Expression Profiles of Cuproptosis-Related Genes Determine Distinct Subtypes of Pancreatic Ductal Adenocarcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Method
2.1. Data Sources and Processing Methods
2.2. Online Tools Exploring Gene Expression Patterns, Genetic Alterations, and Gene Methylation Levels
2.3. Principal Component Analysis (PCA)
2.4. Cancer Subtypes Identified via the Consensus Clustering Method
2.5. Identification of Differentially Expressed Genes (DEGs)
2.6. Gene Set Variation Analysis (GSVA)
2.7. Prediction of Chemotherapeutic Response
2.8. Statistical Analyses
3. Results
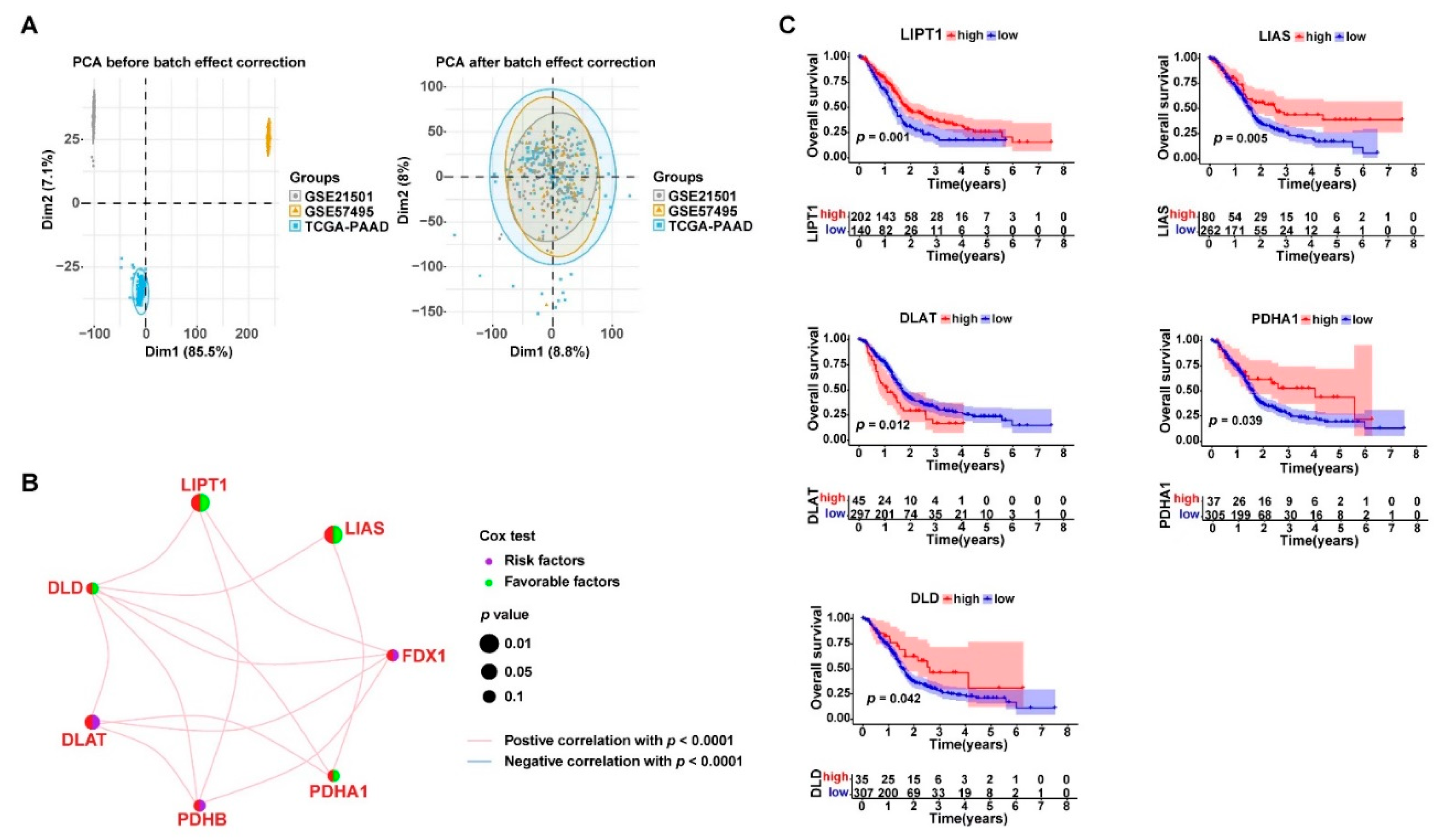
3.1. Gene Expression and Genetic Alterations of Cuproptosis-Related Genes in PDAC
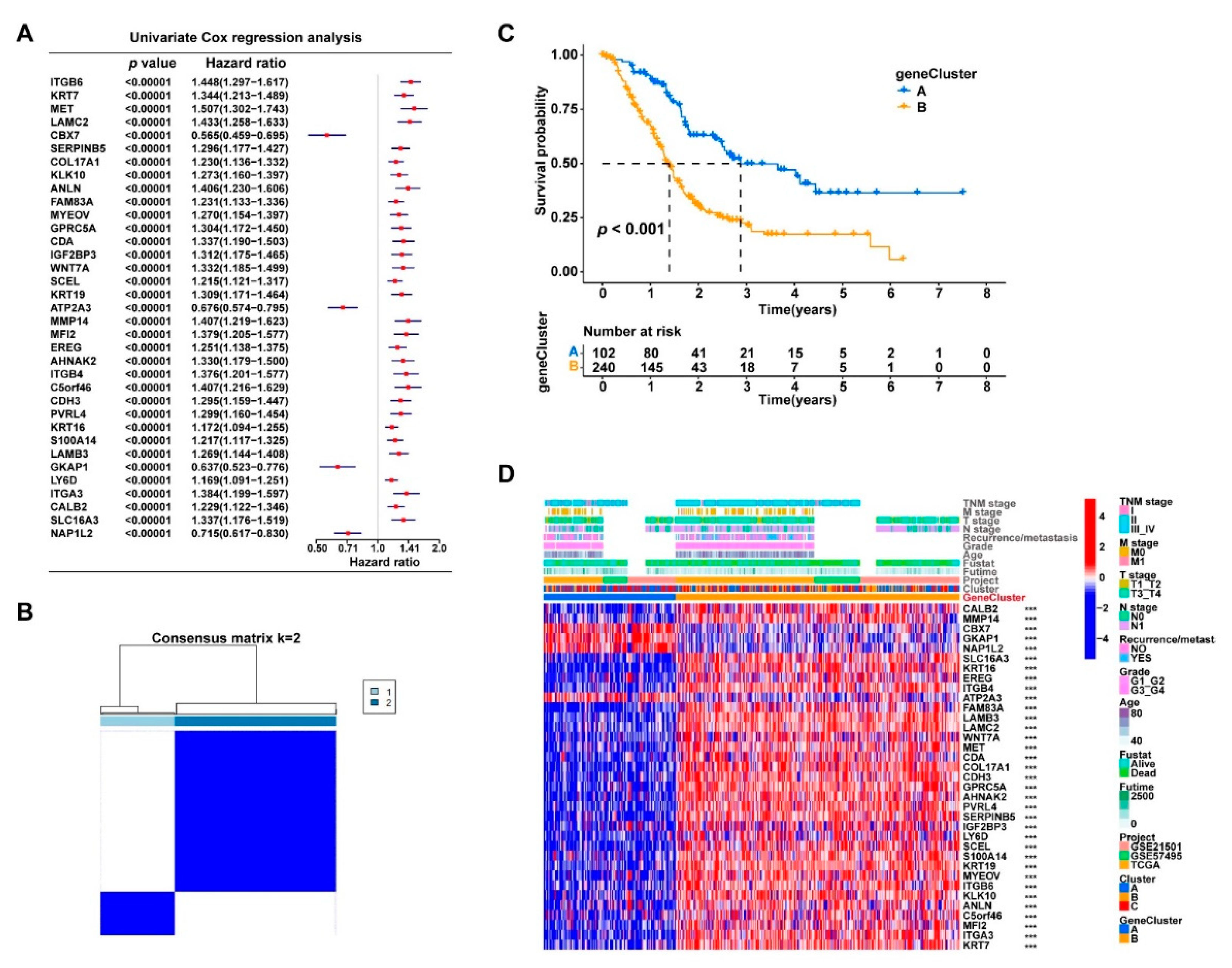
3.2. Cuproptosis-Related Genes Were Significant Prognostic Factors for PDAC
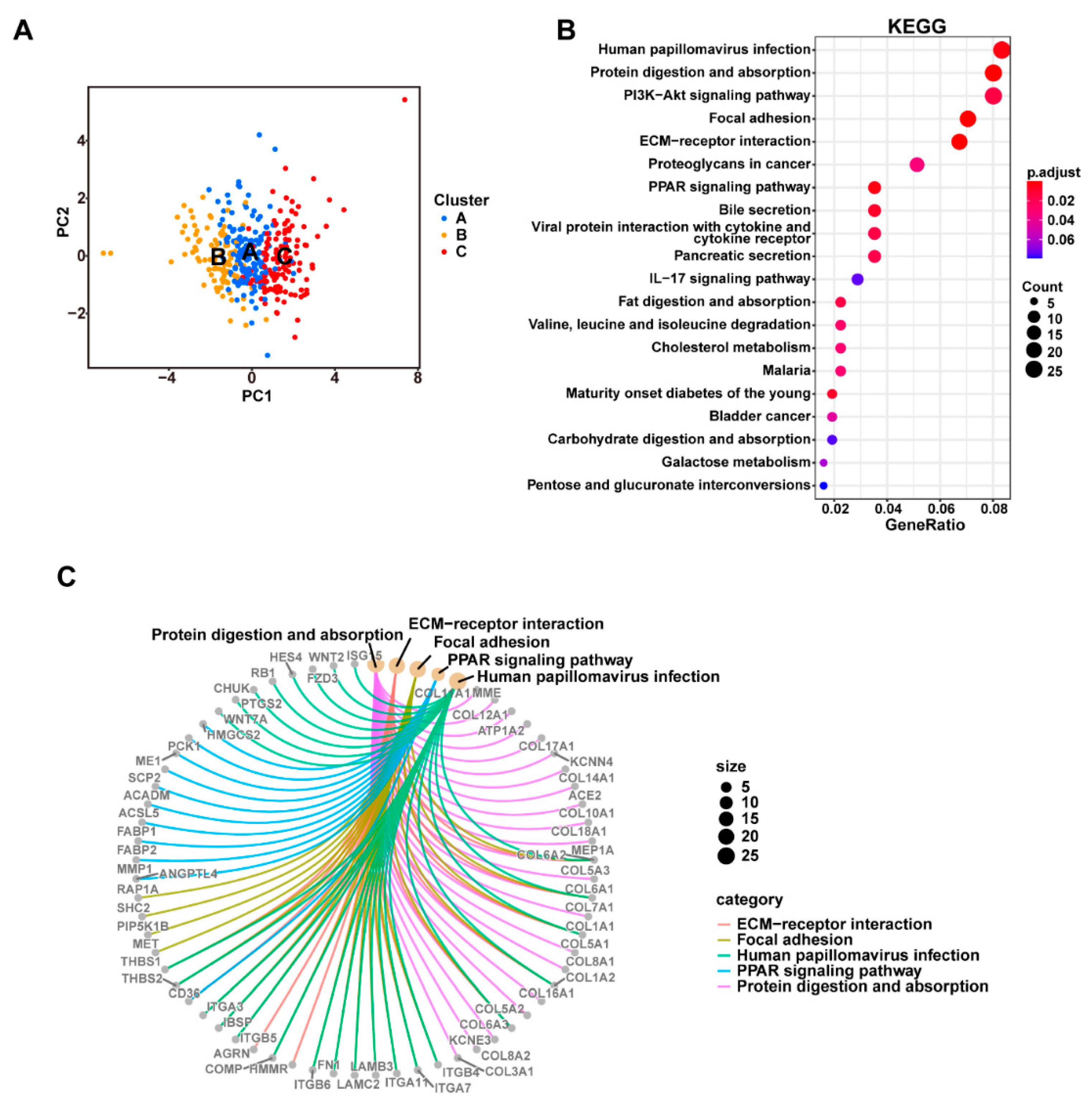
3.3. Consensus Clustering Based on Cuproptosis-Related Genes Expression Identified Three Clusters in PDAC

3.4. The DEGs in the Three Clusters Separated the PDAC Samples into Two Subgroups with Significantly Different Survival and Tumor Immune Status
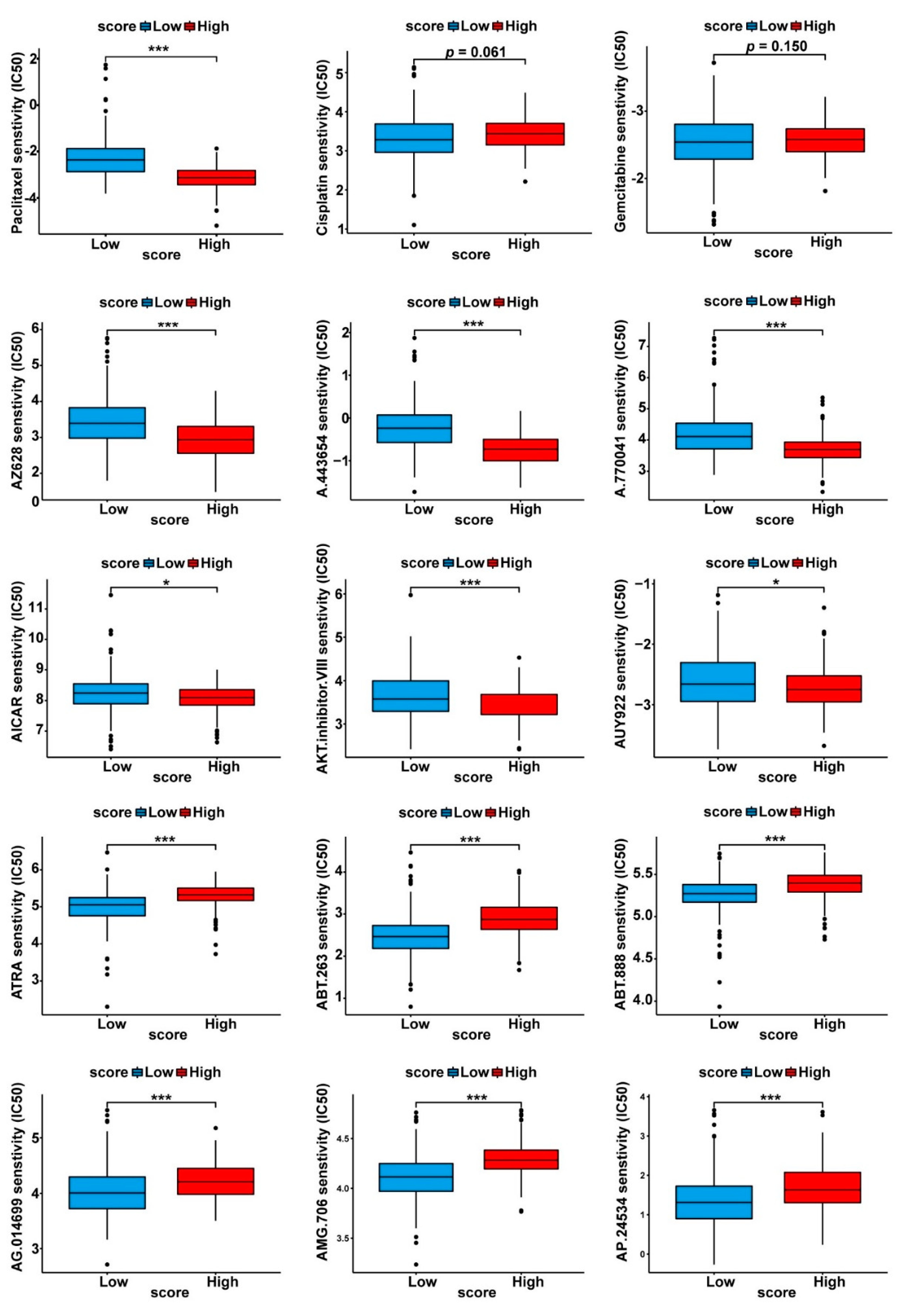
3.5. Drug Sensitivity Profiles of the Two Cuproptosis-Related PDAC Subgroups
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute: Cancer Stat Facts: Pancreatic Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 5 January 2023).
- Jain, T.; Dudeja, V. The war against pancreatic cancer in 2020—Advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; DePinho, R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- O’Hern, C.I.Z.; Djoko, K.Y. Copper cytotoxicity: Cellular casualties of noncognate coordination chemistry. mBio 2022, 13, e0043422. [Google Scholar] [CrossRef]
- Lopez, J.; Ramchandani, D.; Vahdat, L. Copper depletion as a therapeutic strategy in cancer. Met. Ions Life Sci. 2019, 19, 303–330. [Google Scholar]
- Golabek, T.; Darewicz, B.; Borawska, M.; Socha, K.; Markiewicz, R.; Kudelski, J. Copper, zinc, and Cu/Zn ratio in transitional cell carcinoma of the bladder. Urol. Int. 2012, 89, 342–347. [Google Scholar] [CrossRef]
- Gupta, S.K.; Shukla, V.K.; Vaidya, M.P.; Roy, S.K.; Gupta, S. Serum and tissue trace elements in colorectal cancer. J. Surg. Oncol. 1993, 52, 172–175. [Google Scholar] [CrossRef]
- Lener, M.R.; Scott, R.J.; Wiechowska-Kozłowska, A.; Serrano-Fernández, P.; Baszuk, P.; Jaworska-Bieniek, K.; Sukiennicki, G.; Marciniak, W.; Muszyńska, M.; Kładny, J.; et al. Serum concentrations of selenium and copper in patients diagnosed with pancreatic cancer. Cancer Res. Treat. 2016, 48, 1056–1064. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhou, R.; Zhao, Y.; Pan, Y.; Liang, H.; Zhang, J.S.; Tai, S.; Jin, L.; Teng, C.B. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019, 52, e12568. [Google Scholar] [CrossRef] [PubMed]
- Finney, L.; Vogt, S.; Fukai, T.; Glesne, D. Copper and angiogenesis: Unravelling a relationship key to cancer progression. Clin. Exp. Pharmacol. Physiol. 2009, 36, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Gouw, A.M.; LaGory, E.L.; Guo, S.; Attarwala, N.; Tang, Y.; Qi, J.; Chen, Y.S.; Gao, Z.; Casey, K.M.; et al. Mitochondrial copper depletion suppresses triple-negative breast cancer in mice. Nat. Biotechnol. 2021, 39, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Goodman, V.L.; Brewer, G.J.; Merajver, S.D. Copper deficiency as an anti-cancer strategy. Endocr. Relat. Cancer 2004, 11, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Kahlson, M.A.; Dixon, S.J. Copper-induced cell death. Science 2022, 375, 1231–1232. [Google Scholar] [CrossRef]
- Lv, H.; Liu, X.; Zeng, X.; Liu, Y.; Zhang, C.; Zhang, Q.; Xu, J. Comprehensive Analysis of Cuproptosis-Related Genes in Immune Infiltration and Prognosis in Melanoma. Front Pharmacol. 2022, 13, 930041. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, X.; Wu, Y.; Liu, Y.; Zhang, X.; Song, Z. Cuproptosis-related risk score predicts prognosis and characterizes the tumor microenvironment in hepatocellular carcinoma. Front. Immunol. 2022, 13, 925618. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, J.; Zhang, X. A novel Cuproptosis-related LncRNA signature to predict prognosis in hepatocellular carcinoma. Sci. Rep. 2022, 12, 11325. [Google Scholar] [CrossRef]
- Bian, Z.; Fan, R.; Xie, L. A novel cuproptosis-related prognostic gene signature and validation of differential expression in clear cell renal cell carcinoma. Genes 2022, 13, 851. [Google Scholar] [CrossRef]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Hayes, D.N.; Monti, S.; Parmigiani, G.; Gilks, C.B.; Naoki, K.; Bhattacharjee, A.; Socinski, M.A.; Perou, C.; Meyerson, M. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 2006, 24, 5079–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [Green Version]
- Chi Fru, E.; Rodríguez, N.P.; Partin, C.A.; Lalonde, S.V.; Andersson, P.; Weiss, D.J.; El Albani, A.; Rodushkin, I.; Konhauser, K.O. Cu isotopes in marine black shales record the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 2016, 113, 4941–4946. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.H.; Ren, W.Z.; Wang, H.Q.; Gao, W.; Yuan, B. Molecular Subtyping Based on Cuproptosis-Related Genes and Characterization of Tumor Microenvironment Infiltration in Kidney Renal Clear Cell Carcinoma. Front Oncol. 2022, 12, 919083. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.R.; Yang, L.H.; Jin, L.Z.; Yi, L.M.; Bing, P.P.; Zhou, J.; Yang, J.S. Identification of novel cuproptosis-related lncRNA signatures to predict the prognosis and immune microenvironment of breast cancer patients. Front. Oncol. 2022, 12, 988680. [Google Scholar] [CrossRef]
- Jin, L.; Mei, W.; Liu, X.; Sun, X.; Xin, S.; Zhou, Z.; Zhang, J.; Zhang, B.; Chen, P.; Cai, M.; et al. Identification of cuproptosis-related subtypes, the development of a prognosis model, and characterization of tumor microenvironment infiltration in prostate cancer. Front. Immunol. 2022, 13, 974034. [Google Scholar] [CrossRef]
- Jiang, W.; Du, Y.; Zhang, W.; Zhou, W. Construction of a prognostic model based on cuproptosis-related lncRNA signatures in pancreatic cancer. Can. J. Gastroenterol. Hepatol. 2022, 2022, 4661929. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zou, X.; Ma, M.; Liu, Y.; Wang, R.; Dai, Z.; Tashiheng, Y.; Yan, Y.; Yu, X.; Wang, X.; et al. Expression Profiles of Cuproptosis-Related Genes Determine Distinct Subtypes of Pancreatic Ductal Adenocarcinoma. Curr. Oncol. 2023, 30, 1648-1662. https://doi.org/10.3390/curroncol30020126
Chen Y, Zou X, Ma M, Liu Y, Wang R, Dai Z, Tashiheng Y, Yan Y, Yu X, Wang X, et al. Expression Profiles of Cuproptosis-Related Genes Determine Distinct Subtypes of Pancreatic Ductal Adenocarcinoma. Current Oncology. 2023; 30(2):1648-1662. https://doi.org/10.3390/curroncol30020126
Chicago/Turabian StyleChen, Yusheng, Xuan Zou, Mingjian Ma, Yu Liu, Ruijie Wang, Zhengjie Dai, Yesiboli Tashiheng, Yu Yan, Xianjun Yu, Xu Wang, and et al. 2023. "Expression Profiles of Cuproptosis-Related Genes Determine Distinct Subtypes of Pancreatic Ductal Adenocarcinoma" Current Oncology 30, no. 2: 1648-1662. https://doi.org/10.3390/curroncol30020126