
The Identification of RPL4 as a Hub Gene Associated with Goat Litter Size via Weighted Gene Co-Expression Network Analysis
 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Processing
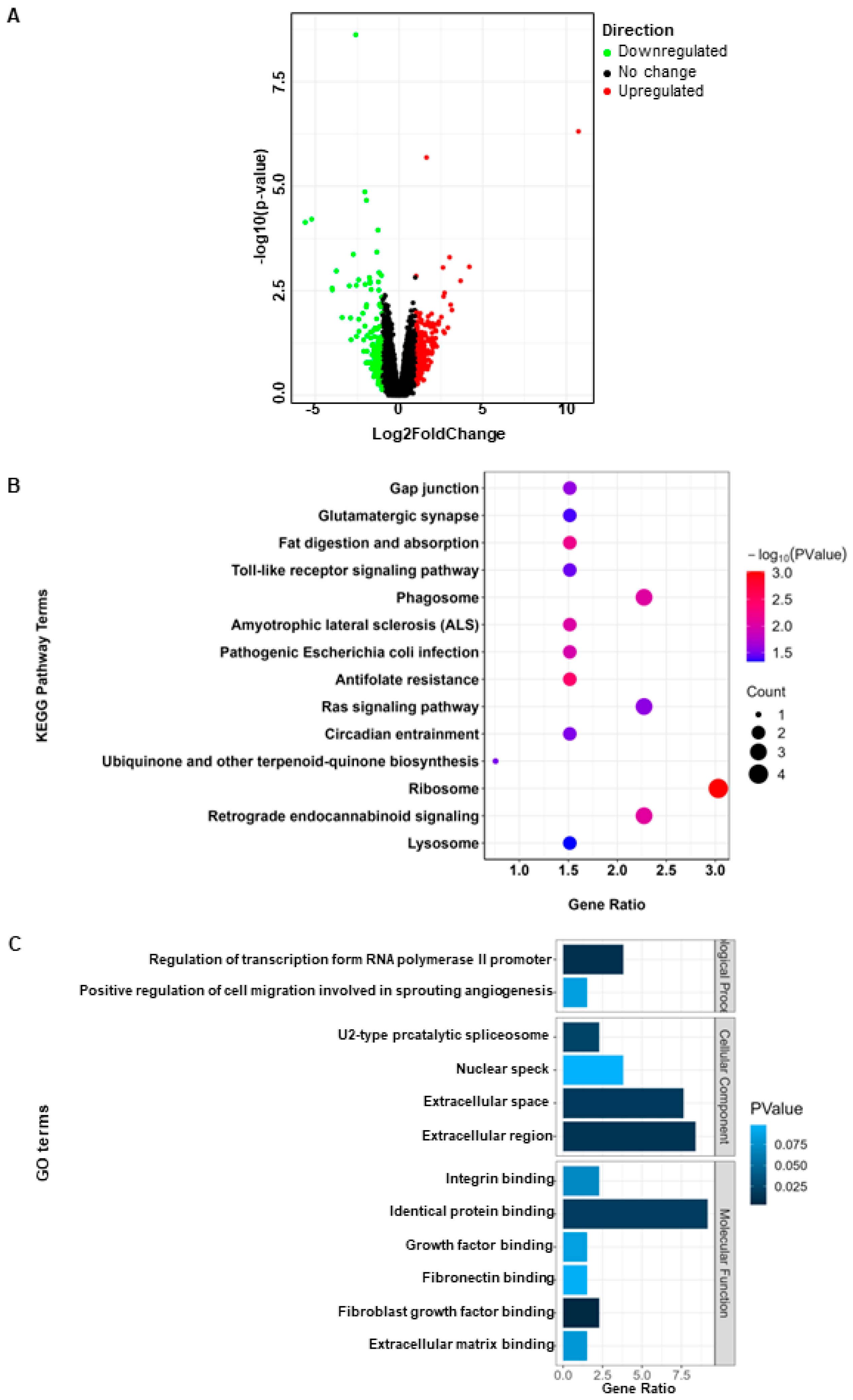
2.2. Identification of DEGs
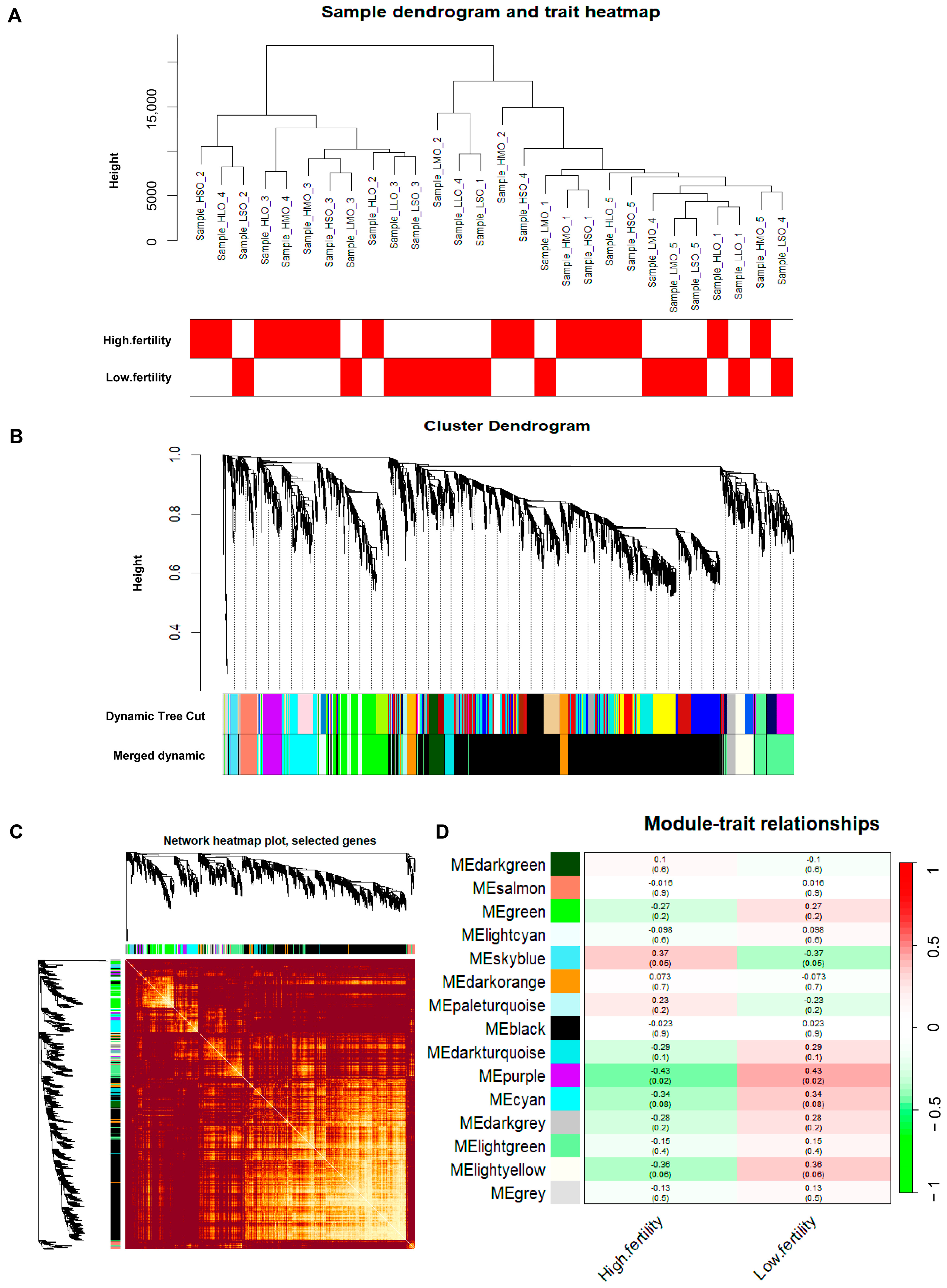
2.3. Construction of the Gene Co-Expression Network
2.4. Construction of Protein–Protein Interaction (PPI) Network
3. Results
3.1. Identification of Litter Size-Related DEGs in Goat Oocytes
3.2. Weighted Gene Co-Expression Network Construction
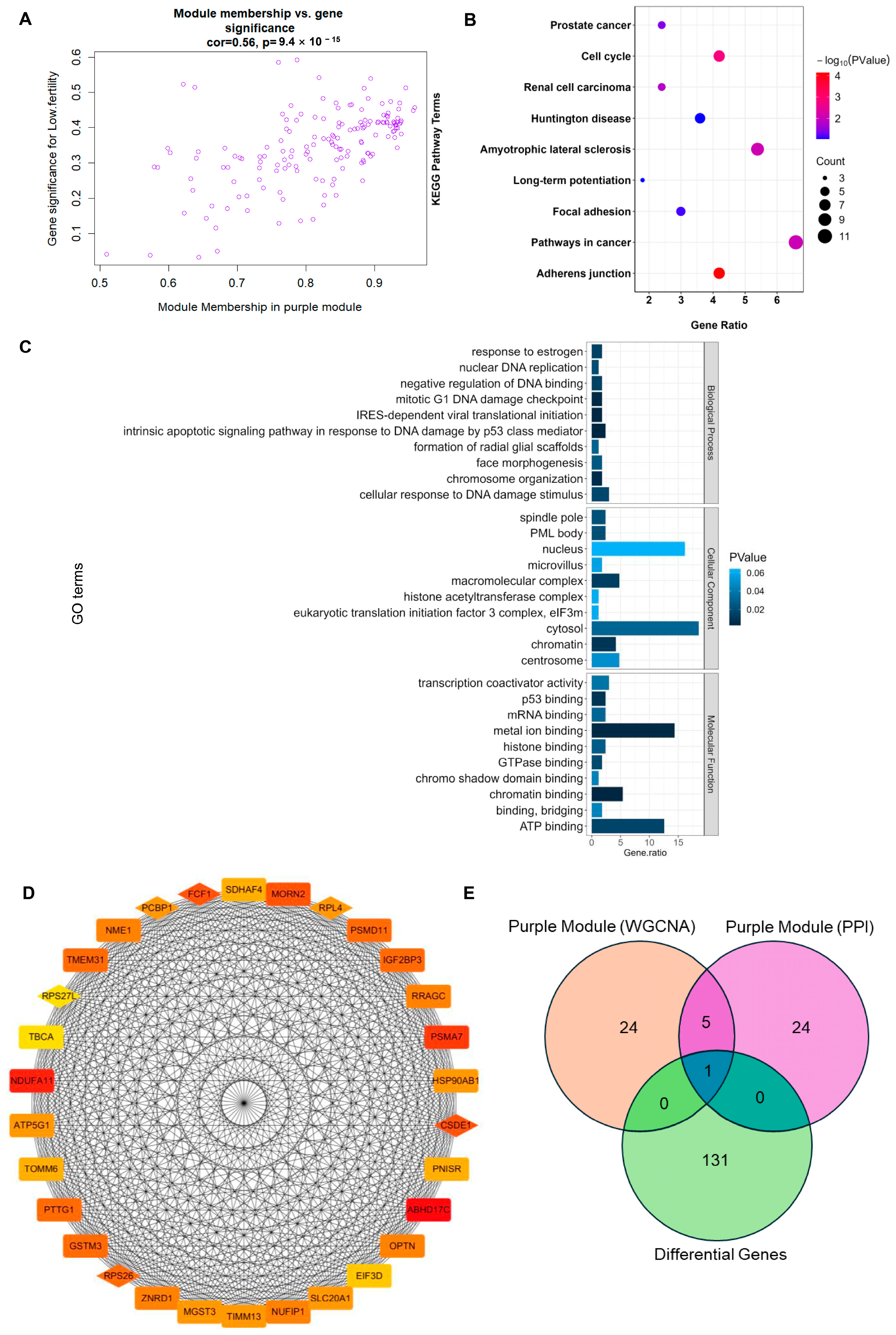
3.3. Screening and Functional Enrichment Analysis of Hub Gene in MEpurple Module
3.4. Identification of RPL4 as a Hub Gene Associated with Goat Litter Size
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alemayehu, G.; Mamo, G.; Alemu, B.; Desta, H.; Wieland, B. Towards objective measurement of reproductive performance of traditionally managed goat flocks in the drylands of Ethiopia. Trop. Anim. Health Prod. 2021, 53, 156. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, M.S.; Asadi Fozi, M.; Gutierrez, J.P.; Notter, D.R. Genetic and phenotypic aspects of early reproductive performance in Raeini Cashmere goats. Trop. Anim. Health Prod. 2019, 51, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Zarazaga, L.A.; Gatica, M.C.; Celi, I.; Guzman, J.L. Reproductive performance is improved during seasonal anoestrus when female and male Murciano-Granadina goats receive melatonin implants and in Payoya goats when females are thus treated. Reprod. Domest. Anim. 2012, 47, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Moaeen-ud-Din, M.; Yand, L.G.; Chen, S.L.; Zhang, Z.R.; Xiao, J.Z.; Wen, Q.Y.; Dai, M. Reproductive performance of Matou goat under sub-tropical monsoonal climate of Central China. Trop. Anim. Health Prod. 2008, 40, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Tiezzi, F.; Tomassone, L.; Mancin, G.; Cornale, P.; Tarantola, M. The Assessment of Housing Conditions, Management, Animal-Based Measure of Dairy Goats’ Welfare and Its Association with Productive and Reproductive Traits. Animals 2019, 9, 893. [Google Scholar] [CrossRef] [PubMed]
- Ejlertsen, M.; Poole, J.; Marshall, K. Traditional breeding objectives and practices of goat, sheep and cattle smallholders in The Gambia and implications in relation to the design of breeding interventions. Trop. Anim. Health Prod. 2013, 45, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.P.; Sreevatsan, S. Global-scale GWAS associates a subset of SNPs with animal-adapted variants in M. tuberculosis complex. BMC Med. Genom. 2023, 16, 260. [Google Scholar] [CrossRef] [PubMed]
- Schroder, J.; Chegwidden, L.; Maj, C.; Gehlen, J.; Speller, J.; Bohmer, A.C.; Borisov, O.; Hess, T.; Kreuser, N.; Venerito, M.; et al. GWAS meta-analysis of 16 790 patients with Barrett’s oesophagus and oesophageal adenocarcinoma identifies 16 novel genetic risk loci and provides insights into disease aetiology beyond the single marker level. Gut 2023, 72, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Royrvik, E.C.; Aranda-Guillen, M.; Berger, A.H.; Landegren, N.; Artaza, H.; Hallgren, A.; Grytaas, M.A.; Strom, S.; Bratland, E.; et al. GWAS for autoimmune Addison’s disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat. Commun. 2021, 12, 959. [Google Scholar] [CrossRef]
- Bai, Y.; Li, J.; Zhu, H.; Liu, J.; Dong, S.; Li, L.; Qu, L.; Chen, H.; Song, X.; Lan, X. Deletion mutation within the goat PPP3CA gene identified by GWAS significantly affects litter size. Reprod. Fertil. Dev. 2021, 33, 476–483. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Zhang, H.; Ma, D.; Zhao, M.; Li, N.; Men, Y.; Zhang, Y.; Chu, H.; Lei, C.; et al. Transcriptome profile of goat folliculogenesis reveals the interaction of oocyte and granulosa cell in correlation with different fertility population. Sci. Rep.-UK 2021, 11, 15698. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Puig, R.R.; Holmas, S.; Mironov, V.; Kuiper, M. Network Building with the Cytoscape BioGateway App Explained in Five Use Cases. Curr. Protoc. Bioinform. 2020, 72, e106. [Google Scholar] [CrossRef] [PubMed]
- Calanni-Pileri, M.; Weitzel, J.M.; Langhammer, M.; Michaelis, M. Higher quality rather than superior quantity of oocytes determine the amount of fertilizable oocytes in two outbred Dummerstorf high-fertility mouse lines. Reprod. Domest. Anim. 2022, 57, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fabjan, J.M.; Locatelli, Y.; Duffard, N.; Corbin, E.; Batista, R.I.; de Figueiredo Freitas, V.J.; Beckers, J.F.; Mermillod, P. Intrinsic quality of goat oocytes already found denuded at collection for in vitro embryo production. Theriogenology 2016, 86, 1989–1998. [Google Scholar] [CrossRef]
- Su, Y.Q.; Wu, X.; O’Brien, M.J.; Pendola, F.L.; Denegre, J.N.; Matzuk, M.M.; Eppig, J.J. Synergistic roles of BMP15 and GDF9 in the development and function of the oocyte-cumulus cell complex in mice: Genetic evidence for an oocyte-granulosa cell regulatory loop. Dev. Biol. 2004, 276, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kumchoo, T.; Mekchay, S. Association of NR4A1 and GNB2L1 genes with reproductive traits in commercial pig breeds. Genet. Mol. Res. 2015, 14, 16276–16284. [Google Scholar] [CrossRef]
- Sbardella, A.P.; Watanabe, R.N.; da Costa, R.M.; Bernardes, P.A.; Braga, L.G.; Baldi Rey, F.S.; Lobo, R.B.; Munari, D.P. Genome-Wide Association Study Provides Insights into Important Genes for Reproductive Traits in Nelore Cattle. Animals 2021, 11, 1386. [Google Scholar] [CrossRef]
- Rekawiecki, R.; Kisielewska, K.; Kowalik, M.K.; Kotwica, J. Methylation of progesterone receptor isoform A and B promoters in the reproductive system of cows. Reprod. Fertil. Dev. 2018, 30, 1634–1642. [Google Scholar] [CrossRef]
- Vieira, I.H.; Carvalho, A.F.; Almeida Reis, S.; Carreira, A.L.; Dias, C.; Fernandes, S.; Ferreira, A.F.; Rodrigues, D.; Sousa, A.P.; Ramalho-Santos, J.; et al. Association Between Follicle-Stimulating Hormone Receptor (FSHR) rs6166 and Estrogen Receptor 1 (ESR1) rs2234693 Polymorphisms and Polycystic Ovary Syndrome Risk, Phenotype, and Reproductive Outcomes in an Infertile Portuguese Population. Cureus 2023, 15, e35690. [Google Scholar] [CrossRef]
- Abd El Fattah, E.M.; Behour, T.S.; Ashour, A.F.; Amin, A.M.S. Association analysis of prolactin and prolactin receptor genes with selected productive and reproductive traits in Egyptian buffalo. Anim. Biotechnol. 2023, 34, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Innos, J.; Koido, K.; Philips, M.A.; Vasar, E. Limbic system associated membrane protein as a potential target for neuropsychiatric disorders. Front. Pharmacol. 2013, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, E.; Krohn, K.; Ovaska, K.; Lindberg, P.; Hayry, V.; Maliniemi, P.; Lintulahti, A.; Korja, M.; Kivisaari, R.; Hussein, S.; et al. Neuron navigator 3 alterations in nervous system tumors associate with tumor malignancy grade and prognosis. Genes Chromosomes Cancer 2013, 52, 191–201. [Google Scholar] [CrossRef]
- Tan, G.; Zhang, G.Y.; Xu, J.; Kang, C.W.; Yan, Z.K.; Lei, M.; Pu, X.B.; Dong, C.C. PLA2G10 facilitates the cell-cycle progression of soft tissue leiomyosarcoma cells at least by elevating cyclin E1/CDK2 expression. Biochem. Biophys. Res. Commun. 2020, 527, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Keane, S.; Ameen, S.; Lindlof, A.; Ejeskar, K. Low DLG2 gene expression, a link between 11q-deleted and MYCN-amplified neuroblastoma, causes forced cell cycle progression, and predicts poor patient survival. Cell Commun. Signal. 2020, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Perez, B.; Hansen, P.J.; Mur-Novales, R.; Garcia-Ispierto, I.; de Sousa, N.M.; Beckers, J.F.; Almeria, S.; Lopez-Gatius, F. Crosstalk between uterine serpin (SERPINA14) and pregnancy-associated glycoproteins at the fetal-maternal interface in pregnant dairy heifers experimentally infected with Neospora caninum. Theriogenology 2016, 86, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp-Demtroder, K.; Mansilla, F.; Sorensen, F.B.; Kruhoffer, M.; Cabezon, T.; Christensen, L.L.; Aaltonen, L.A.; Verspaget, H.W.; Orntoft, T.F. Phosphoprotein Keratin 23 accumulates in MSS but not MSI colon cancers in vivo and impacts viability and proliferation in vitro. Mol. Oncol. 2007, 1, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wei, C.; Wang, C.; Li, F.; Wang, Z.; Xiong, J.; Zhou, Y.; Li, S.; Liu, X.; Yang, G.; et al. TIMP1/CHI3L1 facilitates glioma progression and immunosuppression via NF-kappaB activation. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167041. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, S.; Wang, H.; Zhang, Y.; Feng, T.; Chen, B.; He, Y.; Zeng, Z.; Chen, M. TNFAIP6 is a potential biomarker of disease activity in inflammatory bowel disease. Biomark. Med. 2016, 10, 473–483. [Google Scholar] [CrossRef]
- Kimura, Y.; Katoh, A.; Kaneko, T.; Takahama, K.; Tanaka, H. Two members of the IgLON family are expressed in a restricted region of the developing chick brain and neural crest. Dev. Growth Differ. 2001, 43, 257–263. [Google Scholar] [CrossRef]
- Bazer, F.W.; Roberts, R.M.; Basha, S.M.; Zavy, M.T.; Caton, D.; Barron, D.H. Method for obtaining ovine uterine secretions from unilaterally pregnant ewes. J. Anim. Sci. 1979, 49, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.; Bazer, F.W.; Hansen, P.J.; Chun, P.W.; Roberts, R.M. Purification, secretion and immunocytochemical localization of the uterine milk proteins, major progesterone-induced proteins in uterine secretions of the sheep. Biol. Reprod 1987, 36, 419–430. [Google Scholar] [CrossRef]
- Bauersachs, S.; Ulbrich, S.E.; Gross, K.; Schmidt, S.E.; Meyer, H.H.; Einspanier, R.; Wenigerkind, H.; Vermehren, M.; Blum, H.; Sinowatz, F.; et al. Gene expression profiling of bovine endometrium during the oestrous cycle: Detection of molecular pathways involved in functional changes. J. Mol. Endocrinol. 2005, 34, 889–908. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Bauersachs, S.; Ulbrich, S.E.; Einspanier, R.; Meyer, H.H.; Schmidt, S.E.; Reichenbach, H.D.; Vermehren, M.; Sinowatz, F.; Blum, H.; et al. Monozygotic twin model reveals novel embryo-induced transcriptome changes of bovine endometrium in the preattachment period. Biol. Reprod 2006, 74, 253–264. [Google Scholar] [CrossRef]
- Liu, B.; Xu, Q.; Wang, Q.; Feng, S.; Lai, F.; Wang, P.; Zheng, F.; Xiang, Y.; Wu, J.; Nie, J.; et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 2020, 587, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.S.; Gao, L.; Xie, X.L.; Ren, Y.L.; Shen, Z.Q.; Wang, F.; Shen, M.; Eyϸorsdottir, E.; Hallsson, J.H.; Kiseleva, T.; et al. Genome-Wide Association Analyses Highlight the Potential for Different Genetic Mechanisms for Litter Size Among Sheep Breeds. Front. Genet. 2018, 9, 118. [Google Scholar] [CrossRef]
- Abdoli, R.; Zamani, P.; Mirhoseini, S.Z.; Ghavi Hossein-Zadeh, N.; Nadri, S. A review on prolificacy genes in sheep. Reprod. Domest. Anim. 2016, 51, 631–637. [Google Scholar] [CrossRef]
- Smith, P.; Hudson, N.L.; Corrigan, K.A.; Shaw, L.; Smith, T.; Phillips, D.J.; McNatty, K.P. Effects of the Booroola gene (FecB(B)) on bodymass, testis development and hormone concentrations during fetal life. J. Reprod. Fertil 1996, 108, 253–261. [Google Scholar] [CrossRef]
- Demars, J.; Fabre, S.; Sarry, J.; Rossetti, R.; Gilbert, H.; Persani, L.; Tosser-Klopp, G.; Mulsant, P.; Nowak, Z.; Drobik, W.; et al. Genome-wide association studies identify two novel BMP15 mutations responsible for an atypical hyperprolificacy phenotype in sheep. PLoS Genet. 2013, 9, e1003482. [Google Scholar] [CrossRef]
- Grammont, M. Adherens junction remodeling by the Notch pathway in Drosophila melanogaster oogenesis. J. Cell Biol. 2007, 177, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Liu, C.; Chen, Y.; Cheng, X.; Shen, J.; Zhao, L.; Zhang, J. The mitochondrial ribosomal protein mRpL4 regulates Notch signaling. EMBO Rep. 2023, 24, e55764. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, Y.; Dai, M.S.; Sun, X.X. Ribosomal protein L4 is a novel regulator of the MDM2-p53 loop. Oncotarget 2016, 7, 16217–16226. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | High Litter Size Group vs. Low Litter Size Group |
|---|---|
| Upregulated genes (top 10) | LSAMP; TMEM80; DEFB116; SMCO4; LOC102187672; DLG2; PLA2G10; LOC102179185; GNG11; LOC102170446 |
| Downregulated genes (top 10) | NAV3; ENSCHIG00000010736; KRT23; SLC10A6; TIMP1; THBS1; SERPINA14; TNFAIP6; RPS29; TMSB4X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Tang, X.; Li, D.; Tong, X.; Min, L.; Chen, W.; Ju, X.; Xu, B. The Identification of RPL4 as a Hub Gene Associated with Goat Litter Size via Weighted Gene Co-Expression Network Analysis. Animals 2024, 14, 1470. https://doi.org/10.3390/ani14101470
Zhang Z, Tang X, Li D, Tong X, Min L, Chen W, Ju X, Xu B. The Identification of RPL4 as a Hub Gene Associated with Goat Litter Size via Weighted Gene Co-Expression Network Analysis. Animals. 2024; 14(10):1470. https://doi.org/10.3390/ani14101470
Chicago/Turabian StyleZhang, Zhifei, Xueying Tang, Dagang Li, Xiong Tong, Li Min, Weidong Chen, Xianghong Ju, and Bin Xu. 2024. "The Identification of RPL4 as a Hub Gene Associated with Goat Litter Size via Weighted Gene Co-Expression Network Analysis" Animals 14, no. 10: 1470. https://doi.org/10.3390/ani14101470