
Peptides Targeting HER2-Positive Breast Cancer Cells and Applications in Tumor Imaging and Delivery of Chemotherapeutics
 , ,
, ,
Abstract
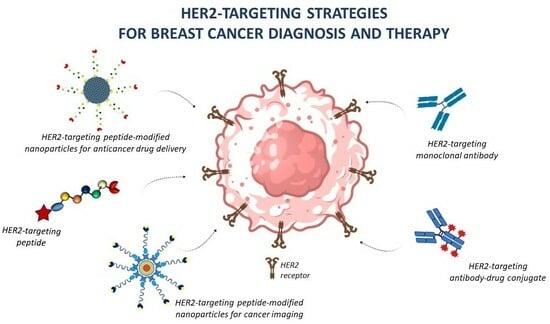
:
1. Introduction
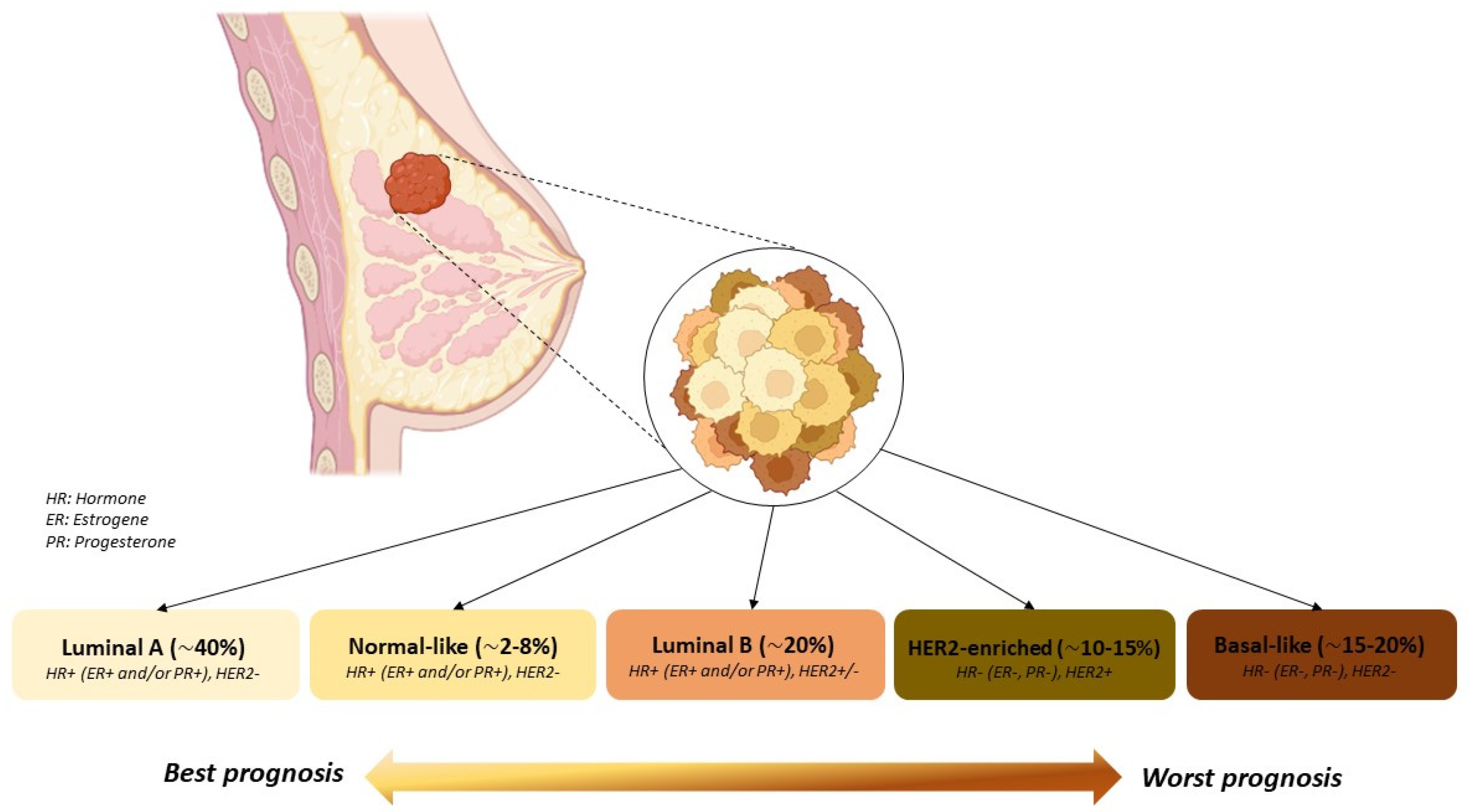
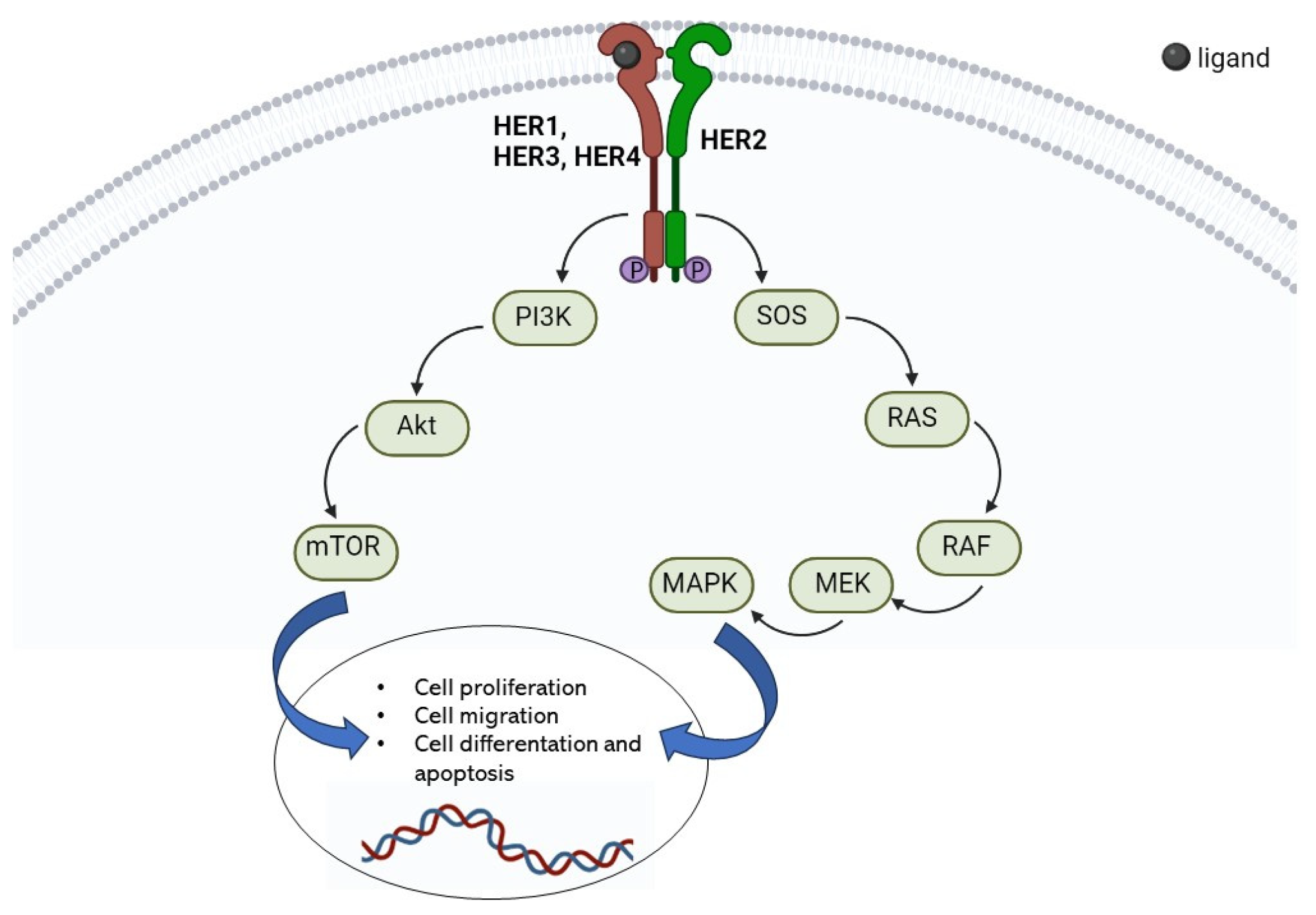
2. HER2 Receptor
3. HER2-Positive Breast Cancer Targeted Therapies
3.1. HER2-Targeting Monoclonal Antibodies
3.2. HER2 Receptor-Targeting Peptides
4. Applications of Peptide-Conjugated Nanoparticles in Breast Cancer Treatment and Imaging
4.1. Application of HER2-Targeted Peptides in Drug Delivery
4.2. Application of HER2-Targeted Peptides for Tumor Imaging
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef]
- Khordadmehr, M.; Shahbazi, R.; Ezzati, H.; Jigari-Asl, F.; Sadreddini, S.; Baradaran, B. Key microRNAs in the biology of breast cancer; emerging evidence in the last decade. J. Cell. Physiol. 2019, 234, 8316–8326. [Google Scholar] [CrossRef]
- Ellis, I.O.; Schnitt, S.J.; Bussolati, G. Tumours of the breast including histological classification. In Pathology and Genetics of Tumours of the Breast and Female Genital Organs; World Health Organization classification of breast tumours; Fattaneh, A., Tavassoli, A., Peter, D., Eds.; IARC Press: Lyon, France, 2005; pp. 9–110. [Google Scholar]
- Eliyatkın, N.; Yalçın, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. Eur. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Page, D.L. Special types of invasive breast cancer, with clinical implications. Am. J. Surg. Pathol. 2003, 27, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer-Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef]
- Guo, F.; Kuo, Y.F.; Shih, Y.C.T.; Giordano, S.H.; Berenson, A.B. Trends in breast cancer mortality by stage at diagnosis among young women in the United States. Cancer 2018, 124, 3500–3509. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Geng, L.; Wang, Z.; Jia, X.; Han, Q.; Xiang, Z.; Li, D.; Yang, X.; Zhang, D.; Bu, X.; Wang, W.; et al. HER2 Targeting Peptides Screening and Applications in Tumor Imaging and Drug Delivery. Theranostics 2016, 6, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Sitia, L.; Sevieri, M.; Signati, L.; Bonizzi, A.; Chesi, A.; Mainini, F.; Corsi, F.; Mazzucchelli, S. HER-2-Targeted Nanoparticles for Breast Cancer Diagnosis and Treatment. Cancers 2022, 14, 2424. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef]
- Olayioye, M.A. Update on HER-2 as a target for cancer therapy: Intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 2001, 3, 385–389. [Google Scholar] [CrossRef]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M.M. HER2 Amplification in Tumors Activates PI3K/Akt Signaling Independent of HER3. Cancer Res. 2018, 78, 3645–3658. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Sheen, M.R.; Marotti, J.D.; Allegrezza, M.J.; Rutkowski, M.; Conejo-Garcia, J.R.; Fiering, S. Constitutively activated PI3K accelerates tumor initiation and modifies histopathology of breast cancer. Oncogenesis 2016, 5, e267. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Di Fiore, P.P.; Pierce, J.H.; Kraus, M.H.; Segatto, O.; King, C.R.; Aaronson, S.A. ErbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science 1987, 237, 178–182. [Google Scholar] [CrossRef]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Burstein, H.J. The distinctive nature of HER2-positive breast cancers. NEJM 2005, 353, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Gallardo, A.; Lerma, E.; Escuin, D.; Tibau, A.; Muñoz, J.; Ojeda, B.; Barnadas, A.; Adrover, E.; Sánchez-Tejada, L.; Giner, D.; et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 2012, 106, 1367–1373. [Google Scholar] [CrossRef]
- Gabos, Z.; Sinha, R.; Hanson, J.; Chauhan, N.; Hugh, J.; Mackey, J.R.; Abdulkarim, B. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J. Clin. Oncol. 2006, 24, 5658–5663. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ichikawa, Y.; Shimizu, D.; Sasaki, T.; Tanabe, M.; Chishima, T.; Takabe, K.; Endo, I. The role of HER-2 in Breast Cancer. J. Surg. Sci. 2014, 2, 4–9. [Google Scholar]
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: State of the art and future directions. Breast Cancer Res. 2021, 23, 84. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 1999, 21, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Greenblatt, K.; Khaddour, K. Trastuzumab. StatPearls: Treasure Island (FL). 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532246/ (accessed on 1 May 2023).
- Spector, N.L.; Blackwell, K.L. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 2009, 27, 5838–5847. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Kute, T.; Stehle, J.R., Jr.; Ornelles, D.; Walker, N.; Delbono, O.; Vaughn, J.P. Understanding key assay parameters that affect measurements of trastuzumab-mediated ADCC against HER2 positive breast cancer cells. Oncoimmunology 2012, 1, 810–821. [Google Scholar] [CrossRef]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Seidman, A.D.; Fornier, M.N.; Esteva, F.J.; Tan, L.; Kaptain, S.; Bach, A.; Panageas, K.S.; Arroyo, C.; Valero, V.; Currie, V.; et al. Weekly trastuzumab and paclitaxel therapy for metastatic breast cancer with analysis of efficacy by HER2 immunophenotype and gene amplification. J. Clin. Oncol. 2001, 19, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Menyhart, O.; Santarpia, L.; Gyorffy, B. A comprehensive outline of trastuzumab resistance biomarkers in HER2 overexpressing breast cancer. Curr. Cancer Drug Targets 2015, 15, 665–683. [Google Scholar] [CrossRef]
- Slamon, D.; Pegram, M. Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin. Oncol. 2001, 28, 13–19. [Google Scholar] [CrossRef]
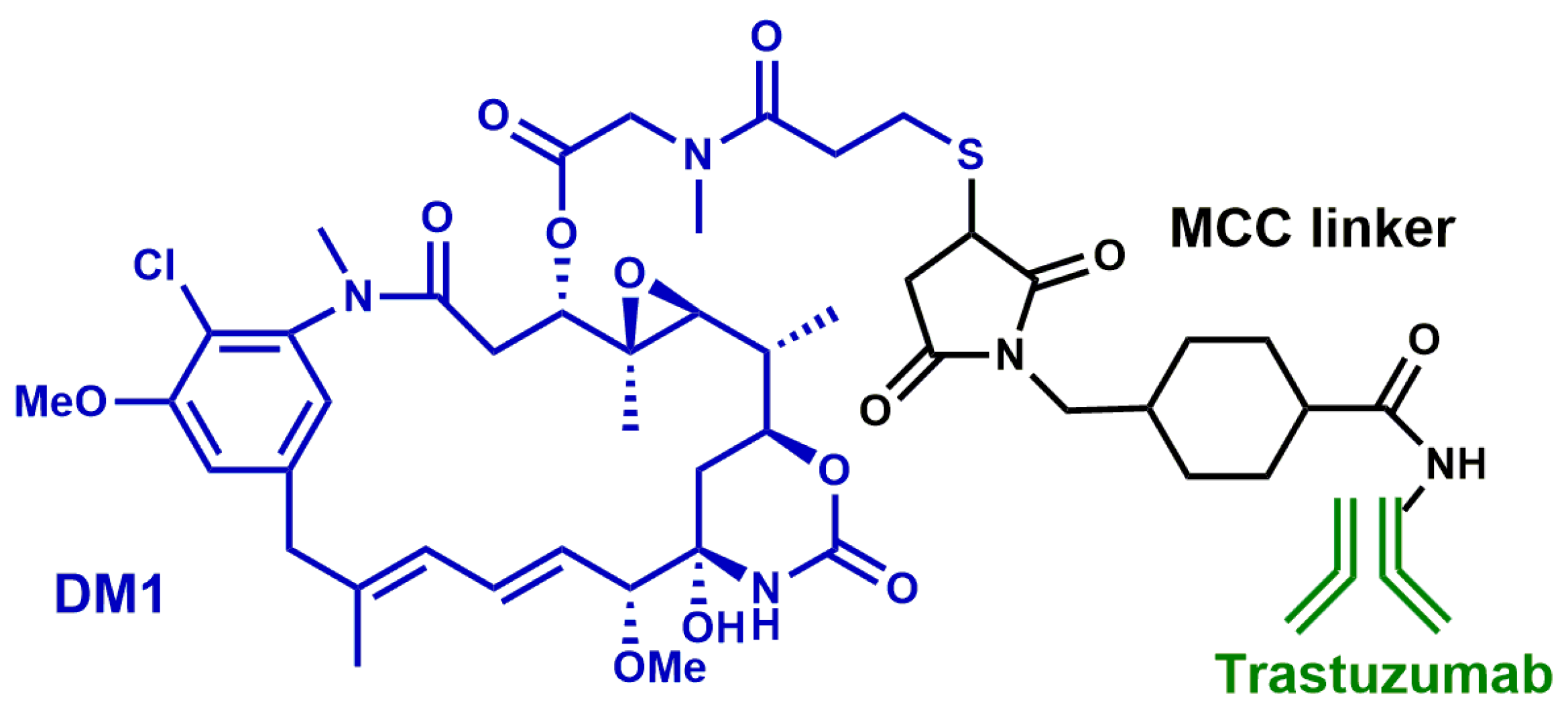
- Ballantyne, A.; Dhillon, S. Trastuzumab emtansine: First global approval. Drugs. 2013, 73, 755–765. [Google Scholar] [CrossRef]
- Eiger, D.; Pondè, N.F.; De Azambujia, E. Pertuzumab in HER2-positive early breast cancer: Current use and perspectives. Future Oncol. 2019, 15, 1823–1843. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Diéras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; Krop, I.E.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomized, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Kim, S.B.; González-Martín, A.; Lo Russo, P.M.; Ferrero, J.M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H. TH3RESA study collaborators. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomized open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Narayan, P.; Osgood, C.L.; Singh, H.; Chiu, H.J.; Ricks, T.K.; Chiu Yuen Chow, E.; Qiu, J.; Song, P.; Yu, J.; Namuswe, F.; et al. FDA Approval Summary: Fam-Trastuzumab Deruxtecan-Nxki for the Treatment of Unresectable or Metastatic HER2-Positive Breast Cancer. Clin Cancer Res. 2021, 27, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Mezni, E.; Vicier, C.; Guerin, M.; Sabatier, R.; Bertucci, F.; Gonçalves, A. New Therapeutics in HER2-Positive Advanced Breast Cancer: Towards a Change in Clinical Practices? Cancers 2020, 12, 1573. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. DESTINY-Breast01 Investigators. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Abstract PD3–06: Updated results from DESTINY-breast01, a phase 2 trial of trastuzumab deruxtecan (T-DXd) in HER2 positive metastatic breast cancer. Cancer Res. 2021, 8, PD3-06. [Google Scholar] [CrossRef]
- Bartsch, R.; Berghoff, A.S.; Furtner, J.; Marhold, M.; Bergen, E.S.; Roider-Schur, S.; Starzer, A.M.; Forstner, H.; Rottenmanner, B.; Dieckmann, K.; et al. Trastuzumab deruxtecan in HER2-positive breast cancer with brain metastases: A single-arm, phase 2 trial. Nat. Med. 2022, 28, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. CLEOPATRA Study Group. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Wedam, S.; Zhang, L.; Tang, S.; Tilley, A.; Ibrahim, A.; Justice, R.; Pazdur, R.; Cortazar, P. First FDA approval of neoadjuvant therapy for breast cancer: Pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin. Cancer Res. 2014, 20, 5359–5364. [Google Scholar] [CrossRef]
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J.; et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011, 13, R123. [Google Scholar] [CrossRef]
- Schlam, I.; Nunes, R.; Lynce, F. Profile of Margetuximab: Evidence to Date in the Targeted Treatment of Metastatic HER2-positive Breast Cancer. Onco Targets Ther. 2022, 15, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Giaccone, G.; Im, S.A.; Oh, D.Y.; Bauer, T.M.; Nordstrom, J.L.; Li, H.; Chichili, G.R.; Moore, P.A.; Hong, S.; et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017, 28, 855–861. [Google Scholar] [CrossRef]
- Rugo, H.S.; Im, S.A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. SOPHIA Study Group. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Verdaguer, H.; Saurí, T.; Acosta, D.A.; Guardiola, M.; Sierra, A.; Hernando, J.; Nuciforo, P.; Miquel, J.M.; Molero, C.; Peiró, S.; et al. ESMO Scale for Clinical Actionability of Molecular Targets Driving Targeted Treatment in Patients with Cholangiocarcinoma. Clin. Cancer Res. 2022, 28, 1662–1671. [Google Scholar] [CrossRef]
- Miglietta, F.; Griguolo, G.; Bottosso, M.; Giarratano, T.; Lo Mele, M.; Fassan, M.; Cacciatore, M.; Genovesi, E.; De Bartolo, D.; Vernaci, G.; et al. HER2-low-positive breast cancer: Evolution from primary tumor to residual disease after neoadjuvant treatment. NPJ Breast Cancer 2022, 8, 66. [Google Scholar] [CrossRef]
- De Luca, S.; Verdoliva, V.; Saviano, M. Peptide Ligands Specifically Targeting HER2 Receptor and the Role Played by a Synthetic Model System of the Receptor Extracellular Domain: Hypothesized Future Perspectives. J. Med. Chem. 2020, 63, 15333–15343. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Eberle, A.N.; Mild, G. Receptor-mediated tumor targeting with radiopeptides. Part 1. General principles and methods. J. Recept. Signal Transduct. Res. 2009, 29, 1–37. [Google Scholar] [CrossRef]
- Spector, N.; Xia, W.; El-Hariry, I.; Yarden, Y.; Bacus, S. HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors. Breast Cancer Res. 2007, 9, 205. [Google Scholar] [CrossRef]
- Di Gioia, M.; Leggio, A.; Liguori, A.; Perri, F. Solid-Phase Synthesis of N-Nosyl- and N-Fmoc-N-Methyl-α-amino Acids. J. Org. Chem. 2007, 72, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Etrych, T.; Chytil, P.; Jelínková, M.; Ríhová, B. HPMA copolymers with pH-controlled release of doxorubicin: In vitro cytotoxicity and in vivo antitumor activity. J. Control. Release 2003, 87, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z.; et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics 2015, 5, 1154–1165. [Google Scholar] [CrossRef]
- Eigenbrot, C.; Ultsch, M.; Dubnovitsky, A.; Abrahmsén, L.; Härd, T. Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc. Natl. Acad. Sci. USA 2010, 107, 15039–15044. [Google Scholar] [CrossRef]
- Ekerljung, L.; Lindborg, M.; Gedda, L.; Frejd, F.Y.; Carlsson, J.; Lennartsson, J. Dimeric HER2-specific affibody molecules inhibit proliferation of the SKBR-3 breast cancer cell line. Biochem. Biophys. Res. Commun. 2008, 377, 489–494. [Google Scholar] [CrossRef]
- Zhou, J.; Zou, Y.; Cai, Y.; Chi, F.; Huang, W.; Shi, W.; Qian, H. A designed cyclic peptide based on Trastuzumab used to construct peptide-drug conjugates for its HER2-targeting ability. Bioorganic Chem. 2021, 117, 105453. [Google Scholar] [CrossRef]
- Langella, E.; Calce, E.; Saviano, M.; De Luca, S. Structural identification of an HER2 receptor model binding pocket to optimize lead compounds: A combined experimental and computational approach. Mol. BioSyst. 2016, 12, 2159–2167. [Google Scholar] [CrossRef]
- Calce, E.; Monfregola, L.; Sandomenico, A.; Saviano, M.; De Luca, S. Fluorescence study for selecting specific ligands toward HER2 receptor: An example of receptor fragment approach. Eur. J. Med. Chem. 2013, 61, 116–121. [Google Scholar] [CrossRef] [PubMed]
- De Luca, S.; Verdoliva, V.; Saviano, M.; Fattorusso, R.; Diana, D. SPR and NMR characterization of the molecular interaction between A9 peptide and a model system of HER2 receptor: A fragment approach for selecting peptide structures specific for their target. J. Pept. Sci. 2020, 26, e3231. [Google Scholar] [CrossRef] [PubMed]
- Honarvar, H.; Calce, E.; Doti, N.; Langella, E.; Orlova, A.; Buijs, J.; D’Amato, V.; Bianco, R.; Saviano, M.; Tolmachev, V.; et al. Evaluation of HER2-specific peptide ligand for its employment as radiolabeled imaging probe. Sci. Rep. 2018, 8, 2998. [Google Scholar] [CrossRef]
- Kohno, M.; Horibe, T.; Haramoto, M.; Yano, Y.; Ohara, K.; Nakajima, O.; Matsuzaki, K.; Kawakami, K. A novel hybrid peptide targeting EGFR-expressing cancers. Eur. J. Cancer 2011, 47, 773–783. [Google Scholar] [CrossRef]
- Kawamoto, M.; Horibe, T.; Kohno, M.; Kawakami, K. HER2-targeted hybrid peptide that blocks HER2 tyrosine kinase disintegrates cancer cell membrane and inhibits tumor growth in vivo. Mol. Cancer Ther. 2013, 12, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Karasseva, N.G.; Glinsky, V.V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Biabani Ardakani, J.; Akhlaghi, M.; Nikkholgh, B.; Hosseinimehr, S.J. Targeting and imaging of HER2 overexpression tumor with a new peptide-based 68Ga-PET radiotracer. Bioorganic Chem. 2021, 106, 104474. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Yu, Y.; Chen, Z.; Yang, Y.; Zhu, L.; Sang, S.; Deng, S. Review: Radionuclide Molecular Imaging Targeting HER2 in Breast Cancer with a Focus on Molecular Probes into Clinical Trials and Small Peptides. Molecules 2021, 26, 6482. [Google Scholar] [CrossRef]
- Chen, K.; Conti, P.S. Target-specific delivery of peptide-based probes for PET imaging. Adv. Drug Deliv. Rev. 2010, 62, 1005–1022. [Google Scholar] [CrossRef]
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled peptides: Valuable tools for the detection and treatment of cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef]
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003, 17, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Mikulová, M.B.; Mikuš, P. Advances in Development of Radiometal Labeled Amino Acid-Based Compounds for Cancer Imaging and Diagnostics. Pharmaceuticals 2021, 14, 167. [Google Scholar] [CrossRef]
- Khodadust, F.; Ahmadpour, S.; Aligholikhamseh, N.; Abedi, S.M.; Hosseinimehr, S.J. An improved 99mTc-HYNIC-(Ser)3-LTVSPWY peptide with EDDA/tricine as co-ligands for targeting and imaging of HER2 overexpression tumor. Eur. J. Med. Chem. 2018, 144, 767–773. [Google Scholar] [CrossRef]
- Sabahnoo, H.; Noaparast, Z.; Abedi, S.M.; Hosseinimehr, S.J. New small 99mTc-labeled peptides for HER2 receptor imaging. Eur. J. Med. Chem. 2017, 127, 1012–1024. [Google Scholar] [CrossRef]
- Kumar, S.R.; Gallazzi, F.A.; Ferdani, R.; Anderson, C.J.; Quinn, T.P.; Deutscher, S.L. In vitro and in vivo evaluation of ⁶⁴Cu-radiolabeled KCCYSL peptides for targeting epidermal growth factor receptor-2 in breast carcinomas. Cancer Biother. Radiopharm. 2010, 25, 693–703. [Google Scholar] [PubMed]
- Larimer, B.M.; Thomas, W.D.; Smith, G.P.; Deutscher, S.L. Affinity maturation of an ERBB2-targeted SPECT imaging peptide by in vivo phage display. Mol. Imaging Biol. 2014, 16, 449–458. [Google Scholar] [CrossRef]
- Kumar, S.R.; Quinn, T.P.; Deutscher, S.L. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor expressing breast carcinomas. Clin. Cancer Res. 2007, 13, 6070–6079. [Google Scholar] [CrossRef] [PubMed]
- Biri-Kovács, B.; Adorján, A.; Szabó, I.; Szeder, B.; Bősze, S.; Mező, G. Structure–Activity Relationship of HER2 Receptor Targeting Peptide and Its Derivatives in Targeted Tumor Therapy. Biomolecules 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Chen, H.; Song, Y.; Qin, Z.; Xu, L.; He, N.; Tan, Y.; Dessie, W. Advancements, challenges and future perspectives on peptide-based drugs: Focus on antimicrobial peptides. Eur. J. Pharm. Sci. 2023, 181, 106363. [Google Scholar] [CrossRef]
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew. Chem. Int. Ed. Engl. 2017, 56, 1525–1529. [Google Scholar] [CrossRef]
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.; Alonso, M.; Brayden, D. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Pichereau, C.; Allary, C. Therapeutic peptides under the spotlight. Eur. Biopharm. 2005, 5, 88–91. [Google Scholar]
- Belsito, E.; Di Gioia, M.L.; Greco, A.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. N-Methyl-N-nosyl-β3-amino Acids. J. Org. Chem. 2007, 72, 4798–4802. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; Malagrinò, F.; Romio, E.; Siciliano, C.; Liguori, A. N-Methylated α-Amino Acids And Peptides: Synthesis And Biological Activity. Mini-Rev. Med. Chem. 2016, 16, 683–690. [Google Scholar] [CrossRef]
- Leggio, A.; Belsito, E.L.; De Marco, R.; Liguori, A.; Perri, F.; Viscomi, M.C. An Efficient Preparation of N-Methyl-α-amino Acids from N-Nosyl-α-amino Acid Phenacyl Esters. J. Org. Chem. 2010, 75, 1386–1392. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int. J. Pept. Res. Ther. 2023, 29, 61. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. Bio. Drugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Freire Haddad, H.; Burke, J.A.; Scott, E.A.; Ameer, G.A. Clinical relevance of pre-existing and treatment-induced anti-poly(ethylene glycol) antibodies. Regen. Eng. Transl. Med. 2021, 8, 32–42. [Google Scholar] [CrossRef]
- Costa, A.R.; Rodrigues, M.E.; Henriques, M.; Oliveira, R.; Azeredo, J. Glycosylation: Impact, control and improvement during therapeutic protein production. Crit. Rev. Biotechnol. 2014, 34, 281–299. [Google Scholar] [CrossRef]
- Van Regenmorte, M.H. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001, 29, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E. Immunogenicity of Protein-Based Therapeutics. U.S. Food and Drug Administration. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics (accessed on 26 August 2023).
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 2014, 190, 15–28. [Google Scholar] [CrossRef]
- Moradi Kashkooli, F.; Soltani, M.; Souri, M. Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies. J. Control. Release 2020, 327, 316–349. [Google Scholar] [CrossRef]
- Gao, J.; Karp, J.M.; Langer, R.; Joshi, N. The Future of Drug Delivery. Chem. Mater. 2023, 35, 359–363. [Google Scholar] [CrossRef]
- Chowdhury, N.K.; Deepika, C.R.; Sonawane, G.A.; Mavinamar, S.; Lyu, X.; Pandey, R.P.; Chang, C.M. Nanoparticles as an effective drug delivery system in COVID-19. Biomed. Pharmacother. 2021, 143, 112162. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.S.A.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N.; et al. Nanoparticles in Drug Delivery: From History to Therapeutic Applications. Nanomaterials 2022, 12, 4494. [Google Scholar] [CrossRef]
- De Santo, M.; Giovinazzo, A.; Fava, M.; Mazzotta, E.; De Napoli, I.E.; Greco, M.; Comandè, A.; Nigro, A.; Argurio, P.; Perrotta, I.; et al. Engineered mesoporous silica-based nanoparticles as smart chemotherapy nanodevice for bortezomib administration. Mater. Chem. Front. 2023, 7, 216–229. [Google Scholar] [CrossRef]
- Chapman, S.; Dobrovolskaia, M.; Farahani, K.; Goodwin, A.; Joshi, A.; Lee, H.; Meade, T.; Pomper, M.; Ptak, K.; Rao, J.; et al. Nanoparticles for cancer imaging: The good, the bad, and the promise. Nano Today 2013, 8, 454–460. [Google Scholar] [CrossRef]
- Mazzotta, E.; De Santo, M.; Lombardo, D.; Leggio, A.; Pasqua, L. Mesoporous silicas in materials engineering: Nanodevices for bionanotechnologies. Mat. Today Bio 2022, 17, 100472. [Google Scholar] [CrossRef]
- Abbasi, M.; Ghoran, S.H.; Niakan, M.H.; Jamali, K.; Moeini, Z.; Jangjou, A.; Izadpanah, P.; Amani, A.M. Mesoporous silica nanoparticle: Heralding a brighter future in cancer nanomedicine. Microporous Mesoporous Mater. 2021, 319, 110967. [Google Scholar] [CrossRef]
- Pasqua, L.; Leggio, A.; Sisci, D.; Andò, S.; Morelli, C. Mesoporous Silica Nanoparticles in Cancer Therapy: Relevance of the Targeting Function. Mini-Rev. Med. Chem. 2016, 16, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Wang, K.; Oppong-Gyebi, A.; Hu, J. Application of Nanotechnology in Cancer Diagnosis and Therapy-A Mini-Review. Int. J. Med. Sci. 2020, 17, 2964–2973. [Google Scholar] [CrossRef]
- Laakkonen, P.; Vuorinen, K. Homing peptides as targeted delivery vehicles. Integr. Biol. 2010, 2, 326–337. [Google Scholar] [CrossRef]
- Singh, M.K.; Pindiprolu, S.K.S.S.; Reddy Sanapalli, B.K.; Yele, V.; Ganesh, G.N.K. Tumor homing peptide modified liposomes of capecitabine for improved apoptotic activity and HER2 targeted therapy in breast cancer: In vitro studies. RSC Adv. 2019, 9, 24987–24994. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Lukyanov, A.N. Peptide and protein drug delivery to and into tumors: Challenges and solutions. Drug Discov. Today 2003, 8, 259–266. [Google Scholar] [CrossRef]
- Tan, M.L.; Choong, P.F.; Dass, C.R. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides 2010, 31, 184–193. [Google Scholar] [CrossRef]
- Pearce, T.R.; Shroff, K.; Kokkoli, E. Peptide targeted lipid nanoparticles for anticancer drug delivery. Adv. Mater. 2012, 24, 3803–3822. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.; Elkhoury, K.; Velot, É.; Bianchi, A.; Acherar, S.; Francius, G.; Tamayol, A.; Grandemange, S.; Arab-Tehrany, E. Functionalized liposomes for targeted breast cancer drug delivery. Bioact. Mater. 2023, 24, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Stefanick, J.F.; Ashley, J.D.; Bilgicer, B. Enhanced cellular uptake of peptide-targeted nanoparticles through increased peptide hydrophilicity and optimized ethylene glycol peptide-linker length. ACS Nano 2013, 7, 8115–8127. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling erbB receptors with rationally designed exocyclic mimetics of antibodies: Structure-function analysis. J. Med. Chem. 2001, 44, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Stefanick, J.F.; Kiziltepe, T.; Bilgicer, B. Improved Peptide-Targeted Liposome Design Through Optimized Peptide Hydrophilicity, Ethylene Glycol Linker Length, and Peptide Density. J. Biomed. Nanotechnol. 2015, 11, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Mizuta, N.; Sakaguchi, K.; Fujiwara, I.; Yoshimori, A.; Takahashi, S.; Takasawa, R.; Tanuma, S. Development of HER2-antagonistic peptides as novel anti-breast cancer drugs by in silico methods. Breast Cancer 2008, 15, 65–72. [Google Scholar] [CrossRef]
- Banappagari, S.; Ronald, S.; Satyanarayanajois, S.D. A conformationally constrained peptidomimetic binds to the extracellular region of HER2 protein. J. Biomol. Struct. Dyn. 2010, 28, 289–308. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Hershman, D.L.; Eisenberger, A.; Wang, J.; Jacobson, J.; Grann, V.; McBride, R.; Tsai, W.; Neugut, A. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3159–3165. [Google Scholar] [CrossRef]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.H.; Kang, S.W.; Seo, J.W.; Hyun, S.W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal Doxorubicin: Review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Kim, B.; Shin, J.; Wu, J.; Omstead, D.T.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. Engineering peptide-targeted liposomal nanoparticles optimized for improved selectivity for HER2-positive breast cancer cells to achieve enhanced in vivo efficacy. J. Control. Release 2020, 322, 530–541. [Google Scholar] [CrossRef]
- Bandekar, A.; Zhu, C.; Gomez, A.; Menzenski, M.Z.; Sempkowski, M.; Sofou, S. Masking and triggered unmasking of targeting ligands on liposomal chemotherapy selectively suppress tumor growth in vivo. Mol. Pharm. 2013, 10, 152–160. [Google Scholar] [CrossRef]
- Karve, S.; Bandekar, A.; Ali, M.R.; Sofou, S. The pH-dependent association with cancer cells of tunable functionalized lipid vesicles with encapsulated doxorubicin for high cell-kill selectivity. Biomaterials 2010, 31, 4409–4416. [Google Scholar] [CrossRef] [PubMed]
- Kempegowda, G.B.; Karve, S.; Bandekar, A.; Adhikari, A.; Khaimchayev, T.; Sofou, S. pH-dependent formation of lipid heterogeneities controls surface topography and binding reactivity in functionalized bilayers. Langmuir 2009, 25, 8144–8151. [Google Scholar] [CrossRef]
- Park, B.W.; Zhang, H.T.; Wu, C.; Berezov, A.; Zhang, X.; Dua, R.; Wang, Q.; Kao, G.; O’Rourke, D.M.; Greene, M.I.; et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat. Biotechnol. 2000, 18, 194–198. [Google Scholar] [CrossRef]
- Zahmatkeshan, M.; Gheybi, F.; Rezayat, S.M.; Jaafari, M.R. Improved drug delivery and therapeutic efficacy of PEgylated liposomal doxorubicin by targeting anti-HER2 peptide in murine breast tumor model. Eur. J. Pharm. Sci. 2016, 86, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Iden, D.L.; Allen, T.M. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 1999, 460, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Ishida, O.; Maruyama, K.; Tanahashi, H.; Iwatsuru, M.; Sasaki, K.; Eriguchi, M.; Yanagie, H. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm. Res. 2001, 18, 1042–1048. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling Receptor Ensembles with Rationally Designed Interface Peptidomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Richter, M.; Song, X.; Berezov, A.; Murali, R.; Greene, M.I.; Zhang, H. AHNP-streptavidin: A tetrameric bacterially produced antibody surrogate fusion protein against p185her2/neu. Oncogene 2006, 25, 7740–7746. [Google Scholar] [CrossRef]
- Kiibanov, A.L.; Huang, L. Long circulating liposomes: Development and perspectives. J. Liposome Res. 2008, 2, 321–334. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.; Huang, L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.S.; Tirrell, M. Bottom-up design of biomimetic assemblies. Adv. Drug Deliv. Rev. 2004, 56, 1537–1563. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, H.; Hong, P.D.; Yu, D.S. Self-assembled proteins and peptides for regenerative medicine. Chem. Rev. 2013, 113, 4837–4861. [Google Scholar] [CrossRef]
- Haburcak, R.; Shi, J.; Du, X.; Yuan, D.; Xu, B. Ligand-receptor Interaction Modulates The Energy Landscape Of Enzyme-instructed Self-assembly Of Small Molecules. J. Am. Chem. Soc. 2016, 138, 15397–15404. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wang, H.; Chen, X.; Xu, B. Self-assembling Ability Determines The Activity Of Enzyme-instructed Self-assembly For Inhibiting Cancer Cells. J. Am. Chem. Soc. 2017, 139, 15377–15384. [Google Scholar] [CrossRef]
- Li, J.; Zhan, Z.; Du, X.; Wang, J.; Hong, B.; Xu, B. Selection of Secondary Structures of Heterotypic Supramolecular Peptide Assemblies by an Enzymatic Reaction. Angew. Chem. Int. Ed. Engl. 2018, 57, 11716–11721. [Google Scholar] [CrossRef]
- He, P.P.; Li, X.D.; Wang, L.; Wang, H. Bispyrene-Based Self-Assembled Nanomaterials: In Vivo Self-Assembly, Transformation, and Biomedical Effects. Acc. Chem. Res. 2019, 52, 367–378. [Google Scholar] [CrossRef]
- Qi, G.B.; Gao, Y.J.; Wang, L.; Wang, H. Self-Assembled Peptide-Based Nanomaterials for Biomedical Imaging and Therapy. Adv. Mater. 2018, 30, e1703444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jing, D.; Jiang, N.; Rojialin, T.; Baehr, A.M.; Zhang, D.; Xiao, W.; Wu, Y.; Cong, Z.; Li, J.J.; et al. Transformable peptide nanoparticles arrest HER2 signalling and cause cancer cell death in vivo. Nat. Nanotechnol. 2020, 15, 145–153. [Google Scholar] [CrossRef]
- Yang, P.P.; Luo, Q.; Qi, G.B.; Gao, Y.J.; Li, B.N.; Zhang, J.P.; Wang, L.; Wang, H. Host Materials Transformable in Tumour Microenvironment for Homing Theranostics. Adv. Mater. 2017, 29, 1605869. [Google Scholar] [CrossRef]
- Tan, J.; Town, T.; Crawford, F.; Mori, T.; Delle Donne, A.; Crescentini, R.; Obregon, D.; Flavell, R.A.; Mullan, M.J. Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat. Neurosci. 2002, 12, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Hock, C.; Konietzko, U.; Papassotiropoulos, A.; Wollmer, A.; Streffer, J.; von Rotz, R.C.; Davey, G.; Moritz, E.; Nitsch, R.M. Generation of Antibodies Specific for Beta-amyloid by Vaccination of Patients with Alzheimer Disease. Nat. Med. 2002, 8, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, H.; Liu, Q.; Murali, R.; Greene, M.I.; Zhang, H. Deoxycholate-based Method to Screen Phage Display Clones for Uninterrupted Open Reading Frames. Biotechniques 2002, 33, 294–296. [Google Scholar] [CrossRef]
- Shivani, T.; Ashu, B.T. New Developments in Breast Cancer Therapy: Role of Iron Oxide Nanoparticles. Adv. Nat. Sci. Nanosci. Nanotechnol. 2017, 8, 023002. [Google Scholar]
- Farzin, A.; Etesami, S.A.; Quint, J.; Memic, A.; Tamayol, A. Magnetic Nanoparticles in Cancer Therapy and Diagnosis. Adv. Healthc. Mater. 2020, 9, e1901058. [Google Scholar] [CrossRef]
- Mu, Q.; Kievit, F.M.; Kant, R.J.; Lin, G.; Jeon, M.; Zhang, M. Anti-HER2/neu Peptide-conjugated Iron Oxide Nanoparticles for Targeted Delivery of Paclitaxel to Breast Cancer Cells. Nanoscale 2015, 7, 18010–18014. [Google Scholar] [CrossRef]
- Fang, C.; Bhattarai, N.; Sun, C.; Zhang, M. Functionalized Nanoparticles with Long-term Stability in Biological Media. Small 2009, 5, 1637–1641. [Google Scholar] [CrossRef]
- Mu, Q.; Jeon, M.; Hsiao, M.H.; Patton, V.K.; Wang, K.; Press, O.W.; Zhang, M. Stable and Efficient Paclitaxel Nnanoparticles for Targeted Glioblastoma Therapy. Adv. Healthc. Mater. 2015, 8, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The Effect of Surface Charge on In Vivo Biodistribution of PEG-oligocholic Acid Based Micellar Nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Han, G.; Toley, B.; Kim, C.K.; Rotello, V.M.; Forbes, N.S. Tuning Payload Delivery in Tumour Cylindroids Using Gold Nanoparticles. Nat. Nanotechnol. 2010, 5, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.C.; Li, H.T.; Li, K.Y. Effects of Shapes of Solute Molecules on Diffusion: A Study of Dependences on Solute Size, Solvent, and Temperature. J. Phys. Chem. B 2015, 119, 15718–15728. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal Clearance of Quantum Dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef]
- Agarwal, R.; Journey, P.; Raythatha, M.; Singh, V.; Sreenivasan, V.; Shi, L. Effect of Shape, Size, and Aspect Ratio on Nanoparticle Penetration and Distribution inside Solid Tissues Using 3D Spheroid Models. Adv. Healthc. Mater. 2015, 4, 2269–2280. [Google Scholar] [CrossRef]
- Ding, H.; Gangalum, P.R.; Galstyan, A.; Fox, I.; Patil, R.; Hubbard, P.; Murali, R.; Ljubimova, J.Y.; Holler, E. HER2-positive Breast Cancer Targeting and Treatment by a Peptide-conjugated Mini Nanodrug. Nanomedicine 2017, 13, 631–639. [Google Scholar] [CrossRef]
- Inoue, S.; Ding, H.; Portilla-Arias, J.; Hu, J.; Konda, B.; Fujita, M.; Espinoza, A.; Suhane, S.; Riley, M.; Gates, M.; et al. Polymalic Acid-based Nanobiopolymer Provides Efficient Systemic Breast Cancer Treatment by Inhibiting both HER2/neu Receptor Synthesis and Activity. Cancer Res. 2011, 71, 1454–1464. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Ding, H.; Portilla-Arias, J.; Patil, R.; Gangalum, P.R.; Chesnokova, A.; Inoue, S.; Rekechenetskiy, A.; Nassoura, T.; Black, K.L.; et al. Polymalic Acid-based Nano Biopolymers for Targeting of Multiple Tumor Markers: An Opportunity for Personalized Medicine? J. Vis. Exp. 2014, 88, 50668. [Google Scholar]
- Askoxylakis, V.; Zitzmann, S.; Mier, W.; Graham, K.; Krämer, S.; von Wegner, F.; Fink, R.H.; Schwab, M.; Eisenhut, M.; Haberkorn, U. Preclinical Evaluation of the Breast Cancer Cell-binding Peptide, p160. Clin. Cancer Res. 2005, 11, 6705–6712. [Google Scholar] [CrossRef]
- Askoxylakis, V.; Mier, W.; Zitzmann, S.; Ehemann, V.; Zhang, J.; Krämer, S.; Beck, C.; Schwab, M.; Eisenhut, M.; Haberkorn, U. Characterization and Development of a Peptide (p160) with Affinity for Neuroblastoma Cells. J. Nucl. Med. 2006, 47, 981–988. [Google Scholar] [PubMed]
- Zhang, J.; Spring, H.; Schwab, M. Neuroblastoma Tumor Cell-binding Peptides Identified through Random Peptide Phage Display. Cancer Lett. 2001, 171, 153–164. [Google Scholar] [PubMed]
- Soudy, R.; Gill, A.; Sprules, T.; Lavasanifar, A.; Kaur, K. Proteolytically Stable Cancer Targeting Peptides with High Affinity for Breast Cancer Cells. J. Med. Chem. 2011, 54, 7523–7534. [Google Scholar]
- Shahin, M.; Soudy, R.; El-Sikhry, H.; Seubert, J.M.; Kaur, K.; Lavasanifar, A. Engineered Peptides for the Development of Actively Tumor Targeted Liposomal Carriers of Doxorubicin. Cancer Lett. 2013, 334, 284–292. [Google Scholar]
- Moreira, J.N.; Ishida, T.; Gaspar, R.; Allen, T.M. Use of the Post-insertion Technique to Insert Peptide Ligands into Pre-formed Stealth Liposomes with Retention of Binding Activity and Cytotoxicity. Pharm. Res. 2002, 19, 265–269. [Google Scholar]
- Allen, T.M.; Sapra, P.; Moase, E. Use of the Post-insertion Method for the Formation of Ligand-coupled Liposomes. Cell. Mol. Biol. Lett. 2002, 3, 889–894. [Google Scholar]
- Elbayoumi, T.A.; Torchilin, V.P. Tumor-specific Anti-nucleosome Antibody Improves Therapeutic Efficacy of Doxorubicin-loaded Long-circulating Liposomes Against Primary and Metastatic Tumor in Mice. Mol. Pharm. 2009, 6, 246–254. [Google Scholar]
- Shahin, M.; Soudy, R.; Aliabadi, H.M.; Kneteman, N.; Kaur, K.; Lavasanifar, A. Engineered Breast Tumor Targeting Peptide Ligand Modified Liposomal Doxorubicin and the Effect of Peptide Density on Anticancer Activity. Biomaterials 2013, 34, 4089–4097. [Google Scholar]
- Kwon, G.S.; Okano, T. Polymeric Micelles as New Drug Carriers. Adv. Drug Deliv. Rev. 1996, 21, 107–116. [Google Scholar]
- Konák, C.; Ulbrich, K. Polymeric Micellar pH-sensitive Drug Delivery System for Doxorubicin. J. Control. Release 2005, 103, 137–148. [Google Scholar]
- Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G.S. Doxorubicin-loaded Poly(ethylene glycol)-poly(beta-benzyl-L-aspartate) Copolymer Micelles: Their Pharmaceutical Characteristics and Biological Significance. J. Control. Release 2000, 64, 143–153. [Google Scholar]
- Nam, J.P.; Lee, K.J.; Choi, J.W.; Yun, C.O.; Nah, J.W. Targeting Delivery of Tocopherol and Doxorubicin Grafted-chitosan Polymeric Micelles for Cancer Therapy: In Vitro and In Vivo Evaluation. Colloids Surf. B Biointerfaces 2015, 133, 254–262. [Google Scholar] [PubMed]
- Chen, L.; Du, Y.; Tian, Z.; Sun, L. Effect of the Degree of Deacetylation and the Substitution of Carboxymethyl Chitosan on its Aggregation Behavior. J. Polym. Sci. B 2005, 43, 296–305. [Google Scholar]
- Chen, X.-G.; Park, H.-J. Chemical Characteristics of O-carboxymethyl Chitosans Related to the Preparation Conditions. Carbohydr. Polym. 2003, 53, 355–359. [Google Scholar]
- Fei Liu, X.; Lin Guan, Y.; Zhi Yang, D.; Li, Z.; De Yao, K. Antibacterial Action of Chitosan and Carboxymethylated Chitosan. J. Appl. Polym. Sci. 2001, 79, 1324–1335. [Google Scholar]
- West, K.R.; Otto, S. Reversible Covalent Chemistry in Drug Delivery. Curr. Drug Discov. Technol. 2005, 2, 123–160. [Google Scholar] [PubMed]
- Lapcík, L., Jr.; Lapcík, L.; De Smedt, S.; Demeester, J.; Chabrecek, P. Hyaluronan: Preparation, Structure, Properties, and Applications. Chem. Rev. 1998, 98, 2663–2684. [Google Scholar]
- Karousou, E.; D’Angelo, M.L.; Kouvidi, K.; Vigetti, D.; Viola, M.; Nikitovic, D.; De Luca, G.; Passi, A. Collagen VI and Hyaluronan: The Common Role in Breast Cancer. Biomed. Res. Int. 2014, 2014, 606458. [Google Scholar]
- Qiu, L.; Li, Z.; Qiao, M.; Long, M.; Wang, M.; Zhang, X.; Tian, C.; Chen, D. Self-assembled pH-responsive Hyaluronic Acid-g-poly((L)-histidine) Copolymer Micelles for Targeted Intracellular Delivery of Doxorubicin. Acta Biomater. 2014, 10, 2024–2035. [Google Scholar]
- Qiu, L.; Qiao, M.; Chen, Q.; Tian, C.; Long, M.; Wang, M.; Li, Z.; Hu, W.; Li, G.; Cheng, L.; et al. Enhanced Effect of pH-sensitive Mixed Copolymer Micelles for Overcoming Multidrug Resistance of Doxorubicin. Biomaterials 2014, 35, 9877–9887. [Google Scholar]
- Chen, Q.; Long, M.; Qiu, L.; Zhu, M.; Li, Z.; Qiao, M.; Hu, H.; Zhao, X.; Chen, D. Decoration of pH-sensitive copolymer micelles with tumor-specific peptide for enhanced cellular uptake of doxorubicin. Int. J. Nanomed. 2016, 11, 5415–5427. [Google Scholar]
- Aloj, L.; Morelli, G. Design, Synthesis and Preclinical Evaluation of Radiolabeled Peptides for Diagnosis and Therapy. Curr. Pharm. Des. 2004, 10, 3009–3031. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Peptide-based Probes for Cancer Imaging. J. Nucl. Med. 2008, 49, 1735–1738. [Google Scholar] [PubMed]
- Orbán, E.; Manea, M.; Marquadt, A.; Bánóczi, Z.; Csík, G.; Fellinger, E.; Bosze, S.; Hudecz, F. A New Daunomycin-peptide Conjugate: Synthesis, Characterization and the Effect on the Protein Expression Profile of HL-60 Cells In Vitro. Bioconjug. Chem. 2011, 22, 2154–2165. [Google Scholar]
- Luo, H.; Lu, L.; Yang, F.; Wang, L.; Yang, X.; Luo, Q.; Zhang, Z. Nasopharyngeal Cancer-specific Therapy Based on Fusion Peptide-functionalized Lipid Nanoparticles. ACS Nano 2014, 8, 4334–4347. [Google Scholar]
- Jie, L.Y.; Cai, L.L.; Wang, L.J.; Ying, X.Y.; Yu, R.S.; Zhang, M.M.; Du, Y.Z. Actively-targeted LTVSPWY peptide-modified magnetic nanoparticles for tumor imaging. Int. J. Nanomed. 2012, 7, 3981–3989. [Google Scholar]
- Ahmad, J.; Garg, A.; Mustafa, G.; Ahmad, M.Z.; Aslam, M.; Mishra, A. Hybrid Quantum Dot as Promising Tools for Theranostic Application in Cancer. Electronics 2023, 12, 972. [Google Scholar]
- Xing, Y.; Chaudry, Q.; Shen, C.; Kong, K.Y.; Zhau, H.E.; Chung, L.W.; Petros, J.A.; O’Regan, R.M.; Yezhelyev, M.V.; Simons, J.W.; et al. Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat. Protoc. 2007, 2, 1152–1165. [Google Scholar]
- Treadway, J.A.; Larson, J.P.; Peale, F.; Bruchez, M.P. Immunofluorescent Labeling of Cancer Marker Her2 and Other Cellular Targets with Semiconductor Quantum Dots. Nat. Biotechnol. 2003, 21, 41–46. [Google Scholar]
- Tada, H.; Higuchi, H.; Wanatabe, T.M.; Ohuchi, N. In Vivo Real-time Tracking of Single Quantum Dots Conjugated with Monoclonal Anti-HER2 Antibody in Tumors of Mice. Cancer Res. 2007, 67, 1138–1144. [Google Scholar]
- Dehghani, N.; Haghiralsadat, F.; Yazdian, F.; Sadeghian-Nodoushan, F.; Ghasemi, N.; Mazaheri, F.; Pourmadadi, M.; Naghib, S.M. Chitosan/silk fibroin/nitrogen-doped carbon quantum dot/α-tricalcium phosphate nanocomposite electrospinned as a scaffold for wound healing application: In vitro and in vivo studies. Int. J. Biol. Macromol. 2023, 238, 124078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Tian, J.; Wang, G.; Luo, W.; Huang, Z.; Huang, Y.; Li, N.; Guo, M.; Fan, X. Tryptophan-sorbitol based carbon quantum dots for theranostics against hepatocellular carcinoma. J. Nanobiotechnol. 2022, 20, 78. [Google Scholar] [CrossRef]
- Kays, J.C.; Saeboe, A.M.; Toufanian, R.; Kurant, D.E.; Dennis, A.M. Shell-Free Copper Indium Sulfide Quantum Dots Induce Toxicity in Vitro and in Vivo. Nano Lett. 2020, 20, 1980–1991. [Google Scholar] [CrossRef]
- Chen, T.; Li, L.; Lin, X.; Yang, Z.; Zou, W.; Chen, Y.; Xu, J.; Liu, D.; Wang, X.; Lin, G. In vitro and in vivo immunotoxicity of PEGylated Cd-free CuInS2/ZnS quantum dots. Nanotoxicology 2020, 14, 372–387. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, P.; Liu, H.; Zhan, H.; Zhang, Q.; Zhao, Y.; Chen, Y. Design of brightnear-infrared-emitting quantum dots capped with different stabilizing ligands for tumor targeting. RSC Adv. 2018, 8, 4221–4229. [Google Scholar] [CrossRef]
- Taya, M.; Nakamura, M. The Development of Cell-adhesive Hydrogel for 3D Printing. Int. J. Bioprinting 2016, 2, 44–53. [Google Scholar]
- Michalska, M.; Aboulaich, A.; Medjahdi, G.; Mahiou, R.; Jurga, S.; Schneider, R. Amine Ligands Control of the Optical Properties and the Shape of Thermally Grown Core/Shell CuInS2/ZnS Quantum Dots. J. Alloys Compd. 2015, 645, 184–192. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Bu, X.; Wei, Z.; Geng, L.; Wu, Y.; Dong, C.; Li, L.; Zhang, D.; Yang, S.; et al. Microarray Based Screening of Peptide Nano Probes for HER2 Positive Tumor. Anal. Chem. 2015, 87, 8367–8372. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence |
|---|---|
| Pep 1 | RIKPRKGYTR |
| Pep 2 | RIKRTNRYTR |
| Pep 3 | RIRPTRRYTR |
| Pep 4 | RIRPRNRYTR |
| Pep 5 | RIRPRKGYTR |
| Sequence | Name |
|---|---|
| CF-KCCYSL-NH2 | P(CC) |
| CF-KCGCYSL-NH2 | P(CGC) |
| CF-KCGGCYSL-NH2 | P(CGCG) |
| CF-KC(Acm)C(Acm)YSL-NH2 | P(C(Acm)C(Acm)) |
| CF-KCSYSL-NH2 | P(CS) |
| CF-KSCYSL-NH2 | P(SC) |
| CF-KSSYSL-NH2 | P(SS) |
| CF-KAAYSL-NH2 | P(AA) |
| CF-GYYNPT-NH2 | P(YY) |
| CF-KAAYSLGYYNPT-NH2 | cP(AA)_P(YY) |
| CF-KSCYSLGYYNPT-NH2 | cP(SC)_P(YY) |
| CF-YSLGYYNPT-NH2 | P(short)_PYY |
| CF-TAKLYPGYANYS-NH2 | scr_P(AA_YY) |
| CF-GYYNPTKAAYSL-NH2 | cP(YY)_P(AA) |
| H-KAAYSLGYYNPT-NH2 | Unlabeled cP(AA)_P(YY) |
| H-KSCYSLGYYNPT-NH2 | Unlabeled cP(SC)_P(YY) |
| Peptide | Delivery System | Chemotherapeutic Agent | Clinical Studies | Ref. |
|---|---|---|---|---|
| WNLPWYYSVSPTC | Liposome | Capecitabine | In vitro studies | [134] |
| Cyclic YCDGFYACYMDV | Liposome | In vitro studies | [135] | |
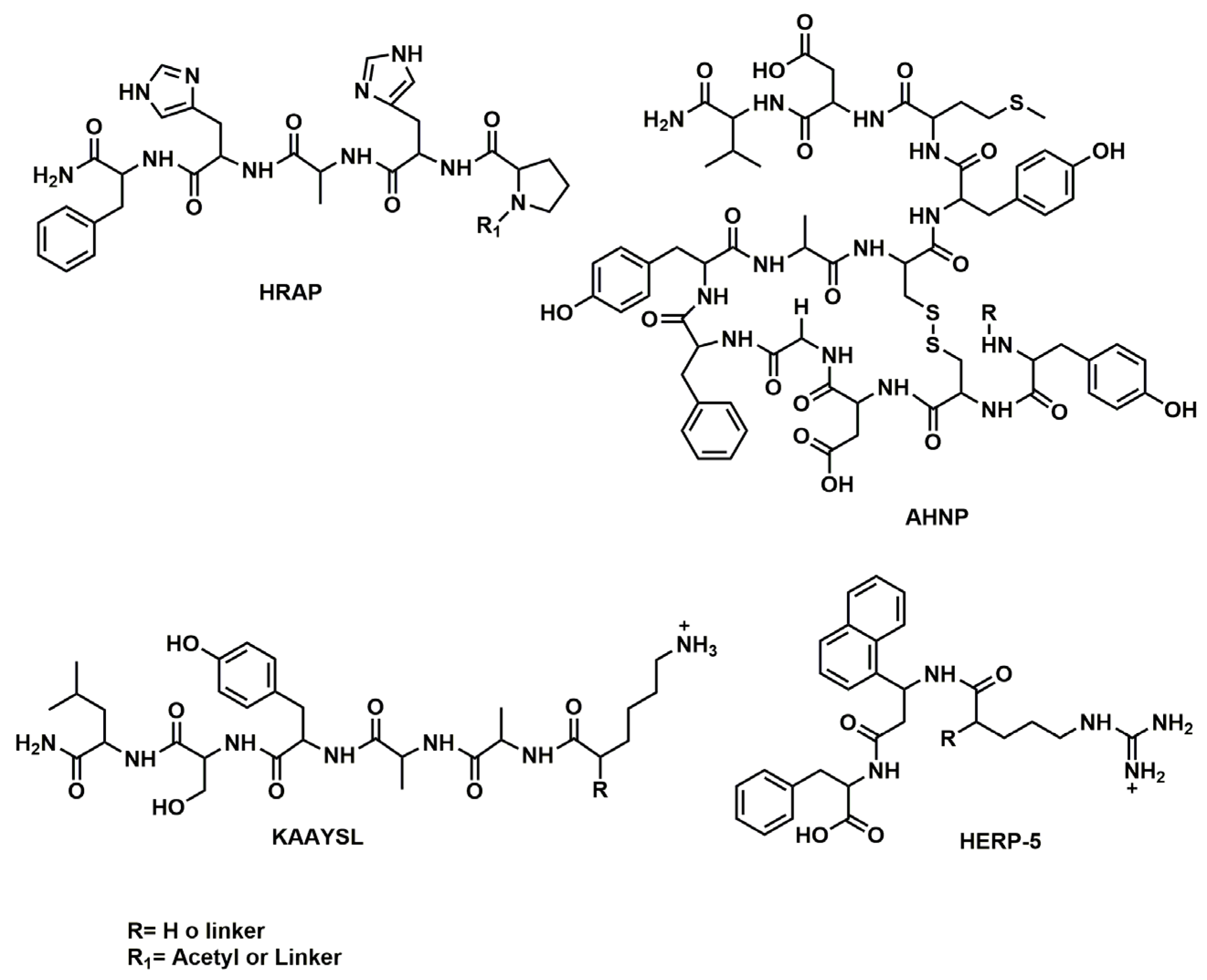
| HERP5 | Liposome | In vitro studies | [136,137,138,139] | |
| HRAP | Liposome | In vitro studies | [136,137,138,139] | |
| KAAYSL | Liposome | In vitro studies | [136,137,138,139] | |
| AHNP | Liposome | In vitro studies | [136,137,138,139] | |
| YCDGFYACYMDV | Liposomal platform | Doxorubicin | In vitro and in vivo studies | [144] |
| GSG-(KCCYSL) | Liposomal vesicle | Doxorubicin | In vitro and in vivo studies | [145] |
| FCDGFYACYADV | Liposome | Doxorubicin | In vitro and in vivo studies | [148,149,153,154] |
| BP-FFVLK-YCDGFYACYMDV | Self-assembled nanoparticle | BP-FFVLK-YCDGFYACYMDV | In vitro and in vivo studies | [165] |
| AHNP | Iron Oxide Nanoparticle | Paclitaxel | In vitro and in vivo studies | [172] |
| YCDGFYACYMDV | Mini nanodrug | Morpholino antisense oligonucleotides (AONs) | In vitro and in vivo studies | [180] |
| WXEAAYQRFL | Liposome | Doxorubicin | In vitro and in vivo studies | [185,186,191] |
| anti-HER2/neu targeting peptide (epitope form, LTVSPWY) | Polymeric micelle | Doxorubicin | In vitro and in vivo studies | [195] |
| YCDGFYACYMDV | Copolymer-based micelle | Doxorubicin | In vitro and in vivo studies | [203] |
| Peptide | Delivery System | Clinical Studies | Ref. |
|---|---|---|---|
| LTVSPWY | Lipid-modified Fe3O4-based magnetic nanoparticles | In vitro and in vivo studies | [209] |
| LTVSPWY | Quantum dot-based probes | In vitro studies | [219] |
| YLFFVFER | Quantum dots | In vivo and ex vivo studies | [221] |
| KLRLEWNR | Quantum dots | In vivo and ex vivo studies | [221] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallaro, P.A.; De Santo, M.; Belsito, E.L.; Longobucco, C.; Curcio, M.; Morelli, C.; Pasqua, L.; Leggio, A. Peptides Targeting HER2-Positive Breast Cancer Cells and Applications in Tumor Imaging and Delivery of Chemotherapeutics. Nanomaterials 2023, 13, 2476. https://doi.org/10.3390/nano13172476
Cavallaro PA, De Santo M, Belsito EL, Longobucco C, Curcio M, Morelli C, Pasqua L, Leggio A. Peptides Targeting HER2-Positive Breast Cancer Cells and Applications in Tumor Imaging and Delivery of Chemotherapeutics. Nanomaterials. 2023; 13(17):2476. https://doi.org/10.3390/nano13172476
Chicago/Turabian StyleCavallaro, Palmira Alessia, Marzia De Santo, Emilia Lucia Belsito, Camilla Longobucco, Manuela Curcio, Catia Morelli, Luigi Pasqua, and Antonella Leggio. 2023. "Peptides Targeting HER2-Positive Breast Cancer Cells and Applications in Tumor Imaging and Delivery of Chemotherapeutics" Nanomaterials 13, no. 17: 2476. https://doi.org/10.3390/nano13172476