
Mechanisms for deNOx and deN2O Processes on FAU Zeolite with a Bimetallic Cu-Fe Dimer in the Presence of a Hydroxyl Group—DFT Theoretical Calculations

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
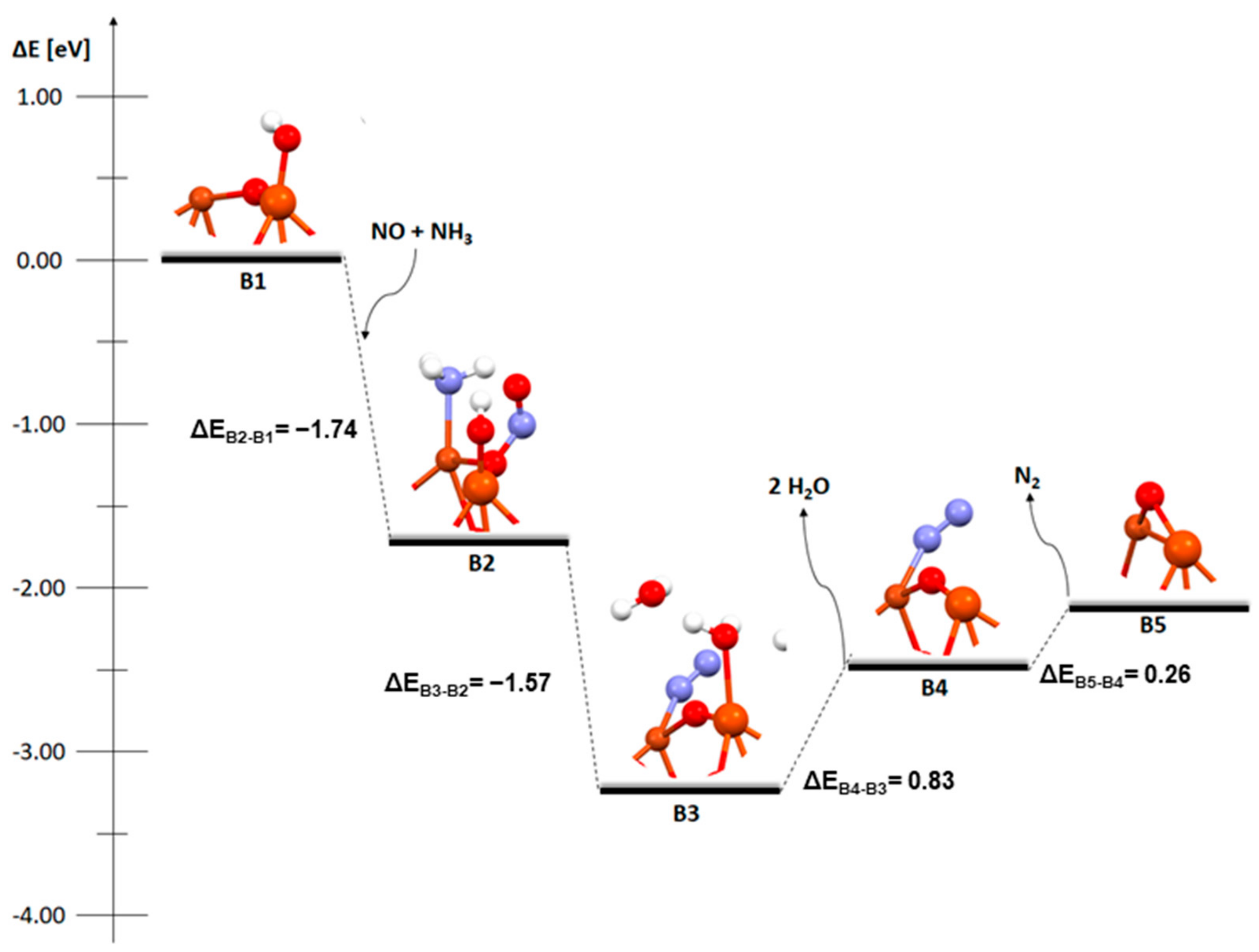
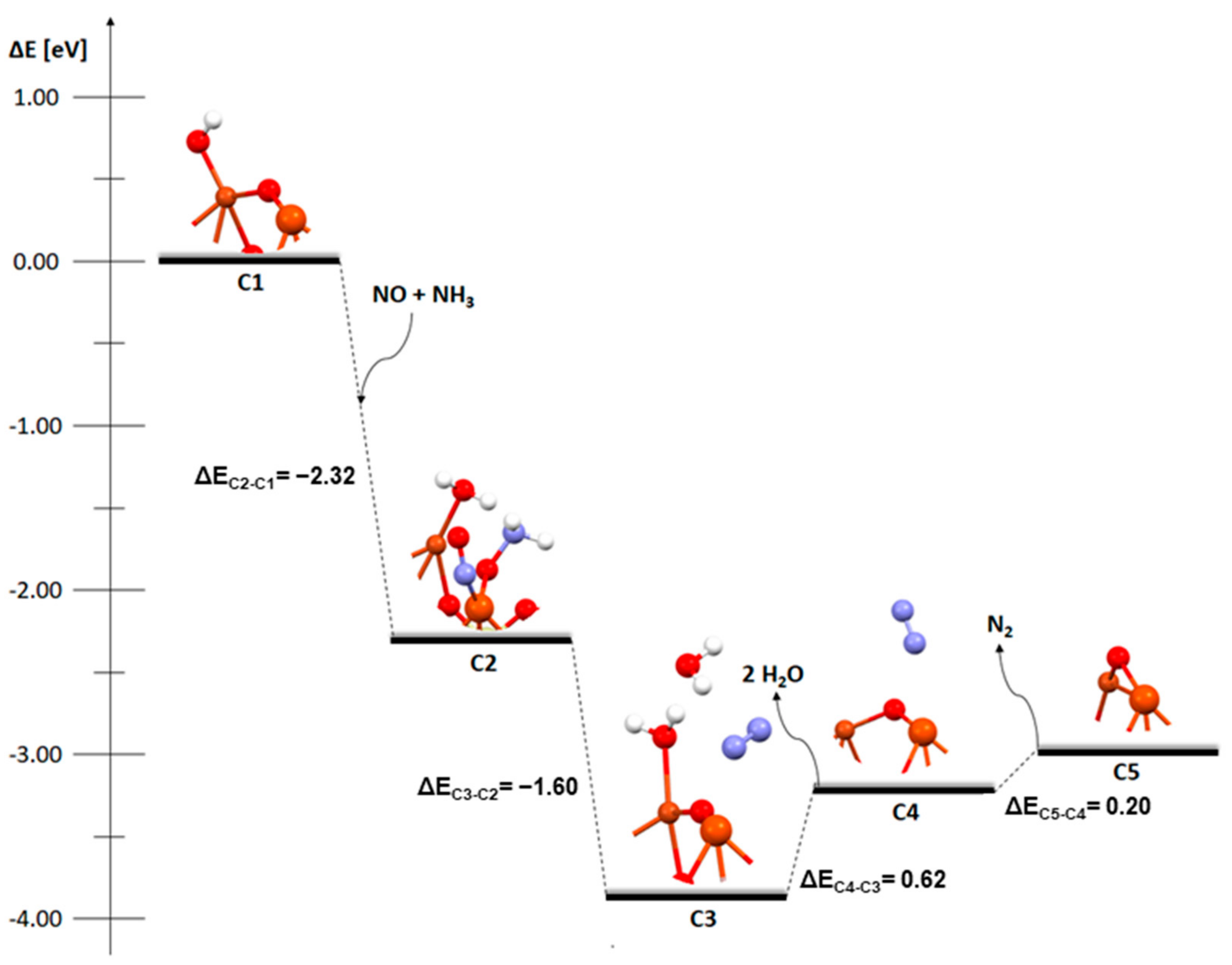
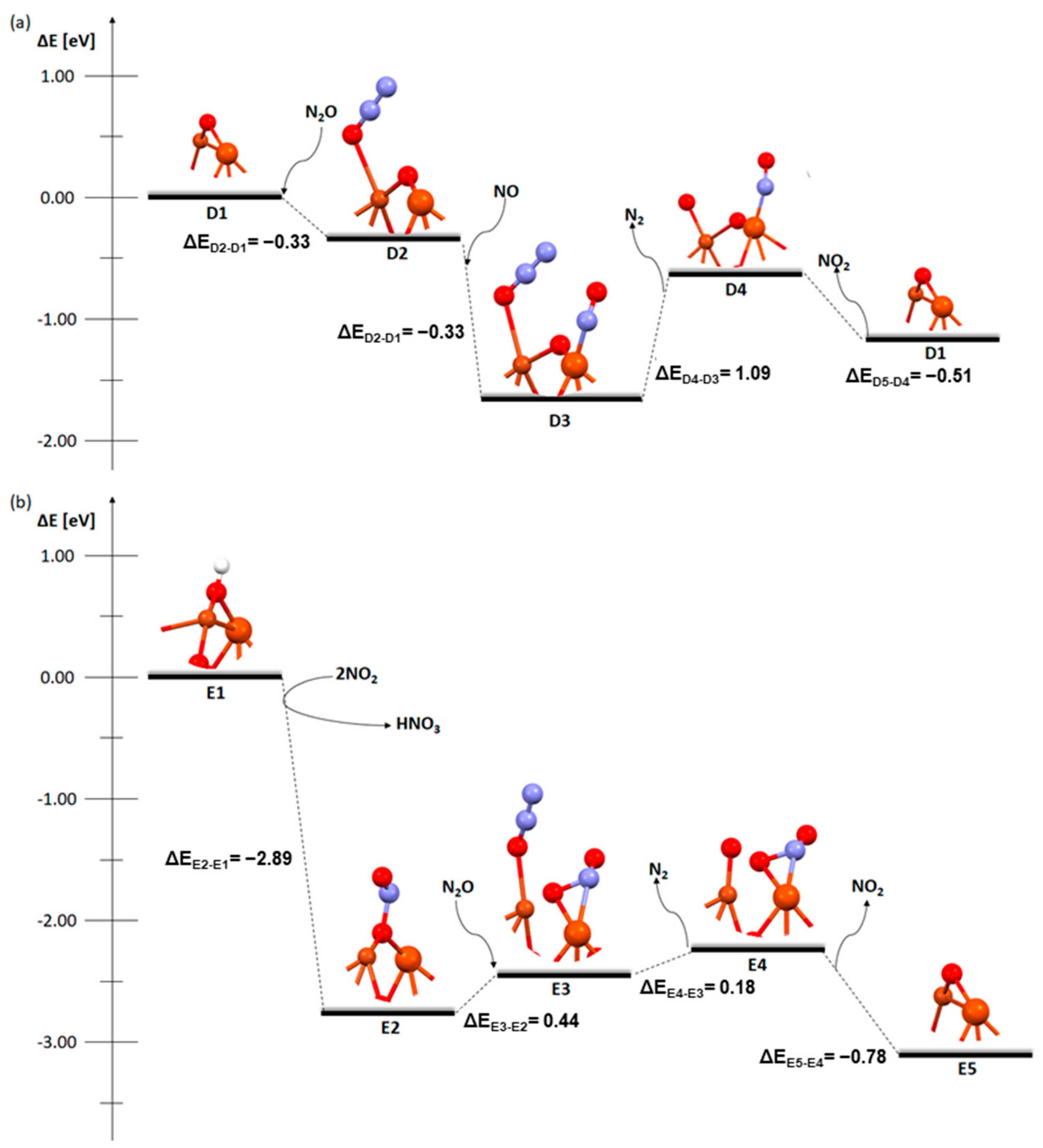
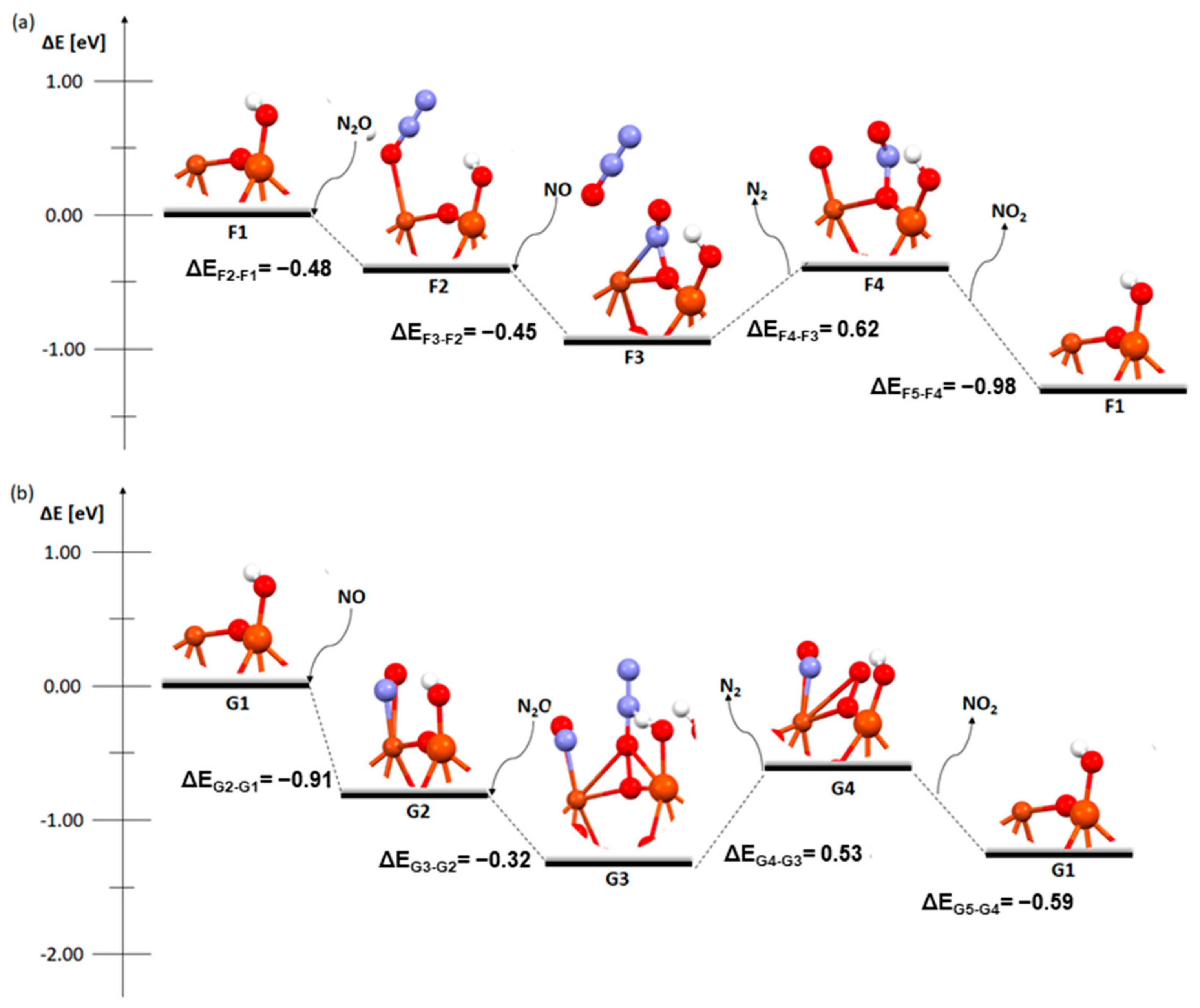
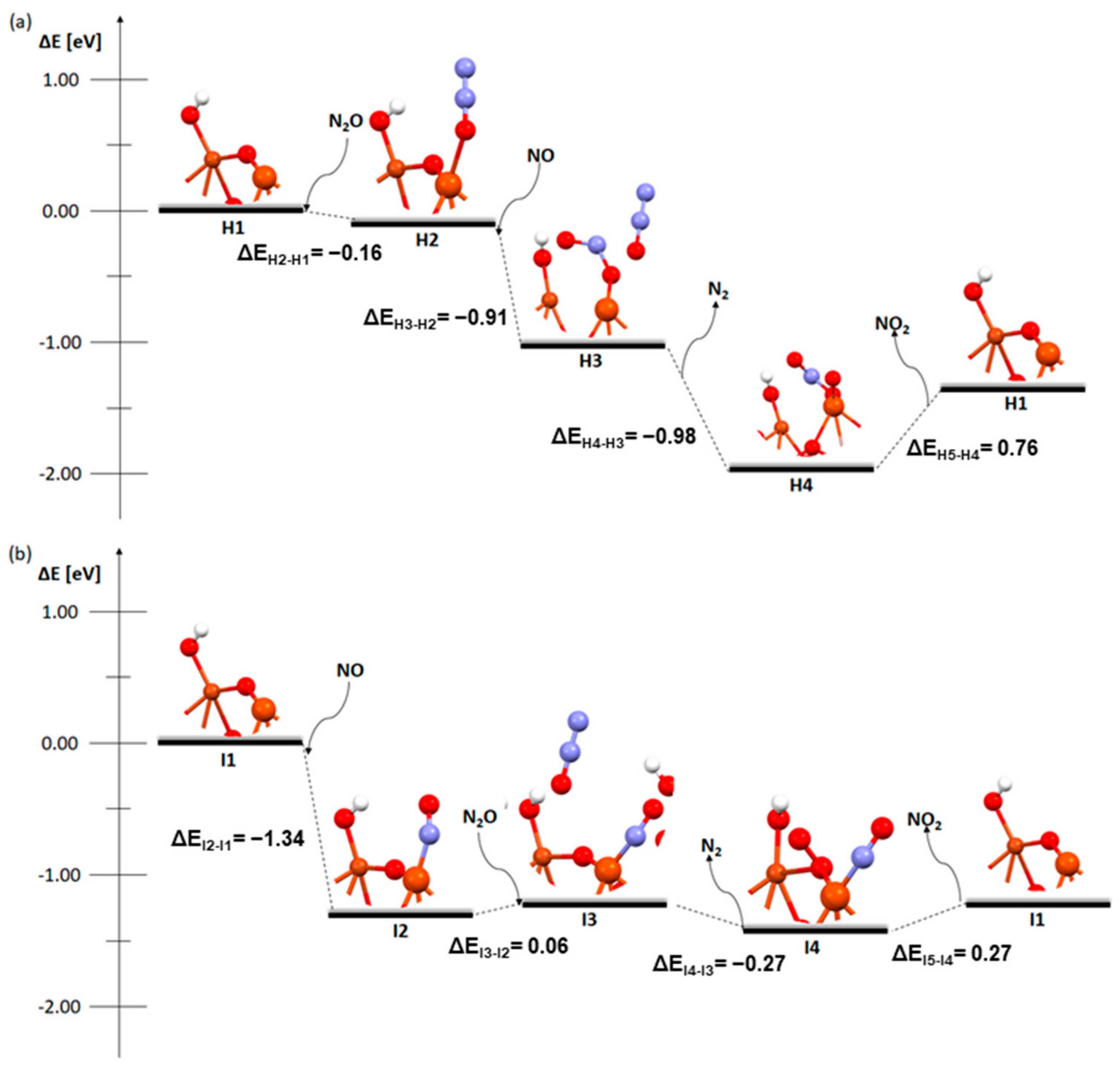
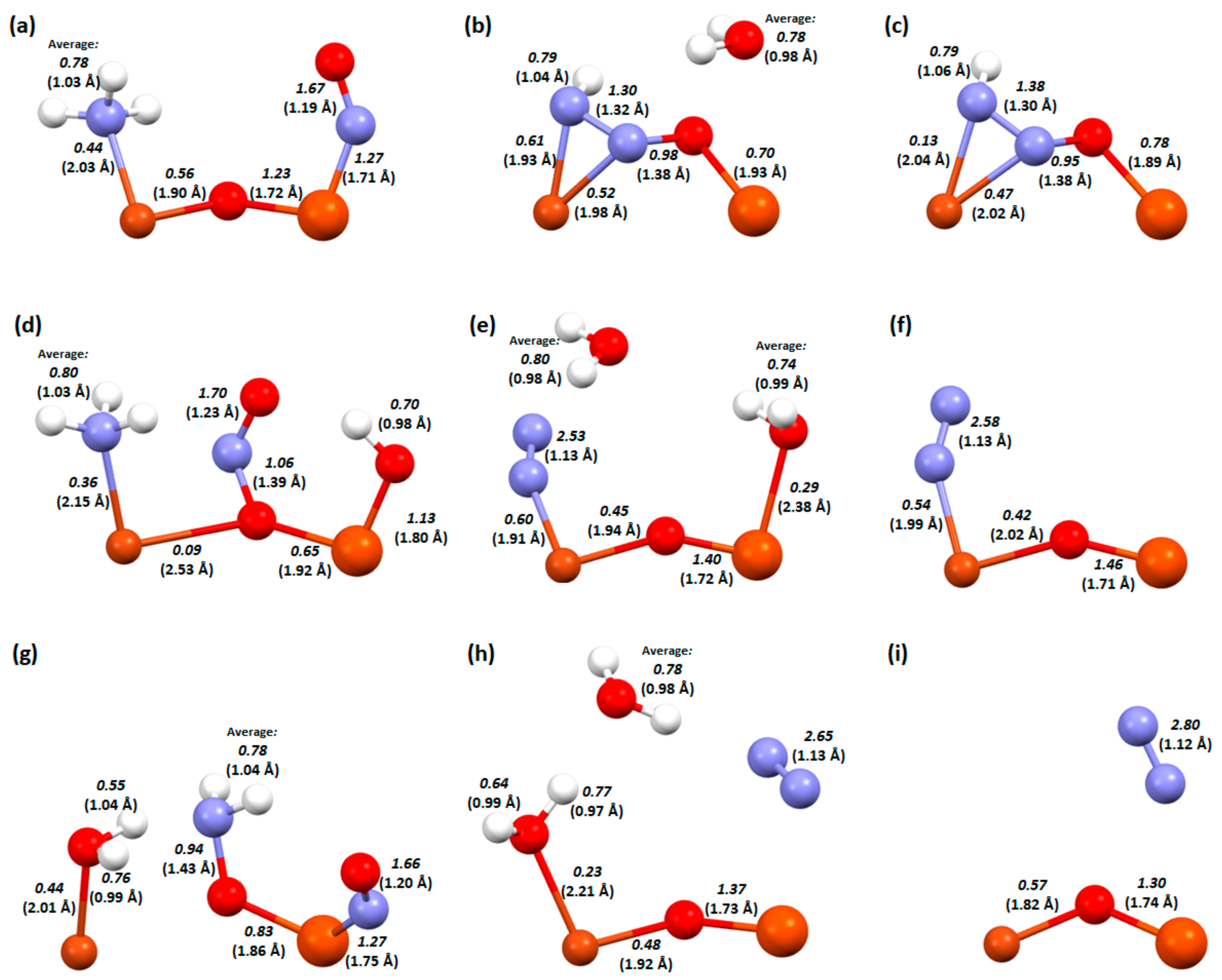
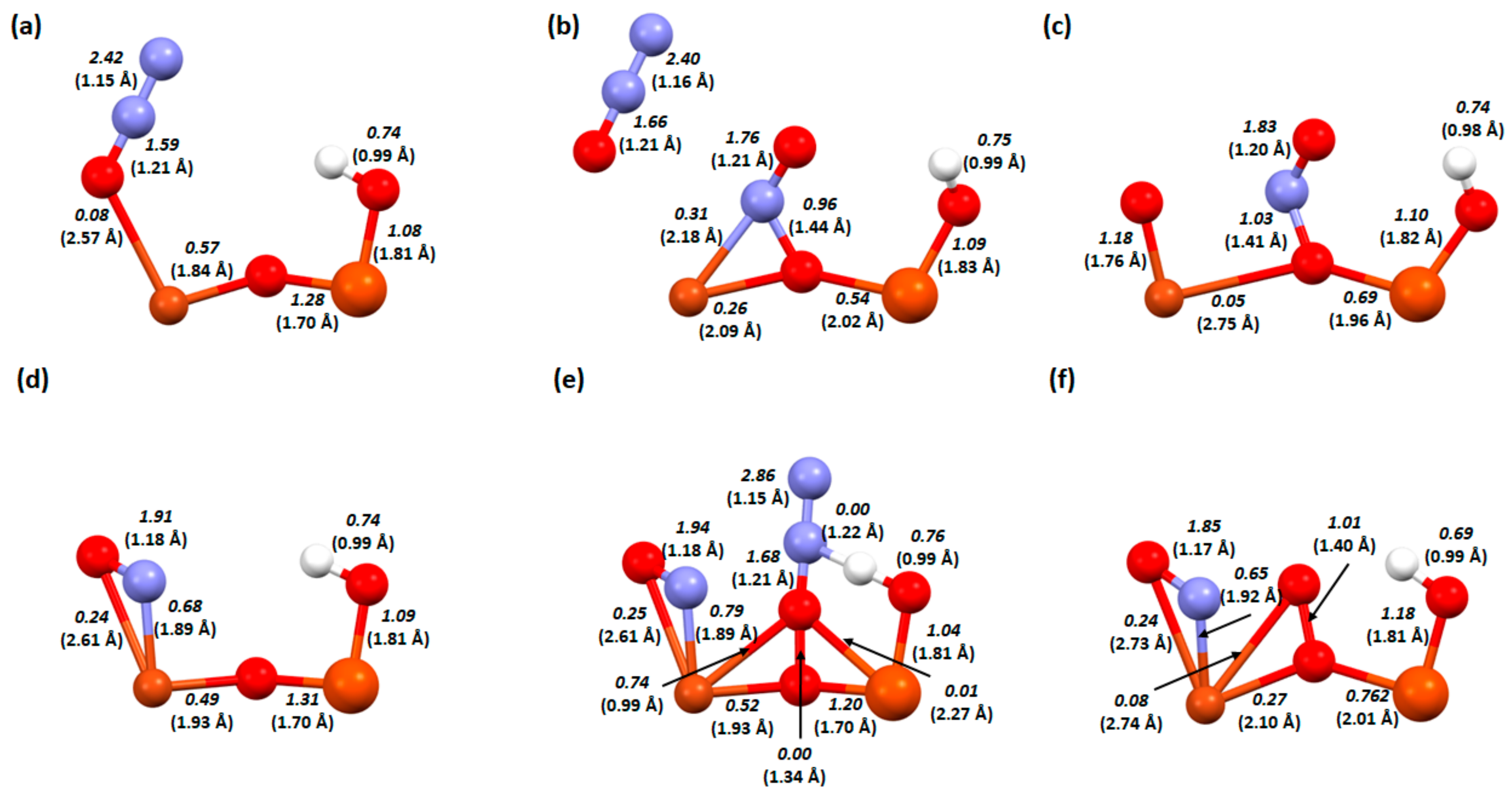
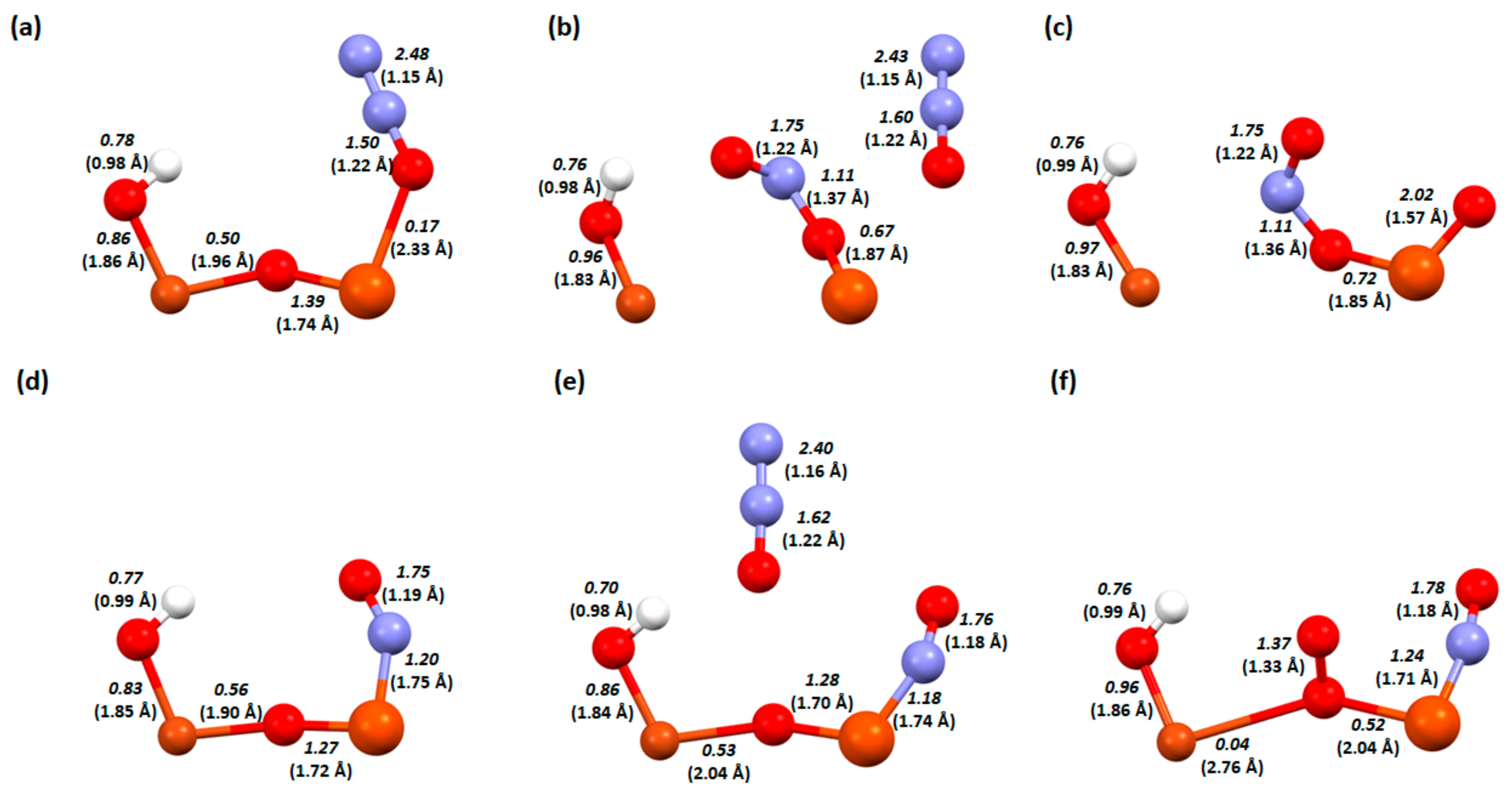
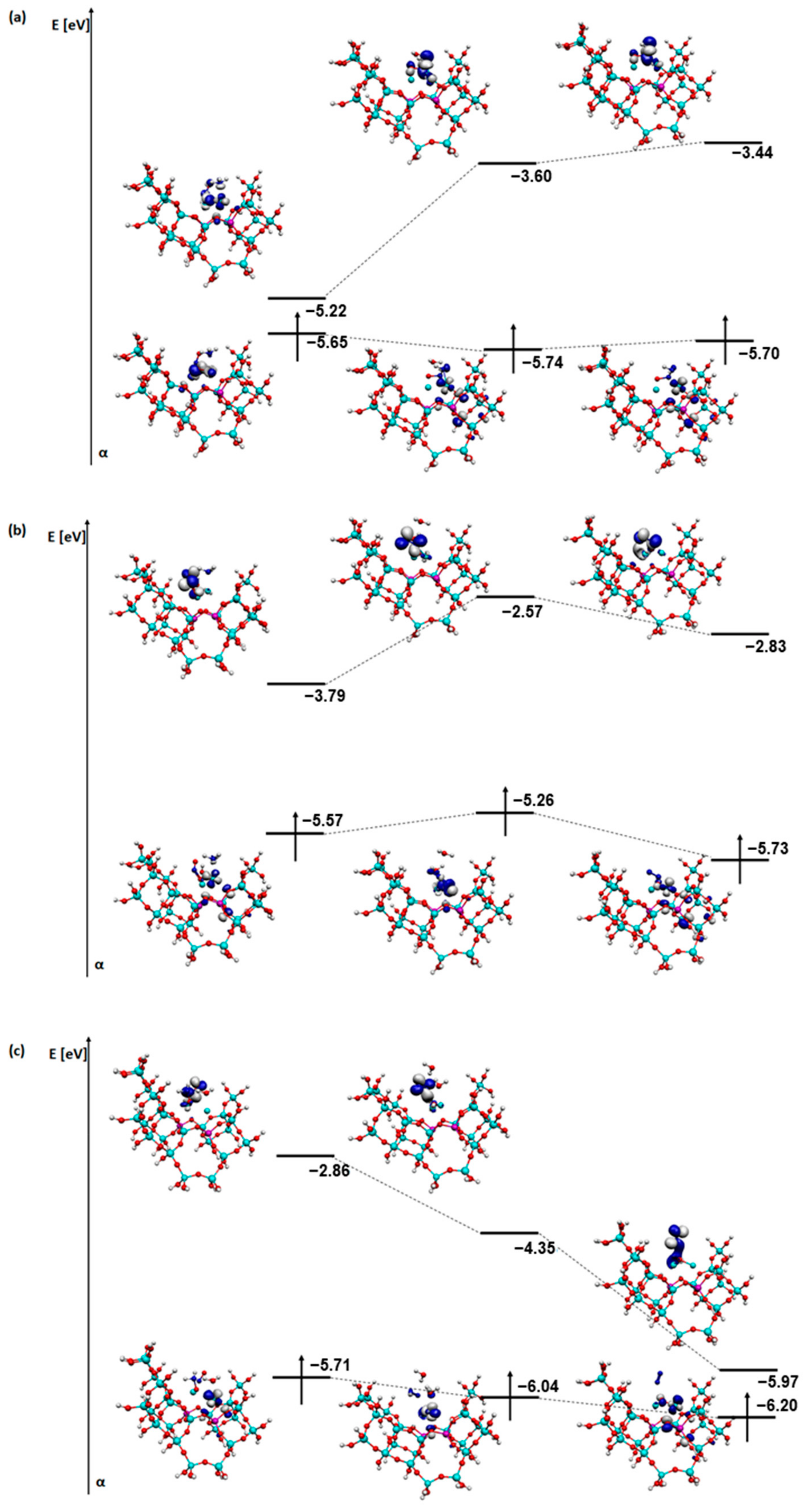
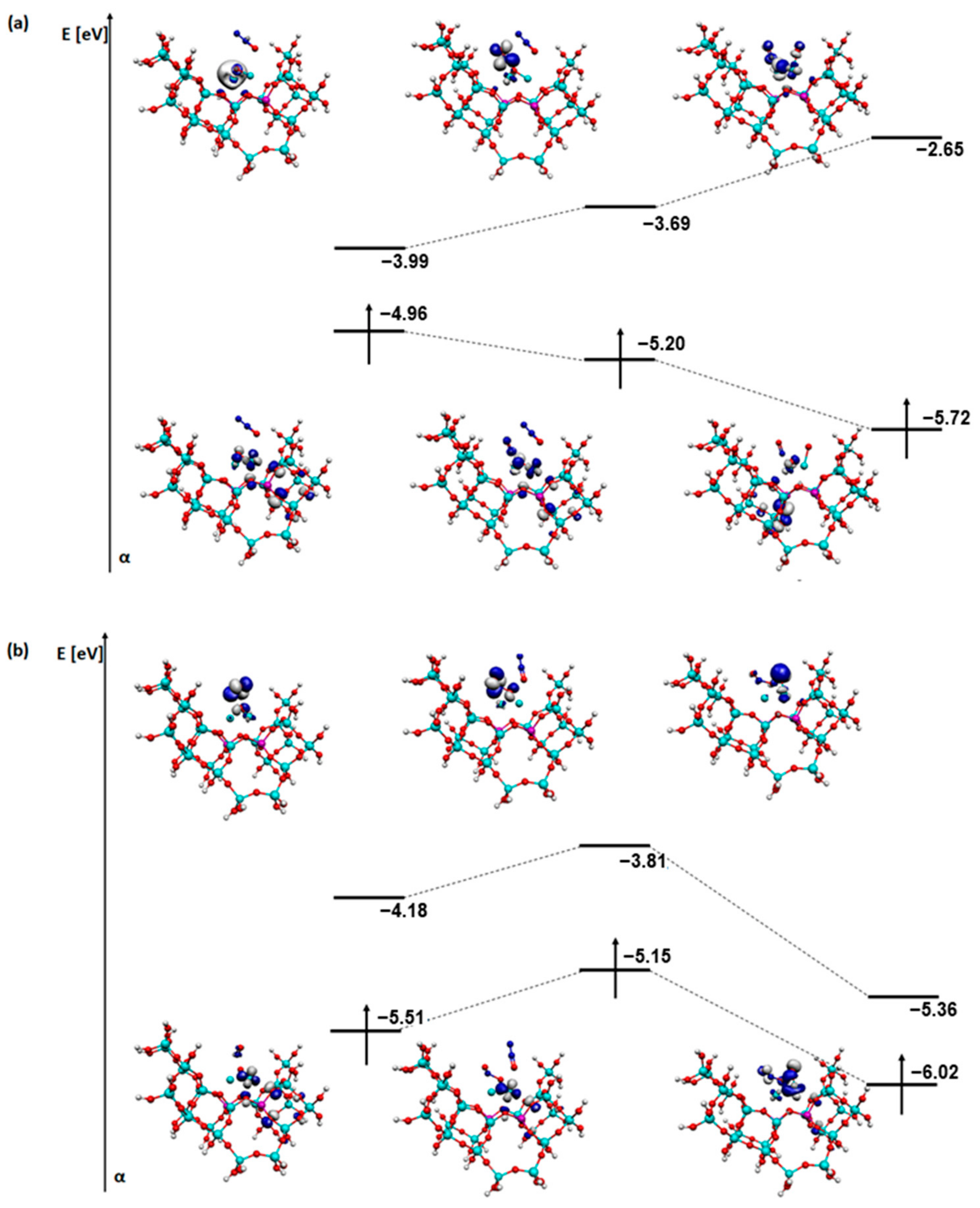
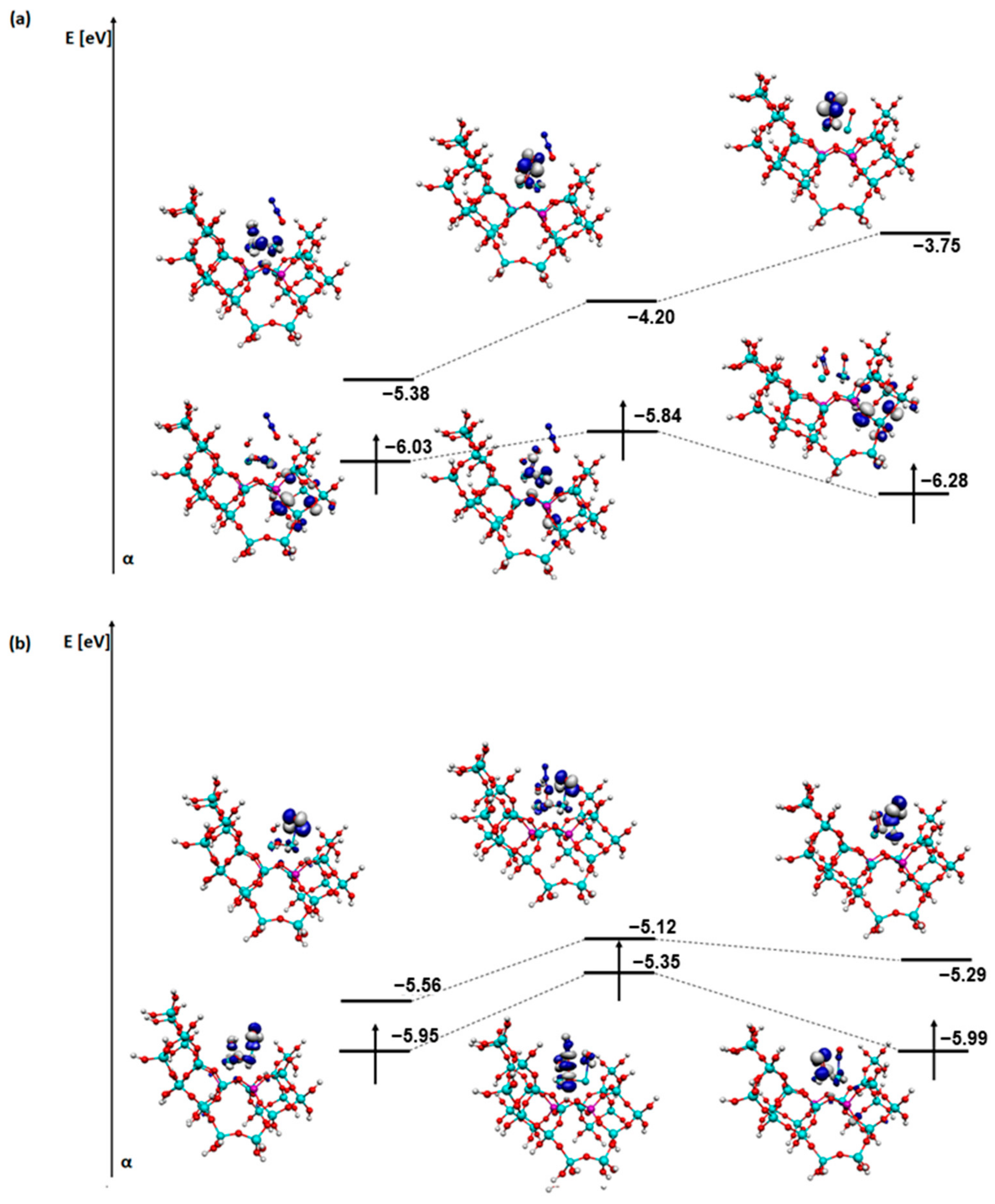
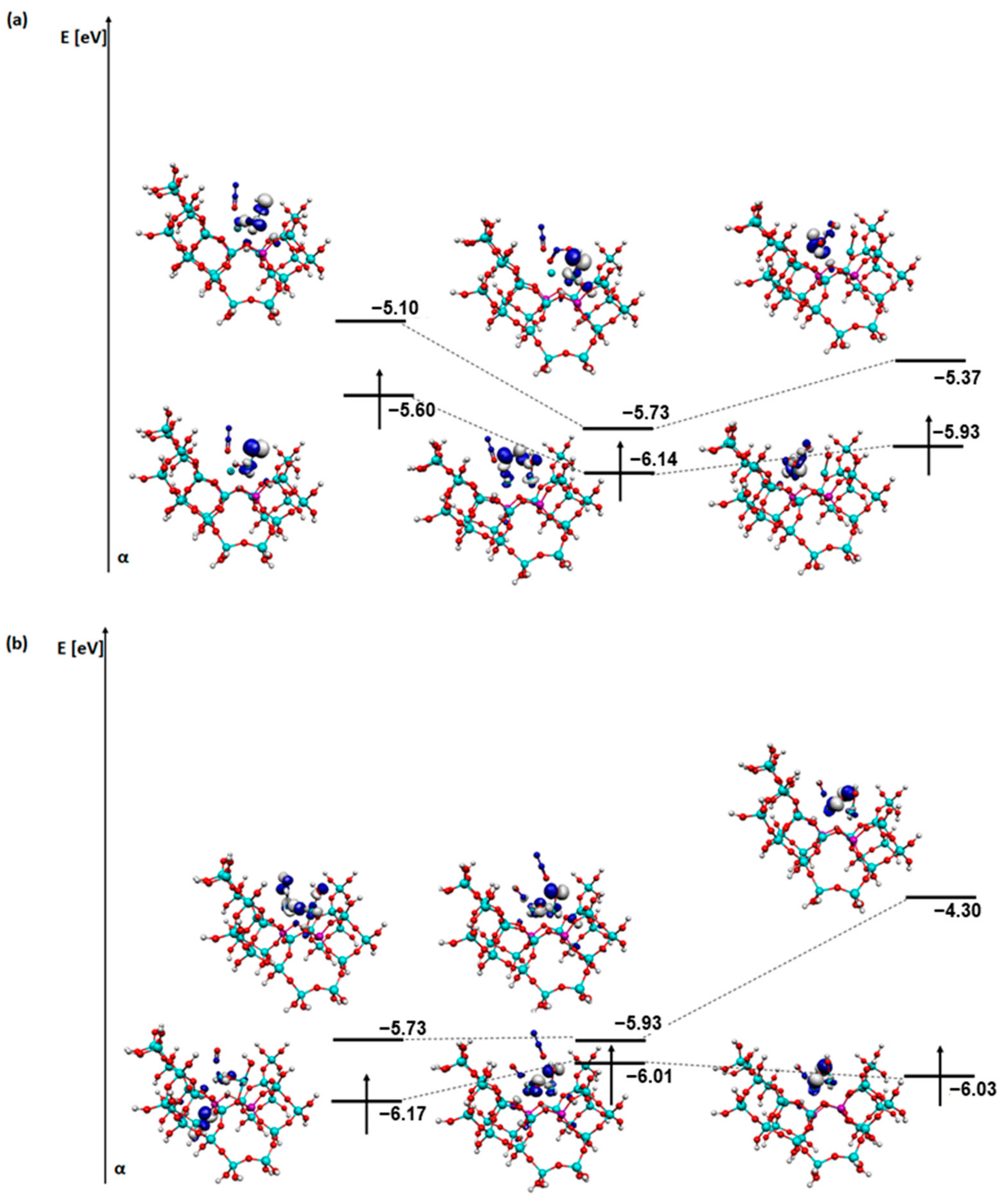
2. Results and Discussion
3. Materials and Methods
3.1. Computational Details
3.2. Geometrical Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pietrzyk, P.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Computational spectroscopy and DFT investigations into nitrogen and oxygen bond breaking and bond making processes in model deNOx and deN2O reactions. Catal. Today 2007, 119, 219–227. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Choi, H.M.; Lee, S.-J.; Moon, S.-H.; Phan, T.N.; Jeon, S.G.; Ko, C.H. Comparison between unsupported mesoporous Co3O4 and supported Co3O4 on mesoporous silica as catalysts for N2O decomposition. Catal. Commun. 2016, 82, 50–54. [Google Scholar] [CrossRef]
- Saramok, M.; Szymaszek, A.; Inger, M.; Antoniak-Jurak, K.; Samojeden, B.; Motak, M. Modified Zeolite Catalyst for a NOx Selective Catalytic Reduction Process in Nitric Acid Plants. Catalysts 2021, 11, 450. [Google Scholar] [CrossRef]
- Reşitoğlu, İ.A.; Altinişik, K.; Keskin, A. The pollutant emissions from diesel-engine vehicles and exhaust aftertreatment systems. Clean Technol. Environ. Policy 2015, 17, 15–27. [Google Scholar] [CrossRef]
- Sun, Y.; Zwolińska, E.; Chmielewski, A.G. Abatement technologies for high concentrations of NOx and SO2 removal from exhaust gases: A review. Crit. Rev. Environ. Sci. Technol. 2016, 46, 119–142. [Google Scholar] [CrossRef]
- Inger, M.; Rajewski, J.; Ruszak, M.; Wilk, M. The influence of NOx presence on the catalytic N2O decomposition over the supported double-promoted cobalt spinel catalyst. Chem. Pap. 2019, 73, 1979–1986. [Google Scholar] [CrossRef]
- Saramok, M.; Inger, M.; Antoniak-Jurak, K.; Szymaszek-Wawryca, A.; Samojeden, B.; Motak, M. Physicochemical Features and NH3-SCR Catalytic Performance of Natural Zeolite Modified with Iron—The Effect of Fe Loading. Catalysts 2022, 12, 731. [Google Scholar] [CrossRef]
- Zhu, H.; Song, L.; Li, K.; Wu, R.; Qiu, W.; He, H. Low-Temperature SCR Catalyst Development and Industrial Applications in China. Catalysts 2022, 12, 341. [Google Scholar] [CrossRef]
- Suharbiansah, R.S.R.; Pyra, K.; Liebau, M.; Poppitz, D.; Góra-Marek, K.; Gläser, R.; Jabłońska, M. Micro-/mesoporous copper-containing zeolite Y applied in NH3-SCR, DeNOx. Microporous Mesoporous Mater. 2022, 334, 111793. [Google Scholar] [CrossRef]
- van den Brink, R.W.; Booneveld, S.; Verhaak, M.J.F.M.; de Bruijn, F.A. Selective catalytic reduction of N2O and NOx in a single reactor in the nitric acid industry. Catal. Today 2002, 75, 227–232. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Kapteijn, F.; Schöffel, K.; Moulijn, J.A. Formation and control of N2O in nitric acid production: Where do we stand today? Appl. Catal. B Environ. 2003, 44, 117–151. [Google Scholar] [CrossRef]
- Isapour, G.; Wang, A.; Han, J.; Feng, Y.; Grönbeck, H.; Creaser, D.; Olsson, L.; Skoglundh, M.; Härelind, H. In situ DRIFT studies on N2O formation over Cu-functionalized zeolites during ammonia-SCR. Catal. Sci. Technol. 2022, 12, 3921–3936. [Google Scholar] [CrossRef]
- Alves, L.; Holz, L.I.V.; Fernandes, C.; Ribeirinha, P.; Mendes, D.; Fagg, D.P.; Mendes, A. A comprehensive review of NOx and N2O mitigation from industrial streams. Renew. Sustain. Energy Rev. 2022, 155, 111916. [Google Scholar] [CrossRef]
- Zhang, Z.; Tian, J.; Li, J.; Cao, C.; Wang, S.; Lv, J.; Zheng, W.; Tan, D. The development of diesel oxidation catalysts and the effect of sulfur dioxide on catalysts of metal-based diesel oxidation catalysts: A review. Fuel Process. Technol. 2022, 233, 107317. [Google Scholar] [CrossRef]
- Bendrich, M.; Scheuer, A.; Hayes, R.E.; Votsmeier, M. Unified mechanistic model for Standard SCR, Fast SCR, and NO2 SCR over a copper chabazite catalyst. Appl. Catal. B Environ. 2018, 222, 76–87. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Zhou, A.; Li, Y.; Yin, P.; Mu, C.; Xu, J. Experimental and kinetic modeling study for N2O formation of NH3-SCR over commercial Cu-zeolite catalyst. Adv. Mech. Eng. 2021, 13, 16878140211010648. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, R.T. N2O Formation Pathways over Zeolite-Supported Cu and Fe Catalysts in NH3-SCR. Energy Fuels 2018, 32, 2170–2182. [Google Scholar] [CrossRef]
- Chen, B.; Liu, N.; Liu, X.; Zhang, R.; Li, Y.; Li, Y.; Sun, X. Study on the direct decomposition of nitrous oxide over Fe-beta zeolites: From experiment to theory. Catal. Today 2011, 175, 245–255. [Google Scholar] [CrossRef]
- Cho, C.; Jung, Y.; Shin, Y.; Pyo, Y.; Jang, J.; Woo, Y.; Ko, A.; Kim, G.; Cho, G. Nitric oxide and nitrous oxide from selective oxidation in a vanadium-based catalytic diesel after-treatment system. Int. J. Energy Res. 2022, 46, 15816–15823. [Google Scholar] [CrossRef]
- Szymaszek, A.; Samojeden, B.; Motak, M. The Deactivation of Industrial SCR Catalysts—A Short Review. Energies 2020, 13, 3870. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zhao, Z.; Liao, J.; Chen, C.; Li, Q. Recent progress of metal-exchanged zeolites for selective catalytic reduction of NOx with NH3 in diesel exhaust. Fuel 2021, 305, 121482. [Google Scholar] [CrossRef]
- Han, L.; Cai, S.; Gao, M.; Hasegawa, J.-y.; Wang, P.; Zhang, J.; Shi, L.; Zhang, D. Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects. Chem. Rev. 2019, 119, 10916–10976. [Google Scholar] [CrossRef] [PubMed]
- Hamoud, H.I.; Valtchev, V.; Daturi, M. Selective catalytic reduction of NOx over Cu- and Fe-exchanged zeolites and their mechanical mixture. Appl. Catal. B Environ. 2019, 250, 419–428. [Google Scholar] [CrossRef]
- Ghasemian, N.; Falamaki, C. Zn2+, Fe2+, Cu2+, Mn2+, H+ Ion-exchanged and Raw Clinoptilolite Zeolite Catalytic Performance in the Propane-SCR-NOx Process: A Comparative Study. Int. J. Chem. React. Eng. 2018, 16, 20160192. [Google Scholar] [CrossRef]
- Shan, Y.; Du, J.; Zhang, Y.; Shan, W.; Shi, X.; Yu, Y.; Zhang, R.; Meng, X.; Xiao, F.-S.; He, H. Selective catalytic reduction of NOx with NH3: Opportunities and challenges of Cu-based small-pore zeolites. Natl. Sci. Rev. 2021, 8, nwab010. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Rizzotto, V.; Guo, A.; Ye, D.; Simon, U.; Chen, P. Recent Understanding of Low-Temperature Copper Dynamics in Cu-Chabazite NH3-SCR Catalysts. Catalysts 2021, 11, 52. [Google Scholar] [CrossRef]
- Liu, Q.; Bian, C.; Ming, S.; Guo, L.; Zhang, S.; Pang, L.; Liu, P.; Chen, Z.; Li, T. The opportunities and challenges of iron-zeolite as NH3-SCR catalyst in purification of vehicle exhaust. Appl. Catal. A Gen. 2020, 607, 117865. [Google Scholar] [CrossRef]
- Gao, F. Fe-Exchanged Small-Pore Zeolites as Ammonia Selective Catalytic Reduction (NH3-SCR) Catalysts. Catalysts 2020, 10, 1324. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, R.; Ren, S.; Chen, L.; Li, X.; Wang, M. Comparative analysis of the dual origins of the N2O byproduct on MnOx, FeOx, and MnFeOx sphere catalysts for a low-temperature SCR of NO with NH3. J. Mater. Chem. A 2022, 10, 21474–21491. [Google Scholar] [CrossRef]
- Ren, S.; Li, X.; He, C.; Chen, L.; Wang, L.; Li, F. Surface tailoring on bifunctional CuOx/MnO2 catalyst to promote the selective catalytic reduction of NO with NH3 and oxidation of CO with O2. Sep. Purif. Technol. 2024, 346, 127471. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, B.; Lv, N.; Wang, T.; Bi, X.; Zhu, H.; Yuan, P.; Bai, Z.; Cui, Q.; Bao, X. Direct Synthesis of Hierarchical FeCu-ZSM-5 Zeolite with Wide Temperature Window in Selective Catalytic Reduction of NO by NH3. ChemCatChem 2019, 11, 4744–4754. [Google Scholar] [CrossRef]
- Denardin, F.G.; Muniz, A.R.; Perez-Lopez, O.W. Experimental and DFT analysis of the acid and reduction properties of Fe-Cu/ZSM-5. Microporous Mesoporous Mater. 2021, 314, 110860. [Google Scholar] [CrossRef]
- Jouini, H.; Mejri, I.; Petitto, C.; Martinez-Ortigosa, J.; Vidal-Moya, A.; Mhamdi, M.; Blasco, T.; Delahay, G. Characterization and NH3-SCR reactivity of Cu-Fe-ZSM-5 catalysts prepared by solid state ion exchange: The metal exchange order effect. Microporous Mesoporous Mater. 2018, 260, 217–226. [Google Scholar] [CrossRef]
- Jodłowski, P.J.; Czekaj, I.; Stachurska, P.; Kuterasiński, Ł.; Chmielarz, L.; Jędrzejczyk, R.J.; Jeleń, P.; Sitarz, M.; Górecka, S.; Mazur, M.; et al. Experimental and Theoretical Studies of Sonically Prepared Cu–Y, Cu–USY and Cu–ZSM-5 Catalysts for SCR deNOx. Catalysts 2021, 11, 824. [Google Scholar] [CrossRef]
- Kurzydym, I.; Czekaj, I. Theoretical Studies on the Mechanism of deNOx Process in Cu–Zn Bimetallic System—Comparison of FAU and MFI Zeolites. Molecules 2022, 27, 300. [Google Scholar] [CrossRef] [PubMed]
- Kurzydym, I.; Czekaj, I. Theoretical studies of DENOx SCR over Cu-, Fe- and Mn-FAU catalysts. Chem. Chem. Technol. 2021, 15, 16–25. [Google Scholar] [CrossRef]
- Battiston, A.A.; Bitter, J.H.; Koningsberger, D.C. Reactivity of binuclear Fe complexes in over-exchanged Fe/ZSM5, studied by in situ XAFS spectroscopy 2. Selective catalytic reduction of NO with isobutane. J. Catal. 2003, 218, 163–177. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Hermann, K.; Pettersson, L.; Casida, M.; Daul, C.; Goursot, A.; Koester, A.; Proynov, E.; St-Amant, A.; Salahub, D.; Carravetta, V.; et al. StoBe-deMon; deMon Software: Stockholm, Sweden; Berlin, Germany, 2005; Available online: http://www.fhi-berlin.mpg.de/KHsoftware/StoBe/ (accessed on 12 May 2024).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Brocławik, E.; Salahub, D.R. Density functional theory and quantum chemistry: Metals and metal oxides. J. Mol. Catal. 1993, 82, 117–129. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. Bond orders and valences: Role of d-orbitals for hypervalent sulphur. J. Mol. Struct. THEOCHEM 1987, 149, 81–89. [Google Scholar] [CrossRef]
- Chiodo, S.; Russo, N.; Sicilia, E. Newly developed basis sets for density functional calculations. J. Comput. Chem. 2005, 26, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Puértolas, B.; Oliveira, C.C.S.; Manayil, J.C.; Barbero, B.; Isaacs, M.; Michailof, C.; Heracleous, E.; Pérez-Ramírez, J.; Lee, A.F.; et al. On the influence of Si:Al ratio and hierarchical porosity of FAU zeolites in solid acid catalysed esterification pretreatment of bio-oil. Biomass Conv. Bioref. 2017, 7, 331–342. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurzydym, I.; Czekaj, I. Mechanisms for deNOx and deN2O Processes on FAU Zeolite with a Bimetallic Cu-Fe Dimer in the Presence of a Hydroxyl Group—DFT Theoretical Calculations. Molecules 2024, 29, 2329. https://doi.org/10.3390/molecules29102329
Kurzydym I, Czekaj I. Mechanisms for deNOx and deN2O Processes on FAU Zeolite with a Bimetallic Cu-Fe Dimer in the Presence of a Hydroxyl Group—DFT Theoretical Calculations. Molecules. 2024; 29(10):2329. https://doi.org/10.3390/molecules29102329
Chicago/Turabian StyleKurzydym, Izabela, and Izabela Czekaj. 2024. "Mechanisms for deNOx and deN2O Processes on FAU Zeolite with a Bimetallic Cu-Fe Dimer in the Presence of a Hydroxyl Group—DFT Theoretical Calculations" Molecules 29, no. 10: 2329. https://doi.org/10.3390/molecules29102329