
A Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred

 , , and
, , and
Abstract
1. Introduction
2. Material and Methods
2.1. Family Enrolment
2.2. Exome and Sanger Sequencing
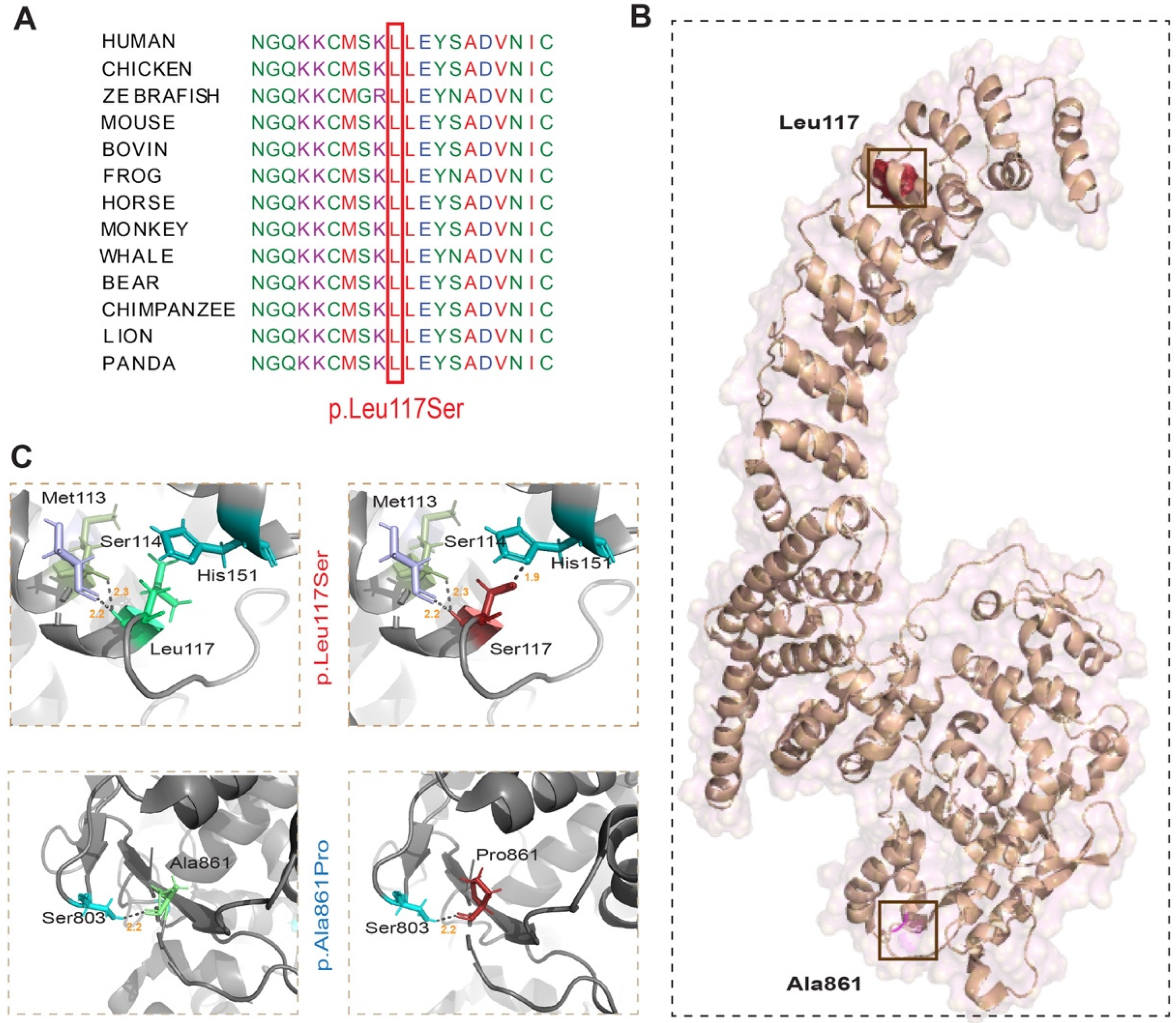
2.3. In-Silico Analysis and 3D Protein Modelling
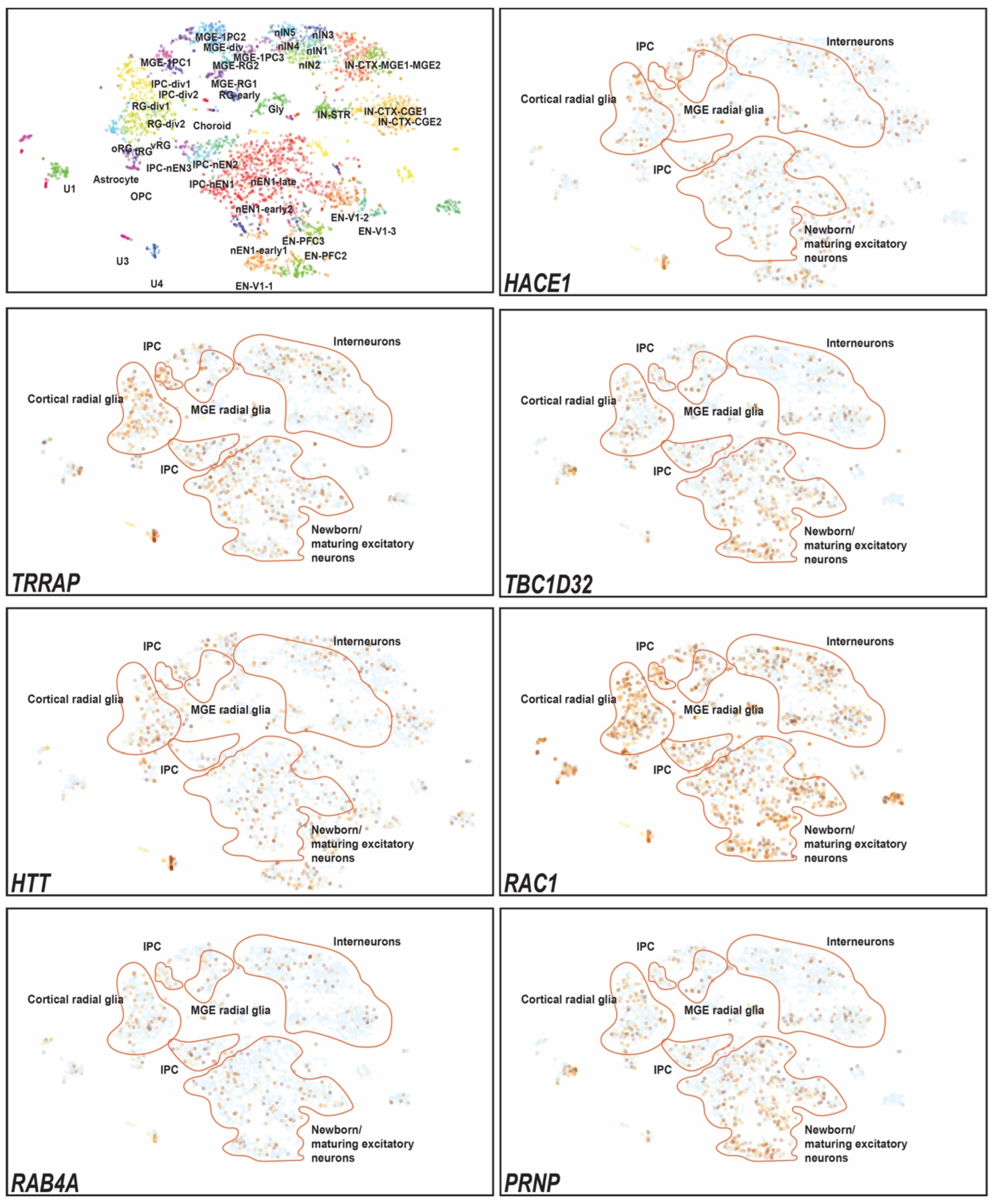
3. Results
3.1. Family PKMR285
3.2. Genetic Studies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, D.R.; Merrick, J. Neurodevelopmental and neurobehavioral disorders. Transl. Pediatr. 2020, 9 (Suppl. S1), S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Francés, L.; Quintero, J.; Fernández, A.; Ruiz, A.; Caules, J.; Fillon, G.; Hervás, A.; Soler, C.V. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: A systematic review in accordance with the PRISMA criteria. Child Adolesc. Psychiatry Ment. Health 2022, 16, 27. [Google Scholar] [CrossRef]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping autosomal recessive intellectual disability: Combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol. Psychiatry 2018, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Jamra, R. Genetics of autosomal recessive intellectual disability. Med. Genetik. 2018, 30, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Turgu, B.; El-Naggar, A.; Kogler, M.; Tortola, L.; Zhang, H.-F.; Hassan, M.; Lizardo, M.M.; Kung, S.H.; Lam, W.; Penninger, J.M.; et al. The HACE1 E3 ligase mediates RAC1-dependent control of mTOR signaling complexes. EMBO Rep. 2023, 24, e56815. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Evdokimova, V.; Melnyk, N.; Zhang, L.; Fernandez, C.V.; Grundy, P.E.; Leach, S.; Marra, M.A.; Brooks-Wilson, A.R.; Penninger, J.; et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Hum. Mol. Genet. 2004, 13, 2061–2074. [Google Scholar] [CrossRef]
- Hollstein, R.; A Parry, D.; Nalbach, L.; Logan, C.V.; Strom, T.M.; Hartill, V.L.; Carr, I.M.; Korenke, G.C.; Uppal, S.; Ahmed, M.; et al. HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J. Med. Genet. 2015, 52, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Mulherkar, S.; Uddin, M.D.; Couvillon, A.D.; Sillitoe, R.V.; Tolias, K.F. The small GTPases RhoA and Rac1 regulate cerebellar development by controlling cell morphogenesis, migration and foliation. Dev. Biol. 2014, 394, 39–53. [Google Scholar] [CrossRef]
- Zang, C.; Liu, H.; Ning, J.; Chen, Q.; Jiang, Y.; Shang, M.; Yang, Y.; Ma, J.; Dong, Y.; Wang, J.; et al. Emerging role and mechanism of HACE1 in the pathogenesis of neurodegenerative diseases: A promising target. Biomed. Pharmacother. 2024, 172, 116204. [Google Scholar] [CrossRef]
- Yu, S.; Levi, L.; Siegel, R.; Noy, N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ). J. Biol. Chem. 2012, 287, 42195–42205. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Leypoldt, F.; Methner, A. Degenerate Suppression PCR Identifies the β2-Adrenergic Receptor as Upregulated by Neuronal Differentiation. Gene Expr. J. Liver Res. 2003, 11, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.; So, P.-L.; Barber, R.D.; Vincent, K.J.; Mazarakis, N.D.; Mitrophanous, K.A.; Kingsman, S.M.; Maden, M. Retinoic acid receptor β2 and neurite outgrowth in the adult mouse spinal cord in vitro. J. Cell Sci. 2002, 115, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Akawi, N.; the DDD study; McRae, J.; Ansari, M.; Balasubramanian, M.; Blyth, M.; Brady, A.F.; Clayton, S.; Cole, T.; Deshpande, C.; et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat. Genet. 2015, 47, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Hollstein, R.; Pai, T.-P.; Herde, M.K.; Buphamalai, P.; Moeseneder, P.; Lenartowicz, E.; Kavirayani, A.; Korenke, G.C.; Kozieradzki, I.; et al. HACE1 deficiency leads to structural and functional neurodevelopmental defects. Neurol. Genet. 2019, 5, e330. [Google Scholar] [CrossRef]
- Ugarteburu, O.; Sánchez-Vilés, M.; Ramos, J.; Barcos-Rodríguez, T.; Garrabou, G.; García-Villoria, J.; Ribes, A.; Tort, F. Physiopathological bases of the disease caused by HACE1 mutations: Alterations in autophagy, mitophagy and oxidative stress response. J. Clin. Med. 2020, 9, 913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.; Sharma, P.; Moon, M.; Sheftel, A.D.; Dawood, F.; Nghiem, M.P.; Wu, J.; Li, R.-K.; Gramolini, A.O.; et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat. Commun. 2014, 5, 3430. [Google Scholar] [CrossRef] [PubMed]
- Tapias, A.; Zhou, Z.-W.; Shi, Y.; Chong, Z.; Wang, P.; Groth, M.; Platzer, M.; Huttner, W.; Herceg, Z.; Yang, Y.-G.; et al. Trrap-dependent histone acetylation specifically regulates cell-cycle gene transcription to control neural progenitor fate decisions. Cell Stem Cell 2014, 14, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, B.; Borday, C.; Erkilic, N.; Mamaeva, D.; Donval, A.; Masson, C.; Parain, K.; Kaminska, K.; Quinodoz, M.; Perea-Romero, I.; et al. TBC1D32 variants disrupt retinal ciliogenesis and cause retinitis pigmentosa. JCI Insight 2023, 8, e169426. [Google Scholar] [CrossRef]
- Tong, Y.; Ha, T.J.; Liu, L.; Nishimoto, A.; Reiner, A.; Goldowitz, D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J. Neurosci. 2011, 31, 14794–14799. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Southwell, A.L.; Sivasubramanian, M.; Qiu, X.; Villanueva, E.B.; Xie, Y.; Waltl, S.; Anderson, L.; Fazeli, A.; Casal, L.; et al. HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum. Mol. Genet. 2018, 27, 239–253. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, A.M.; Clarkson, P.W.; Negri, G.L.; Turgu, B.; Zhang, F.; Anglesio, M.S.; Sorensen, P.H. HACE1 is a potential tumor suppressor in osteosarcoma. Cell Death Dis. 2019, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Nitsch, R.; Razaghi, B.; McDonald, L.; Jarrar, A.; Torrino, S.; Castillo-Lluva, S.; Rotblat, B.; Li, L.; Malliri, A.; et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 2013, 4, 2180. [Google Scholar] [CrossRef]
- White, J.A.; Krzystek, T.J.; Hoffmar-Glennon, H.; Thant, C.; Zimmerman, K.; Iacobucci, G.; Vail, J.; Thurston, L.; Rahman, S.; Gunawardena, S. Excess Rab4 rescues synaptic and behavioral dysfunction caused by defective HTT-Rab4 axonal transport in Huntington’s disease. Acta Neuropathol. Commun. 2020, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Bribián, A.; Fontana, X.; Llorens, F.; Gavín, R.; Reina, M.; García-Verdugo, J.M.; Torres, J.M.; de Castro, F.; del Río, J.A. Role of the cellular prion protein in oligodendrocyte precursor cell proliferation and differentiation in the developing and adult mouse CNS. PLoS ONE. 2012, 7, e33872. [Google Scholar] [CrossRef]
- Ebstein, F.; Küry, S.; Papendorf, J.J.; Krüger, E. Neurodevelopmental Disorders (NDD) Caused by Genomic Alterations of the Ubiquitin-Proteasome System (UPS): The Possible Contribution of Immune Dysregulation to Disease Pathogenesis. Front. Mol. Neurosci. 2021, 14, 733012. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Family | PKMR285 | Family 4 | Family 1 [14] |
|---|---|---|---|
| Origin | Pakistan | United Kingdom | Turkish |
| Affected Individuals | 2 Siblings | 2 Siblings | 3 Siblings |
| Inheritance | Homozygous | Homozygous | Homozygous |
| hg19 Coordinates | 6:105291150 A>G | 6:105178224 C>G | 6:105192058-60del AAG |
| Nucleotide Variant | c.350 T>C | c.2581 G>C | c.2494-96del CTT |
| Amino Acid Substitution | p.Leu117Ser | p.Ala861Pro | p.Leu832del |
| Nucleotide Reference | NM_020771.4 | ||
| ACMG Classification | Pathogenic Strong | Pathogenic Moderate | Pathogenic |
| ACMG Criteria | PP3 | PP3 | NA |
| Meta Score a | 15 | NA | |
| gnomAD Frequencies | Zero | Zero | NA |
| DANN | Uncertain | Uncertain | NA |
| CADD | 26.8 | 24.5 | NA |
| REVEL | Pathogenic Moderate | Benign Moderate | NA |
| M-CAP | Damaging | Tolerated | NA |
| DOGEN2 | Benign Supporting | Benign Moderate | NA |
| PROVEAN | Pathogenic Supporting | Benign Moderate | NA |
| Polyphen-2 HumDiv | Probably Damaging | Probably Damaging | NA |
| Polyphen-2 HumVar | Probably Damaging | Possibly Damaging | NA |
| GERP++ | 5.92 | 4.96 | NA |
| phyloP 100way Vertebrate | 8.904 | 7.455 | NA |
| Mutation Taster | Disease-Causing | Disease-Causing | NA |
| FATHMM | Uncertain | Benign Supporting | NA |
| SIFT | Pathogenic Supporting | Benign Moderate | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usmani, M.A.; Ghaffar, A.; Shahzad, M.; Akram, J.; Majeed, A.I.; Malik, K.; Fatima, K.; Khan, A.A.; Ahmed, Z.M.; Riazuddin, S.; et al. A Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred. Genes 2024, 15, 580. https://doi.org/10.3390/genes15050580
Usmani MA, Ghaffar A, Shahzad M, Akram J, Majeed AI, Malik K, Fatima K, Khan AA, Ahmed ZM, Riazuddin S, et al. A Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred. Genes. 2024; 15(5):580. https://doi.org/10.3390/genes15050580
Chicago/Turabian StyleUsmani, Muhammad A., Amama Ghaffar, Mohsin Shahzad, Javed Akram, Aisha I. Majeed, Kausar Malik, Khushbakht Fatima, Asma A. Khan, Zubair M. Ahmed, Sheikh Riazuddin, and et al. 2024. "A Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred" Genes 15, no. 5: 580. https://doi.org/10.3390/genes15050580
APA StyleUsmani, M. A., Ghaffar, A., Shahzad, M., Akram, J., Majeed, A. I., Malik, K., Fatima, K., Khan, A. A., Ahmed, Z. M., Riazuddin, S., & Riazuddin, S. (2024). A Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred. Genes, 15(5), 580. https://doi.org/10.3390/genes15050580