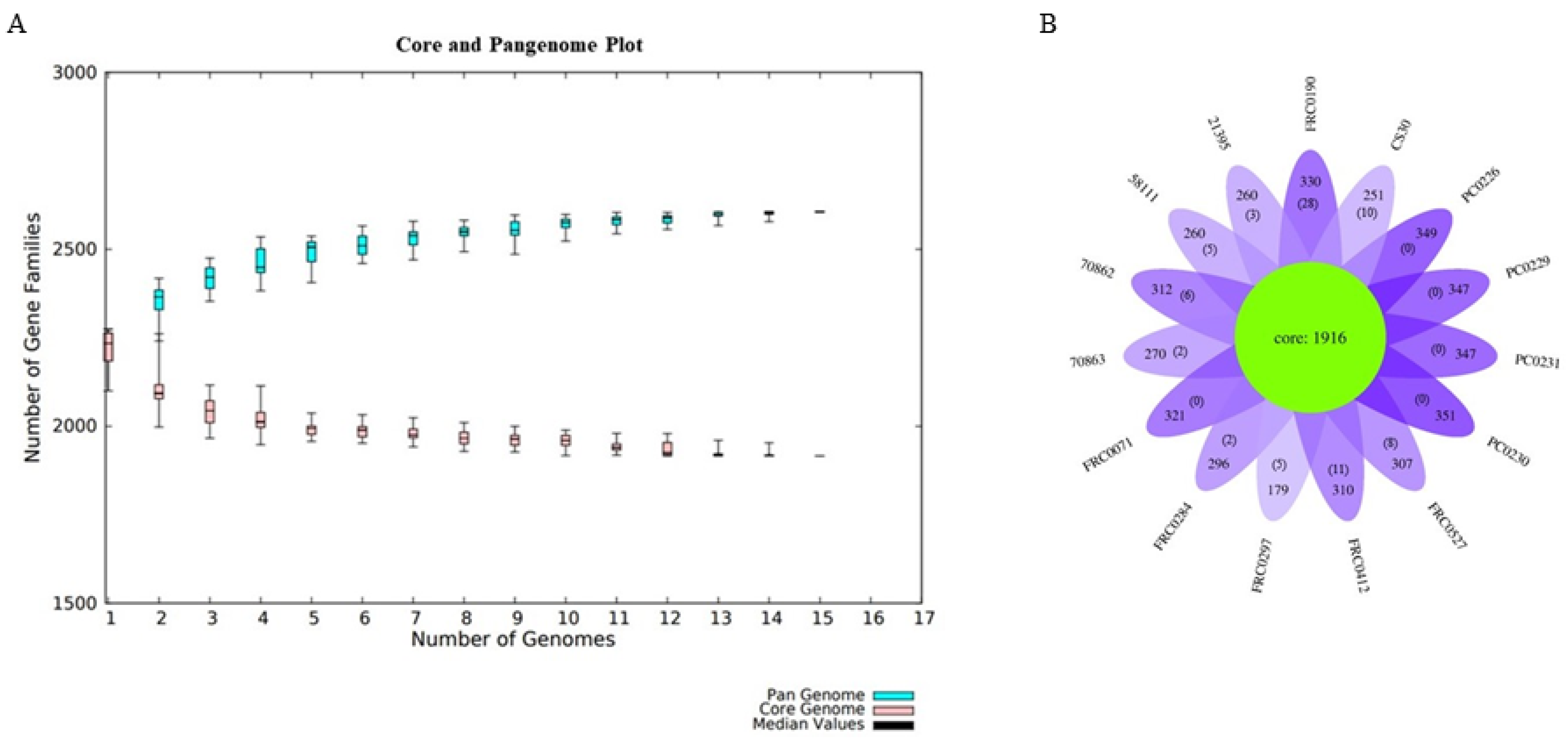
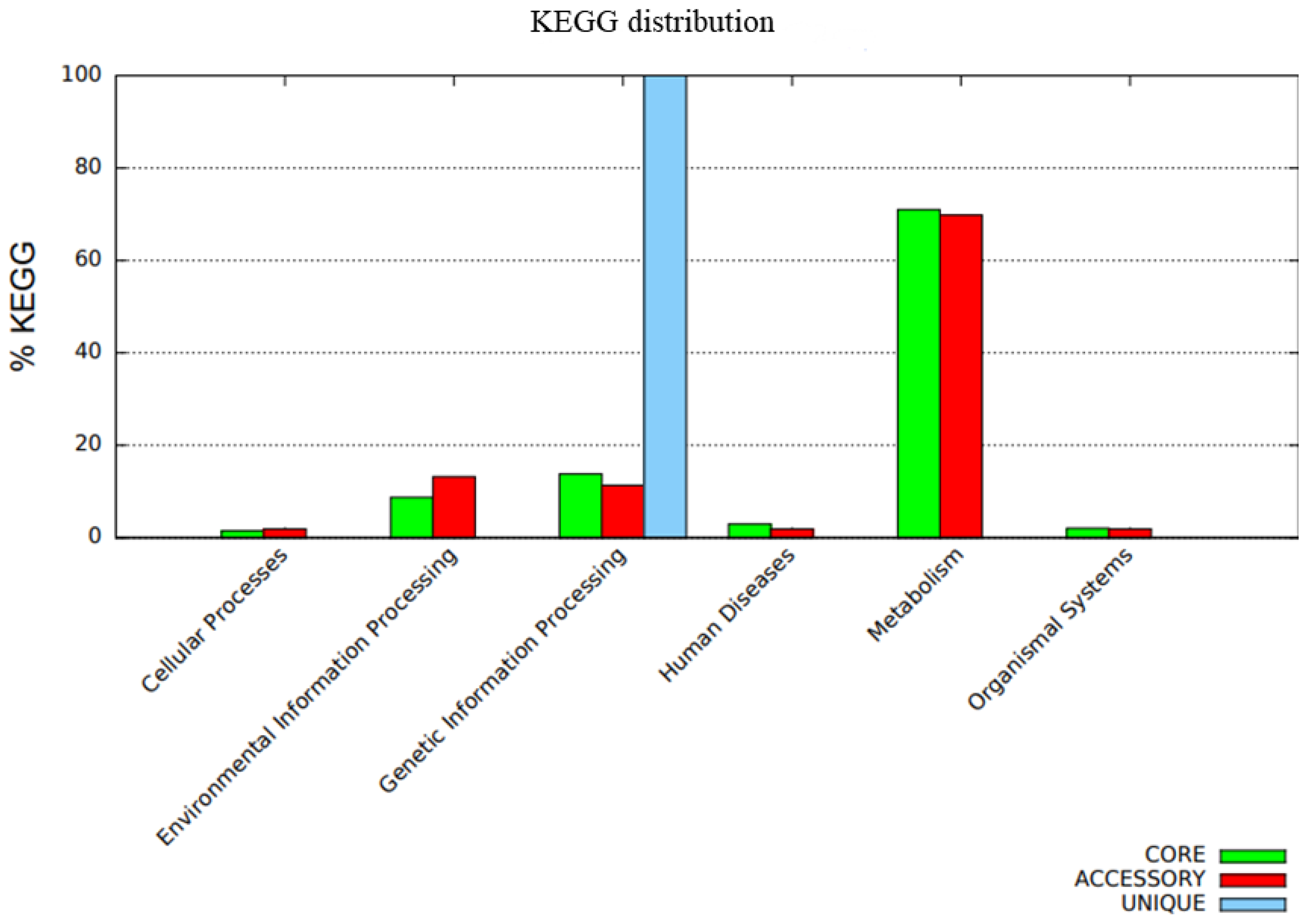
3.5. Pangenome Development and Functional Annotation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat. Rev. Dis. Primers 2019, 81, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Batista Araújo, M.R.; Bernardes Sousa, M.Â.; Seabra, L.F.; Caldeira, L.A.; Faria, C.D.; Bokermann, S.; Sant’Anna, L.O.; Dos Santos, L.S.; Mattos-Guaraldi, A.L. Cutaneous infection by non-diphtheria-toxin producing and penicillin-resistant Corynebacterium diphtheriae strain in a patient with diabetes mellitus. Access Microbiol. 2021, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Araújo, M.R.B.; Ramos, J.N.; de Oliveira Sant’Anna, L.; Bokermann, S.; Santos, M.B.N.; Mattos-Guaraldi, A.L.; Azevedo, V.; Prates, F.D.; Rodrigues, D.L.N.; Aburjaile, F.F.; et al. Phenotypic and molecular characterization and complete genome sequence of a Corynebacterium diphtheriae strain isolated from cutaneous infection in an immunized individual. Braz. J. Microbiol. 2023, 54, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Sant’Anna, L.O.; Ramos, J.N.; Ladeira, E.M.; Stavracakis-Peixoto, R.; Borges, L.L.G.; Santos, C.S.; Napoleão, F.; Camello, T.C.F.; Pereira, G.A. Diphtheria outbreak in Maranhão, Brazil: Microbiological, clinical and epidemiological aspects. Epidemiol. Infect. 2015, 143, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Badenschier, F.; Berger, A.; Dangel, A.; Sprenger, A.; Hobmaier, B.; Sievers, C.; Prins, H.; Dorre, A.; Wagner-Wiening, C.; Kulper-Schiek, W.; et al. Outbreak of imported diphtheria with Corynebacterium diphtheriae among migrants arriving in Germany, 2022. Eurosurveillance 2022, 27, 2200849. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.S.; White, J.M.; Crowcroft, N.S.; De Martin, S.; Mann, G.; Efstratiou, A. Diphtheria in the United Kingdom, 1986–2008: The increasing role of Corynebacterium ulcerans. Epidemiol. Infect. 2010, 138, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Dangel, A.; Berger, A.; Rau, J.; Eisenberg, T.; Kämpfer, P.; Margos, G.; Contzen, M.; Busse, H.J.; Konrad, R.; Peters, M.; et al. Corynebacterium silvaticum sp. nov., a unique group of NTTB corynebacteria in wild boar and roe deer. Int. J. Syst. Evol. Microbiol. 2020, 70, 3614–3624. [Google Scholar] [CrossRef] [PubMed]
- Dazas, M.; Badell, E.; Carmi-Leroy, A.; Criscuolo, A.; Brisse, S. Taxonomic status of Corynebacterium diphtheriae biovar Belfanti and proposal of Corynebacterium belfantii sp. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Cassiday, P.K.; Bernard, K.A.; Bolt, F.; Steigerwalt, A.G.; Bixler, D.; Pawloski, L.C.; Whitney, A.M.; Iwaki, M.; Baldwin, A.; et al. Novel Corynebacterium diphtheriae in Domestic Cats. Emerg. Infect. Dis. 2010, 16, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Badell, E.; Hennart, M.; Rodrigues, C.; Passet, V.; Dazas, M.; Panunzi, L.; Bouchez, V.; Carmi-Leroy, A.; Toubiana, J.; Brisse, S. Corynebacterium rouxii sp. nov., a novel member of the diphtheriae species complex. Res. Microbiol. 2020, 171, 122–127. [Google Scholar] [CrossRef]
- Tyler, R.; Rincon, L.; Weigand, M.R.; Xiaoli, L.; Acosta, A.M.; Kurien, D.; Ju, H.; Lingsweiler, S.; Prot, E.Y. Toxigenic Corynebacterium diphtheriae Infection in Cat, Texas, USA. Emerg. Infect. Dis. 2022, 28, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Schlez, K.; Eisenberg, T.; Rau, J.; Dubielzig, S.; Kornmayer, M.; Wolf, G.; Berger, A.; Dangel, A.; Hoffmann, C.; Ewers, C.; et al. Corynebacterium rouxii, a recently described member of the C. diphtheriae group isolated from three dogs with ulcerative skin lesions. Antonie Van Leeuwenhoek 2021, 114, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, A.; Pampaka, D.; Herrera-León, S.; Peiró, S.; Varona, S.; López-Perea, N.; Masacalles, J.; Herrera-León, L. Molecular and Epidemiological Characterization of Toxigenic and Nontoxigenic Corynebacterium diphtheriae, Corynebacterium belfantii, Corynebacterium rouxii, and Corynebacterium ulcerans Isolates Identified in Spain from 2014 to 2019. J. Clin. Microbiol. 2021, 59, 1–15. [Google Scholar] [CrossRef]
- Ramos, J.N.; Araújo, M.R.B.; Baio, P.V.P.; Sant’Anna, L.O.; Veras, J.F.C.; Vieira, É.M.D.; Sousa, M.A.B.; Camargo, C.H.; Sacchi, C.T.; Campos, K.R.; et al. Molecular characterization and phylogenetic analysis of the first Corynebacterium rouxii strains isolated in Brazil: A recent member of Corynebacterium diphtheriae complex. BMC Genom. Data 2023, 24, 65. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Araya, E.; Muñoz, M.; Rodríguez, C.; Paredes-Sabja, D. FastMLST: A Multi-core Tool for Multilocus Sequence Typing of Draft Genome Assemblies. Bioinform. Biol. Insights 2021, 15, 117793222110592. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Harris, S.R.; Tonkin-Hill, G.; Gladstone, R.A.; Lo, S.W.; Weiser, J.N.; Corander, J.; Bentley, S.D.; Croucher, N.J. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019, 29, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Chiou, C.-S.; Chen, C.-C. PGAdb-builder: A web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 2016, 6, 36213. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Annotation and Classification of CRISPR-Cas Systems. In CRISPR. Methods in Molecular Biology; Lundgre, M., Charpentier, E., Fineran, P., Eds.; Humana Press: New York, NY, USA; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Sangal, V.; Fineran, P.C.; Hoskisson, P.A. Novel configurations of type I and II CRISPR–Cas systems in Corynebacterium diphtheriae. Microbiology 2013, 159 Pt 10, 2118–2126. [Google Scholar] [CrossRef]
- Soares, S.C.; Geyik, H.; Ramos, R.T.J.; de Sá, P.H.C.G.; Barbosa, E.G.V.; Baumbach, J.; Figueiredo, H.C.P.; Miyoshi, A.; Tauch, A.; Silva, A.; et al. GIPSy: Genomic island prediction software. J. Biotechnol. 2016, 232, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vertès, A.A.; Inui, M.; Yukawa, H. Postgenomic Approaches to Using Corynebacteria as Biocatalysts. Annu. Rev. Microbiol. 2012, 66, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.L.N.; Ariute, J.C.; Rodrigues da Costa, F.M.; Benko-Iseppon, A.M.; Barh, D.; Azevedo, V.; Aburjaile, F. PanViTa: Pan Virulence and resisTance analysis. Front. Bioinform. 2023, 3, 1070406. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.V.; Ramos, J.N.; dos Santos, L.S.; Mattos-Guaraldi, A.L. Corynebacterium: Molecular Typing and Pathogenesis of Corynebacterium diphtheriae and Zoonotic Diphtheria Toxin-Producing Corynebacterium Species. In Molecular Typing in Bacterial Infections, Volume I; Springer International Publishing: Cham, 2022; pp. 3–35. [Google Scholar] [CrossRef]
- Bolt, F.; Cassiday, P.; Tondella, M.L.; DeZoysa, A.; Efstratiou, A.; Sing, A.; Zasada, A.; Bernard, K.; Guiso, N.; Badell, E.; et al. Multilocus Sequence Typing Identifies Evidence for Recombination and Two Distinct Lineages of Corynebacterium diphtheriae. J. Clin. Microbiol. 2010, 48, 4177–4185. [Google Scholar] [CrossRef] [PubMed]
- König, C.; Meinel, D.M.; Margos, G.; Konrad, R.; Sing, A. Multilocus Sequence Typing of Corynebacterium ulcerans Provides Evidence for Zoonotic Transmission and for Increased Prevalence of Certain Sequence Types among Toxigenic Strains. J. Clin. Microbiol. 2014, 52, 4318–4324. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, S.M.; Kivistö, R.I.; Rossi, M.; Schott, T.; Kärkkäinen, U.-M.; Tuuminen, T.; Uksila, J.; Rautelin, H.; Hanninen, M.L. Multilocus Sequence Typing (MLST) and Whole-Genome MLST of Campylobacter jejuni Isolates from Human Infections in Three Districts during a Seasonal Peak in Finland. J. Clin. Microbiol. 2014, 52, 4147–4154. [Google Scholar] [CrossRef] [PubMed]
- Hacker, J.; Carniel, E. Ecological fitness, genomic islands and bacterial pathogenicity. EMBO Rep. 2001, 2, 376–381. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Cury, J.; Rocha, E.P.C. The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun. 2017, 8, 841. [Google Scholar] [CrossRef] [PubMed]
- Couturier, M.; Bex, F.; Bergquist, P.L.; Maas, W.K. Identification and classification of bacterial plasmids. Microbiol. Rev. 1988, 52, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Sanchez, Z.K.; Kimbara, K. Genomics of microbial plasmids: Classification and identification based on replication and transfer systems and host taxonomy. Front. Microbiol. 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Serwold-Davis, T.M.; Groman, N.; Rabin, M. Transformation of Corynebacterium diphtheriae, Corynebacterium ulcerans, Corynebacterium glutamicum, and Escherichia coli with the C. diphtheriae plasmid pNG2. Proc. Natl. Acad. Sci. USA 1987, 84, 4964–4968. [Google Scholar] [CrossRef] [PubMed]
- Tauch, A.; Bischoff, N.; Brune, I.; Kalinowski, J. Insights into the genetic organization of the Corynebacterium diphtheriae erythromycin resistance plasmid pNG2 deduced from its complete nucleotide sequence. Plasmid 2003, 49, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Hennart, M.; Panunzi, L.G.; Rodrigues, C.; Gaday, Q.; Baines, S.L.; Barros-Pinkelnig, M.; Carmi-Leroy, A.; Dazas, M.; Wehenkel, A.M.; Didelot, X.; et al. Population genomics and antimicrobial resistance in Corynebacterium diphtheriae. Genome Med. 2020, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Fluit, A.C.; Schmitz, F.J. Class 1 Integrons, Gene Cassettes, Mobility, and Epidemiology. Eur. J. Clin. Microbiol. Infect. Dis. 1999, 18, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, A.A.; Toledo, R.A.; González Pasayo, R.A.; Azevedo, V.; Robles, C.; Paolicchi, F.A.; Belchior, S.G.E. Corynebacterium pseudotuberculosis biovar ovis: Evaluación de la sensibilidad antibiótica in vitro. Rev. Argent. De Microbiol. 2019, 51, 334–338. [Google Scholar] [CrossRef]
- Arcari, G.; Hennart, M.; Badell, E.; Brisse, S. Multidrug-resistant toxigenic Corynebacterium diphtheriae sublineage 453 with two novel resistance genomic islands. Microb. Genom. 2023, 9, 000923. [Google Scholar] [CrossRef] [PubMed]
- Barraud, O.; Badell, E.; Denis, F.; Guiso, N.; Ploy, M.-C. Antimicrobial Drug Resistance in Corynebacterium diphtheriae mitis. Emerg. Infect. Dis. 2011, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, T.; Yamamoto, A.; Komiya, T.; Kenri, T.; Takeuchi, F.; Shibayama, K.; Takahashi, M.; Kuroda, M.; Iwaki, M. Corynebacterium ulcerans 0102 carries the gene encoding diphtheria toxin on a prophage different from the C. diphtheriae NCTC 13129 prophage. BMC Microbiol. 2012, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Yomantas, Y.A.V.; Abalakina, E.G.; Lobanova, J.S.; Mamontov, V.A.; Stoynova, N.V.; Mashko, S.V. Complete nucleotide sequences and annotations of φ673 and φ674, two newly characterised lytic phages of Corynebacterium glutamicum ATCC 13032. Arch. Virol. 2018, 163, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Michel, A.; Clermont, O.; Denamur, E.; Tenaillon, O. Bacteriophage PhiX174′s Ecological Niche and the Flexibility of Its Escherichia coli Lipopolysaccharide Receptor. Appl. Environ. Microbiol. 2010, 76, 7310–7313. [Google Scholar] [CrossRef]
- Leyton-Carcaman, B.; Abanto, M. Beyond to the Stable: Role of the Insertion Sequences as Epidemiological Descriptors in Corynebacterium striatum. Front. Microbiol. 2022, 13, 806576. [Google Scholar] [CrossRef] [PubMed]
- Consuegra, J.; Gaffé, J.; Lenski, R.E.; Hindré, T.; Barrick, J.E.; Tenaillon, O.; Schneider, D. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nat. Commun. 2021, 12, 980. [Google Scholar] [CrossRef] [PubMed]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Leyton, B.; Ramos, J.N.; Baio, P.V.P.; Veras, J.F.C.; Souza, C.; Burkovski, A.; Mattos-Guaraldi, A.L.; Vieira, V.V.; Marin, M.A. Treat Me Well or Will Resist: Uptake of Mobile Genetic Elements Determine the Resistome of Corynebacterium striatum. Int. J. Mol. Sci. 2021, 22, 7499. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Hannon, G.J. The CRISPR System: Small RNA-Guided Defense in Bacteria and Archaea. Mol. Cell 2010, 37, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.-W.; Asmah Hani, A.W.; Nurul Aina Murni, C.A.; Pusparani, R.R.; Chong, C.K.; Verasahib, K.; Yusoff, W.N.W.; Noordin, N.M.; Tee, K.K.; Yin, W.F.; et al. Comparative genomic and phylogenetic analysis of a toxigenic clinical isolate of Corynebacterium diphtheriae strain B-D-16-78 from Malaysia. Infect. Genet. Evol. 2017, 54, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.V.C.; Figueiredo, H.; Ramos, R.; Guimarães, L.C.; Pereira, F.L.; Dorella, F.A.; Selim, S.A.K.; Salaheldean, M.; Silva, A.; Wattam, A.R.; et al. Comparative genomic analysis between Corynebacterium pseudotuberculosis strains isolated from buffalo. PLoS ONE 2017, 12, e0176347. [Google Scholar] [CrossRef] [PubMed]
- Burkovski, A. Proteomics of Toxigenic Corynebacteria. Proteomes 2023, 12, 2. [Google Scholar] [CrossRef]
- Museux, K.; Arcari, G.; Rodrigo, G.; Hennart, M.; Badell, E.; Toubiana, J.; Brisse, S. Corynebacteria of the diphtheriae Species Complex in Companion Animals: Clinical and Microbiological Characterization of 64 Cases from France. Microbiol. Spectr. 2023, 11, e00006-23. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.O.; Mattos-Guaraldi, A.L.; Andrade, A.F.B. Novel lipoarabinomannan-like lipoglycan (CdiLAM) contributes to the adherence of Corynebacterium diphtheriae to epithelial cells. Arch. Microbiol. 2008, 190, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Broadway, M.M.; Rogers, E.A.; Chang, C.; Huang, I.-H.; Dwivedi, P.; Yildirim, S.; Schmitt, M.P.; Das, A.; Ton-That, H. Pilus Gene Pool Variation and the Virulence of Corynebacterium diphtheriae Clinical Isolates during Infection of a Nematode. J. Bacteriol. 2013, 195, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Billington, S.J.; Esmay, P.A.; Songer, J.G.; Jost, B.H. Identification and role in virulence of putative iron acquisition genes from Corynebacterium pseudotuberculosis. FEMS Microbiol. Lett. 2002, 208, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Lee, J.H.; Holmes, R.K. Identification of a DtxR-Regulated Operon That Is Essential for Siderophore-Dependent Iron Uptake in Corynebacterium diphtheriae. J. Bacteriol. 2002, 184, 4846–4856. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, C.A.; Schmitt, M.P. Analysis of a DtxR-Regulated Iron Transport and Siderophore Biosynthesis Gene Cluster in Corynebacterium diphtheriae. J. Bacteriol. 2005, 187, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Lyman, L.R.; Peng, E.D.; Schmitt, M.P. Corynebacterium diphtheriae Iron-Regulated Surface Protein HbpA Is Involved in the Utilization of the Hemoglobin-Haptoglobin Complex as an Iron Source. J. Bacteriol. 2018, 200, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Drazek, E.S.; Hammack, C.A.; Schmitt, M.P. Corynebacterium diphtheriae genes required for acquisition of iron from haemin and haemoglobin are homologous to ABC haemin transporters. Mol. Microbiol. 2000, 36, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bansal, S.; Deb, J.K.; Kundu, B. Interplay between DtxR and nitric oxide reductase activities: A functional genomics approach indicating involvement of homologous protein domains in bacterial pathogenesis. Int. J. Exp. Pathol. 2007, 88, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yellaboina, S.; Ranjan, S.; Chakhaiyar, P.; Hasnain, S.; Ranjan, A. Prediction of DtxR regulon: Identification of binding sites and operons controlled by Diphtheria toxin repressor in Corynebacterium diphtheriae. BMC Microbiol. 2004, 4, 38. [Google Scholar] [CrossRef]
- Allen, C.E.; Schmitt, M.P. Utilization of Host Iron Sources by Corynebacterium diphtheriae: Multiple Hemoglobin-Binding Proteins Are Essential for the Use of Iron from the Hemoglobin-Haptoglobin Complex. J. Bacteriol. 2015, 197, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Chapartegui-González, I.; Fernández-Martínez, M.; Rodríguez-Fernández, A.; Rocha, D.J.P.; Aguiar, E.R.G.R.; Pacheco, L.G.C.; Ramos-Vivas, J.; Calvo, J.; Martínez-Martínez, L.; Navas, J. Antimicrobial Susceptibility and Characterization of Resistance Mechanisms of Corynebacterium urealyticum Clinical Isolates. Antibiotics 2020, 9, 404. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.V.C.; Galdino, J.H.; Profeta, R.; Oliveira, M.; Tavares, L.; de Castro Soares, S.; Carneiro, P.; Wattam, A.R.; Azevedo, V. Analysis of Corynebacterium silvaticum genomes from Portugal reveals a single cluster and a clade suggested to produce diphtheria toxin. PeerJ 2023, 11, e14895. [Google Scholar] [CrossRef]
- Luo, Q.; Luo, H.; Zhang, T.; Liu, X.; Chen, X.; Chen, Q.; Feng, J.; Qu, P.; Chen, C.; Xu, N. Corynebacterium lipophilum sp. nov., a lipophilic bacterium isolated from clinical breast specimens and emended description of the species Corynebacterium pilbarense. Antonie Van Leeuwenhoek 2023, 116, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, J.; Chiba, K.; Kurita, H.; Satoh, H. Contribution of rpoB2 RNA polymerase beta subunit gene to rifampin resistance in Nocardia species. Antimicrob. Agents Chemother. 2006, 50, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Fani, R.; Fondi, M. Defining Orthologs and Pangenome Size Metrics. Methods Mol. Biol. 2015, 1231, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Guimarães, L.C.; Silva, A.; Soares, S.C.; Baraúna, R.A. First Steps in the Analysis of Prokaryotic Pan-Genomes. Bioinform. Biol. Insights 2020, 14, 117793222093806. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.V.C.; Profeta, R.; da Silva, A.L.; Hurtado, R.; Cerqueira, J.C.; Ribeiro, B.F.S.; Almeida, M.O.; Morais-Rodrigues, F.; Soares, S.C.; Oliveira, M.; et al. Taxonomic classification of strain PO100/5 shows a broader geographic distribution and genetic markers of the recently described Corynebacterium silvaticum. PLoS ONE 2020, 15, e0244210. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.C.; Silva, A.; Trost, E.; Blom, J.; Ramos, R.; Carneiro, A.; Ali, A.; Santos, A.R.; Pinto, A.C.; Diniz, C.; et al. The Pan-Genome of the Animal Pathogen Corynebacterium pseudotuberculosis Reveals Differences in Genome Plasticity between the Biovar ovis and equi Strains. PLoS ONE 2013, 8, e53818. [Google Scholar] [CrossRef] [PubMed]
- Trost, E.; Blom, J.; de Castro Soares, S.; Huang, I.-H.; Al-Dilaimi, A.; Schröder, J.; Jaenicke, S.; Dorella, F.A.; Rocha, F.S.; Miyoshi, A.; et al. Pangenomic Study of Corynebacterium diphtheriae That Provides Insights into the Genomic Diversity of Pathogenic Isolates from Cases of Classical Diphtheria, Endocarditis, and Pneumonia. J. Bacteriol. 2012, 194, 3199–3215. [Google Scholar] [CrossRef] [PubMed]
- Ott, L.; Möller, J.; Burkovski, A. Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells. Int. J. Mol. Sci. 2022, 23, 3298. [Google Scholar] [CrossRef] [PubMed]
- Sangal, V.; Burkovski, A.; Hunt, A.C.; Edwards, B.; Blom, J.; Hoskisson, P.A. A lack of genetic basis for biovar differentiation in clinically important Corynebacterium diphtheriae from whole genome sequencing. Infect. Genet. Evol. 2014, 21, 54–57. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Database | Status | Size (Mb) | CDS | GC% | BioSample | Country | Host | Reference Article |
|---|---|---|---|---|---|---|---|---|---|
| FRC0190T | NCBI | Complete | 2,451,019 | 2366 | 53 | SAMEA5992727 | France | Human | [10] |
| 21395 | NCBI | Draft | 2,357,570 | 2238 | 53 | SAMN35995151 | Brazil | Dog | Current study |
| 58111 | NCBI | Draft | 2,354,960 | 2241 | 53 | SAMN35995239 | Brazil | Dog | Current study |
| 70862 | NCBI | Draft | 2,399,341 | 2447 | 53 | SAMN34030355 | Brazil | Cat | [14] |
| 70863 | NCBI | Draft | 2,380,082 | 2397 | 53 | SAMN34030356 | Brazil | Cat | [14] |
| FRC0071 | ENA | Reads | 2,400,198 | 2321 | 53 | SAMEA5992726 | France | Human | [10] |
| FRC0284 | ENA | Reads | 2,381,258 | 2302 | 53 | SAMEA5992728 | France | Human | [10] |
| FRC0297 | ENA | Reads | 2,272,976 | 2165 | 53 | SAMEA5992729 | France | Human | [10] |
| FRC0412 | ENA | Reads | 2,385,950 | 2322 | 53 | SAMEA5992730 | France | Dog | [10] |
| FRC0527 | ENA | Reads | 2,390,401 | 2315 | 53 | SAMEA5992731 | France | Human | [10] |
| PC0230 | ENA | Reads | 2,430,051 | 2362 | 53 | SAMN13343876 | USA | Cat | [11] |
| PC0231 | ENA | Reads | 2,430,200 | 2362 | 53 | SAMN13343877 | USA | Cat | [11] |
| PC0226 | ENA | Reads | 2,430,047 | 2359 | 53 | SAMN13343873 | USA | Cat | [11] |
| PC0229 | ENA | Reads | 2,429,572 | 2364 | 53 | SAMN13343875 | USA | Cat | [11] |
| CS30 | ENA | Reads | 2,345,163 | 2260 | 53 | SAMEA7617347 | Spain | Human | [13] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prates, F.D.; Araújo, M.R.B.; Sousa, E.G.; Ramos, J.N.; Viana, M.V.C.; Soares, S.d.C.; dos Santos, L.S.; Azevedo, V.A.d.C. First Pangenome of Corynebacterium rouxii, a Potentially Toxigenic Species of Corynebacterium diphtheriae Complex. Bacteria 2024, 3, 99-117. https://doi.org/10.3390/bacteria3020007
Prates FD, Araújo MRB, Sousa EG, Ramos JN, Viana MVC, Soares SdC, dos Santos LS, Azevedo VAdC. First Pangenome of Corynebacterium rouxii, a Potentially Toxigenic Species of Corynebacterium diphtheriae Complex. Bacteria. 2024; 3(2):99-117. https://doi.org/10.3390/bacteria3020007
Chicago/Turabian StylePrates, Fernanda Diniz, Max Roberto Batista Araújo, Eduarda Guimarães Sousa, Juliana Nunes Ramos, Marcus Vinícius Canário Viana, Siomar de Castro Soares, Louisy Sanches dos Santos, and Vasco Ariston de Carvalho Azevedo. 2024. "First Pangenome of Corynebacterium rouxii, a Potentially Toxigenic Species of Corynebacterium diphtheriae Complex" Bacteria 3, no. 2: 99-117. https://doi.org/10.3390/bacteria3020007