
Novel Techniques, Biomarkers and Molecular Targets to Address Cardiometabolic Diseases

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
1.1. Novel Techniques and Biomarkers in the Clinical Setting
1.1.1. Epicardial Adipose Tissue
1.1.2. Congestion
1.1.3. Ventricular–Arterial Coupling
1.1.4. Exercise Intolerance and Breath Analysis
1.2. Novel Molecular Targets
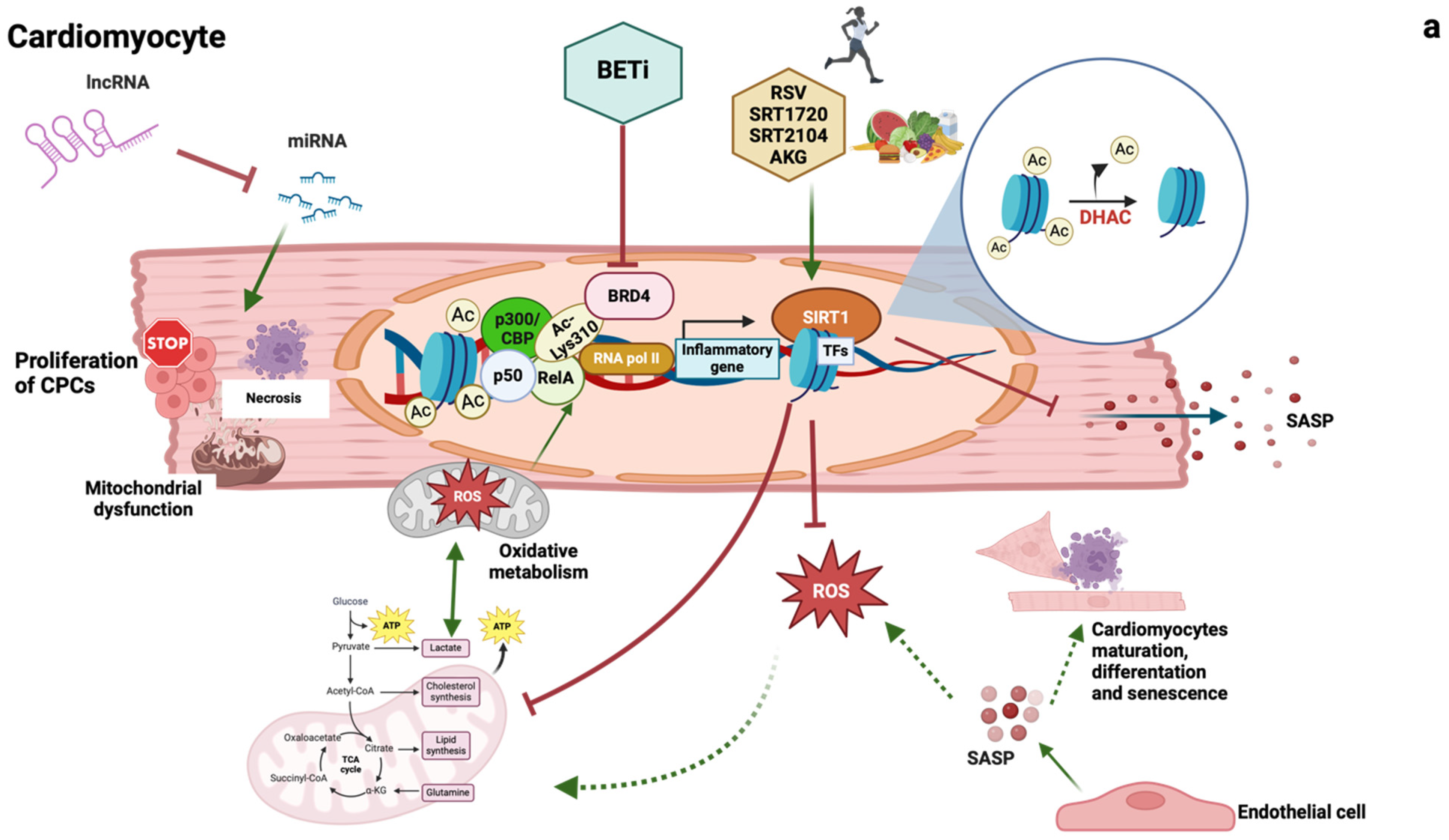
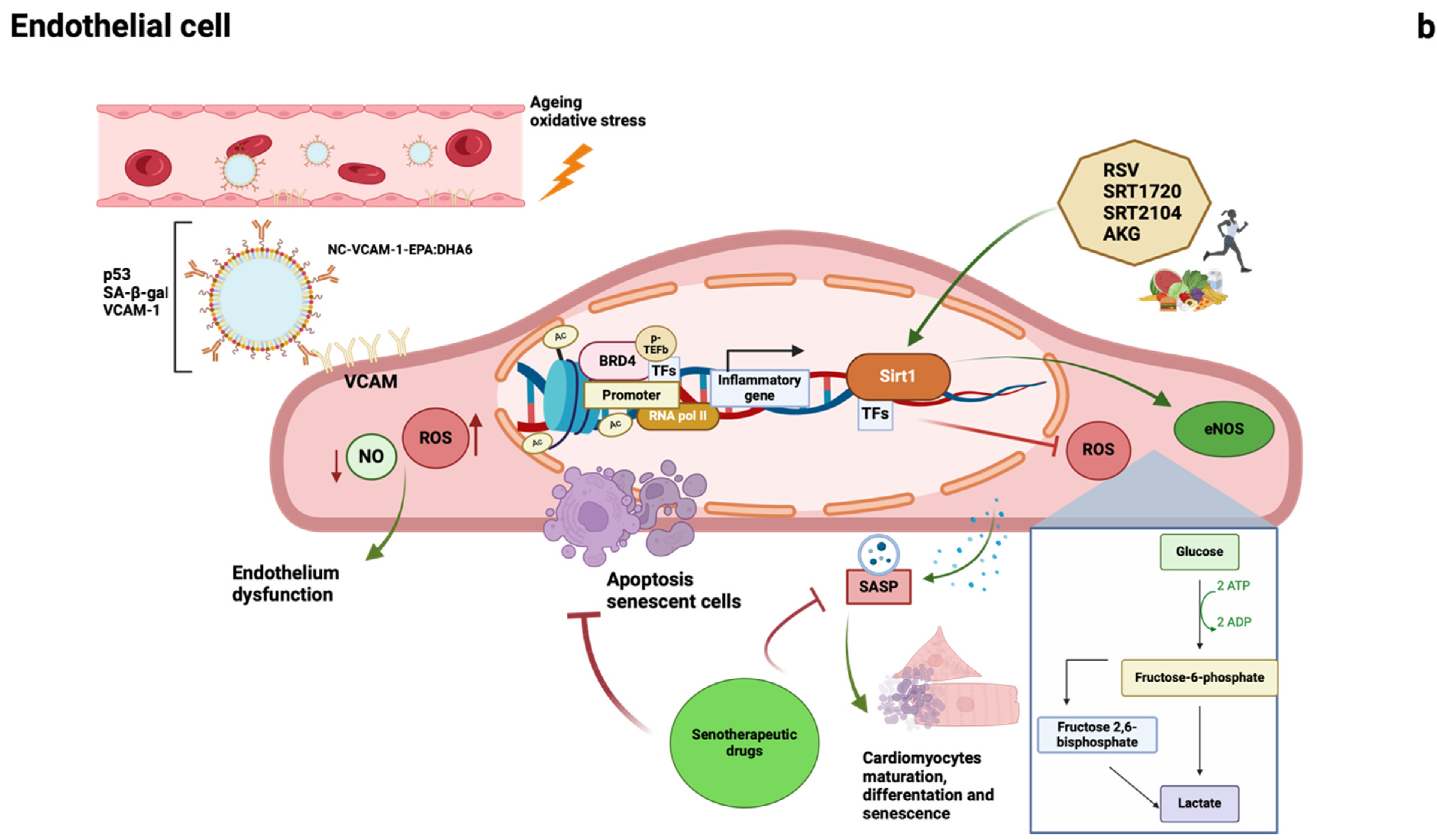
1.2.1. Epigenetic Changes
1.2.2. Long Noncoding RNAs
1.2.3. Sirtuins

{kind=link}
{kind=link}
{kind=link}
| (a) | ||||
| Drug | Model | Disease | Effects | References |
| AKG | Pressure overload-induced mice | Heart failure | Increased mitophagy. Reduction of ferroptosis and cell damage. | [68] |
| rSirt1 | db/db mouse | Metabolic cardiomyopathy | Restoration of Sirt1 levels. Improvement of left ventricular ejection fraction, fractional shortening, and diastolic function. Reduction of medium- and long-chain triacylglycerols containing saturated fatty acids. Increased triacylglycerols containing docosahexaenoic acid. Underregulated lipid trafficking and inflammation genes. | [67] |
| RSV | Diabetic mice + streptozotocin | Diabetic cardiomyopathy | Activation of Sirt1-dependent transcriptional regulatory mechanisms. Improvement of cardiac function. | [69] |
| Accelerated vascular ageing rats | Vascular oxidative stress and inflammation | Protection against the harmful effect of ROS generation, proinflammatory mediators, and endothelial cell apoptosis. Sirt1 activation ameliorated endothelial stiffness by preventing p65-NFκB activation, VCAM-1 upregulation, and decreasing p47phox. | [72] | |
| Mice + high fat/high sucrose diet | Metabolic syndrome | Increased Sirt1 expression and activity, cyclooxygenase-2 expression, and antioxidant enzymes. Restoring vascular homeostasis and endothelial function. Reduction of NFκB acetyl-p65, vascular oxidative stress, and risk of cardiovascular disease. | [73] | |
| SRT1720 | Ageing mice | Vascular endothelial dysfunction. | Increased vascular relaxation and reduced superoxide and inflammation. | [74] |
| Mice + high fat/high sucrose diet | Metabolic syndrome | Anti-inflammatory and antioxidant effects. | [73] | |
| Genetically obese mice | Metabolic syndrome | Increase lifespan and decrease serum glucose. | [61] | |
| Mice + high-fat diet | Metabolic syndrome | Reduced obesity, insulin resistance, hepatic steatosis. Increased longevity. | [61] | |
| (b) | ||||
| Drug | Study population | Effects | References | |
| SRT1720 | Obese and ageing patients | Rescue of endothelial dysfunction by reduction p66Shc expression and modulation of mitochondria respiratory chain. | [62] | |
| RSV | Healthy individuals | Decreased expression of endothelial cells (intercellular adhesion molecule 1, VCAM-1 and interleukin 8) and inflammatory biomarkers (decreased level in plasma, interferon-gamma and insulin). | [75] | |
| Healthy obese men | Reduction in systolic blood pressure and increase in insulin sensitivity. | [61] | ||
| Healthy non-obese men | No effects. | [61] | ||
| Non-alcoholic fatty liver disease | Reduction in LDL, insulin resistance, hepatic steatosis and inflammation | [61] | ||
| Metabolic syndrome patients | Reduction of fat mass. Improvement in diastolic blood pressure. Improvement of insulin sensitivity. | [77] | ||
| T2D | Lowering of glucose levels. Decrease in insulin resistance. | [78] | ||
| Patients with peripheral artery disease | No change in walking performance | [61] | ||
| SRT2104 | T2D | Effects on cardiovascular measures are predominantly neutral. Weight reduction. Deterioration in glycaemic control. | [80] | |
| Healthy older volunteers | Reduction of serum LDL and triglycerides levels. | [61] | ||
| Healthy smokers | Reduction of serum LDL and triglycerides levels. | [61] | ||
1.2.4. Epigenetic Readers: Bromodomain and Extraterminal Domain (BET) Proteins
1.2.5. Cellular Senescence
2. Conclusions: Gap of Knowledge and Next Steps
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKG | alpha-ketoglutarate |
| ANRIL | antisense non-coding RNA in the INK4 Locus |
| BET | bromodomain and extraterminal proteins BETi inhibitors (BETi) |
| BRD4 | bromodomain-containing protein 4 |
| CBP | CREB-binding protein |
| LIPCAR | long intergenic noncoding RNA predicting CARdiac remodelling |
| lncRNA | long noncoding RNA |
| MALAT 1 | metastasis-associated lung adenocarcinoma transcript 1 |
| MCM | metabolic cardiomyopathy |
| ncRNA | noncoding RNA |
| NAD | nicotinamide adenine dinucleotide |
| NC | nano carrier |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| rSirt1 | recombinant Sirt1 |
| RSV | resveratrol |
| RVX-208 | apabetalone |
| SASP | senescent-associated secretory phenotype |
| Sirt1 | silent mating type information regulation 1 homologs |
References
- Di Angelantonio, E.; Kaptoge, S.; Wormser, D.; Willeit, P.; Butterworth, A.S.; Bansal, N.; O’Keeffe, L.M.; Gao, P.; Wood, A.M.; Burgess, S.; et al. Association of cardiometabolic multimorbidity with mortality. JAMA—J. Am. Med. Assoc. 2015, 314, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ma, T.; Ouyang, F.; Zhang, G.; Bai, Y. Trends in the Prevalence of Cardiometabolic Multimorbidity in the United States, 1999–2018. Int. J. Environ. Res. Public Health 2022, 19, 4726. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Organ System Crosstalk in Cardiometabolic Disease in the Age of Multimorbidity. Front. Cardiovasc. Med. 2020, 7, 64. [Google Scholar] [CrossRef]
- Pallarés-Carratalá, V.; Ruiz-García, A.; Serrano-Cumplido, A.; Arranz-Martínez, E.; Divisón-Garrote, J.A.; Moyá-Amengual, A.; Escobar-Cervantes, C.; Barrios, V. Prevalence Rates of Arterial Hypertension According to the Threshold Criteria of 140/90 or 130/80 mmHg and Associated Cardiometabolic and Renal Factors: SIMETAP-HTN Study. Medicina 2023, 59, 1846. [Google Scholar] [CrossRef]
- Redon, J.; Cifkova, R.; Laurent, S.; Nilsson, P.; Narkiewicz, K.; Erdine, S.; Mancia, G. Mechanisms of hypertension in the cardiometabolic syndrome. J. Hypertens. 2009, 27, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Balletti, A.; De Biase, N.; Del Punta, L.; Filidei, F.; Armenia, S.; Masi, F.; Di Fiore, V.; Mazzola, M.; Bacca, A.; Dini, F.L.; et al. Cardiometabolic Phenotyping in Heart Failure: Differences between Patients with Reduced vs. Preserved Ejection Fraction. Diagnostics 2023, 13, 790. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Serio, S.; Pagiatakis, C.; Musolino, E.; Felicetta, A.; Carullo, P.; Laura Frances, J.; Papa, L.; Rozzi, G.; Salvarani, N.; Miragoli, M.; et al. Cardiac Aging Is Promoted by Pseudohypoxia Increasing p300-Induced Glycolysis. Circ. Res. 2023, 133, 687–703. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Iacobellis, G.; Willens, H.J. Echocardiographic Epicardial Fat: A Review of Research and Clinical Applications. J. Am. Soc. Echocardiogr. 2009, 22, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J. Am. Coll. Cardiol. 2018, 71, 2360–2372. [Google Scholar] [CrossRef]
- Bozkurt, B.; Coats, A.J.S.; Tsutsui, H.; Abdelhamid, C.M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Böhm, M.; Butler, J.; et al. Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of heart failure. Eur. J. Heart Fail. 2021, 23, 352–380. [Google Scholar] [CrossRef]
- Pugliese, N.R.; Paneni, F.; Mazzola, M.; De Biase, N.; Del Punta, L.; Gargani, L.; Mengozzi, A.; Virdis, A.; Nesti, L.; Taddei, S.; et al. Impact of epicardial adipose tissue on cardiovascular haemodynamics, metabolic profile, and prognosis in heart failure. Eur. J. Heart Fail. 2021, 23, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Gorter, T.M.; van Woerden, G.; Rienstra, M.; Dickinson, M.G.; Hummel, Y.M.; Voors, A.A.; Hoendermis, E.S.; van Veldhuisen, D.J. Epicardial Adipose Tissue and Invasive Hemodynamics in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2020, 8, 667–676. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Omote, K.; Reddy, Y.N.V.; Sorimachi, H.; Obokata, M.; Borlaug, B.A. Heart failure with preserved ejection fraction in patients with normal natriuretic peptide levels is associated with increased morbidity and mortality. Eur. Heart J. 2022, 43, 1941–1951. [Google Scholar] [CrossRef]
- Núñez, J.; de la Espriella, R.; Rossignol, P.; Voors, A.A.; Mullens, W.; Metra, M.; Chioncel, O.; Januzzi, J.L.; Mueller, C.; Richards, A.M.; et al. Congestion in heart failure: A circulating biomarker-based perspective. A review from the Biomarkers Working Group of the Heart Failure Association, European Society of Cardiology. Eur. J. Heart Fail. 2022, 24, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Damman, K.; Harjola, V.P.; Mebazaa, A.; Brunner-La Rocca, H.P.; Martens, P.; Testani, J.M.; Tang, W.H.H.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Anstrom, K.J.; Adams, K.F.; Ezekowitz, J.A.; Fiuzat, M.; Houston-Miller, N.; Januzzi, J.L.; Mark, D.B.; Piña, I.L.; Passmore, G.; et al. Effect of natriuretic peptide–guided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: A randomized clinical trial. JAMA—J. Am. Med. Assoc. 2017, 318, 713–720. [Google Scholar] [CrossRef]
- Tsutsui, H.; Albert, N.M.; Coats, A.J.S.; Anker, S.D.; Bayes-Genis, A.; Butler, J.; Chioncel, O.; Defilippi, C.R.; Drazner, M.H.; Felker, G.M.; et al. Natriuretic peptides: Role in the diagnosis and management of heart failure: A scientific statement from the Heart Failure Association of the European Society of Cardiology, Heart Failure Society of America and Japanese Heart Failure Society. Eur. J. Heart Fail. 2023, 25, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, N.R.; Pellicori, P.; Filidei, F.; Del Punta, L.; De Biase, N.; Balletti, A.; Di Fiore, V.; Mengozzi, A.; Taddei, S.; Gargani, L.; et al. The incremental value of multi-organ assessment of congestion using ultrasound in outpatients with heart failure. Eur. Heart J.—Cardiovasc. Imaging 2023, 24, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Correale, M.; Fioretti, F.; Tricarico, L.; Croella, F.; Brunetti, N.D.; Inciardi, R.M.; Mattioli, A.V.; Nodari, S. The Role of Congestion Biomarkers in Heart Failure with Reduced Ejection Fraction. J. Clin. Med. 2023, 12, 3834. [Google Scholar] [CrossRef]
- Chubuchny, V.; Pugliese, N.R.; Taddei, C.; Poggianti, E.; Spini, V.; Barison, A.; Formichi, B.; Airò, E.; Bauleo, C.; Prediletto, R.; et al. A novel echocardiographic method for estimation of pulmonary artery wedge pressure and pulmonary vascular resistance. ESC Heart Fail. 2021, 8, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Pellicori, P.; Platz, E.; Dauw, J.; ter Maaten, J.M.; Martens, P.; Pivetta, E.; Cleland, J.G.F.; McMurray, J.J.V.; Mullens, W.; Solomon, S.D.; et al. Ultrasound imaging of congestion in heart failure: Examinations beyond the heart. Eur. J. Heart Fail. 2021, 23, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Prescott, M.F.; Camacho, A.; Iyer, S.R.; Maisel, A.S.; Felker, G.M.; Butler, J.; Piña, I.L.; Ibrahim, N.E.; Abbas, C.; et al. Atrial Natriuretic Peptide and Treatment with Sacubitril/Valsartan in Heart Failure with Reduced Ejection Fraction. JACC Heart Fail. 2021, 9, 127–136. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Aboyans, V.; Blacher, J.; Brodmann, M.; Brutsaert, D.L.; Chirinos, J.A.; De Carlo, M.; Delgado, V.; Lancellotti, P.; Lekakis, J.; et al. The role of ventricular–arterial coupling in cardiac disease and heart failure: Assessment, clinical implications and therapeutic interventions. A consensus document of the European Society of Cardiology Working Group on Aorta & Peripheral Vascular Diseas. Eur. J. Heart Fail. 2019, 21, 402–424. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Melenovsky, V.; Redfield, M.M.; Kessler, K.; Chang, H.J.; Abraham, T.P.; Kass, D.A. Impact of Arterial Load and Loading Sequence on Left Ventricular Tissue Velocities in Humans. J. Am. Coll. Cardiol. 2007, 50, 1570–1577. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Tzortzis, S.; Triantafyllidi, H.; Parissis, J.; Papadopoulos, C.; Venetsanou, K.; Trivilou, P.; Paraskevaidis, I.; Lekakis, J. Association of impaired left ventricular twisting-untwisting with vascular dysfunction, neurohumoral activation and impaired exercise capacity in hypertensive heart disease. Eur. J. Heart Fail. 2015, 17, 1240–1251. [Google Scholar] [CrossRef]
- Vinereanu, D.; Nicolaides, E.; Boden, L.; Payne, N.; Jones, C.J.H.; Fraser, A.G. Conduit arterial stiffness is associated with impaired left ventricular subendocardial function. Heart 2003, 89, 449–450. [Google Scholar] [CrossRef]
- Shah, A.M.; Claggett, B.; Sweitzer, N.K.; Shah, S.J.; Anand, I.S.; Liu, L.; Pitt, B.; Pfeffer, M.A.; Solomon, S.D. Prognostic importance of impaired systolic function in heart failure with preserved ejection fraction and the impact of spironolactone. Circulation 2015, 132, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, N.R.; Balletti, A.; Armenia, S.; De Biase, N.; Faita, F.; Mengozzi, A.; Paneni, F.; Ruschitzka, F.; Virdis, A.; Ghiadoni, L.; et al. Ventricular-Arterial Coupling Derived from Proximal Aortic Stiffness and Aerobic Capacity across the Heart Failure Spectrum. JACC Cardiovasc. Imaging 2022, 15, 1545–1559. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 233–270. [Google Scholar] [CrossRef] [PubMed]
- Del Punta, L.; De Biase, N.; Balletti, A.; Filidei, F.; Pieroni, A.; Armenia, S.; Mengozzi, A.; Mazzola, M.; Di Fiore, V.; Dini, F.L.; et al. Arterial Hypertension and Cardiopulmonary Function: The Value of a Combined Cardiopulmonary and Echocardiography Stress Test. High Blood Press. Cardiovasc. Prev. 2022, 29, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, N.R.; Mazzola, M.; Fabiani, I.; Gargani, L.; De Biase, N.; Pedrinelli, R.; Natali, A.; Dini, F.L. Haemodynamic and metabolic phenotyping of hypertensive patients with and without heart failure by combining cardiopulmonary and echocardiographic stress test. Eur. J. Heart Fail. 2020, 22, 458–468. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Arena, R.; Borlaug, B.A.; Carbone, S.; Canada, J.M.; Kirkman, D.L.; Garten, R.; Rodriguez-Miguelez, P.; Guazzi, M.; Lavie, C.J.; et al. Exercise Intolerance in Patients with Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2209–2225. [Google Scholar] [CrossRef] [PubMed]
- Del Punta, L.; De Biase, N.; Di Fiore, V.; Maremmani, D.; Gargani, L.; Mazzola, M.; De Carlo, M.; Mengozzi, A.; Lomonaco, T.; Galeotti, G.G.; et al. Combining cardiopulmonary exercise testing with echocardiography: A multiparametric approach to the cardiovascular and cardiopulmonary systems. Eur. Heart J.—Imaging Methods Pract. 2023, 1, qyad021. [Google Scholar] [CrossRef]
- Guazzi, M.; Myers, J.; Arena, R. Cardiopulmonary exercise testing in the clinical and prognostic assessment of diastolic heart failure. J. Am. Coll. Cardiol. 2005, 46, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Gargani, L.; Pugliese, N.R.; De Biase, N.; Mazzola, M.; Agoston, G.; Arcopinto, M.; Argiento, P.; Armstrong, W.F.; Bandera, F.; Cademartiri, F.; et al. Exercise Stress Echocardiography of the Right Ventricle and Pulmonary Circulation. J. Am. Coll. Cardiol. 2023, 82, 1973–1985. [Google Scholar] [CrossRef]
- Fabiani, I.; Pugliese, N.R.; Galeotti, G.G.; D’Agostino, A.; Mazzola, M.; Pedrinelli, R.; Dini, F.L. The Added Value of Exercise Stress Echocardiography in Patients with Heart Failure. Am. J. Cardiol. 2019, 123, 1470–1477. [Google Scholar] [CrossRef]
- Nesti, L.; Pugliese, N.R.; Santoni, L.; Armenia, S.; Chiriacò, M.; Sacchetta, L.; De Biase, N.; Del Punta, L.; Masi, S.; Tricò, D.; et al. Distinct effects of type 2 diabetes and obesity on cardiopulmonary performance. Diabetes Obes. Metab. 2024, 26, 351–361. [Google Scholar] [CrossRef]
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic processing in cardiometabolic disease. Atherosclerosis 2019, 281, 150–158. [Google Scholar] [CrossRef]
- Švorcová, J. Transgenerational Epigenetic Inheritance of Traumatic Experience in Mammals. Genes 2023, 14, 120. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2022, 7, 200. [Google Scholar] [CrossRef]
- Biagini, D.; Pugliese, N.R.; Vivaldi, F.M.; Ghimenti, S.; Lenzi, A.; De Angelis, F.; Ripszam, M.; Bruderer, T.; Armenia, S.; Cappeli, F.; et al. Breath analysis combined with cardiopulmonary exercise testing and echocardiography for monitoring heart failure patients: The AEOLUS protocol. J. Breath Res. 2023, 17, 046006. [Google Scholar] [CrossRef]
- Anderson, K.M.; Anderson, D.M. LncRNAs at the heart of development and disease. Mamm. Genome 2022, 33, 354–365. [Google Scholar] [CrossRef]
- Uchida, S.; Dimmeler, S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef]
- Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007, 14, 103–105. [Google Scholar] [CrossRef]
- Huang, Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J. Cell. Mol. Med. 2018, 22, 5768–5775. [Google Scholar] [CrossRef]
- Lozano-Vidal, N.; Bink, D.I.; Boon, R.A. Long noncoding RNA in cardiac aging and disease. J. Mol. Cell Biol. 2019, 11, 860–867. [Google Scholar] [CrossRef]
- Rech, M.; Barandiarán Aizpurua, A.; van Empel, V.; van Bilsen, M.; Schroen, B. Pathophysiological understanding of HFpEF: MicroRNAs as part of the puzzle. Cardiovasc. Res. 2018, 114, 782–793. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Liu, L.; An, X.; Li, Z.; Song, Y.; Li, L.; Zuo, S.; Liu, N.; Yang, G.; Wang, H.; Cheng, X.; et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc. Res. 2016, 111, 56–65. [Google Scholar] [CrossRef]
- Vausort, M.; Wagner, D.R.; Devaux, Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ. Res. 2014, 115, 668–677. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Liu, F.; Zhang, Y.-H.; Li, R.-B.; Zhai, M.; Liu, F.; Zhou, L.-Y.; Liu, C.-Y.; Yan, K.-W.; Dong, Y.-H.; et al. Long Noncoding RNA CPR (Cardiomyocyte Proliferation Regulator) Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2019, 139, 2668–2684. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.-T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Ounzain, S.; Micheletti, R.; Beckmann, T.; Schroen, B.; Alexanian, M.; Pezzuto, I.; Crippa, S.; Nemir, M.; Sarre, A.; Johnson, R.; et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur. Heart J. 2015, 36, 353–368. [Google Scholar] [CrossRef]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Conti, V.; Corbi, G.; Polito, M.V.; Ciccarelli, M.; Manzo, V.; Torsiello, M.; De Bellis, E.; D’Auria, F.; Vitulano, G.; Piscione, F.; et al. Sirt1 Activity in PBMCs as a Biomarker of Different Heart Failure Phenotypes. Biomolecules 2020, 10, 1590. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Mengozzi, A.; Costantino, S.; Paneni, F.; Duranti, E.; Nannipieri, M.; Mancini, R.; Lai, M.; La Rocca, V.; Puxeddu, I.; Antonioli, L.; et al. Targeting SIRT1 Rescues Age- and Obesity-Induced Microvascular Dysfunction in Ex Vivo Human Vessels. Circ. Res. 2022, 131, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, S.G.; Kim, J.-R.; Choi, H.C. Prednisolone suppresses adriamycin-induced vascular smooth muscle cell senescence and inflammatory response via the SIRT1-AMPK signaling pathway. PLoS ONE 2020, 15, e0239976. [Google Scholar] [CrossRef] [PubMed]
- Jęśko, H.; Wencel, P.; Strosznajder, R.P.; Strosznajder, J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2017, 42, 876–890. [Google Scholar] [CrossRef]
- Costantino, S.; Mengozzi, A.; Velagapudi, S.; Mohammed, S.A.; Gorica, E.; Akhmedov, A.; Mongelli, A.; Pugliese, N.R.; Masi, S.; Virdis, A.; et al. Treatment with recombinant Sirt1 rewires the cardiac lipidome and rescues diabetes-related metabolic cardiomyopathy. Cardiovasc. Diabetol. 2023, 22, 312. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Gan, D.; Luo, Z.; Yang, Q.; An, D.; Zhang, H.; Hu, Y.; Ma, Z.; Zeng, Q.; Xu, D.; et al. α-Ketoglutarate improves cardiac insufficiency through NAD(+)-SIRT1 signaling-mediated mitophagy and ferroptosis in pressure overload-induced mice. Mol. Med. 2024, 30, 15. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef]
- Nandave, M.; Acharjee, R.; Bhaduri, K.; Upadhyay, J.; Rupanagunta, G.P.; Ansari, M.N. A pharmacological review on SIRT 1 and SIRT 2 proteins, activators, and inhibitors: Call for further research. Int. J. Biol. Macromol. 2023, 242, 124581. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Podlutsky, A.; Kaminski, P.M.; Wolin, M.S.; Zhang, C.; Mukhopadhyay, P.; Pacher, P.; Hu, F.; de Cabo, R.; et al. Vasoprotective effects of resveratrol and SIRT1: Attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2721–H2735. [Google Scholar] [CrossRef]
- Fry, J.L.; Al Sayah, L.; Weisbrod, R.M.; Van Roy, I.; Weng, X.; Cohen, R.A.; Bachschmid, M.M.; Seta, F. Vascular Smooth Muscle Sirtuin-1 Protects against Diet-Induced Aortic Stiffness. Hypertension 2016, 68, 775–784. [Google Scholar] [CrossRef]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M.J.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef]
- Agarwal, B.; Campen, M.J.; Channell, M.M.; Wherry, S.J.; Varamini, B.; Davis, J.G.; Baur, J.A.; Smoliga, J.M. Resveratrol for primary prevention of atherosclerosis: Clinical trial evidence for improved gene expression in vascular endothelium. Int. J. Cardiol. 2013, 166, 246–248. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Méndez-del Villar, M.; González-Ortiz, M.; Martínez-Abundis, E.; Pérez-Rubio, K.G.; Lizárraga-Valdez, R. Effect of resveratrol administration on metabolic syndrome, insulin sensitivity, and insulin secretion. Metab. Syndr. Relat. Disord. 2014, 12, 497–501. [Google Scholar] [CrossRef]
- Movahed, A.; Nabipour, I.; Lieben Louis, X.; Thandapilly, S.J.; Yu, L.; Kalantarhormozi, M.; Rekabpour, S.J.; Netticadan, T. Antihyperglycemic effects of short term resveratrol supplementation in type 2 diabetic patients. Evid.-Based Complement. Altern. Med. 2013, 2013, 851267. [Google Scholar] [CrossRef]
- Pollack, R.M.; Barzilai, N.; Anghel, V.; Kulkarni, A.S.; Golden, A.; O’Broin, P.; Sinclair, D.A.; Bonkowski, M.S.; Coleville, A.J.; Powell, D.; et al. Resveratrol Improves Vascular Function and Mitochondrial Number but Not Glucose Metabolism in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Noh, R.M.; Venkatasubramanian, S.; Daga, S.; Langrish, J.; Mills, N.L.; Lang, N.N.; Hoffmann, E.; Waterhouse, B.; Newby, D.E.; Frier, B.M. Cardiometabolic effects of a novel SIRT1 activator, SRT2104, in people with type 2 diabetes mellitus. Open Heart 2017, 4, e000647. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef]
- Shankar, D.; Merchand-Reyes, G.; Buteyn, N.J.; Santhanam, R.; Fang, H.; Kumar, K.; Mo, X.; Ganesan, L.P.; Jarjour, W.; Butchar, J.P.; et al. Inhibition of BET Proteins Regulates Fcγ Receptor Function and Reduces Inflammation in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 7623. [Google Scholar] [CrossRef]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef]
- Borck, P.C.; Guo, L.-W.; Plutzky, J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ. Res. 2020, 126, 1190–1208. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef]
- Sun, Y.; Xie, Y.; Du, L.; Sun, J.; Liu, Z. Inhibition of BRD4 attenuates cardiomyocyte apoptosis via NF-κB pathway in a rat model of myocardial infarction. Cardiovasc. Ther. 2018, 36, e12320. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Alexanian, M.; Linares-Saldana, R.; González-Terán, B.; Andreoletti, G.; Huang, Y.; Connolly, A.J.; Kim, W.; Hsu, A.; Duan, Q.; et al. BRD4 (Bromodomain-Containing Protein 4) Interacts with GATA4 (GATA Binding Protein 4) to Govern Mitochondrial Homeostasis in Adult Cardiomyocytes. Circulation 2020, 142, 2338–2355. [Google Scholar] [CrossRef]
- Shahid, S.; Pantakani, M.; Binder, L.; Fischer, A.; Pantakani, K.; Asif, A.R. Small Molecule BRD4 Inhibitors Apabetalone and JQ1 Rescues Endothelial Cells Dysfunction, Protects Monolayer Integrity and Reduces Midkine Expression. Molecules 2022, 27, 7453. [Google Scholar] [CrossRef]
- Mohammed, S.A.; Albiero, M.; Ambrosini, S.; Gorica, E.; Karsai, G.; Caravaggi, C.M.; Masi, S.; Camici, G.G.; Wenzl, F.A.; Calderone, V.; et al. The BET Protein Inhibitor Apabetalone Rescues Diabetes-Induced Impairment of Angiogenic Response by Epigenetic Regulation of Thrombospondin-1. Antioxid. Redox Signal. 2022, 36, 667–684. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Kharenko, O.A.; Stotz, S.C.; Rakai, B.D.; Sarsons, C.D.; Gilham, D.; Wasiak, S.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. Breaking boundaries: Pan BETi disrupt 3D chromatin structure, BD2-selective BETi are strictly epigenetic transcriptional regulators. Biomed. Pharmacother. 2022, 152, 113230. [Google Scholar] [CrossRef]
- Piquereau, J.; Boet, A.; Péchoux, C.; Antigny, F.; Lambert, M.; Gressette, M.; Ranchoux, B.; Gambaryan, N.; Domergue, V.; Mumby, S.; et al. The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats. Int. J. Mol. Sci. 2019, 20, 1527. [Google Scholar] [CrossRef]
- Sun, J.; Gui, Y.; Zhou, S.; Zheng, X.-L. Unlocking the secrets of aging: Epigenetic reader BRD4 as the target to combatting aging-related diseases. J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Napoli, C.; Bontempo, P.; Palmieri, V.; Coscioni, E.; Maiello, C.; Donatelli, F.; Benincasa, G. Epigenetic Therapies for Heart Failure: Current Insights and Future Potential. Vasc. Health Risk Manag. 2021, 17, 247–254. [Google Scholar] [CrossRef]
- Ruscica, M.; Corsini, A.; Ferri, N.; Banach, M.; Sirtori, C.R. Clinical approach to the inflammatory etiology of cardiovascular diseases. Pharmacol. Res. 2020, 159, 104916. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients with Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA—J. Am. Med. Assoc. 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.J.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Rudraraju, R.; et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 2021, 184, 2167–2182. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech. Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Canale, P.; Campolo, J.; Borghini, A.; Andreassi, M.G. Long Telomeric Repeat-Containing RNA (TERRA): Biological Functions and Challenges in Vascular Aging and Disease. Biomedicines 2023, 11, 3211. [Google Scholar] [CrossRef]
- Ogrodnik, M. Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.-H.; Chen, H.-Z. Cardiomyocyte Senescence and Cellular Communications within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Zheng, X.; Diktonaite, K.; Qiu, H. Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential. Biomolecules 2023, 13, 1135. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Salerno, N.; Salerno, L.; Molinaro, C.; Cappetta, D.; Torella, M.; Foti, D.; Sasso, F.C.; Mastroroberto, P.; et al. Diabetes-Induced Cellular Senescence and Senescence-Associated Secretory Phenotype Impair Cardiac Regeneration and Function Independently of Age. Diabetes 2022, 71, 1081–1098. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Lu, Q.; Ren, D.; Sun, X.; Rousselle, T.; Tan, Y.; Li, J. AMPK: A therapeutic target of heart failure-not only metabolism regulation. Biosci. Rep. 2019, 39, BSR20181767. [Google Scholar] [CrossRef]
- Nehlin, J.O. Senolytic and senomorphic interventions to defy senescence-associated mitochondrial dysfunction. Adv. Protein Chem. Struct. Biol. 2023, 136, 217–247. [Google Scholar] [CrossRef]
- Belakova, B.; Wedige, N.K.; Awad, E.M.; Hess, S.; Oszwald, A.; Fellner, M.; Khan, S.Y.; Resch, U.; Lipovac, M.; Šmejkal, K.; et al. Lipophilic Statins Eliminate Senescent Endothelial Cells by inducing Anoikis-Related Cell Death. Cells 2023, 12, 2836. [Google Scholar] [CrossRef]
- Belcastro, E.; Rehman, A.U.; Remila, L.; Park, S.-H.; Gong, D.S.; Anton, N.; Auger, C.; Lefebvre, O.; Goetz, J.G.; Collot, M.; et al. Fluorescent nanocarriers targeting VCAM-1 for early detection of senescent endothelial cells. Nanomedicine 2021, 34, 102379. [Google Scholar] [CrossRef]
- Thapa, R.K.; Nguyen, H.T.; Jeong, J.-H.; Kim, J.R.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Progressive slowdown/prevention of cellular senescence by CD9-targeted delivery of rapamycin using lactose-wrapped calcium carbonate nanoparticles. Sci. Rep. 2017, 7, 43299. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Sardu, C.; Onofrio, N.D.; Torella, M.; Portoghese, M.; Mureddu, S.; Loreni, F.; Ferraraccio, F.; Panarese, I.; Trotta, M.C.; Gatta, G.; et al. Metformin Therapy Effects on the Expression of Pericoronary Fat Excised from Pre-Diabetic Patients with Acute Myocardial Infarction. Biomedicines 2021, 9, 904. [Google Scholar] [CrossRef]
- Onofrio, N.D.; Sardu, C.; Trotta, M.C.; Scisciola, L.; Turriziani, F.; Ferraraccio, F.; Panarese, I.; Petrella, L.; Fanelli, M.; Modugno, P.; et al. Sodium-glucose co-transporter2 expression and in fl ammatory activity in diabetic atherosclerotic plaques: Effects of sodium-glucose co-transporter2 inhibitor treatment. Mol. Metab. 2021, 54, 101337. [Google Scholar] [CrossRef]
- Nesti, L.; Pugliese, N.R.; Sciuto, P.; Natali, A. Type 2 diabetes and reduced exercise tolerance: A review of the literature through an integrated physiology approach. Cardiovasc. Diabetol. 2020, 19, 134. [Google Scholar] [CrossRef]
- Guazzi, M.; Bandera, F.; Ozemek, C.; Systrom, D.; Arena, R. Cardiopulmonary Exercise Testing What Is its Value? J. Am. Coll. Cardiol. 2017, 70, 1618–1636. [Google Scholar] [CrossRef]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic opportunities for senolysis in cardiovascular disease. FEBS J. 2023, 290, 1235–1255. [Google Scholar] [CrossRef]

| Main Group | Traditional Biomarkers | Novel Biomarkers | Value and New Perspectives |
|---|---|---|---|
| Epicardial adiposity |
|
|
|
| Congestion |
|
|
|
| Ventricular–arterial coupling |
|
|
|
| Exercise capacity |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Fiore, V.; Cappelli, F.; Del Punta, L.; De Biase, N.; Armenia, S.; Maremmani, D.; Lomonaco, T.; Biagini, D.; Lenzi, A.; Mazzola, M.; et al. Novel Techniques, Biomarkers and Molecular Targets to Address Cardiometabolic Diseases. J. Clin. Med. 2024, 13, 2883. https://doi.org/10.3390/jcm13102883
Di Fiore V, Cappelli F, Del Punta L, De Biase N, Armenia S, Maremmani D, Lomonaco T, Biagini D, Lenzi A, Mazzola M, et al. Novel Techniques, Biomarkers and Molecular Targets to Address Cardiometabolic Diseases. Journal of Clinical Medicine. 2024; 13(10):2883. https://doi.org/10.3390/jcm13102883
Chicago/Turabian StyleDi Fiore, Valerio, Federica Cappelli, Lavinia Del Punta, Nicolò De Biase, Silvia Armenia, Davide Maremmani, Tommaso Lomonaco, Denise Biagini, Alessio Lenzi, Matteo Mazzola, and et al. 2024. "Novel Techniques, Biomarkers and Molecular Targets to Address Cardiometabolic Diseases" Journal of Clinical Medicine 13, no. 10: 2883. https://doi.org/10.3390/jcm13102883