
The Applications of Nanopore Sequencing Technology in Animal and Human Virus Research


Abstract
1. Introduction
2. Nanopore Sequencing Technology
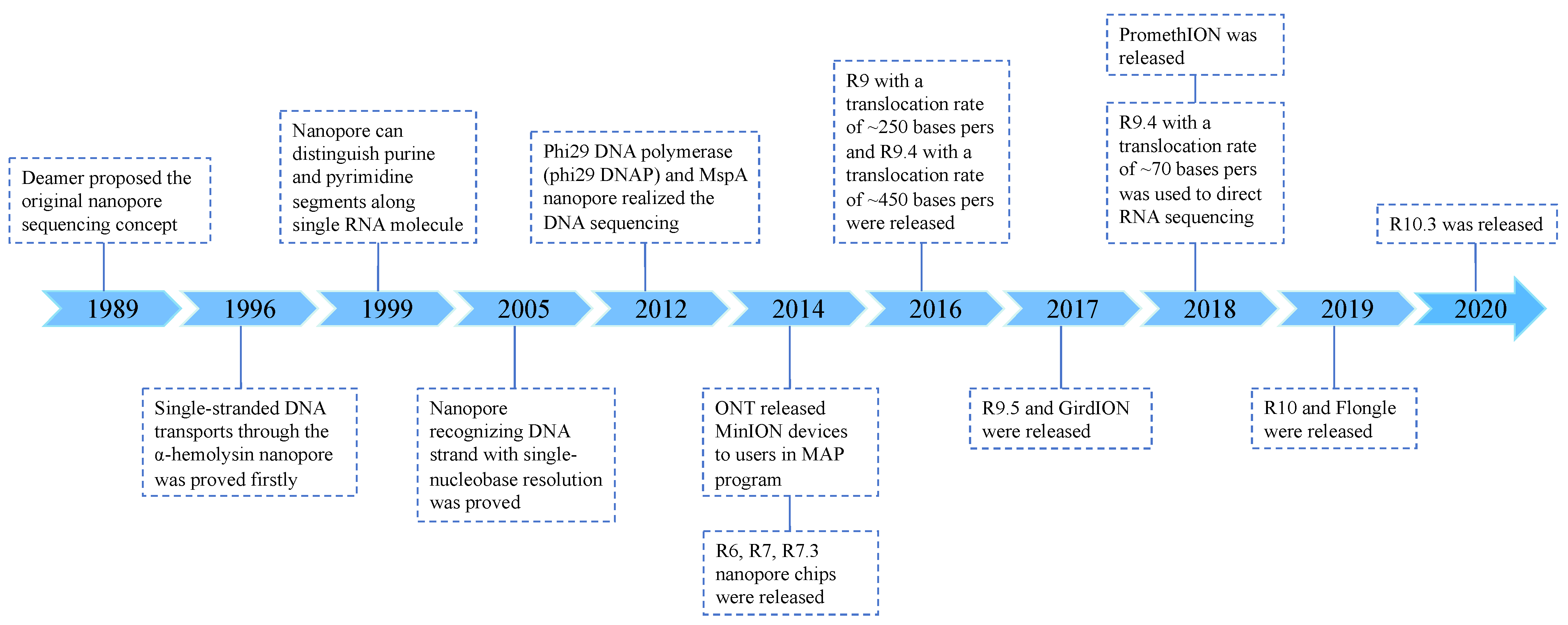
2.1. Development
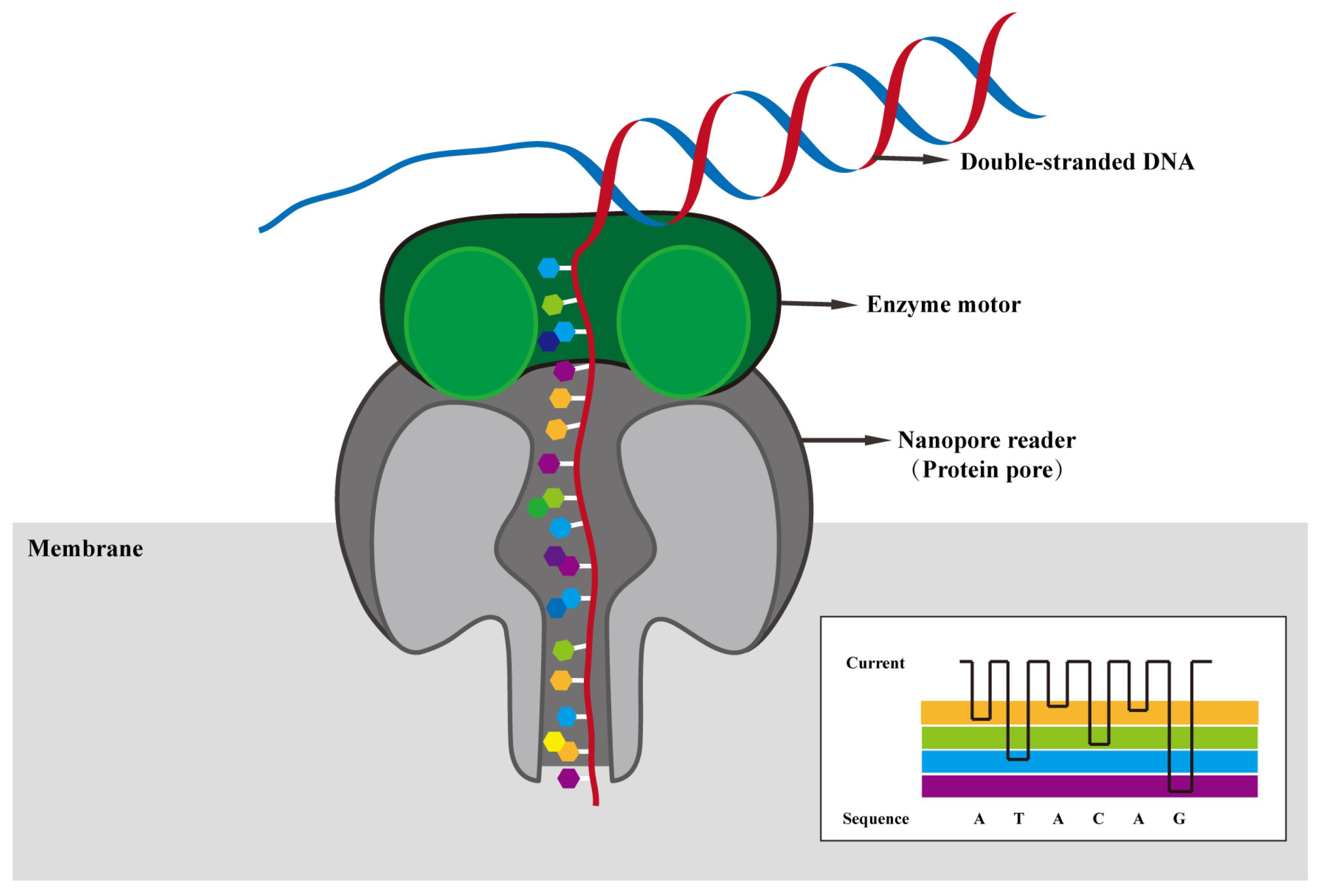
2.2. Principle
2.3. Methods
2.3.1. DNA Sequencing
2.3.2. RNA Sequencing
2.3.3. Bioinformatics Analysis
2.4. Advantages
3. Applications
3.1. Diagnosis and Monitoring of Viruses
3.2. Identification of Unknown Viruses
3.3. Transcriptome Assembly and Detection of Novel Transcript and Splice Variants
3.4. Analysis of Chemical Modifications
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef]
- Lunney, J.K.; Benfield, D.A.; Rowland, R.R. Porcine reproductive and respiratory syndrome virus: An update on an emerging and re-emerging viral disease of swine. Virus Res. 2010, 154, 1–6. [Google Scholar] [CrossRef]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef]
- Sun, R.Q.; Cai, R.J.; Chen, Y.Q.; Liang, P.S.; Chen, D.K.; Song, C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012, 18, 161–163. [Google Scholar] [CrossRef]
- Dimitrov, K.M.; Ramey, A.M.; Qiu, X.; Bahl, J.; Afonso, C.L. Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect. Genet. Evol. 2016, 39, 22–34. [Google Scholar] [CrossRef]
- Raj, G.D.; Jones, R.C. Infectious bronchitis virus: Immunopathogenesis of infection in the chicken. Avian Pathol. 1997, 26, 677–706. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, S.; Li, S.H.; Yu, S.; Wang, Q.; Zhang, K.; Qu, L.; Sun, Y.; Bi, Y.; Tang, F.; et al. Transcriptome profiling in swine macrophages infected with African swine fever virus at single-cell resolution. Proc. Natl. Acad. Sci. USA 2022, 119, e2201288119. [Google Scholar] [CrossRef]
- te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- Hutchinson, E.C. Influenza Virus. Trends Microbiol. 2018, 26, 809–810. [Google Scholar] [CrossRef]
- Tali, S.H.S.; LeBlanc, J.J.; Sadiq, Z.; Oyewunmi, O.D.; Camargo, C.; Nikpour, B.; Armanfard, N.; Sagan, S.M.; Jahanshahi-Anbuhi, S. Tools and Techniques for Severe Acute Respiratory Syndrome. Clin. Microbiol. Rev. 2021, 34, e00228-20. [Google Scholar] [CrossRef]
- Younis, M.C. Evaluation of deep learning approaches for identification of different corona-virus species and time series prediction. Comput. Med. Imaging Graph. 2021, 90, 101921. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, P.J.; Hatta, M.; Chiba, S.; Maemura, T.; Fan, S.; Takeda, M.; Kinoshita, N.; Hattori, S.-i.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; et al. Transmission of SARS-CoV-2 in Domestic Cats. N. Engl. J. Med. 2020, 383, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Sit, T.H.C.; Brackman, C.J.; Ip, S.M.; Tam, K.W.S.; Law, P.Y.T.; To, E.M.W.; Yu, V.Y.T.; Sims, L.D.; Tsang, D.N.C.; Chu, D.K.W.; et al. Canine SARS-CoV-2 infection. Nature 2020, 586, 776–778. [Google Scholar] [CrossRef] [PubMed]
- McAloose, D.; Laverack, M.; Wang, L.; Killian, M.L.; Caserta, L.C.; Yuan, F.; Mitchell, P.K.; Queen, K.; Mauldin, M.R.; Cronk, B.D.; et al. From People toPanthera: Natural SARS-CoV-2 Infection in Tigers and Lions at the Bronx Zoo. mBio 2020, 11, e02220-20. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-I.; Kim, S.-G.; Kim, S.-M.; Kim, E.-H.; Park, S.-J.; Yu, K.-M.; Chang, J.-H.; Kim, E.J.; Lee, S.; Casel, M.A.B.; et al. Infection and Rapid Transmission of SARS-CoV-2 in Ferrets. Cell Host Microbe 2020, 27, 704–709.e2. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Haas, J.L.; Corriveau, E.J.; Liem, A.T.; Willis, K.L.; Kadavy, D.R.; Rosenzweig, C.N.; Minot, S.S. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. GigaScience 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7, 99. [Google Scholar] [CrossRef]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, T.; Jenjaroenpun, P.; Taylor, M.K.; Lee, J.; Tolardo, A.L.; Parvathareddy, J.; Kandel, S.; Wadley, T.D.; Kaewnapan, B.; Athipanyasilp, N.; et al. Rapid Sequencing of Multiple RNA Viruses in Their Native Form. Front. Microbiol. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Hayer, K.E.; McIntyre, A.B.R.; Gokhale, N.S.; Abebe, J.S.; Della Fera, A.N.; Mason, C.E.; Horner, S.M.; Wilson, A.C.; Depledge, D.P.; et al. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat. Commun. 2020, 11, 6016. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Quick, J.; Claro, I.M.; Thézé, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.-A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree Lab: Portable genomics for Early Detection of Plant Viruses and Pests in Sub-Saharan Africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Roach, N.P.; Sadowski, N.; Alessi, A.F.; Timp, W.; Taylor, J.; Kim, J.K. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res. 2020, 30, 299–312. [Google Scholar] [CrossRef]
- Euskirchen, P.; Bielle, F.; Labreche, K.; Kloosterman, W.P.; Rosenberg, S.; Daniau, M.; Schmitt, C.; Masliah-Planchon, J.; Bourdeaut, F.; Dehais, C.; et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017, 134, 691–703. [Google Scholar] [CrossRef]
- De Roeck, A.; De Coster, W.; Bossaerts, L.; Cacace, R.; De Pooter, T.; Van Dongen, J.; D’Hert, S.; De Rijk, P.; Strazisar, M.; Van Broeckhoven, C.; et al. NanoSatellite: Accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019, 20, 239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315-19. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed]
- Akeson, M.; Branton, D.; Kasianowicz, J.J.; Brandin, E.; Deamer, D.W. Microsecond Time-Scale Discrimination Among Polycytidylic Acid, Polyadenylic Acid, and Polyuridylic Acid as Homopolymers or as Segments Within Single RNA Molecules. Biophys. J. 1999, 77, 3227–3233. [Google Scholar] [CrossRef] [PubMed]
- Ashkenasy, N.; Sánchez-Quesada, J.; Bayley, H.; Ghadiri, M.R. Recognizing a single base in an individual DNA strand: A step toward DNA sequencing in nanopores. Angew. Chem. Int. Ed. Engl. 2005, 44, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, D.; Heron, A.J.; Mikhailova, E.; Maglia, G.; Bayley, H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc. Natl. Acad. Sci. USA 2009, 106, 7702–7707. [Google Scholar] [CrossRef]
- Stoddart, D.; Heron, A.J.; Klingelhoefer, J.; Mikhailova, E.; Maglia, G.; Bayley, H. Nucleobase Recognition in ssDNA at the Central Constriction of the α-Hemolysin Pore. Nano Lett. 2010, 10, 3633–3637. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, D.; Maglia, G.; Mikhailova, E.; Heron, A.J.; Bayley, H. Multiple Base-Recognition Sites in a Biological Nanopore: Two Heads are Better than One. Angew. Chem. Int. Ed. 2010, 49, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Manrao, E.A.; Derrington, I.M.; Laszlo, A.H.; Langford, K.W.; Hopper, M.K.; Gillgren, N.; Pavlenok, M.; Niederweis, M.; Gundlach, J.H. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 2012, 30, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; E Olsen, H.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Zwolak, M.; Di Ventra, M. Electronic Signature of DNA Nucleotides via Transverse Transport. Nano Lett. 2005, 5, 421–424. [Google Scholar] [CrossRef]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.M.; Iyer, R.; Howard, K.R.; McPike, M.P.; Borer, P.N.; Movileanu, L. Engineering a Rigid Protein Tunnel for Biomolecular Detection. J. Am. Chem. Soc. 2012, 134, 9521–9531. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J.; et al. Metagenomic Nanopore Sequencing of Influenza Virus Direct. J. Clin. Microbiol. 2019, 58, e00963-19. [Google Scholar] [CrossRef]
- Brown, E.; Freimanis, G.; Shaw, A.E.; Horton, D.L.; Gubbins, S.; King, D. Characterising Foot-and-Mouth Disease Virus in Clinical Samples Using Nanopore Sequencing. Front. Vet. Sci. 2021, 8, 656256. [Google Scholar] [CrossRef]
- Hu, Z.L.; Huo, M.Z.; Ying, Y.L.; Long, Y.T. Biological Nanopore Approach for Single-Molecule Protein Sequencing. Angew. Chem. Int. Ed. 2021, 60, 14738–14749. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zhou, C.; Ding, Y.; Liu, B.; Lu, L.; Zhu, F.; Duan, S. Nanopore sequencing technology and its applications. MedComm 2023, 4, e316. [Google Scholar] [CrossRef]
- Kilianski, A.; Roth, P.A.; Liem, A.T.; Hill, J.M.; Willis, K.L.; Rossmaier, R.D.; Marinich, A.V.; Maughan, M.N.; Karavis, M.A.; Kuhn, J.H.; et al. Use of Unamplified RNA/cDNA-Hybrid Nanopore Sequencing for Rapid Detection and Characterization of RNA Viruses. Emerg. Infect. Dis. 2016, 22, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.; Ulm, S.-L.; Uddin, S.; Förster, S.; Seifert, D.; Oehme, R.; Corty, M.; Schaade, L.; Michel, J.; Nitsche, A. AmpliCoV: Rapid Whole-Genome Sequencing Using Multiplex PCR Amplification and Real-Time Oxford Nanopore MinION Sequencing Enables Rapid Variant Identification of SARS-CoV-2. Front. Microbiol. 2021, 12, 651151. [Google Scholar] [CrossRef]
- Lee, R.S.; Pai, M. Real-Time Sequencing of Mycobacterium tuberculosis: Are We There Yet? J. Clin. Microbiol. 2017, 55, 1249–1254. [Google Scholar] [CrossRef]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.T.; Alcantara, L.C.J.; Loman, N.J.; Pybus, O.G. Mobile real-time surveillance of Zika virus in Brazil. Genome Med. 2016, 8, 97. [Google Scholar] [CrossRef]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, D.J.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci. Rep. 2017, 7, 18022. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.B.R.; Rizzardi, L.; Yu, A.M.; Alexander, N.; Rosen, G.L.; Botkin, D.J.; E Stahl, S.; John, K.K.; Castro-Wallace, S.L.; McGrath, K.; et al. Nanopore sequencing in microgravity. NPJ Microgravity 2016, 2, 16035. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M.; Birol, I. BulkVis: A graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wong, C.-H.; Cheng, W.-C.; Tjong, H.; Menghi, F.; Ngan, C.Y.; Liu, E.T.; Wei, C.-L. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat. Methods 2018, 15, 455–460. [Google Scholar] [CrossRef]
- Stancu, M.C.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-Inclan, J.E.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, D.; Jayadas, T.T.P.; Jayathilaka, D.; Jeewandara, C.; Dissanayake, O.; Guruge, D.; Ariyaratne, D.; Gunasinghe, D.; Gomes, L.; Wijesinghe, A.; et al. Comparison of different sequencing techniques for identification of SARS-CoV-2 variants of concern with multiplex real-time PCR. PLoS ONE 2022, 17, e0265220. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Parker, J.; Helmstetter, A.J.; Devey, D.; Wilkinson, T.; Papadopulos, A.S.T. Field-based species identification of closely-related plants using real-time nanopore sequencing. Sci. Rep. 2017, 7, 8345. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Debbonaire, A.R.; Nicholls, S.M.; Rassner, S.M.E.; Sattler, B.; Cook, J.M.; Davy, T.; Soares, A.; Mur, L.A.J.; Hodson, A.J. In-field metagenome and 16S rRNA gene amplicon nanopore sequencing robustly characterize glacier microbiota. bioRxiv 2016, 10, 073965. [Google Scholar]
- Pomerantz, A.; Penafiel, N.; Arteaga, A.; Bustamante, L.; Pichardo, F.; Coloma, L.A.; Barrio-Amoros, C.L.; Salazar-Valenzuela, D.; Prost, S. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. GigaScience 2018, 7, giy033. [Google Scholar] [CrossRef]
- Pomerantz, A.; Sahlin, K.; Vasiljevic, N.; Seah, A.; Lim, M.; Humble, E.; Kennedy, S.; Krehenwinkel, H.; Winter, S.; Ogden, R.; et al. Rapid in situ identification of biological specimens via DNA amplicon sequencing using miniaturized laboratory equipment. Nat. Protoc. 2022, 17, 1415–1443. [Google Scholar] [CrossRef]
- Egeter, B.; Veríssimo, J.; Lopes-Lima, M.; Chaves, C.; Pinto, J.; Riccardi, N.; Beja, P.; Fonseca, N. Speeding up the detection of invasive bivalve species using environmental DNA: A Nanopore and Illumina sequencing comparison. Mol. Ecol. Resour. 2022, 22, 2232–2247. [Google Scholar] [CrossRef] [PubMed]
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2021, 22, 119–131. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hao, H.; Ma, L.; Zhang, Y.; Hu, X.; Chen, Z.; Liu, D.; Yuan, J.; Hu, Z.; Guan, W. Methyltransferase-like 3 Modulates Severe Acute Respiratory Syndrome Coronavirus-2 RNA N6-Methyladenosine Modification and Replication. mBio 2021, 12, e0106721. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; et al. The SARS-CoV-2 subgenome landscape and its novel regulatory features. Mol. Cell 2021, 81, 2135–2147.e5. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; De La Torre, A.; Dixon, L.; Gallardo, C.; Jori, F.; Laddomada, A.; Martins, C.; Parkhouse, R.M.; Revilla, Y.; Rodriguez, F.a.J.-M.; et al. Approaches and Perspectives for Development of African Swine Fever Virus Vaccines. Vaccines 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Jiang, M.; Wu, K.; Hu, J.; Wang, Y.; Quan, W.; Hao, M.; Liu, H.; Wei, H.; Fan, W.; et al. Nanopore sequencing of African swine fever virus. Sci. China Life Sci. 2020, 63, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hübner, A.; Olesen, A.S.; Höper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.K.; Grau, F.R.; Mayr, G.A.; Samayoa, T.L.S.; Dodd, K.A.; Barrette, R.W. Rapid Sequence-Based Characterization of African Swine Fever Virus by Use of the Oxford Nanopore MinION Sequence Sensing Device and a Companion Analysis Software Tool. J. Clin. Microbiol. 2019, 58, e01104-19. [Google Scholar] [CrossRef] [PubMed]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef]
- Luna, N.; Muñoz, M.; Bonilla-Aldana, D.K.; Patiño, L.H.; Kasminskaya, Y.; Paniz-Mondolfi, A.; Ramírez, J.D. Monkeypox virus (MPXV) genomics: A mutational and phylogenomic analyses of B.1 lineages. Travel Med. Infect. Dis. 2023, 52, 102551. [Google Scholar] [CrossRef] [PubMed]
- Luna, N.; Ramírez, A.L.; Muñoz, M.; Ballesteros, N.; Patiño, L.H.; Castañeda, S.A.; Bonilla-Aldana, D.K.; Paniz-Mondolfi, A.; Ramírez, J.D. Phylogenomic analysis of the monkeypox virus (MPXV) 2022 outbreak: Emergence of a novel viral lineage? Travel Med. Infect. Dis. 2022, 49, 102402. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Acharya, A.; Gendelman, H.E.; Byrareddy, S.N. The 2022 outbreak and the pathobiology of the monkeypox virus. J. Autoimmun. 2022, 131, 102855. [Google Scholar] [CrossRef] [PubMed]
- Kakuk, B.; Dormo, A.; Csabai, Z.; Kemenesi, G.; Holoubek, J.; Ruzek, D.; Prazsak, I.; Dani, V.E.; Denes, B.; Torma, G.; et al. In-depth Temporal Transcriptome Profiling of Monkeypox and Host Cells using Nanopore Sequencing. Sci. Data 2023, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Bosmeny, M.S.; White, A.A.; Pater, A.A.; Crew, J.; Geltz, J.; Gagnon, K.T. Global mpox lineage discovery and rapid outbreak tracking with nanopore sequencing. Virol. J. 2023, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Webby, R.J.; Webster, R.G. Are we ready for pandemic influenza? Science 2023, 302, 1519–1522. [Google Scholar] [CrossRef]
- Xu, Y.; Lewandowski, K.; Downs, L.O.; Kavanagh, J.; Hender, T.; Lumley, S.; Jeffery, K.; Foster, D.; Sanderson, N.D.; Vaughan, A.; et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: Diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Eurosurveillance 2021, 26, 2000004. [Google Scholar] [CrossRef] [PubMed]
- Rambo-Martin, B.L.; Keller, M.W.; Wilson, M.M.; Nolting, J.M.; Anderson, T.K.; Vincent, A.L.; Bagal, U.R.; Jang, Y.; Neuhaus, E.B.; Davis, C.T.; et al. Influenza A Virus Field Surveillance at a Swine-Human Interface. mSphere 2020, 5, e00822-19. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.G.S.; Snell, L.B.; Alder, C.; Charalampous, T.; Alcolea-Medina, A.; Sehmi, J.K.; Al-Yaakoubi, N.; Humayun, G.; Miah, S.; Lackenby, A.; et al. Feasibility and clinical utility of local rapid Nanopore influenza A virus whole genome sequencing for integrated outbreak management, genotypic resistance detection and timely surveillance. Microb. Genom. 2023, 9, 001083. [Google Scholar] [CrossRef]
- Yip, C.C.-Y.; Chan, W.-M.; Ip, J.D.; Seng, C.W.-M.; Leung, K.-H.; Poon, R.W.-S.; Ng, A.C.-K.; Wu, W.-L.; Zhao, H.; Chan, K.-H.; et al. Nanopore Sequencing Reveals Novel Targets for Detection and Surveillance of Human and Avian Influenza A Viruses. J. Clin. Microbiol. 2020, 58, e02127-19. [Google Scholar] [CrossRef]
- Xu, Y.; Lewandowski, K.; Jeffery, K.; Downs, L.O.; Foster, D.; Sanderson, N.D.; Kavanagh, J.; Vaughan, A.; Salvagno, C.; Vipond, R.; et al. Nanopore metagenomic sequencing to investigate nosocomial transmission of human metapneumovirus from a unique genetic group among haematology patients in the United Kingdom. J. Infect. 2020, 80, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Groen, K.; van Nieuwkoop, S.; Meijer, A.; van der Veer, B.; van Kampen, J.J.A.; Fraaij, P.L.; Fouchier, R.A.M.; van den Hoogen, B.G. Emergence and Potential Extinction of Genetic Lineages of Human Metapneumovirus between 2005 and 2021. mBio 2022, 14, e0228022. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H.; Mao, L.; Yu, H.; Yu, X.; Sun, Z.; Qian, X.; Cheng, S.; Chen, S.; Chen, J.; et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci. Rep. 2020, 10, 17492. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.B.; Alcolea-Medina, A.; Charalampous, T.; Alder, C.; Williams, T.G.S.; Flaviani, F.; Batra, R.; Bakrania, P.; Thangarajah, R.; Neil, S.J.D.; et al. Real-Time Whole Genome Sequencing to Guide Patient-Tailored Therapy of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Clin. Infect. Dis. 2023, 76, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Løvestad, A.H.; Jørgensen, S.B.; Handal, N.; Ambur, O.H.; Aamot, H.V. Investigation of intra-hospital SARS-CoV-2 transmission using nanopore whole-genome sequencing. J. Hosp. Infect. 2021, 111, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Charre, C.; Regue, H.; Dény, P.; Josset, L.; Chemin, I.; Zoulim, F.; Scholtes, C. Improved hepatitis delta virus genome characterization by single molecule full-length genome sequencing combined with VIRiONT pipeline. J. Med. Virol. 2023, 95, e28634. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef]
- Prazsák, I.; Moldován, N.; Balázs, Z.; Tombácz, D.; Megyeri, K.; Szűcs, A.; Csabai, Z.; Boldogkői, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genom. 2018, 19, 873. [Google Scholar] [CrossRef]
- Zheng, H.H.; Fu, P.F.; Chen, H.Y.; Wang, Z.Y. Pseudorabies Virus: From Pathogenesis to Prevention Strategies. Viruses 2022, 14, 1638. [Google Scholar] [CrossRef] [PubMed]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 2017, 8, 2708. [Google Scholar] [CrossRef]
- Rocholl, C.; Gerber, K.; Daly, J.; Pavia, A.T.; Byington, C.L. Adenoviral infections in children: The impact of rapid diagnosis. Pediatrics 2004, 113, e51–e56. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Fishbein, M.; Echavarria, M. Adenovirus. Semin. Respir. Crit. Care Med. 2011, 32, 494–511. [Google Scholar] [CrossRef]
- Berciaud, S.; Rayne, F.; Kassab, S.; Jubert, C.; Faure-Della Corte, M.; Salin, F.; Wodrich, H.; Lafon, M.E. Adenovirus infections in Bordeaux University Hospital 2008–2010: Clinical and virological features. J. Clin. Virol. 2012, 54, 302–307. [Google Scholar] [CrossRef]
- Savón, C.; Acosta, B.; Valdés, O.; Goyenechea, A.; Gonzalez, G.; Piñón, A.; Más, P.; Rosario, D.; Capó, V.; Kourí, V.; et al. A myocarditis outbreak with fatal cases associated with adenovirus subgenera C among children from Havana City in 2005. J. Clin. Virol. 2008, 43, 152–157. [Google Scholar] [CrossRef]
- Westergren Jakobsson, A.; Segerman, B.; Wallerman, O.; Bergström Lind, S.; Zhao, H.; Rubin, C.-J.; Pettersson, U.; Akusjärvi, G.; Parrish, C.R. The Human Adenovirus 2 Transcriptome: An Amazing Complexity of Alternatively Spliced mRNAs. J. Virol. 2021, 95, e01869-20. [Google Scholar] [CrossRef]
- Price, A.M.; Steinbock, R.T.; Lauman, R.; Charman, M.; Hayer, K.E.; Kumar, N.; Halko, E.; Lum, K.K.; Wei, M.; Wilson, A.C.; et al. Novel viral splicing events and open reading frames revealed by long-read direct RNA sequencing of adenovirus transcripts. PLoS Pathog. 2022, 18, e1010797. [Google Scholar] [CrossRef]
- Ugolini, C.; Mulroney, L.; Leger, A.; Castelli, M.; Criscuolo, E.; Williamson, M.K.; Davidson, A.D.; Almuqrin, A.; Giambruno, R.; Jain, M.; et al. Nanopore ReCappable sequencing maps SARS-CoV-2 5′ capping sites and provides new insights into the structure of sgRNAs. Nucleic Acids Res. 2022, 50, 3475–3489. [Google Scholar] [CrossRef]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; Horner, S.M. RNA modifications go viral. PLoS Pathog. 2017, 13, e1006188. [Google Scholar] [CrossRef]
- Courtney, D.G. Post-Transcriptional Regulation of Viral RNA through Epitranscriptional Modification. Cells 2021, 10, 1129. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Zhang, Z.; Wang, D.P.; Liang, F.; Miao, Y.; Xiao, C.L.; Luo, F.; Wang, J. DeepSignal: Detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 2019, 35, 4586–4595. [Google Scholar] [CrossRef]
- Moss, B.; Gershowitz, A.; Stringer, J.R.; E Holland, L.; Wagner, E.K. 5′-Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J. Virol. 1977, 23, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, K.P.; Depledge, D.P.; Abebe, J.S.; Rice, S.A.; Mohr, I.; Wilson, A.C. Widespread remodeling of the m6A RNA-modification landscape by a viral regulator of RNA processing and export. Proc. Natl. Acad. Sci. USA 2021, 118, e2104805118. [Google Scholar] [CrossRef]
- Burgess, H.M.; Depledge, D.P.; Thompson, L.; Srinivas, K.P.; Grande, R.C.; Vink, E.I.; Abebe, J.S.; Blackaby, W.P.; Hendrick, A.; Albertella, M.R.; et al. Targeting the m6A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, P.; Ma, X.; Wu, Y.; Luo, C.; Qiu, L.; Zeshan, B.; Yang, Z.; Zhou, Y.; Wang, X. Nanopore-Based Direct RNA-Sequencing Reveals a High-Resolution Transcriptional Landscape of Porcine Reproductive and Respiratory Syndrome Virus. Viruses 2021, 13, 2531. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 sequence variation: Drift, shift, and attenuation. Cell 2001, 104, 469–472. [Google Scholar] [CrossRef]
- Khatchikian, D.; Orlich, M.; Rott, R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 1989, 340, 156–157. [Google Scholar] [CrossRef]
- Hon, C.-C.; Lam, T.-Y.; Shi, Z.-L.; Drummond, A.J.; Yip, C.-W.; Zeng, F.; Lam, P.-Y.; Leung, F.C.-C. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and Its Implications on the Direct Ancestor of SARS Coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef]
- Ashton, P.M.; Nair, S.; Dallman, T.; Rubino, S.; Rabsch, W.; Mwaigwisya, S.; Wain, J.; O’Grady, J. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 2014, 33, 296–300. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lewandowski, K.; Lumley, S.; Pullan, S.; Vipond, R.; Carroll, M.; Foster, D.; Matthews, P.C.; Peto, T.; Crook, D. Detection of Viral Pathogens With Multiplex Nanopore MinION Sequencing: Be Careful With Cross-Talk. Front. Microbiol. 2018, 9, 2225. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Lärkeryd, A.; Sjödin, A.; Forsman, M.; Stenberg, P. Scaffolding of a bacterial genome using MinION nanopore sequencing. Sci. Rep. 2015, 5, 11996. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Ryan, J.; Holmes, I. Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing. Genome Biol. 2021, 22, 38. [Google Scholar] [CrossRef] [PubMed]
- Grünberger, F.; Ferreira-Cerca, S.; Grohmann, D. Nanopore sequencing of RNA and cDNA molecules in Escherichia coli. RNA 2022, 28, 400–417. [Google Scholar] [CrossRef]
- Cocquet, J.; Chong, A.; Zhang, G.; Veitia, R.A. Reverse transcriptase template switching and false alternative transcripts. Genomics 2006, 88, 127–131. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sequencing Kit | Advantage | Preparation Time | Input Type | Fragmentation | Parallel Quantities | Application |
|---|---|---|---|---|---|---|
| Ligation Sequencing Kit | Higher output | 60 min | gDNA Amplified-DNA | Optional | 24/96 | Whole-genome sequencing/ Methylation/ Adaptive sampling |
| Rapid Sequencing Kit | Higher speed | 10 min | gDNA | Transposase- based | 24/96 | Whole-genome sequencing/ Methylation/ Adaptive sampling |
| Rapid PCR Barcoding Kit | Lower input | 15 min + PCR | gDNA | Transposase- based | 24 | Whole-genome sequencing/ Adaptive sampling |
| Ultra-Long DNA Sequencing Kit | Ultra-long reads | 200 min + 1xO/N incubation | Cells | Transposase- based | - | Whole-genome sequencing/ Methylation/ Adaptive sampling |
| 16S Barcoding Kit | Targeted sequencing | 10 min + PCR | gDNA | - | 24 | 16S sequencing/ Genus-level bacterial identification |
| Cas9 Sequencing Kit | 110 min | gDNA | Cas9-dependent cleavage | - | Target-site sequencing/ Methylation |
| Type | Sequencing Kit | Preparation Time | Input Type | Fragmentation | Parallel Quantities | Application |
|---|---|---|---|---|---|---|
| Direct RNA | Direct RNA Sequencing Kit | 105 min | 500 ng total RNA 50 ng poly(A)+ RNA | Optional | - | Characterize and quantify full-length RNA transcripts, splice variants, and fusions Methylation |
| cDNA | cDNA-PCR Sequencing Kit | 210 min + PCR | 200 ng total RNA 4 ng poly(A)+ RNA | 24 | Characterize and quantify full-length RNA transcripts, splice variants, and fusions |
| Type | Software | Functions | Availability |
|---|---|---|---|
| Sequencing | MinKNOW (Oxford Nanopore Technologies, Oxford, England) | MinKNOW is unique control software from ONT. It is used for sequencing, real-time data monitoring and simple analysis. | https://nanoporetech.com (accessed on 1 January 2024) |
| Basecalling | Guppy (Oxford Nanopore Technologies, Oxford, England) | Basecalling | https://nanoporetech.com (accessed on 1 January 2024) |
| Basecalling | Dorado (Oxford Nanopore Technologies, Oxford, England) | Dorado is a high-performance, easy-to-use, open source basecaller for Oxford Nanopore reads. It is designed for duplex basecalling, simplex barcode classification, initial support for poly(A) tail estimation, and so on. | https://github.com/nanoporetech/dorado (accessed on 1 January 2024) |
| Filter | Porechop version 0.2.4 | Porechop is used for adapter removal and demultiplexing of nanopore reads. | https://github.com/rrwick/Porechop (accessed on 1 January 2024) |
| Assemble | Spades version 3.15.5 | SPAdes is an assembly toolkit containing various assembly pipelines. Spades is used for bacteria, fungal, virus, and other small genomes. | https://github.com/ablab/spades (accessed on 1 January 2024) |
| Assemble | Canu version 2.2 | Canu is an assembly toolkit designed for high-noise single-molecule sequencing. Canu includes four steps: (1) detecting overlaps in high-noise sequences, (2) generating corrected sequence consensus, (3) trimming corrected sequences, and (4) assembling trimmed corrected sequences. | https://github.com/marbl/canu (accessed on 1 January 2024) |
| Error correction | Medaka version 1.11.3 (Oxford Nanopore Technologies, Oxford, England) | A toolkit for polishing consensus sequences and variant calling. | https://github.com/nanoporetech/medaka (accessed on 1 January 2024) |
| Error correction | Pilon version 1.24 | Pilon is an automated genome assembly improvement and variant detection tool. | https://github.com/broadinstitute/pilon (accessed on 1 January 2024) |
| Alignment | Minimap2 version 2.28 | Minimap2 is a versatile sequence alignment program that aligns genomic and spliced nucleotide sequences against a large reference database. | https://github.com/lh3/minimap2 (accessed on 1 January 2024) |
| Alignment | Megalodon (Oxford Nanopore Technologies, Oxford, England) | Megalodon is a research command line tool for reference genome/transcriptome anchored modified base and variant analysis. | https://github.com/nanoporetech/megalodon (accessed on 1 January 2024) |
| Massaging | Samtools version 1.20 | A tool for dealing with SAM, BAM, and CRAM files. These file formats convert to each other. | https://github.com/samtools/samtools (accessed on 1 January 2024) |
| Taxonomy | Centrifuge version 0.32.2 | Centrifuge is a classifier for metagenomic sequence, enabling rapid, accurate, and sensitive labeling of reads and quantification of species. | https://github.com/DaehwanKimLab/centrifuge (accessed on 1 January 2024) |
| Taxonomy | Emu | An approach that employs an expectation-maximization (EM) algorithm to generate taxonomic abundance profiles from full-length 16S rRNA reads. This software can reduce the number of false positives and distinguish between genomically similar species. | https://github.com/treangenlab/emu (accessed on 1 January 2024) |
| Variant analysis | NanoVar version 1.6.2 (Oxford Nanopore Technologies, Oxford, England) | NanoVar is a genomic structural variant (SV) caller that utilizes low-depth long-read sequencing such as ONT. It characterizes six classes of SVs including novel-sequence insertion, deletion, inversion, tandem duplication, sequence transposition (TPO), and translocation (TRA), and requires 4x and 8x sequencing depth for detecting homozygous and heterozygous SVs, respectively. | https://github.com/cytham/nanovar (accessed on 1 January 2024) |
| Methylation analysis | Tombo version 1.5.1 (Oxford Nanopore Technologies, Oxford, England) | Tombo is a suite of tools primarily for the identification and visualization of modified nucleotides from raw nanopore sequencing data. | https://nanoporetech.github.io/tombo (accessed on 1 January 2024) |
| Methylation analysis | Nanopolish (Oxford Nanopore Technologies, Oxford, England) | Nanopolish is a comprehensive tool. It can calculate an improved consensus sequence for a genome assembly, detect base modifications, and call SNPs and indels with respect to a reference genome. | https://github.com/jts/nanopolish (accessed on 1 January 2024) |
| Methylation analysis | DRUMMER | DRUMMER is a tool to identify RNA modifications at nucleotide-level resolution on distinct transcript isoforms through the comparative analysis of basecall errors in nanopore direct RNA sequencing (DRS) datasets. | https://github.com/DepledgeLab/DRUMMER (accessed on 1 January 2024) |
| Sequence splitting and merging | Ont_fast5_api (Oxford Nanopore Technologies, Oxford, England) | The ont_fast5_api provides a simple interface to convert between files in the Oxford Nanopore single_read and multi_read. fast5 file formats. | https://github.com/nanoporetech/ont_fast5_api (accessed on 1 January 2024) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C.-M.; Feng, X.-Y.; Huang, Y.-W.; Chen, R.-A. The Applications of Nanopore Sequencing Technology in Animal and Human Virus Research. Viruses 2024, 16, 798. https://doi.org/10.3390/v16050798
Ji C-M, Feng X-Y, Huang Y-W, Chen R-A. The Applications of Nanopore Sequencing Technology in Animal and Human Virus Research. Viruses. 2024; 16(5):798. https://doi.org/10.3390/v16050798
Chicago/Turabian StyleJi, Chun-Miao, Xiao-Yin Feng, Yao-Wei Huang, and Rui-Ai Chen. 2024. "The Applications of Nanopore Sequencing Technology in Animal and Human Virus Research" Viruses 16, no. 5: 798. https://doi.org/10.3390/v16050798
APA StyleJi, C.-M., Feng, X.-Y., Huang, Y.-W., & Chen, R.-A. (2024). The Applications of Nanopore Sequencing Technology in Animal and Human Virus Research. Viruses, 16(5), 798. https://doi.org/10.3390/v16050798