Advances in Catalysts for Urea Electrosynthesis Utilizing CO2 and Nitrogenous Materials: A Mechanistic Perspective

 ,
,
Abstract
:1. Introduction
2. Molecular Catalyst Interaction Mechanism Achieving Reactant Targeted Adsorption
2.1. Bimetallic Catalyst
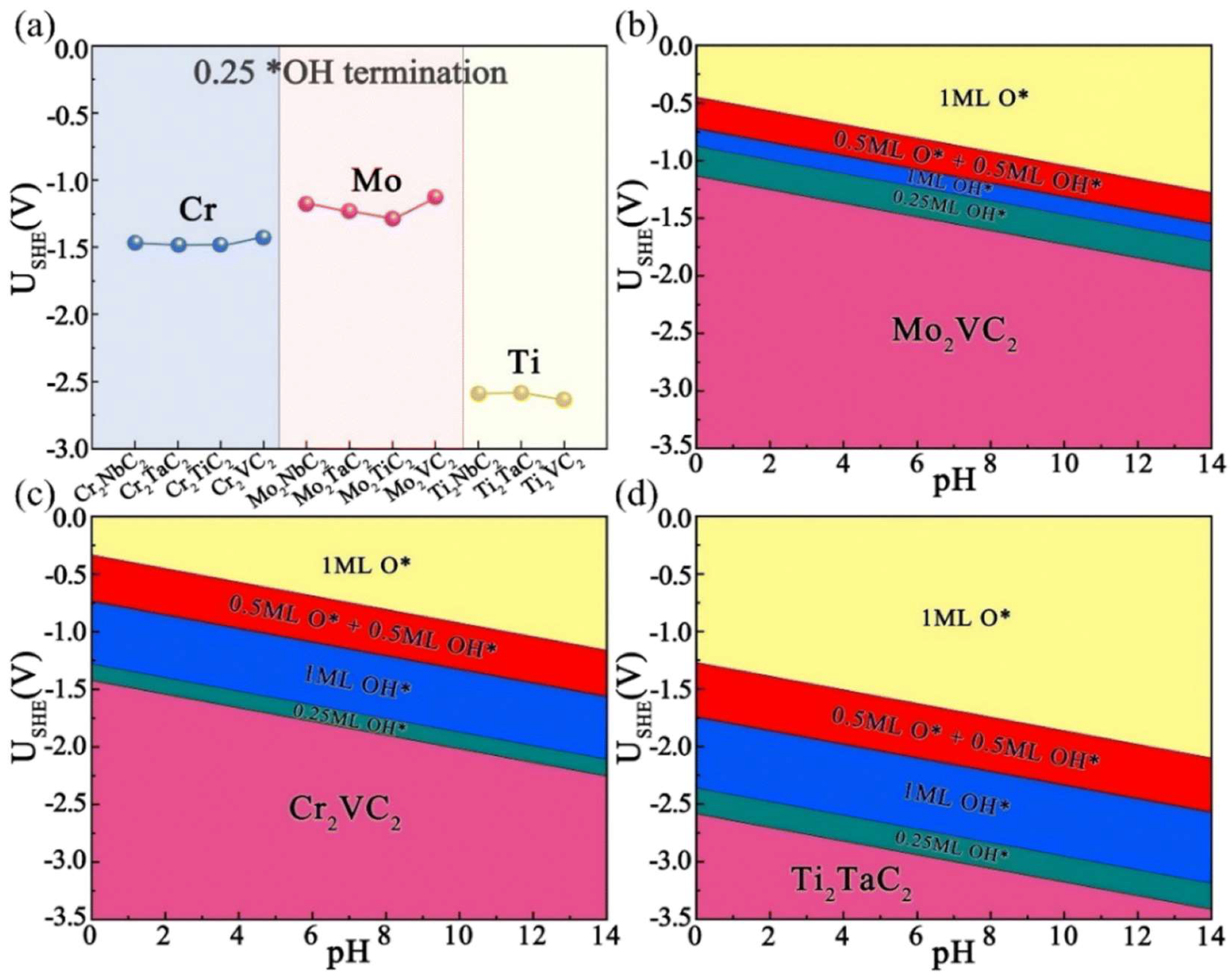
2.1.1. Double Transition-Metal MXenes
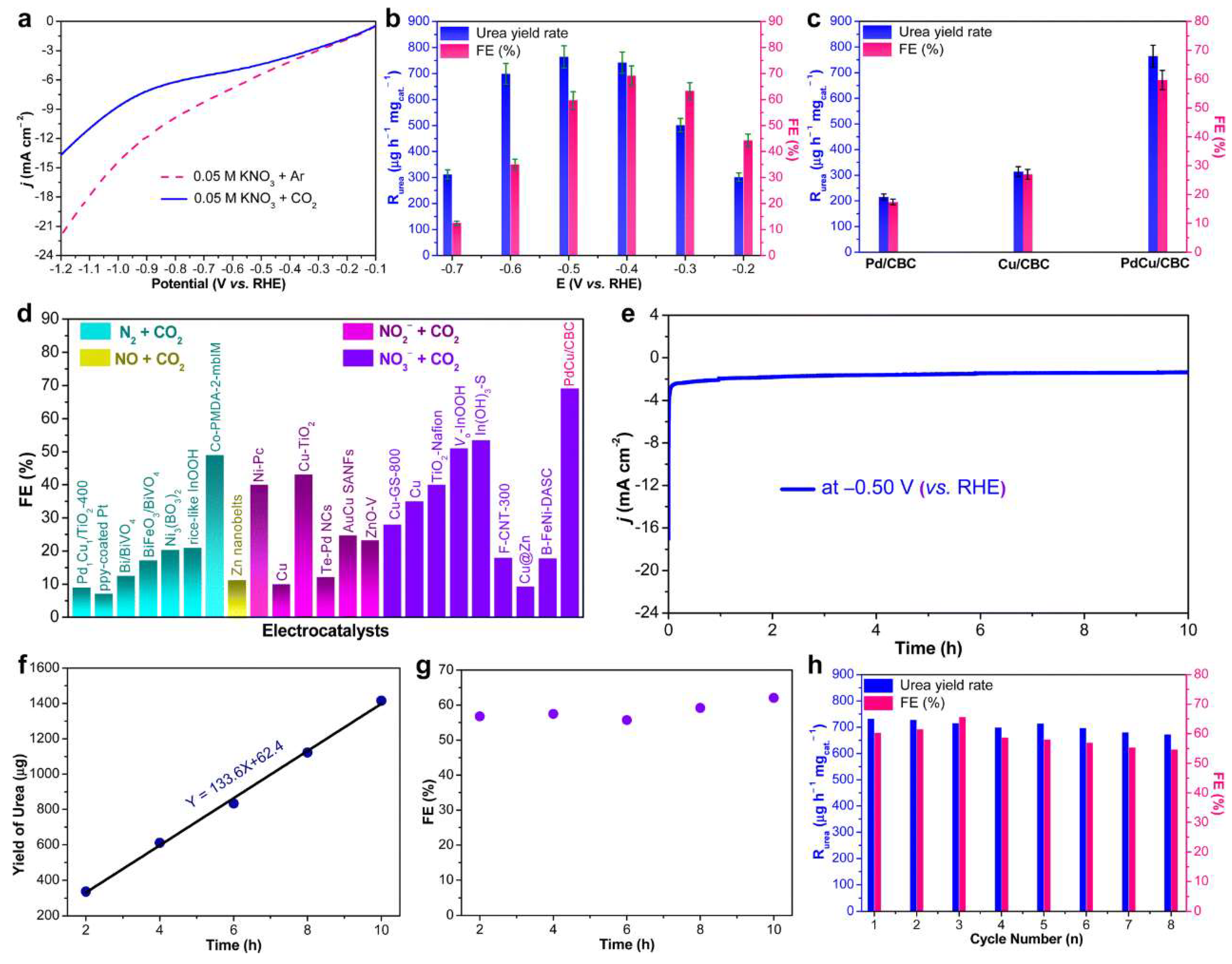
2.1.2. Pd–Cu Bimetallic Catalyst
2.2. Heterogeneous Interface-Rich Catalysts
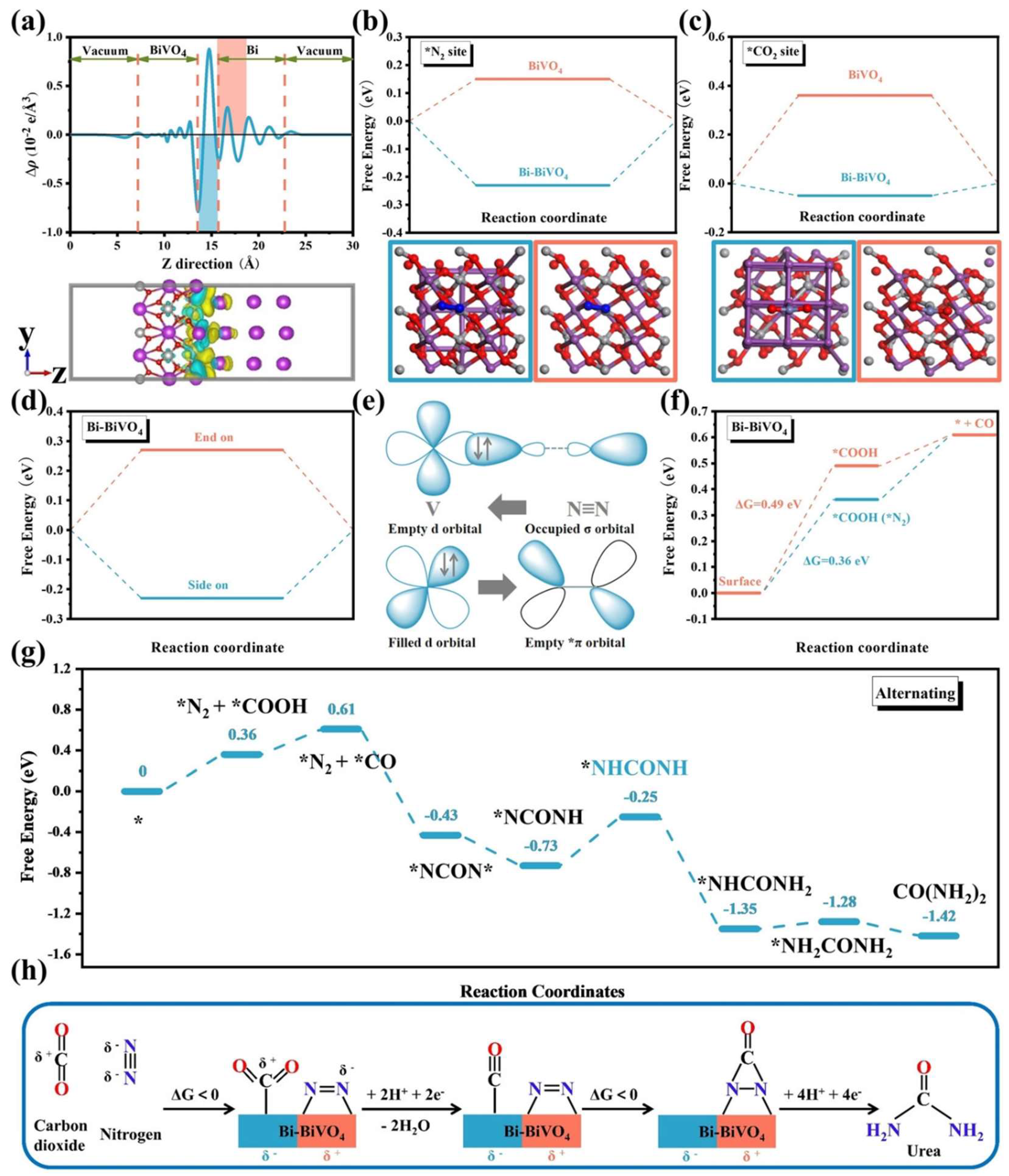
2.2.1. Perovskite Hybrids BiFeO3/BiVO4
2.2.2. Mott–Schottky Heterostructure Bi-BiVO4
2.3. Frustrated Lewis Pairs (FLPs)
2.3.1. InOOH Nanoparticles
2.3.2. Flower-Like Ni3(BO3)2 Nanocrystals
3. Mechanism for Breaking Chemical Bonds and Directional Coupling: Achieving C–N Bond Coupling
3.1. Theoretical Prediction of Catalysts
3.1.1. Double Transition-Metal MXenes
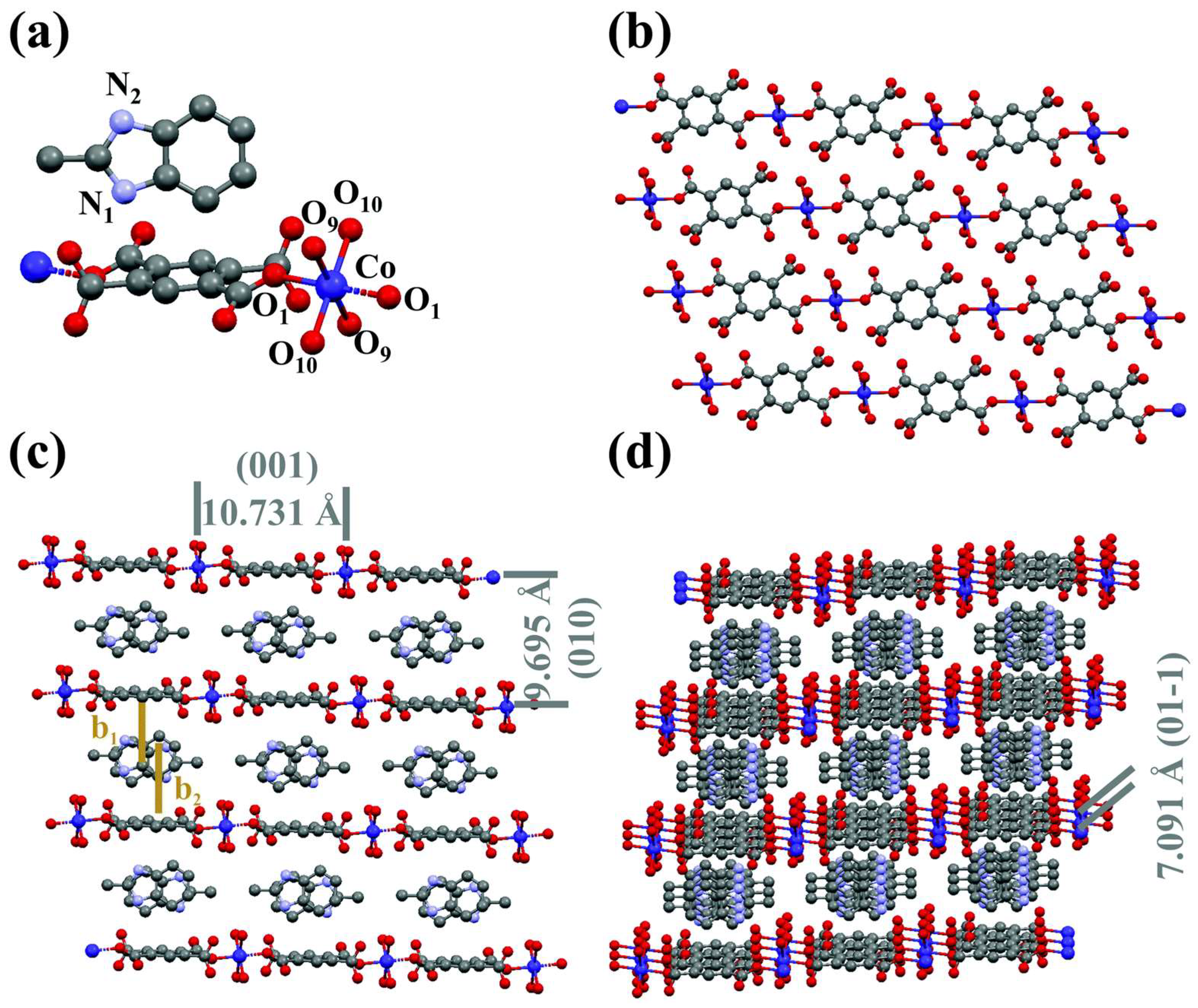
3.1.2. Conductive MOF Co–PMDA–2-mbIM (PMDA = pyromellitic dianhydride; 2-mbIM = 2-methyl benzimidazole)
3.2. Defective Catalysts
3.2.1. Oxygen Vacancy-Enriched CeO2
3.2.2. PdCu-TiO2
4. Discussion
- (1)
- Compared to CO2, the adsorption and activation of inert and non-polar N2 molecules is significantly problematic when CO2 and N2 are utilized as the feedstock for urea electrosynthesis. Hence, when designing catalysts, priority should be given to enhancing the catalytic activity of N2. The investigation of nitrogen reduction reactions and electrocatalytic urea synthesis should complement each other.
- (2)
- To obtain more insight into the electrocatalytic process, the advanced in situ operational characterization can contribute to analyzing the chemical and electronic structures of the catalytic sites, as well as the essential intermediates and the critical steps in the catalytic reaction. Theoretical calculations can be applied to optimize the electrocatalytic behavior of the catalyst. Furthermore, in situ operational characterization can uncover the exact conformational relationships of the catalyst by monitoring the structural transformation of the active site during the electrocatalytic process.
- (3)
- Multiphase catalytic processes occur at the two- or three-phase interface, and the adsorption, dissolution and diffusion of intermediates as well as the generated products governed by the composition of the electrodes and membranes take place. Simultaneously, diffusion is dramatically impacted by the structure of the electrode and membrane assembly in the catalyst layer. Fluid dynamics can be controlled by optimizing the equipment structure, e.g., by tweaking the batch flow rate. The specificity of the products of the cascade reaction can be adjusted by controlling the fluid dynamics, including the mass velocity and volume pressure of the gas and liquid phases. Meanwhile, the selectivity of cascade reaction can be modulated by evaluating the device structure. A proper electrocatalytic device design can effectively regulate the product selectivity, stability and energy efficiency of electrocatalytic reactions, representing a robust tool for high-performance devices that can achieve superior urea electrosynthesis performance.
- (4)
- Molecular–catalyst interaction mechanisms as well as chemical bond breakage and directional coupling mechanisms have explained the mechanism of electrocatalytic synthesis of urea from different microscopic perspectives, contributing to solving the difficult problems of adsorption, activation and the coupling of reactant molecules in urea synthesis. Meanwhile, this mechanism could be applied in other directions, such as the molecular catalyst interaction mechanisms in electrocatalytic nitrogen reduction, which could enhance the electrophilicity of catalyst surfaces through the carrier effect and inhibit proton reduction. Similarly, the chemical bond breaking and directional coupling mechanisms enhance the efficiency of ammonia synthesis by accelerating the breaking of chemical bonds through electron feeding. Furthermore, this mechanism will be of considerable interest in other areas.
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Qing, G.; Ghazfar, R.; Jackowski, S.T.; Habibzadeh, F.; Ashtiani, M.M.; Chen, C.-P.; Smith, M.R., III; Hamann, T.W. Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020, 120, 5437–5516. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Quan, Y.; Guan, A.; Yang, C.; Ji, Y.; Zhang, L.; Zheng, G. Oxygen Vacancies Enhanced Cooperative Electrocatalytic Reduction of Carbon Dioxide and Nitrite Ions to Urea. J. Colloid Interface Sci. 2020, 577, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhu, M.; Wang, M.; He, Y.; Luo, X.; Wu, C.; Zhang, L.; Jin, Z. Review on Electrocatalytic Coreduction of Carbon Dioxide and Nitrogenous Species for Urea Synthesis. ACS Nano 2023, 17, 3209–3224. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, X.; Wen, X.; Zhou, Y.; Zhou, L.; Li, H.; Tao, L.; Li, Q.; Du, S.; Liu, T.; et al. Coupling N2 and CO2 in H2O to Synthesize Urea under Ambient Conditions. Nat. Chem. 2020, 12, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, X.; Ye, L. A Novel Elastic Urea–Melamine–Formaldehyde Foam: Structure and Properties. Ind. Eng. Chem. Res. 2016, 55, 8743–8750. [Google Scholar] [CrossRef]
- Yuan, M.; Chen, J.; Xu, Y.; Liu, R.; Zhao, T.; Zhang, J.; Ren, Z.; Liu, Z.; Streb, C.; He, H.; et al. Highly Selective Electroreduction of N2 and CO2 to Urea over Artificial Frustrated Lewis pairs. Energy Environ. Sci. 2021, 14, 6605–6615. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, X.; Jing, Y.; Li, Y. Electrochemical Synthesis of Urea on MBenes. Nat. Commun. 2021, 12, 4080. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Chen, J.; Bai, Y.; Liu, Z.; Zhang, J.; Zhao, T.; Wang, Q.; Li, S.; He, H.; Zhang, G. Unveiling Electrochemical Urea Synthesis by Co-Activation of CO2 and N2 with Mott–Schottky Heterostructure Catalysts. Angew. Chem. Int. Ed. 2021, 60, 10910–10918. [Google Scholar] [CrossRef]
- Gurrola, M.P.; Cruz, J.C.; Espinosa-Lagunes, F.I.; Martínez-Lázaro, A.; Ledesma-García, J.; Arriaga, L.G.; Escalona-Villalpando, R.A. Perspective of Use of Pd/rGO in a Direct Urea Microfluidic Fuel Cell. Catalysts 2023, 13, 788. [Google Scholar] [CrossRef]
- Lv, C.; Lee, C.; Zhong, L.; Liu, H.; Liu, J.; Yang, L.; Yan, C.; Yu, W.; Hng, H.H.; Qi, Z.; et al. A Defect Engineered Electrocatalyst that Promotes High-Efficiency Urea Synthesis under Ambient Conditions. ACS Nano 2022, 16, 8213–8222. [Google Scholar] [CrossRef]
- Lv, C.; Zhong, L.; Liu, H.; Fang, Z.; Yan, C.; Chen, M.; Kong, Y.; Lee, C.; Liu, D.; Li, S.; et al. Selective Electrocatalytic Synthesis of Urea with Nitrate and Carbon Dioxide. Nat. Sustain. 2021, 4, 868–876. [Google Scholar] [CrossRef]
- Tao, Z.; Rooney, C.L.; Liang, Y.; Wang, H. Accessing Organonitrogen Compounds via C–N Coupling in Electrocatalytic CO2 Reduction. J. Am. Chem. Soc. 2021, 143, 19630–19642. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Lee, C.; Liu, J.; Hu, Y.; Han, D.; Niu, L.; Yan, Q. Booming Electrocatalysts for Urea Synthesis via Nitrogen-Integrated Carbon Dioxide Reduction Reaction. Mater. Today Catal. 2023, 2, 100011. [Google Scholar] [CrossRef]
- Geng, J.; Ji, S.; Jin, M.; Zhang, C.; Xu, M.; Wang, G.; Liang, C.; Zhang, H. Ambient Electrosynthesis of Urea with Nitrate and Carbon Dioxide over Iron-Based Dual-Sites. Angew. Chem. Int. Ed. 2022, 62, e202210958. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Chen, J.; Bai, Y.; Liu, Z.; Zhang, J.; Zhao, T.; Shi, Q.; Li, S.; Wang, X.; Zhang, G. Electrochemical C–N Coupling with Perovskite Hybrids toward Efficient Urea Synthesis. Chem. Sci. 2021, 12, 6048–6058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, D.; Chen, C.; Wang, S. Electrocatalytic Urea Synthesis via C–N Coupling from CO2 and Nitrogenous Species. Acc. Chem. Res. 2024, 57, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ding, Y.; Li, W.; Liu, C.; Li, Y.; Zhao, Z.; Shan, Y.; Li, F.; Sun, L.; Li, F. Efficient Urea Electrosynthesis from Carbon Dioxide and Nitrate via Alternating Cu–W Bimetallic C–N Coupling Sites. Nat. Commun. 2023, 14, 4491. [Google Scholar] [CrossRef]
- Jin, S.; Shi, Z.; Jing, H.; Wang, L.; Hu, Q.; Chen, D.; Li, N.; Zhou, A. Mo2C-MXene/CdS Heterostructures as Visible-Light Photocatalysts with an Ultrahigh Hydrogen Production Rate. ACS Appl. Energy Mater. 2021, 4, 12754–12766. [Google Scholar] [CrossRef]
- Verger, L.; Xu, C.; Natu, V.; Cheng, H.-M.; Ren, W.; Barsoum, M.W. Overview of the synthesis of MXenes and Other Ultrathin 2D Transition Metal Carbides and Nitrides. Curr. Opin. Solid State Mater. Sci. 2019, 23, 149–163. [Google Scholar] [CrossRef]
- Sápi, A.; Rajkumar, T.; Kiss, J.; Kukovecz, Á.; Kónya, Z.; Somorjai, G.A. Metallic Nanoparticles in Heterogeneous Catalysis. Catal. Lett. 2021, 151, 2153–2175. [Google Scholar] [CrossRef]
- Chen, C.; He, N.; Wang, S. Electrocatalytic C–N Coupling for Urea Synthesis. Small Sci. 2021, 1, 2100070. [Google Scholar] [CrossRef]
- Meng, N.; Huang, Y.; Liu, Y.; Yu, Y.; Zhang, B. Electrosynthesis of Urea from Nitrite and CO2 over Oxygen Vacancy-Rich ZnO Porous Nanosheets. Cell Rep. Phys. Sci. 2021, 2, 100378. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, H.; Zhang, Y.; Huang, X.; Li, L.; Cheng, T.; Shao, Q. Te-Doped Pd Nanocrystal for Electrochemical Urea Production by Efficiently Coupling Carbon Dioxide Reduction with Nitrite Reduction. Nano Lett. 2020, 20, 8282–8289. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, D.; Song, J.; Lee, S.; Hur, N.H.; Shin, W. Electrocatalytic Conversion of Carbon Dioxide and Nitrate Ions to Urea by a Titania–Nafion Composite Electrode. ChemSusChem 2017, 10, 3999–4003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, L.; Du, J.; Hou, Y. Double Metal Synergistic Synthetic Urea: An Electrocatalytic Study. New J. Chem. 2022, 46, 5278–5287. [Google Scholar] [CrossRef]
- Liu, J.; Guo, X.; Frauenheim, T.; Gu, Y.; Kou, L. Urea Electrosynthesis Accelerated by Theoretical Simulations. Adv. Funct. Mater. 2024, 34, 2313420. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef]
- Li, N.; Zeng, Z.; Zhang, Y.; Chen, X.; Kong, Z.; Arramel; Li, Y.; Zhang, P.; Nguyen, B.-S. Double Transition Metal Carbides MXenes (D-MXenes) as Promising Electrocatalysts for Hydrogen Reduction Reaction: Ab Initio Calculations. ACS Omega 2021, 6, 23676–23682. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, Z.; Liu, J.; Sun, F.; Yang, P.; Qiu, J. A Hierarchically Porous and Hydrophilic 3D Nickel–Iron/MXene Electrode for Accelerating Oxygen and Hydrogen Evolution at High Current Densities. Nano Energy 2019, 63, 103880. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, S.; Wang, Z.; Zhao, J.; Qiu, J. Boosting Electrocatalytic Oxygen Evolution by Synergistically Coupling Layered Double Hydroxide with MXene. Nano Energy 2018, 44, 181–190. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, Z.; Han, J.; Jin, Z.; Han, Y.; Wang, F.; Niu, Y.; Wu, Y.; Xu, Y. High-Performance Electrocatalytic Conversion of N2 to NH3 Using Oxygen-Vacancy-Rich TiO2 In Situ Grown on Ti3C2Tx MXene. Adv. Energy Mater. 2019, 9, 1803406. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, J.; Shi, Z.; Zhang, P.; Arramel, A.; Li, N. Unveiling the Key Intermediates in Electrocatalytic Synthesis of Urea with CO2 and N2 Coupling Reactions on Double Transition-Metal MXenes. J. Mater. Chem. A 2023, 11, 6428–6439. [Google Scholar] [CrossRef]
- Fan, J.; Du, H.; Zhao, Y.; Wang, Q.; Liu, Y.; Li, D.; Feng, J. Recent Progress on Rational Design of Bimetallic Pd Based Catalysts and Their Advanced Catalysis. ACS Catal. 2020, 10, 13560–13583. [Google Scholar] [CrossRef]
- Ribeirinha, P.; Mateos-Pedrero, C.; Boaventura, M.; Sousa, J.; Mendes, A. CuO/ZnO/Ga2O3 Catalyst for Low Temperature MSR Reaction: Synthesis, Characterization and Kinetic Model. Appl. Catal. B Environ. 2018, 221, 371–379. [Google Scholar] [CrossRef]
- Azenha, C.S.R.; Mateos-Pedrero, C.; Queirós, S.; Concepción, P.; Mendes, A. Innovative ZrO2-Supported CuPd Catalysts for the Selective Production of Hydrogen from Methanol Steam Reforming. Appl. Catal. B Environ. 2017, 203, 400–407. [Google Scholar] [CrossRef]
- Zhang, S.; Geng, J.; Zhao, Z.; Jin, M.; Li, W.; Ye, Y.; Li, K.; Wang, G.; Zhang, Y.; Yin, H.; et al. High-Efficiency Electrosynthesis of Urea over Bacterial Cellulose Regulated Pd–Cu Bimetallic Catalyst. EES Catal. 2023, 1, 45–53. [Google Scholar] [CrossRef]
- Zhang, S.; Shi, T.; Li, K.; Sun, Q.; Lin, Y.; Zheng, L.R.; Wang, G.; Zhang, Y.; Yin, H.; Zhang, H. Ambient Electrochemical Nitrogen Fixation over a Bifunctional Mo–(O–C2)4 Site Catalyst. J. Phys. Chem. C 2022, 126, 965–973. [Google Scholar] [CrossRef]
- Zhang, S.; Jin, M.; Shi, T.; Han, M.; Sun, Q.; Lin, Y.; Ding, Z.; Zheng, L.R.; Wang, G.; Zhang, Y.; et al. Electrocatalytically Active Fe-(O-C2)4 Single-Atom Sites for Efficient Reduction of Nitrogen to Ammonia. Angew. Chem. Int. Ed. 2020, 59, 13423–13429. [Google Scholar] [CrossRef]
- Hsu, J.; Houache, M.S.E.; Abu-Lebdeh, Y.; Patton, R.A.; Guzman, M.I.; Al-Abadleh, H.A. In Situ Electrochemistry of Formate on Cu Thin Films Using ATR-FTIR Spectroscopy and X-ray Photoelectron Spectroscopy. Langmuir 2024, 40, 2377–2384. [Google Scholar] [CrossRef]
- Cui, H.; Li, B.; Zhang, Y.; Zheng, X.; Li, X.; Li, Z.; Xu, S. Constructing Z-Scheme Based CoWO4/CdS Photocatalysts with Enhanced Dye Degradation and H2 Generation Performance. Int. J. Hydrog. Energy 2018, 43, 18242–18252. [Google Scholar] [CrossRef]
- Tsvetkov, N.; Lu, Q.; Sun, L.; Crumlin, E.J.; Yildiz, B. Improved Chemical and Electrochemical Stability of Perovskite Oxides with Less Reducible Cations at the Surface. Nat. Mater. 2016, 15, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kang, Y.; Zhao, H.; Dai, X.; Cui, M.; Luan, X.; Zhang, X.; Nie, F.; Ren, Z.; Song, W. An Interfacial Electron Transfer on Tetrahedral NiS2/NiSe2 Heterocages with Dual-Phase Synergy for Efficiently Triggering the Oxygen Evolution Reaction. Small 2020, 16, 1905083. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, K.; Liu, S.; Chen, X.; Cheng, Y.; Cheong, W.-C.; Chen, Z.; Zheng, L.; Zhang, J.; Li, X.; et al. Construction of CoP/NiCoP Nanotadpoles Heterojunction Interface for Wide pH Hydrogen Evolution Electrocatalysis and Supercapacitor. Adv. Energy Mater. 2019, 9, 1901213. [Google Scholar] [CrossRef]
- Yuan, M.; Bai, Y.; Zhang, J.; Zhao, T.; Li, S.; He, H.; Liu, Z.; Wang, Z.; Zhang, G. Work Function Regulation of Nitrogen-Doped Carbon Nanotubes Triggered by Metal Nanoparticles for Efficient Electrocatalytic Nitrogen Fixation. J. Mater. Chem. A 2020, 8, 26066–26074. [Google Scholar] [CrossRef]
- Yuan, M.; Dipazir, S.; Wang, M.; Sun, Y.; Gao, D.; Bai, Y.; Zhang, M.; Lu, P.; He, H.; Zhu, X.; et al. Polyoxometalate-Assisted Formation of CoSe/MoSe2 Heterostructures with Enhanced Oxygen Evolution Activity. J. Mater. Chem. A 2019, 7, 3317–3326. [Google Scholar] [CrossRef]
- Skúlason, E.; Bligaard, T.; Gudmundsdóttir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jónsson, H.; Nørskov, J.K. A Theoretical Evaluation of Possible Transition Metal Electro-Catalysts for N2 Reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef]
- Xi, C.; Zhu, G.; Liu, Y.; Shen, X.; Zhu, W.; Ji, Z.; Kong, L. Belt-Like Nickel Hydroxide Carbonate/Reduced Graphene Oxide Hybrids: Synthesis and Performance as Supercapacitor Electrodes. Colloids Surf. A Physicochem. Eng. Asp. 2018, 538, 748–756. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, M.; Lu, P.; Sun, Y.; Dipazir, S.; Zhang, J.; Li, S.; Zhang, G. Tuning Carbon Nanotube-Grafted Core-Shell-Structured Cobalt Selenide@Carbon Hybrids for Efficient Oxygen Evolution Reaction. J. Colloid Interface Sci. 2019, 533, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.-H.; Su, H.; Yu, Q.-Y.; Zhang, B.; Wang, H.-H.; Li, X.-H.; Chen, J.-S. Janus Co/CoP Nanoparticles as Efficient Mott–Schottky Electrocatalysts for Overall Water Splitting in Wide Ph Range. Adv. Energy Mater. 2017, 7, 1602355. [Google Scholar] [CrossRef]
- He, K.; Tsega, T.T.; Liu, X.; Zai, J.; Li, X.-H.; Liu, X.; Li, W.; Ali, N.; Qian, X. Utilizing the Space-Charge Region of the FeNi-LDH/CoP p-n Junction to Promote Performance in Oxygen Evolution Electrocatalysis. Angew. Chem. Int. Ed. 2019, 58, 11903–11909. [Google Scholar] [CrossRef]
- Geng, Z.; Liu, Y.; Kong, X.; Li, P.; Li, K.; Liu, Z.; Du, J.; Shu, M.; Si, R.; Zeng, J. Achieving a Record-High Yield Rate of 120.9 for N2 Electrochemical Reduction over Ru Single-Atom Catalysts. Adv. Mater. 2018, 30, 1803498. [Google Scholar] [CrossRef]
- Wang, J.; Yu, L.; Hu, L.; Chen, G.; Xin, H.; Feng, X. Ambient Ammonia Synthesis via Palladium-Catalyzed Electrohydrogenation of Dinitrogen at Low Overpotential. Nat. Commun. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Suryanto, B.H.R.; Du, H.-L.; Wang, D.; Chen, J.; Simonov, A.N.; MacFarlane, D.R. Challenges and Prospects in the Catalysis of Electroreduction of Nitrogen to Ammonia. Nat. Catal. 2019, 2, 290–296. [Google Scholar] [CrossRef]
- Cheng, H.; Cui, P.; Wang, F.; Ding, L.-X.; Wang, H. High Efficiency Electrochemical Nitrogen Fixation Achieved with a Lower Pressure Reaction System by Changing the Chemical Equilibrium. Angew. Chem. Int. Ed. 2019, 58, 15541–15547. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, M.; He, Z.; Gu, B.; Xiao, C.; Zhou, T.; Guo, Z.; Liu, J.; He, H.; Ye, B.; et al. Pothole-rich Ultrathin WO3 Nanosheets that Trigger N≡N Bond Activation of Nitrogen for Direct Nitrate Photosynthesis. Angew. Chem. Int. Ed. 2019, 58, 731–735. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis Pairs: From Concept to Catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhang, H.; Xu, Y.; Liu, R.; Wang, R.; Zhao, T.; Zhang, J.; Liu, Z.; He, H.; Yang, C.; et al. Artificial Frustrated Lewis Pairs Facilitating the Electrochemical N2 and CO2 Conversion to Urea. Chem Catal. 2022, 2, 309–320. [Google Scholar] [CrossRef]
- Ghuman, K.K.; Hoch, L.B.; Wood, T.E.; Mims, C.; Singh, C.V.; Ozin, G.A. Surface Analogues of Molecular Frustrated Lewis Pairs in Heterogeneous CO2 Hydrogenation Catalysis. ACS Catal. 2016, 6, 5764–5770. [Google Scholar] [CrossRef]
- Hoch, L.B.; Szymanski, P.; Ghuman, K.K.; Hea, L.; Liao, K.; Qiao, Q.; Reyes, L.M.; Zhu, Y.; El-Sayed, M.A.; Singh, C.V.; et al. Carrier Dynamics and the Role of Surface Defects: Designing a Photocatalyst for Gas-Phase CO2 Reduction. Proc. Natl. Acad. Sci. United States Am. 2016, 113, E8011–E8020. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, K.K.; Hoch, L.B.; Szymanski, P.; Loh, J.Y.Y.; Kherani, N.P.; El-Sayed, M.A.; Ozin, G.A.; Singh, C.V. Photoexcited Surface Frustrated Lewis Pairs for Heterogeneous Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2016, 138, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Barzagli, F.; Mani, F.; Peruzzini, M. From Greenhouse Gas to Feedstock: Formation of Ammonium Carbamate from CO2 and NH3 in Organic Solvents and Its Catalytic Conversion into Urea under Mild Conditions. Green Chem. 2011, 13, 1267–1274. [Google Scholar] [CrossRef]
- Wei, X.; Wen, X.; Liu, Y.; Chen, C.; Xie, C.; Wang, D.; Qiu, M.; He, N.; Zhou, P.; Chen, W.; et al. Oxygen Vacancy-Mediated Selective C–N Coupling toward Electrocatalytic Urea Synthesis. J. Am. Chem. Soc. 2022, 144, 11530–11535. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, X.; Wang, Z.; Liu, B.; Zhang, P.; Li, X.; Li, N. Uncovering the Mechanism for Urea Electrochemical Synthesis by Coupling N2 and CO2 on Mo2C-MXene. Chin. J. Struct. Chem. 2022, 41, 152–162. [Google Scholar] [CrossRef]
- Nasr Allah, T.; Ponsard, L.; Nicolas, E.; Cantat, T. Catalytic Challenges and Strategies for the Carbonylation of σ-Bonds. Green Chem. 2021, 23, 723–739. [Google Scholar] [CrossRef]
- Wang, X.-L.; Dong, L.-Z.; Qiao, M.; Tang, Y.-J.; Liu, J.; Li, Y.; Li, S.-L.; Su, J.-X.; Lan, Y.-Q. Exploring the Performance Improvement of the Oxygen Evolution Reaction in a Stable Bimetal–Organic Framework System. Angew. Chem. Int. Ed. 2018, 57, 9660–9664. [Google Scholar] [CrossRef] [PubMed]
- Narayan, T.C.; Miyakai, T.; Seki, S.; Dincă, M. High Charge Mobility in a Tetrathiafulvalene-Based Microporous Metal–Organic Framework. J. Am. Chem. Soc. 2012, 134, 12932–12935. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Pfeffermann, M.; Liang, H.; Zheng, Z.; Zhu, X.; Zhang, J.; Feng, X. Large-Area, Free-Standing, Two-Dimensional Supramolecular Polymer Single-Layer Sheets for Highly Efficient Electrocatalytic Hydrogen Evolution. Angew. Chem. Int. Ed. 2015, 54, 12058–12063. [Google Scholar] [CrossRef]
- Le Ouay, B.; Boudot, M.; Kitao, T.; Yanagida, T.; Kitagawa, S.; Uemura, T. Nanostructuration of PEDOT in Porous Coordination Polymers for Tunable Porosity and Conductivity. J. Am. Chem. Soc. 2016, 138, 10088–10091. [Google Scholar] [CrossRef]
- Yuan, M.; Chen, J.; Zhang, H.; Li, Q.; Zhou, L.; Yang, C.; Liu, R.; Liu, Z.; Zhang, S.; Zhang, G. Host–Guest Molecular Interaction Promoted Urea Electrosynthesis over a Precisely Designed Conductive Metal–Organic Framework. Energy Environ. Sci. 2022, 15, 2084–2095. [Google Scholar] [CrossRef]
- Shen, G.; Pan, L.; Zhang, R.; Sun, S.; Hou, F.; Zhang, X.; Zou, J.-J. Low-Spin-State Hematite with Superior Adsorption of Anionic Contaminations for Water Purification. Adv. Mater. 2020, 32, 1905988. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tong, Y.; Chen, P.; Xu, K.; Zhao, J.; Lin, Y.; Chu, W.; Peng, Z.; Wu, C.; Xie, Y. Engineering the Electronic State of a Perovskite Electrocatalyst for Synergistically Enhanced Oxygen Evolution Reaction. Adv. Mater. 2015, 27, 5989–5994. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Miao, X.; Zhao, X.; Ma, C.; Qiu, Y.; Hu, Z.; Zhao, J.; Shi, L.; Zeng, J. Engineering Electrocatalytic Activity in Nanosized Perovskite Cobaltite through Surface Spin-State Transition. Nat. Commun. 2016, 7, 11510. [Google Scholar] [CrossRef] [PubMed]
- Zobel, C.; Kriener, M.; Bruns, D.; Baier, J.; Grüninger, M.; Lorenz, T.; Reutler, P.; Revcolevschi, A. Evidence for a Low-Spin to Intermediate-Spin State Transition in LaCoO3. Phys. Rev. B 2002, 66, 020402. [Google Scholar] [CrossRef]
- Chitsiga, T.; Daramola, M.O.; Wagner, N.; Ngoy, J. Effect of the Presence of Water-Soluble Amines on the Carbon Dioxide (CO2) Adsorption Capacity of Amine-Grafted Poly-Succinimide (PSI) Adsorbent during CO2 Capture. Energy Procedia 2016, 86, 90–105. [Google Scholar] [CrossRef]
- Xie, C.; Yan, D.; Chen, W.; Zou, Y.; Chen, R.; Zang, S.; Wang, Y.; Yao, X.; Wang, S. Insight into the Design of Defect Electrocatalysts: From Electronic Structure to Adsorption Energy. Mater. Today 2019, 31, 47–68. [Google Scholar] [CrossRef]
- Xie, C.; Yan, D.; Li, H.; Du, S.; Chen, W.; Wang, Y.; Zou, Y.; Chen, R.; Wang, S. Defect Chemistry in Heterogeneous Catalysis: Recognition, Understanding, and Utilization. ACS Catal. 2020, 10, 11082–11098. [Google Scholar] [CrossRef]
- Devadas, A.; Vasudevan, S.; Epron, F. Nitrate Reduction in Water: Influence of the Addition of a Second Metal on the Performances of the Pd/CeO2 Catalyst. J. Hazard. Mater. 2011, 185, 1412–1417. [Google Scholar] [CrossRef]
- Epron, F.; Gauthard, F.; Barbier, J. Catalytic Reduction of Nitrate in Water on a Monometallic Pd/CeO2 Catalyst. J. Catal. 2002, 206, 363–367. [Google Scholar] [CrossRef]
- Kiss, J.; Solymosi, F. The Effect of Adsorbed Oxygen on the Stability of NCO on Rh(111) Studied by Reflection Absorption Infrared Spectroscopy. J. Catal. 1998, 179, 277–282. [Google Scholar] [CrossRef]
- Yan, D.; Li, H.; Chen, C.; Zou, Y.; Wang, S. Defect Engineering Strategies for Nitrogen Reduction Reactions under Ambient Conditions. Small Methods 2019, 3, 1800331. [Google Scholar] [CrossRef]
- Wan, J.; Chen, W.; Jia, C.; Zheng, L.; Dong, J.; Zheng, X.; Wang, Y.; Yan, W.; Chen, C.; Peng, Q.; et al. Defect Effects on TiO2 Nanosheets: Stabilizing Single Atomic Site Au and Promoting Catalytic Properties. Adv. Mater. 2018, 30, 1705369. [Google Scholar] [CrossRef] [PubMed]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia Synthesis from First-Principles Calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, J.-C.; Xu, M.; Zhao, Y.; Ma, X.-L.; Dong, J.; Zheng, X.; Zheng, J.; Allen, C.S.; Danaie, M.; et al. Molecular Nitrogen Promotes Catalytic Hydrodeoxygenation. Nat. Catal. 2019, 2, 1078–1087. [Google Scholar] [CrossRef]
- Ma , S.; Lan , Y.; Perez, G.M.J.; Moniri, S.; Kenis , P.J.A. Silver Supported on Titania as an Active Catalyst for Electrochemical Carbon Dioxide Reduction. ChemSusChem 2014, 7, 866–874. [Google Scholar] [CrossRef]
- Chieng, B.W.; Ibrahim, N.A.; Yunus, W.M.Z.W.; Hussein, M.Z. Poly(Lactic acid)/Poly(Ethylene Glycol) Polymer Nanocomposites: Effects of Graphene Nanoplatelets. Polymers 2014, 6, 93–104. [Google Scholar] [CrossRef]





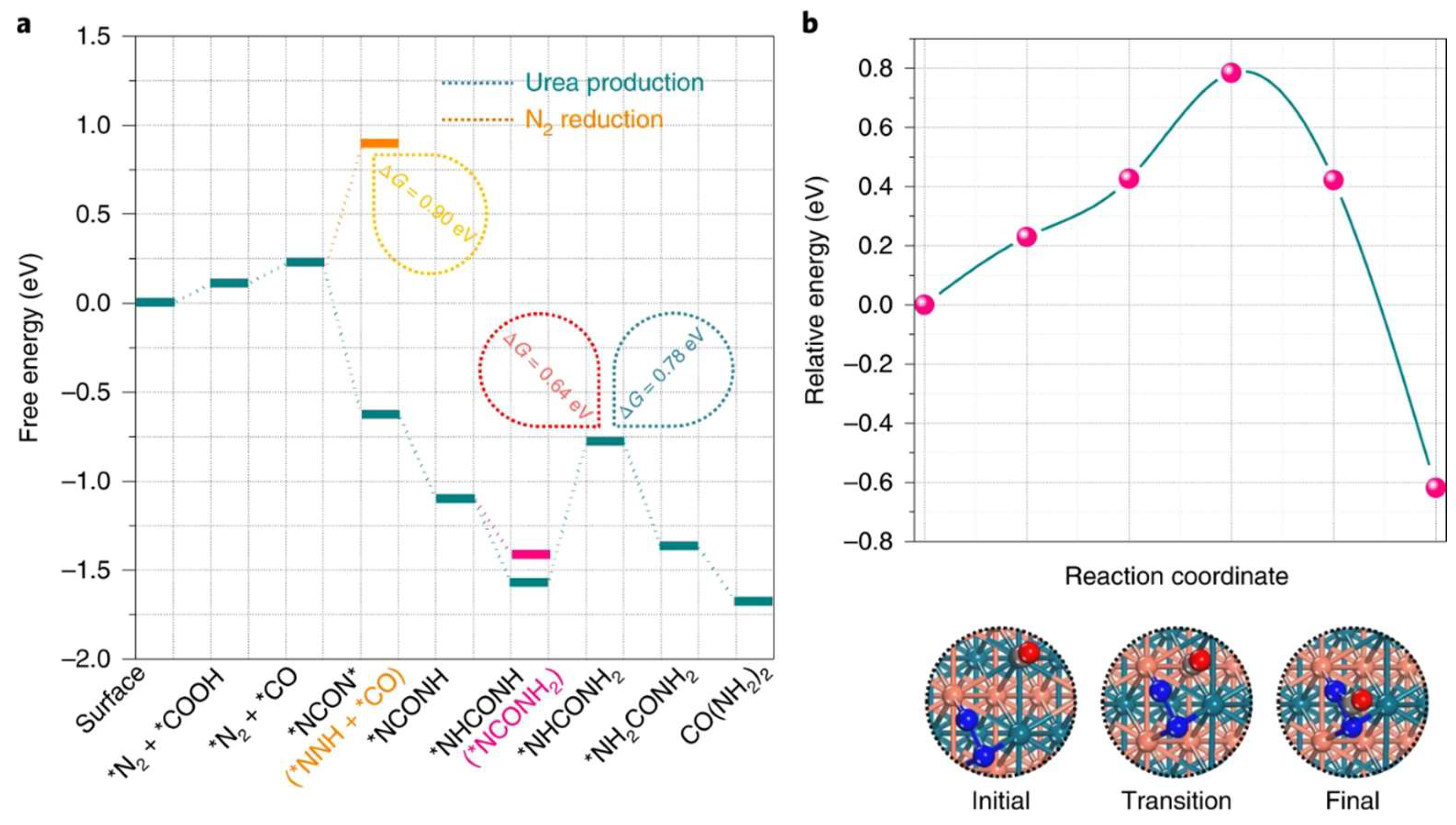

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Synthesis Methods | Faraday Efficiency/Yield Rate |
|---|---|---|
| MXenes [27] | Theoretical calculations | -- |
| Pd–Cu [31] | Wet chemistry impregnation + carbonization fixation process | 69.1 ± 3.8% |
| BiFeO3/BiVO4 [13] | Hydrothermal method | 17.18% |
| Bi-BiVO4 [8] | Hydrothermal method + NaBH4 reduction | 12.55% |
| InOOH [53] | Hydrothermal method + annealing treatment | 20.97% |
| Ni3(BO3)2 [6] | Wet chemistry + low-temperature annealing | 20.36% |
| Co–PMDA–2-mbIM [65] | Hydrothermal method | 48.97% |
| CeO2 [58] | Hydrothermal method + annealing treatment | 943.6 mg h−1 g−1 |
| PdCu-TiO2 [4] | High temperature reduction | 8.92% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Feng, T.; Che, X.; Wang, Y.; Wang, P.; Chai, M.; Yuan, M. Advances in Catalysts for Urea Electrosynthesis Utilizing CO2 and Nitrogenous Materials: A Mechanistic Perspective. Materials 2024, 17, 2142. https://doi.org/10.3390/ma17092142
Zhang M, Feng T, Che X, Wang Y, Wang P, Chai M, Yuan M. Advances in Catalysts for Urea Electrosynthesis Utilizing CO2 and Nitrogenous Materials: A Mechanistic Perspective. Materials. 2024; 17(9):2142. https://doi.org/10.3390/ma17092142
Chicago/Turabian StyleZhang, Mengfei, Tianjian Feng, Xuanming Che, Yuhan Wang, Pengxian Wang, Mao Chai, and Menglei Yuan. 2024. "Advances in Catalysts for Urea Electrosynthesis Utilizing CO2 and Nitrogenous Materials: A Mechanistic Perspective" Materials 17, no. 9: 2142. https://doi.org/10.3390/ma17092142