
Polyglycerol Sebacate Elastomer: A Critical Overview of Synthetic Methods and Characterisation Techniques







Abstract
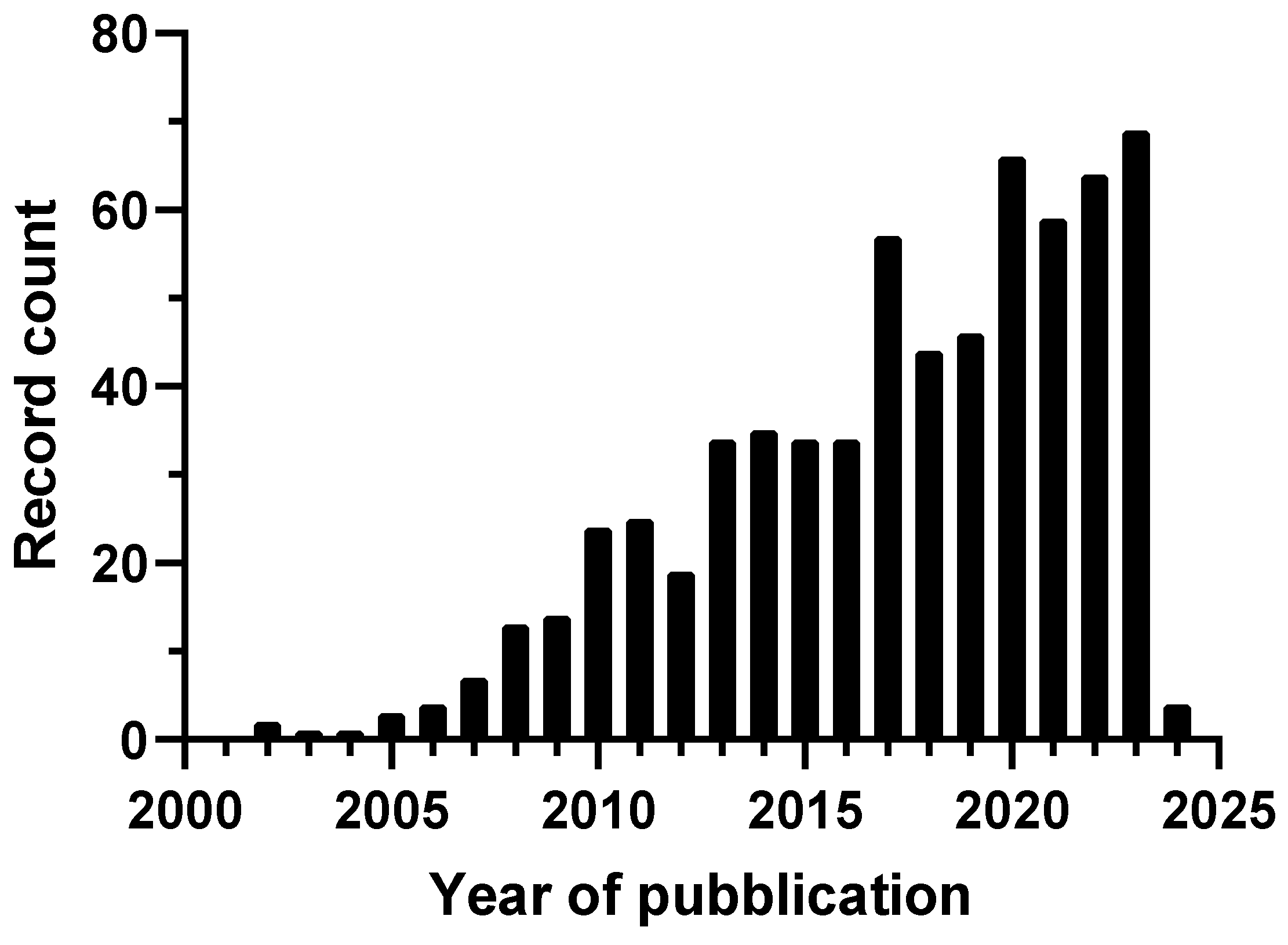
1. Introduction
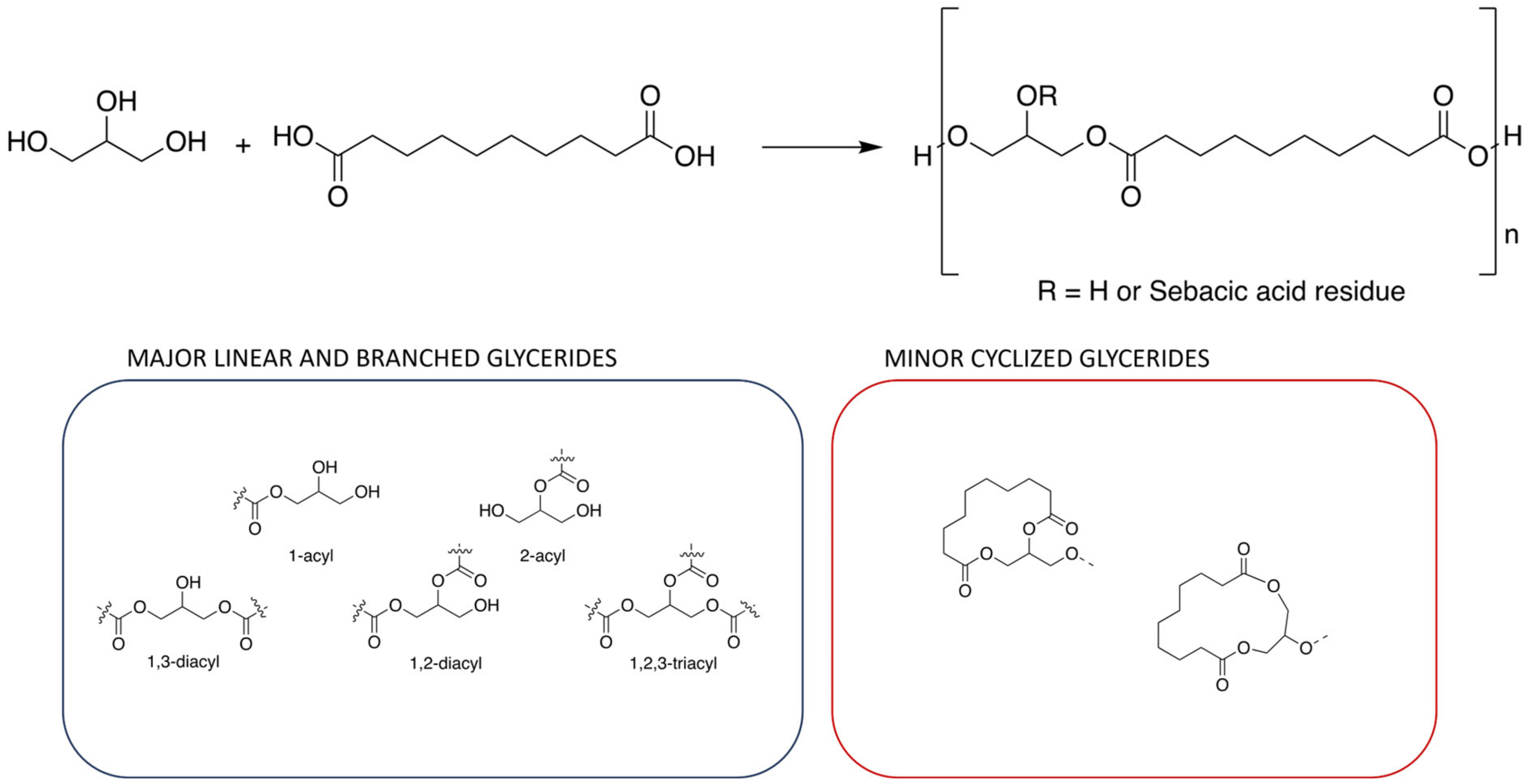
2. Classic Synthesis
Thermal Polycondensation
3. Alternative Synthesis
3.1. Microwave-Assisted Polycondensation
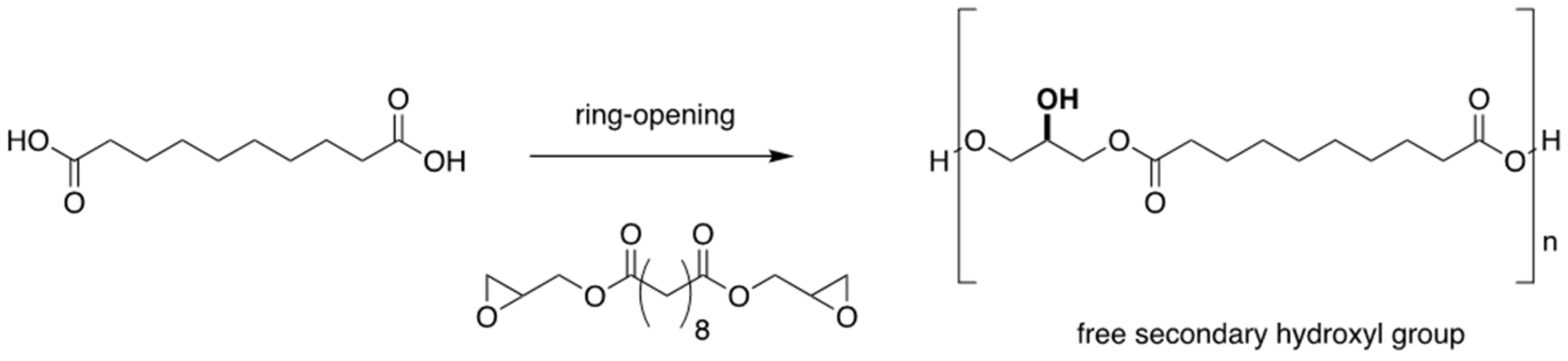
3.2. Ring-Opening Polymerisation
3.3. Enzyme-Catalysed Synthesis
3.4. Ultraviolet Light-Driven Photopolymerization
3.5. Hybrid Chemical-Physical Crosslinking
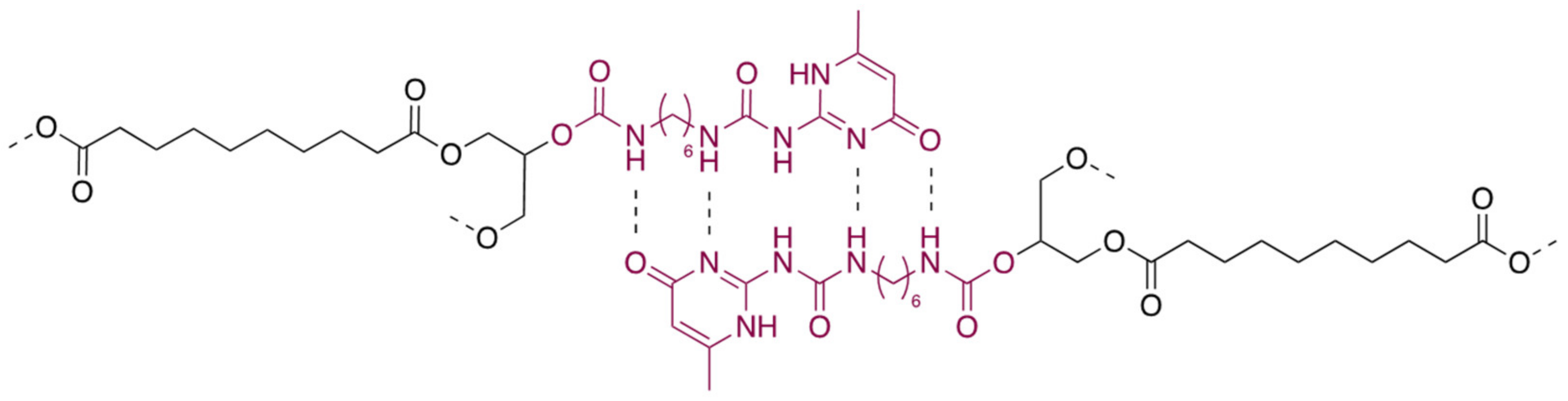
3.6. Supramolecular Crosslinking
3.7. PGS Co-Polymers
4. Polymer Characterisation
5. Polymer Tailoring
5.1. Reagents Ratio
5.2. Reaction Temperature and Time
5.3. Atmosphere Influence
5.4. Purification of PGS Pre-Polymer and Elastomer
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bonferoni, M.C.; Caramella, C.; Catenacci, L.; Conti, B.; Dorati, R.; Ferrari, F.; Genta, I.; Modena, T.; Perteghella, S.; Rossi, S.; et al. Biomaterials for Soft Tissue Repair and Regeneration: A Focus on Italian Research in the Field. Pharmaceutics 2021, 13, 1341. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, M.C.; Farè, S.; Candiani, G. Chapter 8—Advanced Applications. In Foundations of Biomaterials Engineering; Tanzi, M.C., Farè, S., Candiani, G., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 471–545. ISBN 978-0-08-101034-1. [Google Scholar]
- Raia, N.R.; McGill, M.; Marcet, T.; Vidal Yucha, S.E.; Kaplan, D.L. Soft Tissue Engineering. In Biomaterials Science; Academic Press: Cambridge, MA, USA, 2020; pp. 1399–1414. [Google Scholar] [CrossRef]
- Góra, A.; Pliszka, D.; Mukherjee, S.; Ramakrishna, S. Tubular Tissues and Organs of Human Body-Challenges in Regenerative Medicine. J. Nanosci. Nanotechnol. 2016, 16, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Shen, Y.I.; Harmon, J.W. Engineering Pro-Regenerative Hydrogels for Scarless Wound Healing. Adv. Healthc. Mater. 2018, 7, e1800016. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W.; Woodfield, T.B.F.; Dalton, P.D. Scaffold Design and Fabrication, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; ISBN 9780124201453. [Google Scholar]
- Raeisdasteh Hokmabad, V.; Davaran, S.; Ramazani, A.; Salehi, R. Design and Fabrication of Porous Biodegradable Scaffolds: A Strategy for Tissue Engineering. J. Biomater. Sci. Polym. Ed. 2017, 28, 1797–1825. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Liang, S.; Thouas, G.A. Elastomeric Biomaterials for Tissue Engineering. Prog. Polym. Sci. 2013, 38, 584–671. [Google Scholar] [CrossRef]
- Tanzi, M.C.; Farè, S.; Candiani, G. Organization, Structure, and Properties of Materials. In Foundations of Biomaterials Engineering; Academic Press: Cambridge, MA, USA, 2019; pp. 3–103. ISBN 9780081010341. [Google Scholar]
- Owczarzy, A.; Kurasiński, R.; Kulig, K.; Rogóż, W.; Szkudlarek, A.; Maciążek-Jurczyk, M. Collagen—Structure, Properties and Application. Eng. Biomater. 2020, 23, 17–23. [Google Scholar] [CrossRef]
- Kristensen, J.H.; Karsdal, M.A. Elastin. In Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 197–201. ISBN 9780128098998. [Google Scholar]
- Panja, S.; Siehr, A.; Sahoo, A.; Siegel, R.A.; Shen, W. Biodegradable Elastomers Enabling Thermoprocessing below 100 °C. Biomacromolecules 2022, 23, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ameer, G.A.; Sheppard, B.J.; Langer, R. A Tough Biodegradable Elastomer. Nat. Biotechnol. 2002, 20, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Gull, M.; Pasek, M.A. The Role of Glycerol and Its Derivatives in the Biochemistry of Living Organisms, and Their Prebiotic Origin and Significance in the Evolution of Life. Catalysts 2021, 11, 86. [Google Scholar] [CrossRef]
- Yamaga, M.; Tani, H.; Yamaki, A.; Tatefuji, T.; Hashimoto, K. Metabolism and Pharmacokinetics of Medium Chain Fatty Acids after Oral Administration of Royal Jelly to Healthy Subjects. RSC Adv. 2019, 9, 15392–15401. [Google Scholar] [CrossRef]
- Chen, Q.; Bismarck, A.; Hansen, U.; Junaid, S.; Tran, M.Q. Characterisation of a Soft Elastomer Poly (Glycerol Sebacate) Designed to Match the Mechanical Properties of Myocardial Tissue. Biomaterials 2008, 29, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Engelmayr, G.C., Jr.; Cheng, M.; Bettinger, C.J.; Borenstein, J.T.; Langer, R.; Freed, L.E. Accordion-Like Honeycombs for Tissue Engineering of Cardiac Anisotropy. Nat. Mater. 2008, 7, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Flaig, F.; Ragot, H.; Simon, A.; Revet, G.; Kitsara, M.; Kitasato, L.; Hébraud, A.; Agbulut, O.; Schlatter, G. Design of Functional Electrospun Scaffolds Based on Poly(Glycerol Sebacate) Elastomer and Poly(Lactic Acid) for Cardiac Tissue Engineering. ACS Biomater. Sci. Eng. 2020, 6, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.H.; Rai, R.; Dippold, D.; Roether, J.E.; Schubert, D.W.; Rosellini, E.; Barbani, N.; Boccaccini, A.R. Development and Characterization of Novel Electrically Conductive PANI-PGS Composites for Cardiac Tissue Engineering Applications. Acta Biomater. 2014, 10, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lei, D.; Huang, S.; Yang, Q.; Song, B.; Guo, Y.; Shen, A.; Yuan, Z.; Li, S.; Qing, F.L.; et al. Elastic 3D-Printed Hybrid Polymeric Scaffold Improves Cardiac Remodeling after Myocardial Infarction. Adv. Healthc. Mater. 2019, 8, e1900065. [Google Scholar] [CrossRef] [PubMed]
- Crapo, P.M.; Wang, Y. Physiologic Compliance in Engineered Small-Diameter Arterial Constructs Based on an Elastomeric Substrate. Biomaterials 2010, 31, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Crapo, P.; Nerem, R.; Wang, Y. Co-Expression of Elastin and Collagen Leads to Highly Compliant Engineered Blood Vessels. J. Biomed. Mater. Res. A 2008, 85, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Kharazi, A.Z.; Atari, M.; Vatankhah, E.; Javanmard, S.H. A Nanofibrous Bilayered Scaffold for Tissue Engineering of Small-Diameter Blood Vessels. Polym. Adv. Technol. 2018, 29, 3151–3158. [Google Scholar] [CrossRef]
- Khosravi, R.; Best, C.A.; Allen, R.A.; Stowell, C.E.T.; Onwuka, E.; Zhuang, J.J.; Lee, Y.-U.; Yi, T.; Bersi, M.R.; Shinoka, T.; et al. Long-Term Functional Efficacy of a Novel Electrospun Poly(Glycerol Sebacate)-Based Arterial Graft in Mice. Ann. Biomed. Eng. 2016, 44, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kai, D.; Ye, H.; Tian, L.; Ding, X.; Ramakrishna, S.; Loh, X.J. Electrospinning of Poly(Glycerol Sebacate)-Based Nanofibers for Nerve Tissue Engineering. Mater. Sci. Eng. C 2017, 70, 1089–1094. [Google Scholar] [CrossRef]
- Singh, D.; Harding, A.J.; Albadawi, E.; Boissonade, F.M.; Haycock, J.W.; Claeyssens, F. Additive Manufactured Biodegradable Poly(Glycerol Sebacate Methacrylate) Nerve Guidance Conduits. Acta Biomater. 2018, 78, 48–63. [Google Scholar] [CrossRef]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility Analysis of Poly(Glycerol Sebacate) as a Nerve Guide Material. Biomaterials 2005, 26, 5454–5464. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Guo, B.; Shao, Y.; Ma, P.X. Electroactive Biodegradable Polyurethane Significantly Enhanced Schwann Cells Myelin Gene Expression and Neurotrophin Secretion for Peripheral Nerve Tissue Engineering. Biomaterials 2016, 87, 18–31. [Google Scholar] [CrossRef]
- Keirouz, A.; Fortunato, G.; Zhang, M.; Callanan, A.; Radacsi, N. Nozzle-Free Electrospinning of Polyvinylpyrrolidone/Poly (Glycerol Sebacate) Fibrous Scaffolds for Skin Tissue Engineering Applications. Med. Eng. Physiscs 2019, 71, 56–67. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Yang, D.; Nie, J.; Ma, G. Electrospun Core–Shell Fibrous 2D Scaffold with Biocompatible Poly(Glycerol Sebacate) and Poly-l-Lactic Acid for Wound Healing. Adv. Fiber Mater. 2020, 2, 105–117. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, C.; Qiao, X.; Liu, T.; Sun, K. Porous Poly(Glycerol Sebacate) (PGS) Elastomer Scaffolds for Skin Tissue Engineering. Polym. Test. 2016, 54, 118–125. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Guo, B.; Dong, R.; Qiu, Y.; Ma, P.X. Antibacterial Anti-Oxidant Electroactive Injectable Hydrogel as Self-Healing Wound Dressing with Hemostasis and Adhesiveness for Cutaneous Wound Healing. Biomaterials 2017, 122, 34–47. [Google Scholar] [CrossRef]
- Kemppainen, J.M.; Hollister, S.J. Tailoring the Mechanical Properties of 3D-Designed Poly(Glycerol Sebacate) Scaffolds for Cartilage Applications. J. Biomed. Mater. Res. A 2010, 94, 9–18. [Google Scholar] [CrossRef]
- Lin, D.; Cai, B.; Wang, L.; Cai, L.; Wang, Z.; Xie, J.; Lv, Q.X.; Yuan, Y.; Liu, C.; Shen, S.G. A Viscoelastic PEGylated Poly(Glycerol Sebacate)-Based Bilayer Scaffold for Cartilage Regeneration in Full-Thickness Osteochondral Defect. Biomaterials 2020, 253, 120095. [Google Scholar] [CrossRef]
- Silva, J.C.; Udangawa, R.N.; Chen, J.; Mancinelli, C.D.; Garrudo, F.F.F.; Mikael, P.E.; Cabral, J.M.S.; Ferreira, F.C.; Linhardt, R.J. Kartogenin-Loaded Coaxial PGS/PCL Aligned Nanofibers for Cartilage Tissue Engineering. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 107, 110291. [Google Scholar] [CrossRef]
- Pomerantseva, I.; Krebs, N.; Hart, A.; Neville, C.M.; Huang, A.Y.; Sundback, C.A. Degradation Behavior of Poly(Glycerol Sebacate). J. Biomed. Mater. Res. 2008, 91A, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cook, W.D.; Moorhoff, C.; Huang, W.C.; Chen, Q.Z. Synthesis, Characterization and Properties of Biocompatible Poly(Glycerol Sebacate) Pre-Polymer and Gel. Polym. Int. 2013, 62, 534–547. [Google Scholar] [CrossRef]
- Mahdavi, A.; Ferreira, L.; Sundback, C.; Nichol, J.W.; Chan, E.P.; Carter, D.J.D.; Bettinger, C.J.; Patanavanich, S.; Chignozha, L.; Ben-Joseph, E.; et al. A Biodegradable and Biocompatible Gecko-Inspired Tissue Adhesive. Proc. Natl. Acad. Sci. USA 2008, 105, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Z.; Ishii, H.; Thouas, G.A.; Lyon, A.R.; Wright, J.S.; Blaker, J.J.; Chrzanowski, W.; Boccaccini, A.R.; Ali, N.N.; Knowles, J.C.; et al. An Elastomeric Patch Derived from Poly(Glycerol Sebacate) for Delivery of Embryonic Stem Cells to the Heart. Biomaterials 2010, 31, 3885–3893. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jin, K.; Wang, L.; Fan, Y. A Review: Optimization for Poly(Glycerol Sebacate) and Fabrication Techniques for Its Centered Scaffolds. Macromol. Biosci. 2021, 21, 2100022. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, M.; Liu, L.; Mithieux, S.M.; Weiss, A.S. Polyglycerol Sebacate-Based Elastomeric Materials for Arterial Regeneration. J. Biomed. Mater. Res. A 2024, 112, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Vogt, L.; Ruther, F.; Salehi, S.; Boccaccini, A.R. Poly(Glycerol Sebacate) in Biomedical Applications—A Review of the Recent Literature. Adv. Healthc. Mater. 2021, 10, 2002026. [Google Scholar] [CrossRef]
- Maliger, R.; Halley, P.J.; Cooper-White, J.J. Poly(Glycerol-Sebacate) Bioelastomers-Kinetics of Step-Growth Reactions Using Fourier Transform (FT)-Raman Spectroscopy. J. Appl. Polym. Sci. 2012, 127, 3980–3986. [Google Scholar] [CrossRef]
- Kafouris, D.; Kossivas, F.; Constantinides, C.; Nguyen, N.Q.; Wesdemiotis, C.; Patrickios, C.S. Biosourced Amphiphilic Degradable Elastomers of Poly(Glycerol Sebacate): Synthesis and Network and Oligomer Characterization. Macromolecules 2013, 46, 622–630. [Google Scholar] [CrossRef]
- Maliger, R.B.; Halley, P.J.; Cooper-White, J.J. Poly (Glycerol-Sebacate) Bioelastomers: 2. Synthesis Using Brabender Plasticoder® as a Batch Reactor. J. Appl. Polym. Sci. 2016, 133, 1–8. [Google Scholar] [CrossRef]
- Gao, J.; Crapo, P.M.; Wang, Y. Macroporous Elastomeric Scaffolds with Extensive Micropores for Soft Tissue Engineering. Tissue Eng. 2006, 12, 917–925. [Google Scholar] [CrossRef]
- Bettinger, C.J.; Orrick, B.; Misra, A.; Langer, R.; Borenstein, J.T. Microfabrication of Poly (Glycerol-Sebacate) for Contact Guidance Applications. Biomaterials 2006, 27, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Yi, F.C.; Cai, W. Synthesis and Shape Memory Effect of Poly (Glycerol-Sebacate) Elastomer. Adv. Mater. Res. 2012, 476–478, 2141–2144. [Google Scholar] [CrossRef]
- Motlagh, D.; Yang, J.; Lui, K.Y.; Webb, A.R.; Ameer, G.A. Hemocompatibility Evaluation of Poly(Glycerol-Sebacate) in Vitro for Vascular Tissue Engineering. Biomaterials 2006, 27, 4315–4324. [Google Scholar] [CrossRef]
- Conejero-García, Á.; Gimeno, H.R.; Sáez, Y.M.; Vilariño-Feltrer, G.; Ortuño-Lizarán, I.; Vallés-Lluch, A. Correlating Synthesis Parameters with Physicochemical Properties of Poly(Glycerol Sebacate). Eur. Polym. J. 2017, 87, 406–419. [Google Scholar] [CrossRef]
- Guo, X.L.; Lu, X.L.; Dong, D.L.; Sun, Z.J. Characterization and Optimization of Glycerol/Sebacate Ratio in Poly(Glycerol-Sebacate) Elastomer for Cell Culture Application. J. Biomed. Mater. Res. A 2014, 102, 3903–3907. [Google Scholar] [CrossRef]
- Salehi, S.; Fathi, M.; Javanmard, S.; Barneh, F.; Moshayedi, M. Fabrication and Characterization of Biodegradable Polymeric Films as a Corneal Stroma Substitute. Adv. Biomed. Res. 2015, 4, 9. [Google Scholar] [CrossRef]
- Aydin, H.M.; Salimi, K.; Rzayev, Z.M.O.; Piskin, E. Microwave-Assisted Rapid Synthesis of Poly(Glycerol-Sebacate) Elastomers. Biomater. Sci. 2013, 1, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hong, A.T.L.; Naskar, N.; Chung, H.J. Criteria for Quick and Consistent Synthesis of Poly(Glycerol Sebacate) for Tailored Mechanical Properties. Biomacromolecules 2015, 16, 1525–1533. [Google Scholar] [CrossRef]
- Atya, A.M.N.; Tevlek, A.; Almemar, M.; Gökcen, D.; Aydin, H.M. Fabrication and Characterization of Carbon Aerogel/Poly(Glycerol-Sebacate) Patches for Cardiac Tissue Engineering. Biomed. Mater. 2021, 16, 065027. [Google Scholar] [CrossRef]
- Lau, C.C.; Bayazit, M.K.; Knowles, J.C.; Tang, J. Tailoring Degree of Esterification and Branching of Poly(Glycerol Sebacate) by Energy Efficient Microwave Irradiation. Polym. Chem. 2017, 8, 3937–3947. [Google Scholar] [CrossRef]
- Lee, K.W.; Gade, P.S.; Dong, L.; Zhang, Z.; Aral, A.M.; Gao, J.; Ding, X.; Stowell, C.E.T.; Nisar, M.U.; Kim, K.; et al. A Biodegradable Synthetic Graft for Small Arteries Matches the Performance of Autologous Vein in Rat Carotid Arteries. Biomaterials 2018, 181, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Bodakhe, S.; Verma, S.; Samal, S.K.; Sharma, S.S.; Kumar, N. Injectable Photocrosslinkable Nanocomposite Based on Poly (Glycerol Sebacate) Fumarate and Hydroxyapatite: Development, Biocompatibility and Bone Regeneration in a Rat Calvarial Bone Defect Model. Nanomedicine 2013, 8, 1777–1795. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Ouyang, L.; Highley, C.B.; Burdick, J.A. Norbornene-Modified Poly(Glycerol Sebacate) as a Photocurable and Biodegradable Elastomer. Polym. Chem. 2017, 8, 5091–5099. [Google Scholar] [CrossRef]
- Zhu, C.; Kustra, S.R.; Bettinger, C.J. Photocrosslinkable Biodegradable Elastomers Based on Cinnamate- Functionalized Polyesters. Acta Biomater. 2013, 9, 7362–7370. [Google Scholar] [CrossRef]
- Godinho, B.; Gama, N.; Barros-Timmons, A.; Ferreira, A. Enzymatic Synthesis of Poly(Glycerol Sebacate) Pre-Polymer with Crude Glycerol, by-Product from Biodiesel Prodution. AIP Conf. Proc. 2018, 1981, 020031. [Google Scholar] [CrossRef]
- Ning, Z.; Lang, K.; Xia, K.; Linhardt, R.J.; Gross, R.A. Lipase-Catalyzed Synthesis and Characterization of Poly(Glycerol Sebacate). Biomacromolecules 2022, 23, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Perin, G.B.; Felisberti, M.I. Enzymatic Synthesis of Poly(Glycerol Sebacate): Kinetics, Chain Growth, and Branching Behavior. Macromolecules 2020, 53, 7925–7935. [Google Scholar] [CrossRef] [PubMed]
- Ebner, C.; Bodner, T.; Stelzer, F.; Wiesbrock, F. One Decade of Microwave-Assisted Polymerizations: Quo Vadis? Macromol. Rapid Commun. 2011, 32, 254–288. [Google Scholar] [CrossRef]
- Gawande, M.B.; Shelke, S.N.; Zboril, R.; Varma, R.S. Microwave-Assisted Chemistry: Synthetic Applications for Rapid Assembly of Nanomaterials and Organics. Acc. Chem. Res. 2014, 47, 1338–1348. [Google Scholar] [CrossRef]
- Kharissova, O.V.; Kharisov, B.I.; González, C.M.O.; Méndez, Y.P.; López, I. Greener Synthesis of Chemical Compounds and Materials. R. Soc. Open Sci. 2019, 6, 191378. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Cherdron, H.; Rehahn, M.; Ritter, H.; Voit, B. Methods and Techniques for Synthesis, Characterization, Processing, and Modification of Polymers. In Polymer Synthesis: Theory and Practice: Fundamentals, Methods, Experiments; Springer: Berlin/Heidelberg, Germany, 2013; pp. 33–147. ISBN 978-3-642-28980-4. [Google Scholar]
- You, Z.; Bi, X.; Wang, Y. Fine Control of Polyester Properties via Epoxide ROP Using Monomers Carrying Diverse Functional Groups. Macromol. Biosci. 2012, 12, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.K.; Kokaz, S.F.; Abed, S.N.; Paradkar, A.; Tekade, R.K. Chapter 6—Pharmaceutical and Biomedical Applications of Polymers. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2019; pp. 203–267. ISBN 978-0-12-817909-3. [Google Scholar]
- You, Z.; Cao, H.; Gao, J.; Shin, P.H.; Day, B.W.; Wang, Y. A Functionalizable Polyester with Free Hydroxyl Groups and Tunable Physiochemical and Biological Properties. Biomaterials 2010, 31, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, L.; Zhou, X.; Guo, Y.; Song, J.; Qian, S.; Liu, Z.; Guan, Q.; Meade Jeffries, E.; Liu, W.; et al. Mechanically and Biologically Skin-like Elastomers for Bio-Integrated Electronics. Nat. Commun. 2020, 11, 1107. [Google Scholar] [CrossRef] [PubMed]
- Hevilla, V.; Sonseca, A.; Echeverría, C.; Muñoz-Bonilla, A.; Fernández-García, M. Enzymatic Synthesis of Polyesters and Their Bioapplications: Recent Advances and Perspectives. Macromol. Biosci. 2021, 21, 2100156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Loos, K. Enzymatic Synthesis of Biobased Polyesters and Polyamides. Polymers 2016, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, L.; Feng, N.; Jiang, W.; Jin, Y.; Li, X. Recent Advances in the Synthesis of Biodegradable Polyesters by Sustainable Polymerization: Lipase-Catalyzed Polymerization. RSC Adv. 2020, 10, 36230–36240. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.S.; Gao, W.; Gross, R.A. Glycerol Copolyesters: Control of Branching and Molecular Weight Using a Lipase Catalyst. Macromolecules 2005, 38, 3193–3204. [Google Scholar] [CrossRef]
- Taresco, V.; Creasey, R.G.; Kennon, J.; Mantovani, G.; Alexander, C.; Burley, J.C.; Garnett, M.C. Variation in Structure and Properties of Poly(Glycerol Adipate) via Control of Chain Branching during Enzymatic Synthesis. Polymer 2016, 89, 41–49. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, W.; Cai, J.; Hou, Y.; Ouyang, S.; Xie, W.; Gross, R.A. Poly(Oleic Diacid-Co-Glycerol): Comparison of Polymer Structure Resulting from Chemical and Lipase Catalysis. Macromolecules 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Jagtap, A.; More, A. A Review on Self-Initiated and Photoinitiator-Free System for Photopolymerization. Polym. Bull. 2022, 79, 8057–8091. [Google Scholar] [CrossRef]
- Bagheri, A.; Jin, J. Photopolymerization in 3D Printing. ACS Appl. Polym. Mater. 2019, 1, 593–611. [Google Scholar] [CrossRef]
- Lim, K.S.; Galarraga, J.H.; Cui, X.; Lindberg, G.C.J.; Burdick, J.A.; Woodfield, T.B.F. Fundamentals and Applications of Photo-Cross-Linking in Bioprinting. Chem. Rev. 2020, 120, 10662–10694. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, A.; Dutta, S.D.; Ganguly, K.; Patel, D.K.; Patil, T.V.; Lim, K.T. Recent Advances in 3D Printing of Photocurable Polymers: Types, Mechanism, and Tissue Engineering Application. Macromol. Biosci. 2022, 23, 2200278. [Google Scholar] [CrossRef] [PubMed]
- Nijst, C.L.E.; Bruggeman, J.P.; Karp, J.M.; Ferreira, L.; Zumbuehl, A.; Bettinger, C.J.; Langer, R. Synthesis and Characterization of Photocurable Elastomers from Poly(Glycerol-Co-Sebacate). Biomacromolecules 2007, 8, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Padera, R.F.; Burdick, J.A. Biodegradable and Radically Polymerized Elastomers with Enhanced Processing Capabilities. Biomed. Mater. 2008, 3, 034104. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Devlin, J.J.; Eng, G.; Martens, T.P.; Vunjak-Novakovic, G.; Burdick, J.A. Biodegradable Fibrous Scaffolds with Tunable Properties Formed from Photo-Cross-Linkable Poly(Glycerol Sebacate). ACS Appl. Mater. Interfaces 2009, 1, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Gerecht, S.; Townsend, S.A.; Pressler, H.; Zhu, H.; Nijst, C.L.E.; Bruggeman, J.P.; Nichol, J.W.; Langer, R. A Porous Photocurable Elastomer for Cell Encapsulation and Culture. Biomaterials 2007, 28, 4826–4835. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Wu, K.; Mauck, R.L.; Burdick, J.A. The Influence of Fibrous Elastomer Structure and Porosity on Matrix Organization. PLoS ONE 2010, 5, e15717. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Highley, C.B.; Ouyang, L.; Burdick, J.A. 3D Printing of Photocurable Poly(Glycerol Sebacate) Elastomers. Biofabrication 2016, 8, 045004. [Google Scholar] [CrossRef]
- Pashneh-Tala, S.; Owen, R.; Bahmaee, H.; Rekštyte, S.; Malinauskas, M.; Claeyssens, F. Synthesis, Characterization and 3D Micro-Structuring via 2-Photon Polymerization of Poly(Glycerol Sebacate)-Methacrylate-an Elastomeric Degradable Polymer. Front. Phys. 2018, 6, 41. [Google Scholar] [CrossRef]
- Wang, M.; Lei, D.; Liu, Z.; Chen, S.; Sun, L.; Lv, Z.; Huang, P.; Jiang, Z.; You, Z. A Poly(Glycerol Sebacate) Based Photo/Thermo Dual Curable Biodegradable and Biocompatible Polymer for Biomedical Applications. J. Biomater. Sci. Polym. Ed. 2017, 28, 1728–1739. [Google Scholar] [CrossRef]
- Ding, X.; Wu, Y.-L.; Gao, J.; Wells, A.; Lee, K.-W.; Wang, Y. Tyramine Functionalization of Poly(Glycerol Sebacate) Increases the Elasticity of the Polymer. J. Mater. Chem. B 2017, 5, 6097–6109. [Google Scholar] [CrossRef]
- Voorhaar, L.; Diaz, M.M.; Leroux, F.; Rogers, S.; Abakumov, A.M.; Van Tendeloo, G.; Van Assche, G.; Van Mele, B.; Hoogenboom, R. Supramolecular Thermoplastics and Thermoplastic Elastomer Materials with Self-Healing Ability Based on Oligomeric Charged Triblock Copolymers. NPG Asia Mater. 2017, 9, e385. [Google Scholar] [CrossRef]
- Pereira, M.J.N.; Ouyang, B.; Sundback, C.A.; Lang, N.; Friehs, I.; Mureli, S.; Pomerantseva, I.; McFadden, J.; Mochel, M.C.; Mwizerwa, O.; et al. A Highly Tunable Biocompatible and Multifunctional Biodegradable Elastomer. Adv. Mater. 2013, 25, 1209–1215. [Google Scholar] [CrossRef]
- Kajita, T.; Noro, A.; Matsushita, Y. Design and Properties of Supramolecular Elastomers. Polymer 2017, 128, 297–310. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Zhao, X.; Hou, S.; Guo, B.; Ma, P.X. Self-Healing Supramolecular Bioelastomers with Shape Memory Property as a Multifunctional Platform for Biomedical Applications via Modular Assembly. Biomaterials 2016, 104, 18–31. [Google Scholar] [CrossRef]
- Yoon, S.; Chen, B. Elastomeric and PH-Responsive Hydrogels Based on Direct Crosslinking of the Poly(Glycerol Sebacate) Pre-Polymer and Gelatin. Polym. Chem. 2018, 9, 3727–3740. [Google Scholar] [CrossRef]
- Patel, A.; Gaharwar, A.K.; Iviglia, G.; Zhang, H.; Mukundan, S.; Mihaila, S.M.; Demarchi, D.; Khademhosseini, A. Highly Elastomeric Poly(Glycerol Sebacate)-Co-Poly(Ethylene Glycol) Amphiphilic Block Copolymers. Biomaterials 2013, 34, 3970–3983. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, W.; Zhou, X.; Nie, W.; Chen, L.; He, C. Synthesis and Characterization of Poly(Glycerol Sebacate)-Based Elastomeric Copolyesters for Tissue Engineering Applications. Polym. Chem. 2016, 7, 2553–2564. [Google Scholar] [CrossRef]
- Tang, B.-C.; Yao, C.-L.; Xieh, K.-Y.; Hong, S.-G. Improvement of Physical Properties of Poly(Glycerol Sebacate) by Copolymerization with Polyhydroxybutyrate-Diols. J. Polym. Res. 2017, 24, 215. [Google Scholar] [CrossRef]
- Wilson, R.; Divakaran, A.; Kiran, S.; Varyambath, A.; Kumaran, A.; Sivaram, S.; Ragupathy, L. Poly(Glycerol Sebacate)-Based Polyester–Polyether Copolymers and Their Semi-Interpenetrated Networks with Thermoplastic Poly(Ester–Ether) Elastomers: Preparation and Properties. ACS Omega 2018, 3, 18714–18723. [Google Scholar] [CrossRef]
- Cai, W.; Liu, L. Shape-Memory Effect of Poly (Glycerol-Sebacate) Elastomer. Mater. Lett. 2008, 62, 2171–2173. [Google Scholar] [CrossRef]
- Krook, N.M.; Jaafar, I.H.; Sarkhosh, T.; LeBlon, C.; Coulter, J.P.; Jedlicka, S.S. In Vitro Examination of Poly(Glycerol Sebacate) Degradation Kinetics: Effects of Porosity and Cure Temperature. Int. J. Polym. Mater. Polym. Biomater. 2020, 69, 535–543. [Google Scholar] [CrossRef]
- Martín-Cabezuelo, R.; Vilariño-Feltrer, G.; Vallés-Lluch, A. Influence of Pre-Polymerisation Atmosphere on the Properties of Pre- and Poly(Glycerol Sebacate). Mater. Sci. Eng. C 2021, 119, 111429. [Google Scholar] [CrossRef]
- Sun, Z.; Sun, C.; Sun, B.; Lu, X.; Dong, D.-L. The Polycondensing Temperature Rather than Time Determines the Degradation and Drug Release of Poly (Glycerol-Sebacat) Doped with 5-Fluorouracil. J. Biomater. Sci. 2012, 23, 833–841. [Google Scholar] [CrossRef]
- Moorhoff, C.; Li, Y.; Cook, W.D.; Braybrook, C.; Chen, Q.-Z. Characterization of the Prepolymer and Gel of Biocompatible Poly(Xylitol Sebacate) in Comparison with Poly(Glycerol Sebacate) Using a Combination of Mass Spectrometry and Nuclear Magnetic Resonance. Polym. Int. 2015, 64, 668–688. [Google Scholar] [CrossRef]
- Jaafar, I.H.; Ammar, M.M.; Jedlicka, S.S.; Pearson, R.A.; Coulter, J.P. Spectroscopic Evaluation, Thermal, and Thermomechanical Characterization of Poly (Glycerol-Sebacate) with Variations in Curing Temperatures and Durations. J. Mater. Sci. 2010, 45, 2525–2529. [Google Scholar] [CrossRef]
- Tevlek, A.; Topuz, B.; Akbay, E.; Aydin, H.M. Surface Channel Patterned and Endothelialized Poly(Glycerol Sebacate) Based Elastomers. J. Biomater. Appl. 2022, 37, 287–302. [Google Scholar] [CrossRef]
- ASTM D412-98a; Standard Test Methods for Vulcanized Rubber and Thermoplastic Elastomers—Tension. American Society for Testing and Materials: West Conshohocken, PA, USA, 2017. [CrossRef]
- Riaz, R.; Abbas, S.R.; Iqbal, M. Synthesis, Rheological Characterization, and Proposed Application of Pre-Polyglycerol Sebacate as Ultrasound Contrast Agent Based on Theoretical Estimation. J. Appl. Polym. Sci. 2022, 139, 51963. [Google Scholar] [CrossRef]
- Wright, T.P.; Song, C.; Sears, S.; Petters, M.D. Thermodynamic and Kinetic Behavior of Glycerol Aerosol. Aerosol Sci. Technol. 2016, 50, 1385–1396. [Google Scholar] [CrossRef]
- Gadomska-Gajadhur, A.; Wrzecionek, M.; Matyszczak, G.; Piętowski, P.; Więcław, M.; Ruśkowski, P. Optimization of Poly(Glycerol Sebacate) Synthesis for Biomedical Purposes with the Design of Experiments. Org. Process Res. Dev. 2018, 22, 1793–1800. [Google Scholar] [CrossRef]
- Barham, J.P.; Koyama, E.; Norikane, Y.; Ohneda, N.; Yoshimura, T. Microwave Flow: A Perspective on Reactor and Microwave Configurations and the Emergence of Tunable Single-Mode Heating Toward Large-Scale Applications. Chem. Rec. 2019, 19, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Zhao, L.; Zhang, Y.; Li, Z.-T.; Huang, F. Multiple Hydrogen Bonding Driven Supramolecular Architectures and Their Biomedical Applications. Chem. Soc. Rev. 2024, 53, 1592–1623. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Sharma, M. Green Solvents for the Dissolution and Processing of Biopolymers. Curr. Opin. Green. Sustain. Chem. 2019, 18, 72–78. [Google Scholar] [CrossRef]
- Joseph, B.; Krishnan, S.; Kavil, S.V.; Pai, A.R.; James, J.; Kalarikkal, N.; Thomas, S. Green Chemistry Approach for Fabrication of Polymer Composites. Sustain. Chem. 2021, 2, 254–270. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PGS-Copolymer | Crosslinking Conditions | Young Modulus (MPa) | UTS (MPa) | Max. Elongation (%) | Ref. |
|---|---|---|---|---|---|
| PGS-co-gelatine | 120 °C/24 h | 0.16–0.62 | 0.27–0.7 | 121–252 | [95] |
| PGS-co-PEG | 130 °C/48 h | 0.04–0.15 | 0.03–0.4 | 192 | [96] |
| PGS-co-LLA-co-PEG | 150 °C/24–96 h | 0.07–1.94 | 0.07–0.86 | 83–450 | [97] |
| PGS-co-PHB-diol | 130 °C/48 h | 0.72–1.89 | 0.58–1.54 | 82–106 | [98] |
| PGS-PTMO | 170 °C/8 h | - | 0.36–0.75 | 85–737 | [99] |
| Investigated Property | Characterization Method | Findings | Ref. |
|---|---|---|---|
| Chemical structure | Fourier-Transform InfraRed Spectroscopy (FTIR) |
| [13,50,54,63,100,101,102,103] |
| 1H-Nuclear Magnetic Resonance (1H-NMR) |
| [37,44,63,82,104] | |
| 13C-Nuclear Magnetic Resonance (13C-NMR) |
| [37,63,104] | |
| Mass Spectroscopy (MS) |
| [44,104] | |
| Thermal behaviour | Differential Scanning Calorimetry (DSC) |
| [13,50,100,101,102,105] |
| Degree of esterification /crosslinking | Ester groups titration |
| [37,54,102] |
| Swelling behaviour (Flory-Rehner molecular theory of rubber elasticity) |
| [16,105] | |
| Mechanical method (rubbers theory) |
| [13,16,36,106] | |
| Molecular weight | Size exclusion chromatography (SEC)/Gel permeation chromatography (GPC) |
| [44,63,82] |
| Mass Spectroscopy (MS) | See above in “Chemical structure” section. | [44,104] | |
| Mechanical properties | Tensile testing |
| [13,36,37,54,106,107] |
| Compressive testing |
| [50,101,102] | |
| In vitro biodegradability | Mass loss |
| [36,50,51,54] |
| Size exclusion chromatography (SEC)/Gel permeation chromatography (GPC) | Decrease in MW and increase in PDI. See above in “Molecular weight” section. | [44] | |
| Tensile testing | Decrease in tensile resistance is related to increased polymer degradation. See above in “Mechanical properties” | [16] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosalia, M.; Rubes, D.; Serra, M.; Genta, I.; Dorati, R.; Conti, B. Polyglycerol Sebacate Elastomer: A Critical Overview of Synthetic Methods and Characterisation Techniques. Polymers 2024, 16, 1405. https://doi.org/10.3390/polym16101405
Rosalia M, Rubes D, Serra M, Genta I, Dorati R, Conti B. Polyglycerol Sebacate Elastomer: A Critical Overview of Synthetic Methods and Characterisation Techniques. Polymers. 2024; 16(10):1405. https://doi.org/10.3390/polym16101405
Chicago/Turabian StyleRosalia, Mariella, Davide Rubes, Massimo Serra, Ida Genta, Rossella Dorati, and Bice Conti. 2024. "Polyglycerol Sebacate Elastomer: A Critical Overview of Synthetic Methods and Characterisation Techniques" Polymers 16, no. 10: 1405. https://doi.org/10.3390/polym16101405
APA StyleRosalia, M., Rubes, D., Serra, M., Genta, I., Dorati, R., & Conti, B. (2024). Polyglycerol Sebacate Elastomer: A Critical Overview of Synthetic Methods and Characterisation Techniques. Polymers, 16(10), 1405. https://doi.org/10.3390/polym16101405