

Additive Cytotoxic and Colony-Formation Inhibitory Effects of Aspirin and Metformin on PI3KCA-Mutant Colorectal Cancer Cells

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
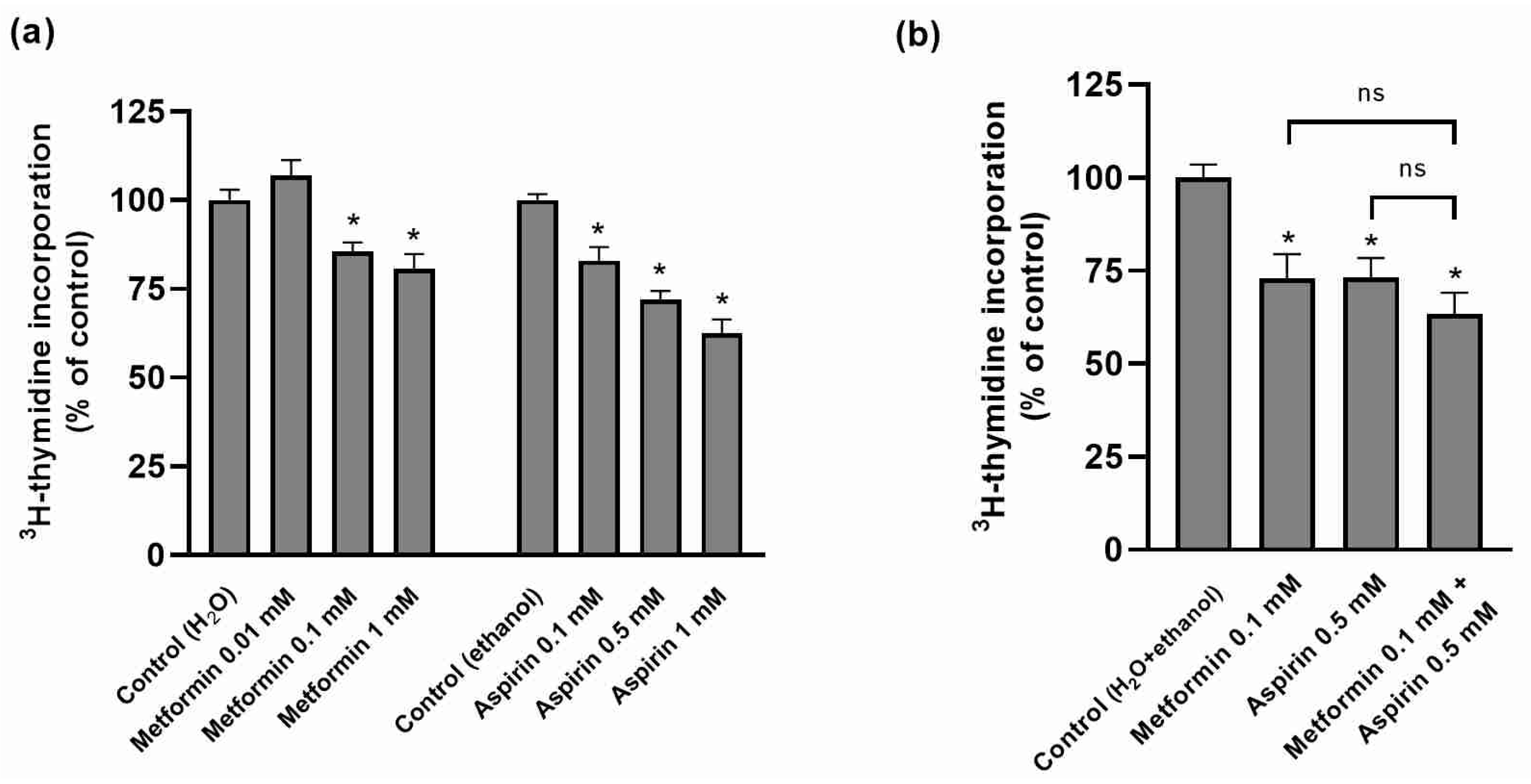
2.1. Effect of Metformin (MET) and/or Aspirin (ASP) on HT-29 Cellular Proliferation
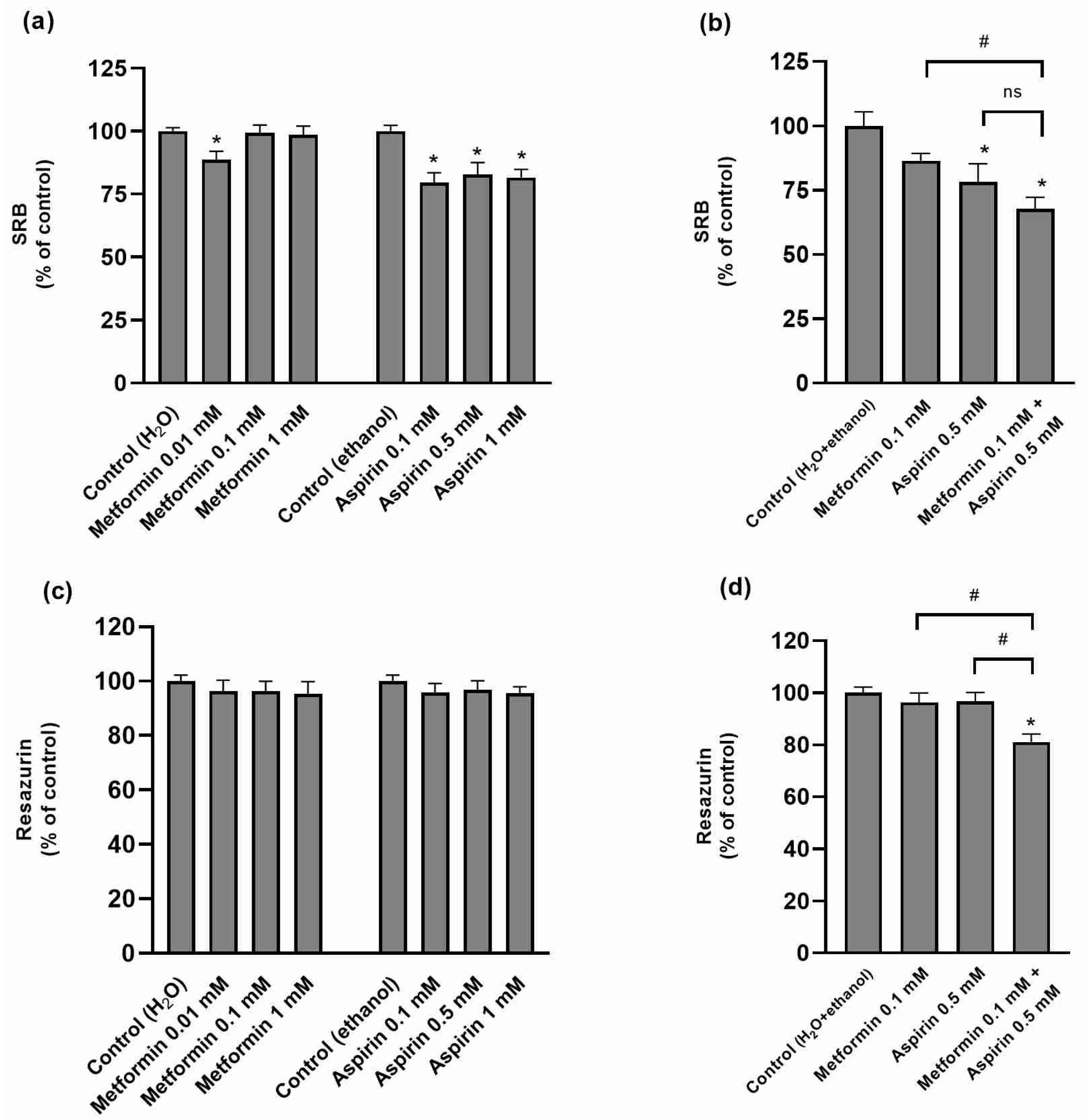
2.2. Effect of Metformin (MET) and/or Aspirin (ASP) on HT-29 Cell Viability
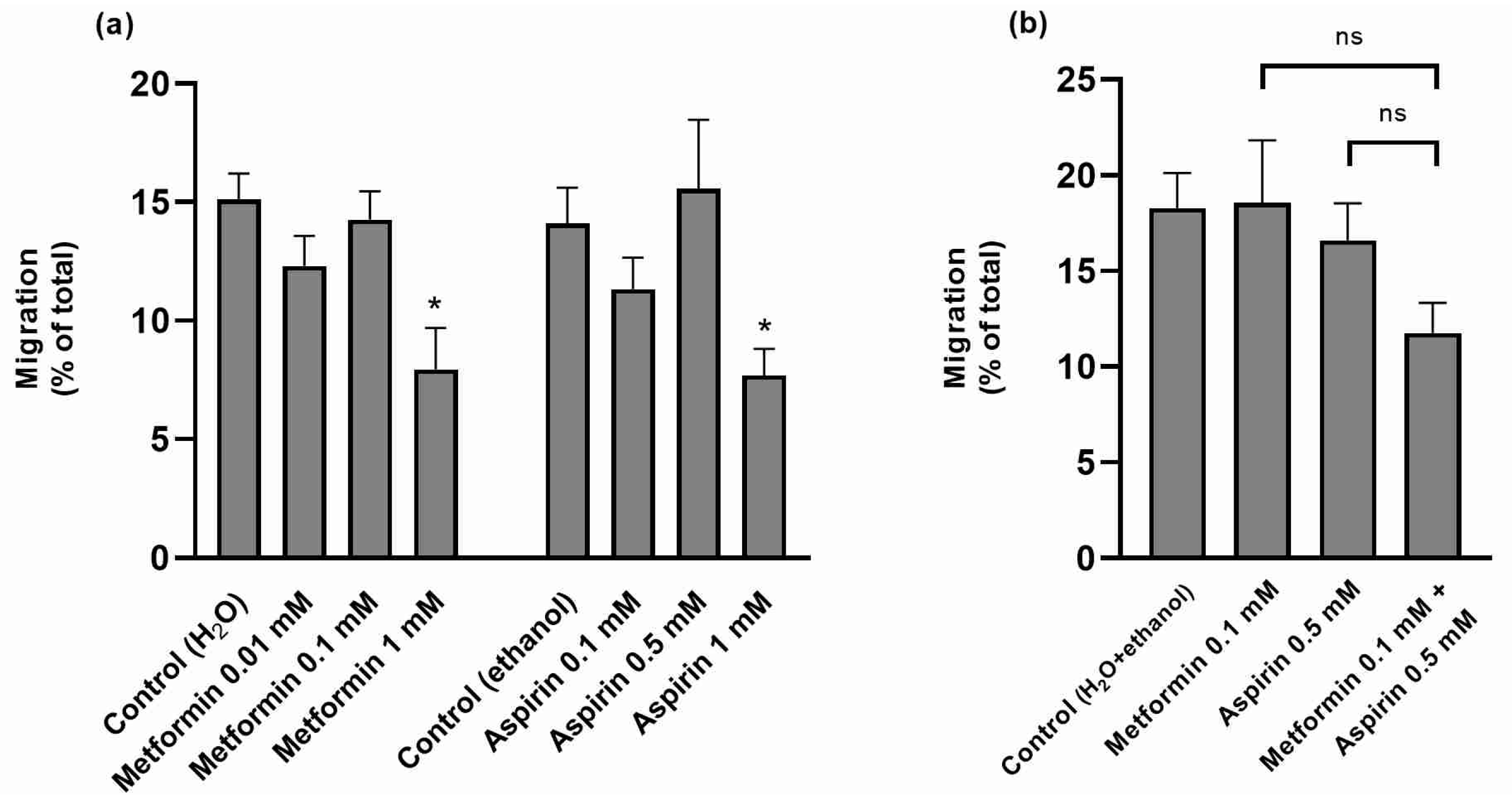
2.3. Effect of Metformin (MET) and/or Aspirin (ASP) on HT-29 Cell Migration Rates
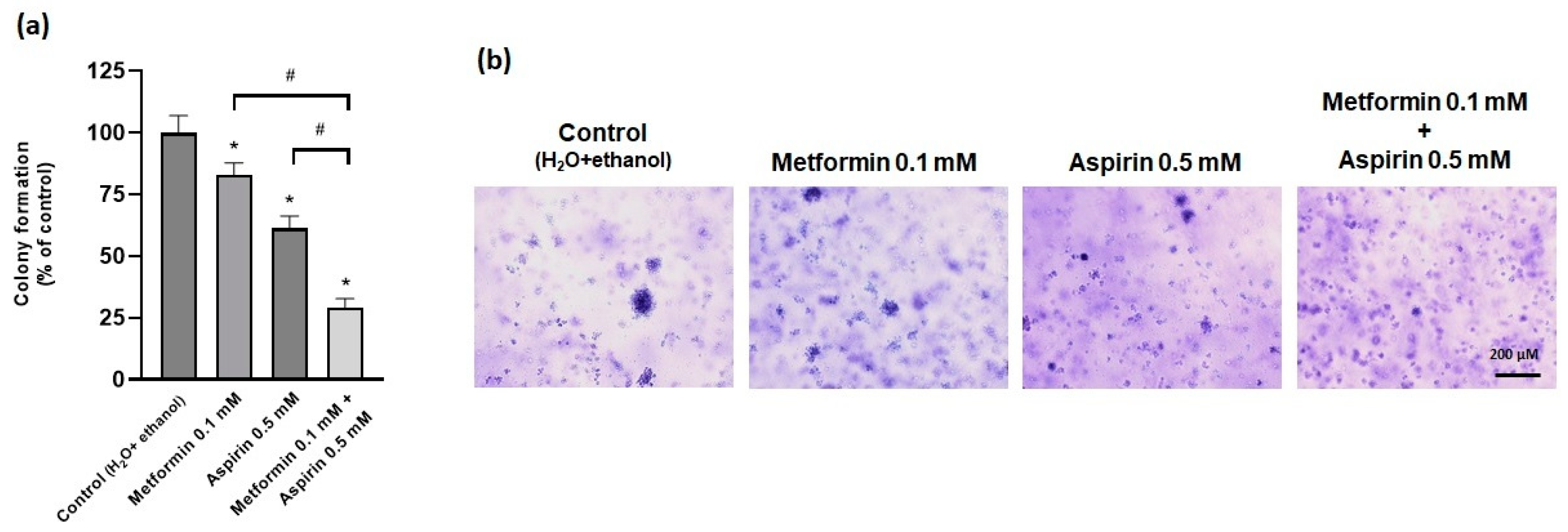
2.4. Effect of Metformin (MET) and/or Aspirin (ASP) on Anchorage-Ndependent Growth Ability
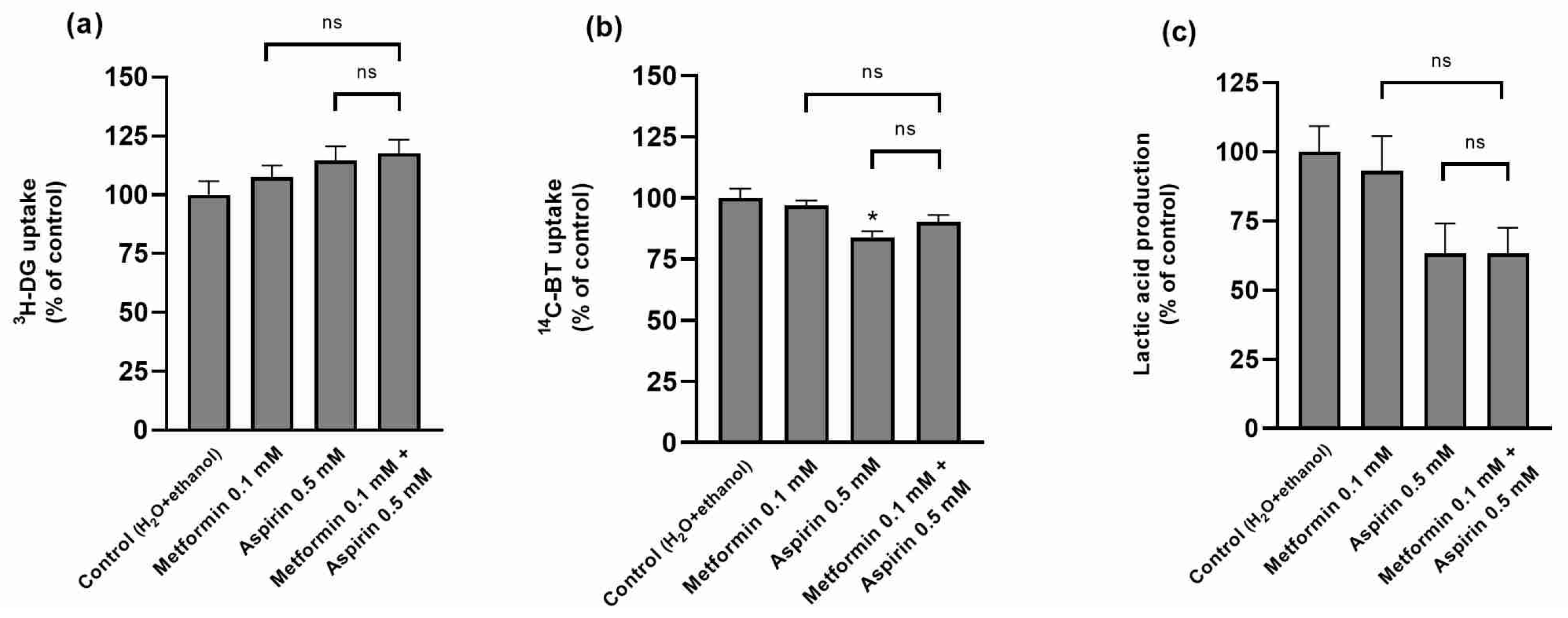
2.5. Effect of Metformin (MET) and/or Aspirin (ASP) on HT-29 Nutrient Uptake and Metabolism
2.6. Effect of Metformin (MET) and/or Aspirin (ASP) on Caco-2 Cell Proliferation, Viability, Migration Rates, Anchorage-Independent Growth Ability, and Nutrient Uptake
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Treatment of the Cells
4.4. Determination of Cell Proliferation Rates
Incorporation of 3H-Thymidine Assay
4.5. Determination of Cell Viability
4.5.1. SRB Assay
4.5.2. Resazurin Reduction Assay
4.5.3. Extracellular LDH Activity Assay
4.6. Determination of Cell Migration Rates
Scratch Injury Assay
4.7. Determination of Anchorage-Independent Growth Ability
Soft Agar Colony Formation Assay
4.8. Determination of 3H-Deoxy-d-glucose (3H-DG) and 3H-Butyrate (14C-BT) Uptake
4.9. Determination of Glucose Metabolism
Quantification of Lactate Metabolism
4.10. Protein Determination
4.11. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2021, 68, 258–278. [Google Scholar] [CrossRef] [PubMed]
- Antoszczak, M.; Markowska, A.; Markowska, J.; Huczynski, A. Old wine in new bottles: Drug repurposing in oncology. Eur. J. Pharmacol. 2020, 866, 172784. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Forde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Rao, P.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Higurashi, T.; Hosono, K.; Takahashi, H.; Komiya, Y.; Umezawa, S.; Sakai, E.; Uchiyama, T.; Taniguchi, L.; Hata, Y.; Uchiyama, S.; et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: A multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016, 17, 475–483. [Google Scholar] [CrossRef]
- Heer, E.; Ruan, Y.; Mah, B.; Nguyen, T.; Lyons, H.; Poirier, A.; Boyne, D.J.; O’Sullivan, D.E.; Heitman, S.J.; Hilsden, R.J.; et al. The efficacy of chemopreventive agents on the incidence of colorectal adenomas: A systematic review and network meta-analysis. Prev. Med. 2022, 162, 107169. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef]
- Meng, F.; Song, L.; Wang, W. Metformin improves overall survival of colorectal cancer patients with diabetes: A meta-analysis. J. Diabetes Res. 2017, 2017, 5063239. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, C.H.; Eun, C.S.; Park, D.I.; Han, D.S. Metformin use and the risk of colorectal adenoma: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2017, 32, 957–965. [Google Scholar] [CrossRef]
- Liu, F.; Yan, L.; Wang, Z.; Lu, Y.; Chu, Y.; Li, X.; Liu, Y.; Rui, D.; Nie, S.; Xiang, H. Metformin therapy and risk of colorectal adenomas and colorectal cancer in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Oncotarget 2017, 8, 16017–16026. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, S.; Higurashi, T.; Komiya, Y.; Arimoto, J.; Horita, N.; Kaneko, T.; Iwasaki, M.; Nakagama, H.; Nakajima, A. Chemoprevention of colorectal cancer: Past, present, and future. Cancer Sci. 2019, 110, 3018–3026. [Google Scholar] [CrossRef]
- Veettil, S.K.; Lim, K.G.; Ching, S.M.; Saokaew, S.; Phisalprapa, P.; Chaiyakunapruk, N. Effects of aspirin and non-aspirin nonsteroidal anti-inflammatory drugs on the incidence of recurrent colorectal adenomas: A systematic review with meta-analysis and trial sequential analysis of randomized clinical trials. BMC Cancer 2017, 17, 763. [Google Scholar] [CrossRef] [PubMed]
- Garcia Rodriguez, L.A.; Soriano-Gabarro, M.; Bromley, S.; Lanas, A.; Cea Soriano, L. New use of low-dose aspirin and risk of colorectal cancer by stage at diagnosis: A nested case-control study in UK general practice. BMC Cancer 2017, 17, 637. [Google Scholar] [CrossRef]
- Joharatnam-Hogan, N.; Cafferty, F.; Hubner, R.; Swinson, D.; Sothi, S.; Gupta, K.; Falk, S.; Patel, K.; Warner, N.; Kunene, V.; et al. Aspirin as an adjuvant treatment for cancer: Feasibility results from the Add-Aspirin randomised trial. Lancet Gastroenterol. Hepatol. 2019, 4, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Tomimoto, A.; Endo, H.; Sugiyama, M.; Fujisawa, T.; Hosono, K.; Takahashi, H.; Nakajima, N.; Nagashima, Y.; Wada, K.; Nakagama, H.; et al. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008, 99, 2136–2141. [Google Scholar] [CrossRef]
- Kilari, R.S.; Bashir, A.I.J.; Devitt, A.; Perry, C.J.; Safrany, S.T.; Nicholl, I.D. The cytotoxicity and synergistic potential of aspirin and aspirin analogues towards oesophageal and colorectal cancer. Curr. Clin. Pharmacol. 2019, 14, 141–151. [Google Scholar] [CrossRef]
- Bordini, H.P.; Kremer, J.L.; Fagundes, T.R.; Melo, G.P.; Conchon-Costa, I.; da Silva, S.S.; Cecchini, A.L.; Panis, C.; Luiz, R.C. Protective effect of metformin in an aberrant crypt foci model induced by 1,2-dimethylhydrazine: Modulation of oxidative stress and inflammatory process. Mol. Carcinog. 2017, 56, 913–922. [Google Scholar] [CrossRef]
- Shiff, S.J.; Koutsos, M.I.; Qiao, L.; Rigas, B. Nonsteroidal antiinflammatory drugs inhibit the proliferation of colon adenocarcinoma cells: Effects on cell cycle and apoptosis. Exp. Cell Res. 1996, 222, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Papadimitriou, N.; Markozannes, G.; Cividini, S.; Kakourou, A.; Gill, D.; Rizos, E.C.; Monori, G.; Ward, H.A.; Kyrgiou, M.; et al. Type 2 diabetes and cancer: An umbrella review of observational and mendelian randomization studies. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Worldwide burden of cancer attributable to diabetes and high body-mass index: A comparative risk assessment. Lancet Diabetes Endocrinol. 2018, 6, e6–e15. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.; Ho, J.M.; Lam, A.S.; Yau, S.T.; Tsoi, K.K. Use of metformin and aspirin is associated with delayed cancer incidence. Cancer Epidemiol. 2020, 69, 101808. [Google Scholar] [CrossRef] [PubMed]
- Shami, J.J.P.; Yan, V.K.C.; Wei, Y.; Alwafi, H.; Blais, J.E.; Wan, E.; Wong, C.K.H.; Cheung, K.S.; Leung, W.K.; Wong, M.C.S.; et al. Low-dose aspirin does not lower the risk of colorectal cancer in patients with type 2 diabetes taking metformin. J. Intern. Med. 2023, 293, 371–383. [Google Scholar] [CrossRef]
- Wang, C.H.; Huang, C.W.; Nguyen, P.A.; Lin, M.C.; Yeh, C.Y.; Islam, M.M.; Rahmanti, A.R.; Yang, H.C. Chemopreventive effects of concomitant or individual use of statins, aspirin, metformin, and angiotensin drugs: A study using claims data of 23 million individuals. Cancers 2022, 14, 1211. [Google Scholar] [CrossRef] [PubMed]
- De Monte, A.; Brunetti, D.; Cattin, L.; Lavanda, F.; Naibo, E.; Malagoli, M.; Stanta, G.; Bonin, S. Metformin and aspirin treatment could lead to an improved survival rate for Type 2 diabetic patients with stage II and III colorectal adenocarcinoma relative to non-diabetic patients. Mol. Clin. Oncol. 2018, 8, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Petrera, M.; Paleari, L.; Clavarezza, M.; Puntoni, M.; Caviglia, S.; Briata, I.M.; Oppezzi, M.; Mislej, E.M.; Stabuc, B.; Gnant, M.; et al. The ASAMET trial: A randomized, phase II, double-blind, placebo-controlled, multicenter, 2 × 2 factorial biomarker study of tertiary prevention with low-dose aspirin and metformin in stage I-III colorectal cancer patients. BMC Cancer 2018, 18, 1210. [Google Scholar] [CrossRef]
- Higurashi, T.; Arimoto, J.; Ashikari, K.; Takatsu, T.; Misawa, N.; Yoshihara, T.; Matsuura, T.; Fuyuki, A.; Ohkubo, H.; Nakajima, A. The efficacy of aspirin and metformin combination therapy in patients with rectal aberrant crypt foci: A double-blinded randomized controlled trial. BMC Cancer 2020, 20, 1043. [Google Scholar] [CrossRef]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef]
- Palazzolo, G.; Mollica, H.; Lusi, V.; Rutigliani, M.; Di Francesco, M.; Pereira, R.C.; Filauro, M.; Paleari, L.; DeCensi, A.; Decuzzi, P. Modulating the distant spreading of patient-derived colorectal cancer cells via aspirin and metformin. Transl. Oncol. 2020, 13, 100760. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M. The human colon carcinoma cell lines HT-29 and Caco-2: Two in vitro models for the study of intestinal differentiation. Biochimie 1986, 68, 1035–1040. [Google Scholar] [CrossRef]
- de Both, N.J.; Vermey, M.; Dinjens, W.N.; Bosman, F.T. A comparative evaluation of various invasion assays testing colon carcinoma cell lines. Br. J. Cancer 1999, 81, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Ponce de Leon-Rodriguez, M.D.C.; Guyot, J.P.; Laurent-Babot, C. Intestinal in vitro cell culture models and their potential to study the effect of food components on intestinal inflammation. Crit. Rev. Food Sci. Nutr. 2019, 59, 3648–3666. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Hinoue, T.; Weisenberger, D.J.; Pan, F.; Campan, M.; Kim, M.; Young, J.; Whitehall, V.L.; Leggett, B.A.; Laird, P.W. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS ONE 2009, 4, e8357. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Uchiyama, T.; Suzuki, K.; Nozaki, Y.; Yoneda, K.; Fujita, K.; Yoneda, M.; et al. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog. 2010, 49, 662–671. [Google Scholar] [CrossRef]
- Din, F.V.; Dunlop, M.G.; Stark, L.A. Evidence for colorectal cancer cell specificity of aspirin effects on NF kappa B signalling and apoptosis. Br. J. Cancer 2004, 91, 381–388. [Google Scholar] [CrossRef]
- Tsakiridis, E.E.; Broadfield, L.; Marcinko, K.; Biziotis, O.D.; Ali, A.; Mekhaeil, B.; Ahmadi, E.; Singh, K.; Mesci, A.; Zacharidis, P.G.; et al. Combined metformin-salicylate treatment provides improved anti-tumor activity and enhanced radiotherapy response in prostate cancer; drug synergy at clinically relevant doses. Transl. Oncol. 2021, 14, 101209. [Google Scholar] [CrossRef]
- Qi, L.; Ye, J.W.; Xue, W.H.; Tian, X.; Zhang, H.J. The mechanism of aspirin combined with metformin induced apoptosis of thyroid cancer TPC-1 cells. Zhonghua Zhong Liu Za Zhi 2019, 41, 276–281. [Google Scholar]
- Abdelmonsif, D.A.; Sultan, A.S.; El-Hadidy, W.F.; Abdallah, D.M. Targeting AMPK, mTOR and beta-Catenin by combined metformin and aspirin therapy in HCC: An appraisal in egyptian HCC Patients. Mol. Diagn. Ther. 2018, 22, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.E.A.; Nery, L.R.; Leite, C.E.; de Azevedo Junior, W.F.; Campos, M.M. Pre-clinical effects of metformin and aspirin on the cell lines of different breast cancer subtypes. Investig. New Drugs 2018, 36, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Motafeghi, F.; Shahsavari, R.; Mortazavi, P.; Babaei, A.; SamadiMojaveri, P.; Khojasteh, O.A.; Shokrzadeh, M. Metformin and Aspirin: Anticancer effects on A549 and PC3 cancer cells and the mechanisms of action. Toxicol. Res. 2023, 12, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.G.; Lockery, J.E.; Broder, J.C.; Ernst, M.E.; Espinoza, S.; Gibbs, P.; Wolfe, R.; Polekhina, G.; Zoungas, S.; Loomans-Kropp, H.A.; et al. Association of metformin, aspirin, and cancer incidence with mortality risk in adults with diabetes. JNCI Cancer Spectr. 2023, 7, pkad017. [Google Scholar] [CrossRef] [PubMed]
- Mussin, N.; Oh, S.C.; Lee, K.W.; Park, M.Y.; Seo, S.; Yi, N.J.; Kim, H.; Yoon, K.C.; Ahn, S.W.; Kim, H.S.; et al. Sirolimus and metformin synergistically inhibits colon cancer in vitro and in vivo. J. Korean Med. Sci. 2017, 32, 1385–1395. [Google Scholar] [CrossRef]
- Farrash, W.F.; Aslam, A.; Almaimani, R.; Minshawi, F.; Almasmoum, H.; Alsaegh, A.; Iqbal, M.S.; Tabassum, A.; Elzubier, M.E.; El-Readi, M.Z.; et al. Metformin and thymoquinone co-treatment enhance 5-fluorouracil cytotoxicity by suppressing the PI3K/mTOR/HIF1alpha pathway and increasing oxidative stress in colon cancer cells. Biofactors 2023, 49, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Lin, C.T.; Chen, C.N.; Chang, S.F.; Chang, H.I.; Lee, K.C. Metformin increases the cytotoxicity of oxaliplatin in human DLD-1 colorectal cancer cells through down-regulating HMGB1 expression. J. Cell Biochem. 2018, 119, 6943–6952. [Google Scholar] [CrossRef] [PubMed]
- Ruberte, A.C.; Gonzalez-Gaitano, G.; Sharma, A.K.; Aydillo, C.; Encio, I.; Sanmartin, C.; Plano, D. New formulation of a methylseleno-aspirin analog with anticancer ativity towards colon cancer. Int. J. Mol. Sci. 2020, 21, 9017. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Zhou, Z.; Fang, J.; Sun, Z.; He, M.; He, B.; Chen, Q.; Paek, C.; Chen, P.; Zhou, J.; et al. Aspirin induces immunogenic cell death and enhances cancer immunotherapy in colorectal cancer. Int. Immunopharmacol. 2023, 121, 110350. [Google Scholar] [CrossRef]
- Goncalves, P.; Martel, F. Butyrate and colorectal cancer: The role of butyrate transport. Curr. Drug Metab. 2013, 14, 994–1008. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Zhang, X.; Ogihara, T.; Zhu, M.; Gantumur, D.; Li, Y.; Mizoi, K.; Kamioka, H.; Tsushima, Y. Effect of metformin on (18)F-fluorodeoxyglucose uptake and positron emission tomographic imaging. Br. J. Radiol. 2022, 95, 20200810. [Google Scholar] [CrossRef] [PubMed]
- Collura, A.; Marisa, L.; Trojan, D.; Buhard, O.; Lagrange, A.; Saget, A.; Bombled, M.; Mechighel, P.; Ayadi, M.; Muleris, M.; et al. Extensive characterization of sphere models established from colorectal cancer cell lines. Cell Mol. Life Sci. 2013, 70, 729–742. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. The landscape of PIK3CA mutations in colorectal cancer. Clin. Color. Cancer 2021, 20, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Anwer, S.T.; Mobashir, M.; Fantoukh, O.I.; Khan, B.; Imtiyaz, K.; Naqvi, I.H.; Rizvi, M.M.A. Synthesis of silver nano particles using myricetin and the in-vitro assessment of anti-colorectal cancer Activity: In-silico integration. Int. J. Mol. Sci. 2022, 23, 11024. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef] [PubMed]
- Shiovitz, S.; Grady, W.M. Molecular markers predictive of chemotherapy response in colorectal cancer. Curr. Gastroenterol. Rep. 2015, 17, 431. [Google Scholar] [CrossRef]
- Ala, M.; Ala, M. Metformin for cardiovascular protection, inflammatory bowel disease, osteoporosis, periodontitis, polycystic ovarian syndrome, neurodegeneration, cancer, inflammation and senescence: What Is Next? ACS Pharmacol. Transl. Sci. 2021, 4, 1747–1770. [Google Scholar] [CrossRef]
- Daugan, M.; Dufay Wojcicki, A.; d’Hayer, B.; Boudy, V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol. Res. 2016, 113 Pt A, 675–685. [Google Scholar] [CrossRef]
- Hall, D.C.N.; Benndorf, R.A. Aspirin sensitivity of PIK3CA-mutated colorectal cancer: Potential mechanisms revisited. Cell Mol. Life Sci. 2022, 79, 393. [Google Scholar] [CrossRef]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K.; et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef]
- Domingo, E.; Church, D.N.; Sieber, O.; Ramamoorthy, R.; Yanagisawa, Y.; Johnstone, E.; Davidson, B.; Kerr, D.J.; Tomlinson, I.P.; Midgley, R. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J. Clin. Oncol. 2013, 31, 4297–4305. [Google Scholar] [CrossRef]
- Reimers, M.S.; Bastiaannet, E.; Langley, R.E.; van Eijk, R.; van Vlierberghe, R.L.; Lemmens, V.E.; van Herk-Sukel, M.P.; van Wezel, T.; Fodde, R.; Kuppen, P.J.; et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern. Med. 2014, 174, 732–739. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.; Xia, G.; Adilijiang, N.; Li, Y.; Hou, Z.; Fan, Z.; Li, J. Recent advances in targeted drug delivery strategy for enhancing oncotherapy. Pharmaceutics 2023, 15, 2233. [Google Scholar] [CrossRef] [PubMed]
- Amaral, I.; Silva, C.; Correia-Branco, A.; Martel, F. Effect of metformin on estrogen and progesterone receptor-positive (MCF-7) and triple-negative (MDA-MB-231) breast cancer cells. Biomed. Pharmacother. 2018, 102, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.R.; Goncalves, P.; Catarino, T.A.; Martel, F. The effect of inflammatory status on butyrate and folate uptake by tumoral (Caco-2) and non-tumoral (IEC-6) intestinal epithelial cells. Cell J. 2017, 19 (Suppl. 1), 96–105. [Google Scholar] [PubMed]
- Griffiths, M.; Sundaram, H. Drug design and testing: Profiling of antiproliferative agents for cancer therapy using a cell-based methyl-[3H]-thymidine incorporation assay. Methods Mol. Biol. 2011, 731, 451–465. [Google Scholar]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. Bio Protoc. 2016, 6, e1984. [Google Scholar] [CrossRef]
- Petiti, J.; Revel, L.; Divieto, C. Standard operating procedure to optimize resazurin-based viability assays. Biosensors 2024, 14, 156. [Google Scholar] [CrossRef]
- Bergmeyer, H.B.E. Lactate dehydrogenase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic Press: London, UK, 1963. [Google Scholar]
- Cory, G. Scratch-wound assay. Methods Mol. Biol. 2011, 769, 25–30. [Google Scholar]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, J.; Pinto, S.; Carmo, F.; Silva, C.; Andrade, N.; Martel, F. Additive Cytotoxic and Colony-Formation Inhibitory Effects of Aspirin and Metformin on PI3KCA-Mutant Colorectal Cancer Cells. Int. J. Mol. Sci. 2024, 25, 5381. https://doi.org/10.3390/ijms25105381
Gonçalves J, Pinto S, Carmo F, Silva C, Andrade N, Martel F. Additive Cytotoxic and Colony-Formation Inhibitory Effects of Aspirin and Metformin on PI3KCA-Mutant Colorectal Cancer Cells. International Journal of Molecular Sciences. 2024; 25(10):5381. https://doi.org/10.3390/ijms25105381
Chicago/Turabian StyleGonçalves, Joana, Sara Pinto, Francisca Carmo, Cláudia Silva, Nelson Andrade, and Fátima Martel. 2024. "Additive Cytotoxic and Colony-Formation Inhibitory Effects of Aspirin and Metformin on PI3KCA-Mutant Colorectal Cancer Cells" International Journal of Molecular Sciences 25, no. 10: 5381. https://doi.org/10.3390/ijms25105381
APA StyleGonçalves, J., Pinto, S., Carmo, F., Silva, C., Andrade, N., & Martel, F. (2024). Additive Cytotoxic and Colony-Formation Inhibitory Effects of Aspirin and Metformin on PI3KCA-Mutant Colorectal Cancer Cells. International Journal of Molecular Sciences, 25(10), 5381. https://doi.org/10.3390/ijms25105381