Abstract
β-halogenated enol esters and ethers are versatile building blocks in organic synthesis, which has attracted increasing attention. In this study, we report the facile trans-oxyiodination and oxychlorination of alkynes, leading to the direct construction of versatile halogenated enol esters and ethers. This transformation features an easy operation, optimal atomic economy, a strong functional group tolerance, broad substrate scope, and excellent trans-selectivity. Employing highly electrophilic bifunctional N–X (halogen) reagents was the key to achieving broad reaction generality. To our knowledge, this transformation represents the first oxyhalogenation system employing N–X (halogen) reagents as both oxylation and halogenation sources.
1. Introduction
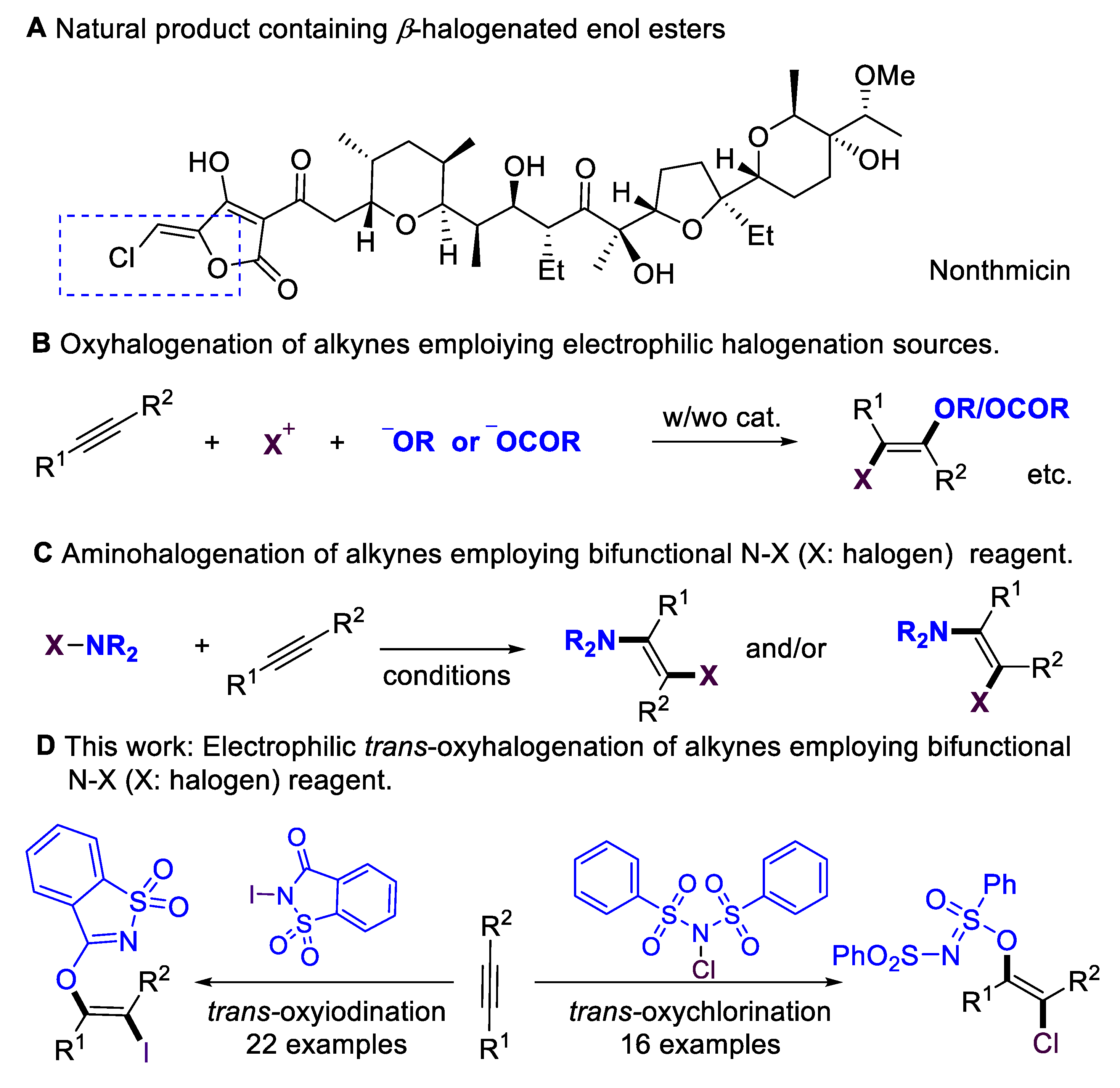
Multi-substituted enol esters and ethers have been widely applied in natural products and bioactive molecules (Scheme 1A) [1,2,3,4]; they also serve as versatile building blocks [5,6,7,8,9,10,11] in organic synthesis, medicinal chemistry, and polymer chemistry. Alkynes are fundamental and easy-to-access starting materials in organic synthesis. The stereoselective installation of O-centered functional groups in alkynes has become an attractive alternative for synthesizing enol esters/ethers. Despite the significance of hydroalkoxylation [12,13], the scope of alkynes has mainly been focused on terminal alkynes, which lead to disubstituted enol esters/ethers. Introducing halogen atoms into organic molecules is an important step in organic synthesis [14,15,16,17,18,19,20,21], as halogen groups could serve as versatile synthetic handles for further transformations [22,23,24]. The oxyhalogenation of alkynes is a straightforward route to β-halogenated enol esters and ethers by simultaneously installing O-centered groups and halogen groups into C–C triple bonds. Considerable progress has been achieved in the intramolecular oxyhalogenation of alkynes that were initiated through nucleophilic cyclization, leading to the formation of halogenated O-containing heterocycles. However, there are only a few existing examples of the intermolecular oxyhalogenation of alkynes [25,26,27,28,29,30,31,32] via the employment of electrophilic halogenation reagents and additional acid, alcohol, or phenol nucleophiles (Scheme 1B). Furthermore, intermolecular oxyhalogenation systems without additional nucleophiles have remained largely underdeveloped. This core challenge could be attributed to a lack of efficient bifunctional oxyhalogenation reagents. The development of novel bifunctional reagents, or exploring new reaction modes of existing halogenation reagents, is highly desirable.
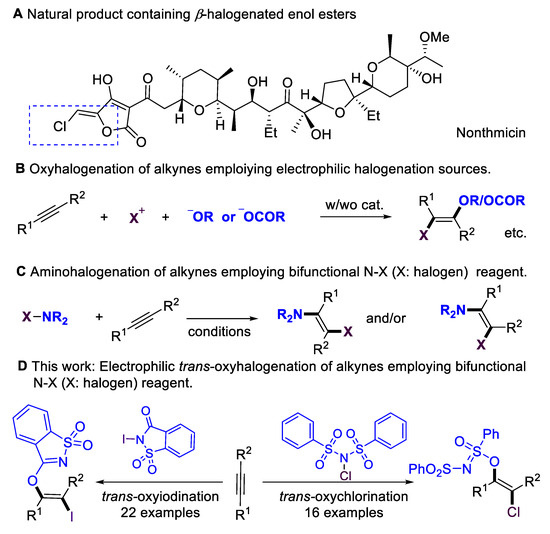
Scheme 1.
Motivation for oxyhalogenation of alkynes.
On the other hand, N–X (halogen) reagents [14,15,16,17], such as NBS, NIS, or NFSI, are the most significant electrophilic halogenation reagents with extremely important applications in organic synthesis. They also serve as bifunctional reagents in addition to alkynes, delivering amino-halogenation products (Scheme 1C) [33,34,35,36]. In 1999, the Wille and Lüning group [34] achieved the radical amino-bromination of alkynes. In 2011, the Liu group [35] achieved the copper-catalyzed chloramination of terminal alkynes, leading to the regio- and stereoselective formation of (E)-β-Chloro-enesulfonamides. In 2014, the Liang and Zhang group [36] accomplished the cis-amino-halogenation of terminal alkynes with N-haloimides via alkynyl halide intermediates. N-iodosaccharin is a highly active electrophilic iodination source [37,38,39,40,41,42] due to the saccharin anion that is released; however, in previous reports, this reagent has never been employed as a bifunctional reagent. Inspired by reports that employ saccharin anions as O-centered nucleophilic source [43,44], here, we evaluate the possibility of employing N-iodosaccharins as novel bifunctional oxyiodination reagents. Based on our previous works [45,46,47,48,49,50,51,52] on the development of sustainable transformations, we now report on the intermolecular oxyiodination and oxychlorination of alkynes that employ N-iodosaccharin or N-chlorobenzenesulfonimide (NCBSI) [53,54,55] as bifunctional reagents (Scheme 1D).
2. Results and Discussions
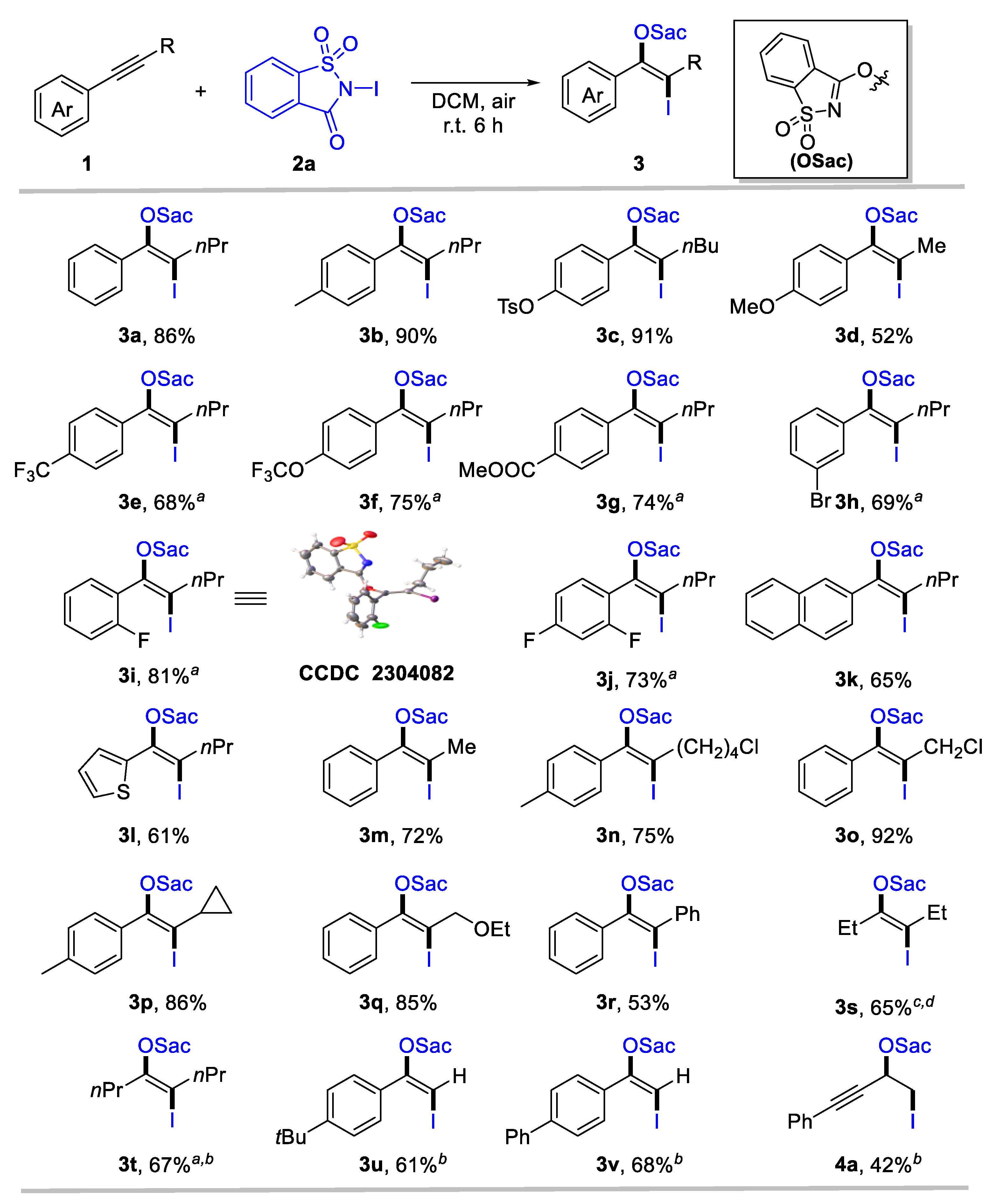
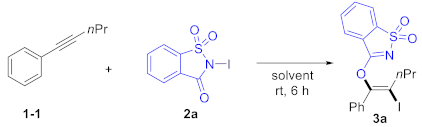
We utilized 1-phenyl-1-pentyne (1a) and N-iodosaccharin (2a) as model substrates to test the designed transformation. The oxyiodination of alkyne proceeded when 1a (0.2 equiv) and 2a (1.0 equiv) in dichloromethane (DCM) were employed as the solvent, yielding the corresponding product 3a in a 68% yield (Table 1, Entry 1). A simple adjustment in the ratio of 1a and 2a improved the yield of product 3a by 86% (Entry 2). However, further increasing the amount of 2a had no noticeable effect on the yield (Entry 3). Alternative solvents were tested (Entries 4–8), and we found that dichloroethane (DCE) and perfluorotoluene (PhCF3) exhibited a similar efficiency to DCM, while toluene and acetonitrile (CH3CN) provided substantially reduced yields. Methanol (MeOH) proved unsuitable for this transformation. Importantly, the reaction could be conducted under ambient conditions, providing oxyiodination products 3a in 86% yields (Entry 9). Remarkably, exclusive regioselectivity and stereoselectivity were achieved in all cases.

Table 1.
Optimization of intermolecular oxyiodination of alkynes.
Considering the easy operation, Table 1 Entry 9 was identified as the standard condition to investigate the generality and limitation of the oxyiodination system. First, the substituent effect of the aryl group for aryl-substituted internal alkynes was investigated. The top section of Scheme 2 reveals that various aromatic internal alkynes bearing electron-donating (e.g., alkyl, tosylate, and methoxy), electron-withdrawing (such as trifluoromethyl, trifluoromethoxy, and ester carbonyl), and halogen groups were well tolerated, affording the desired iodinated enol ethers 3a-3j in moderate to high yields (52–91%). Notably, electron-donating groups in para-substituted internal aryl alkynes 1-2 and 1-3 exhibited high reactivity (3b, 90%; 3c, 91%). However, strong electron-donating 1-methoxy-4-(prop-1-yn-1-yl)benzene 1-4 provides 3d only in a moderate yield. Moreover, internal alkynes with an electron-withdrawing capacity at aromatic rings could deliver β-iodinated enol ethers with acceptable yields (3e-3g, 68–75%). This indicates a broad substrate scope for alkynes. Meta- and ortho-substituted aryl alkynes were tolerated, forming 3h and 3i in 69% and 81% yield, respectively. The structure of 3i was confirmed through X-ray single crystal diffraction (CCDC 2304082). Even di-substituted internal aryl alkyne was a suitable substrate for this transformation, affording enol ethers 3j in 73% yields. Naphthalene and heteroaromatics were compatible for this transformation, as identified by the formation of 3k and 3l. Then, the scope of aliphatic groups for aryl-substituted internal alkynes was evaluated, revealing the compatibility of alkyl (3m), chloride (3n, 3o), strained rings (3p), and ether (3q). These substrates provided the corresponding products with 72–92% yields. In all cases, specific regio- and stereo-specificity were observed. In some cases, such as diaryl (3r) or dialkyl-substituted alkynes (3s, 3t), a reduced reaction efficiency was noted, despite their tolerance. In addition, terminal alkynes were used to explore the scope of this system further, delivering the desired enol ethers 3u and 3v in 61% and 68% yields, respectively. For 1,3-enynes, oxyiodination was preferred to occur at olefin units, as identified by the formation of 4a. However, N-chlorosaccharin and N-bromosaccharin failed to deliver the desired oxyhalogenation products under standard conditions, likely owing to their considerably low electrophilic reactivity.
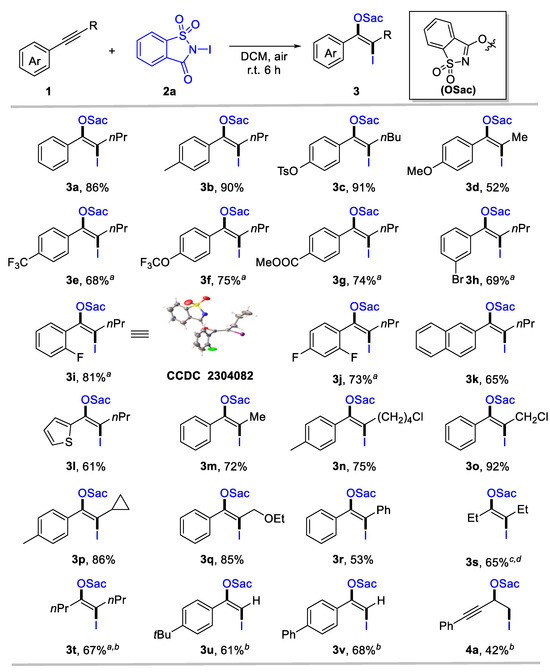
Scheme 2.
Scope for anti-oxyiodination of alkynes. Reaction conditions: 1 (0.2 mmol), N-iodosaccharin 2a (1.2 equiv), DCM (2 mL), r.t., 6 h. Yield of the isolated product. a 24 h. b 2a (0.2 mmol) and 1 (1.2 equiv) was employed, and yield was calculated based on 2a.
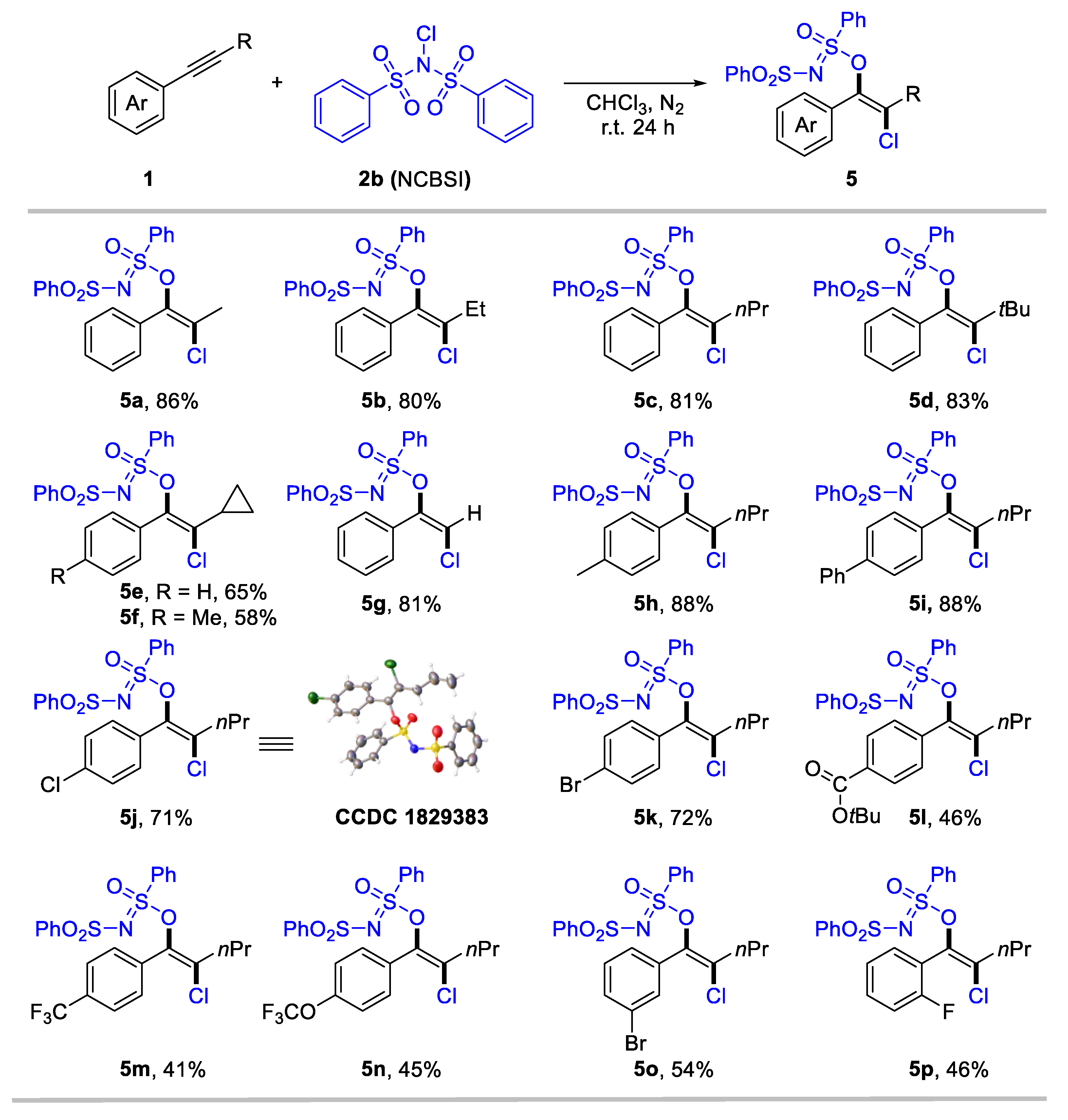
We consider that the highly electron-deficient bisphenylsulfonimide may improve the electrophilic activity of N–X (X: halogen) reagents, thereby promoting the formation of halogenated products. Gratefully, when employing N-chlorobenzenesulfonimide (NCBSI) as the chlorination reagent, we obtained trans-oxychlorination products in chloroform under a N2 atmosphere (details for optimization conditions see Supplementary Materials, Table S1). The scope of the oxychlorination of alkynes was subsequently examined, and results are summarized in Scheme 3. The desired enol sulfinimidates (5a-5p) were obtained in moderate-to-high yields with specific anti-selectivity. Internal aryl alkynes bearing different aliphatic substituents, such as alkyl (5a-5f), bulky tertiary butyl (5d), and cyclopropyl (5e, 5f), were tolerated, delivering anti-oxychlorination products in 58–86% yields. Terminal aryl alkyne was also suitable for this conversion, with 5g achieving an 81% yield. We next investigate the substituent effect of the aryl rings in internal alkynes for oxychlorination system. Aryl alkynes bearing electron-donating (5h), phenyl (5i), and halogen (5j-5k) groups at the para-position, performed effectively (71–88%). Notably, the structure of 5j was confirmed through X-ray single crystal diffraction (CCDC 1829383). Meanwhile, the electron-withdrawing group, including ester carbonyl (5l), trifluoromethyl (5m), and trifluoromethoxy (5n), that substituted aryl acetylenes generated a slightly low yield (41–46%). Moreover, meta- and ortho-substituted aryl alkynes were applicable for the reaction, delivering 5o and 5p in 54% and 46% yield, respectively.
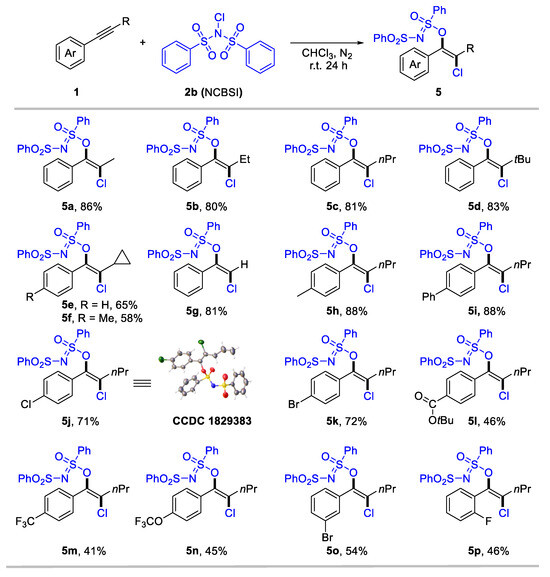
Scheme 3.
Subtract scope for anti-oxychlorination of alkynes. Reaction conditions: 1 (0.2 mmol), NCBSI 2b (2.0 equiv), CHCl3 (2 mL), r.t. for 24 h under nitrogen atmosphere. Yield of isolated products.
3. Mechanistic Investigation
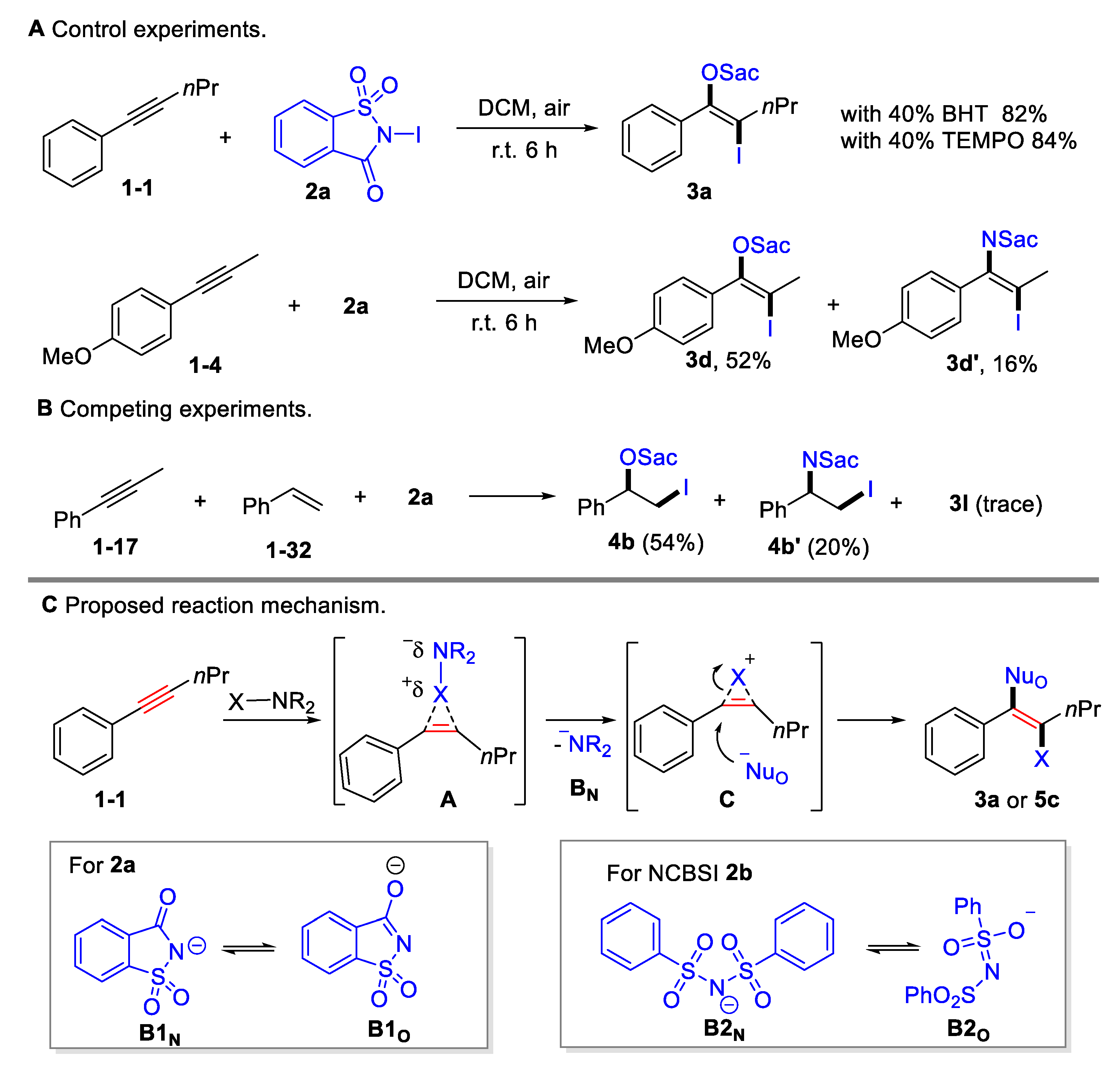
Several preliminary mechanistic studies were conducted in order to shed insight onto this transformation (Scheme 4A upper). The addition of radical scavengers, such as butylated hydroxytoluene (BHT; 40 mol%) or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO; 40 mol%), showed no considerable impact on the yields, indicating that the oxyiodination may not proceed via a radical pathway. Competitive amino iodination products were obtained for 1-methoxy-4-(prop-1-yn-1-yl)benzene 1-4 or styrene 1-32 (Scheme 4A lower, Scheme 4B), indicating that there was equilibration between N-centered and O-centered nucleophile. Moreover, it was found that styrene 1-32 exhibited a higher reactivity than alkynes 1-17, which is consistent with their electrophilic reactivity (Scheme 4B). A plausible mechanism was proposed based on the control experiment and on previous reports [45] (Scheme 4C). The process begins with the selective coordination of alkyne triple bonds and halogenating reagents, generating a cyclic halonium onium intermediate A by releasing a saccharin anion B1N or benzenesulfonamide anion B2N. The released B1N or B2N could resonate to the O-centered nucleophiles B1O or B2O. Owing to its steric hindrance, halonium onium intermediate preferred to be opened by O-centered nucleophiles, resulting in anti-selective oxyhalogenation.
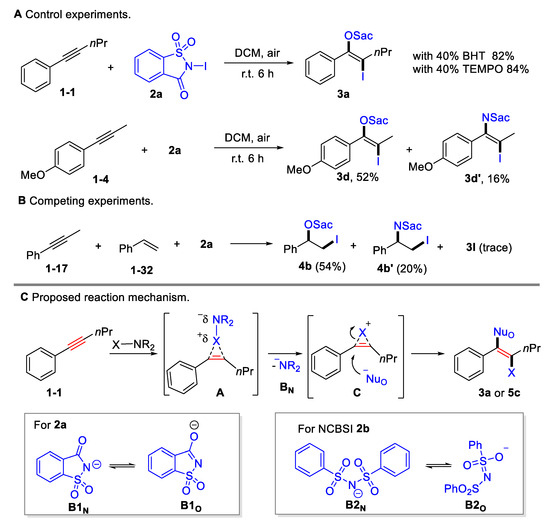
Scheme 4.
Mechanistic investigation and proposed reaction pathway.
4. Materials and Methods
4.1. Materials and Instruments
All chemicals were obtained from commercial sources and were used as they were received, unless otherwise noted. All reactions were carried out using a test tube or a pressure tube. The reactions were monitored with the aid of thin-layer chromatography (TLC) on 0.25 mm precoated silica gel plates. Visualization was carried out with a UV light and a aqueous potassium permanganate stain. Melting points were measured on Büchi B-540 apparatus. NMR spectra were recorded on a 500 or 600 MHz NMR spectrometer in the solvent indicated. The chemical shift is given in dimensionless δ values and is frequency-referenced, relative to TMS in 1H and 13C NMR spectroscopy. HRMS data were obtained on a Bruck microtof. Column chromatography was performed on silica gel (200–300 mesh) using ethyl acetate/hexanes.
4.2. The General Procedure for the Synthesis of 3
The reaction tube equipped with a magnetic stir bar was charged with 2a (0.24 mmol, 74.1 mg), DCM (2 mL), and 1 (0.2 mmol). The test tube was then sealed off with a screw cap, and the reaction mixture was stirred at room temperature for 6.0 or 24 h. After the reaction was completed, as indicated by TLC analysis, the mixture was extracted using DCM (2 × 10 mL). The combined organic phases were dried over anhydrous Na2SO4, and the solvent was evaporated under a vacuum. The residue was purified via column chromatography (petroleum ether/ethyl acetate 10:1 (v/v)) to provide the corresponding product 3.
- (E)-3-((2-iodo-1-phenylpent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3a
1-1 (0.2 mmol, 28.8 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3a (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow solid (83.7 mg, 86%), crystallization in CDCl3, mp. 140–142 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.2 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.77 (td, J = 7.2, 1.2 Hz, 1H), 7.73 (td, J = 7.8, 1.2 Hz, 1H), 7.60–7.58 (m, 2H), 7.39–7.35 (m, 3H), 2.57 (t, J = 7.2 Hz, 2H), 1.72–1.65 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 167.3, 147.0, 143.9, 135.3, 134.3, 133.6, 130.3, 129.8, 128.2, 126.2, 123.3, 122.1, 98.7, 39.8, 22.3, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C18H16INNaO3S+ ([M + Na]+), 475.9788, found, 475.9790.
- (E)-3-((2-iodo-1-(p-tolyl)pent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3b
1-2 (0.2 mmol, 31.6 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3b (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (84.5 mg, 90%), crystallization in CDCl3, mp. 106–108 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.85 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.76 (td, J = 7.2, 1.2 Hz, 1H), 7.72 (td, J = 7.8, 1.2 Hz, 1H), 7.48 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 2.57–2.55 (m, 2H), 2.35 (s, 3H), 1.70–1.64 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 147.1, 143.9, 139.9, 134.3, 133.5, 132.4, 130.2, 128.9, 126.2, 123.3, 122.1, 98.3, 39.9, 22.3, 21.4, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C19H18INNaO3S+ ([M + Na]+), 489.9944, found, 489.9950.
- (E)-4-(1-((1,1-dioxidobenzo[d]isothiazol-3-yl)oxy)-2-iodohex-1-en-1-yl)phenyl 4-methylbenzenesulfonate 3c
1-3 (0.2 mmol, 65.6 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3c (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (116.3 mg, 91%), crystallization in CDCl3, mp. 109–111 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.2 Hz, 1H), 7.80 (dt, J = 7.2, 4.2 Hz, 2H), 7.75 (td, J = 7.2, 0.6 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.55–7.50 (m, 2H), 7.29 (d, J = 7.8 Hz, 2H), 7.00–6.93 (m, 2H), 2.63–2.55 (m, 2H), 2.42 (s, 3H), 1.63–1.58 (m, 2H), 1.41–1.31 (m, 2H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.2, 150.2, 145.6, 145.4, 143.8, 134.5, 134.1, 133.7, 131.9, 129.8, 128.5, 125.8, 123.3, 122.3, 122.1, 99.7, 37.7, 30.9, 21.7, 21.6, 13.8. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C26H24INNaO2S2+, ([M + Na]+), 659.9982, found, 659.9990.
- (E)-3-((2-iodo-1-(4-methoxyphenyl)prop-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3d
1-4 (0.2 mmol, 29.2 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3d (PE/EtOAc = 5:1, Rf = 0.28) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), colorless oil (47.2 mg, 52%). NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 7.3 Hz, 1H), 7.83 (d, J = 7.4 Hz, 1H), 7.80–7.75 (m, 1H), 7.73 (td, J = 7.5, 1.1 Hz, 1H), 7.53 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H), 2.57 (s, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 166.8 (C=N), 160.5, 147.2, 143.4, 134.3, 133.6, 131.7, 127.3, 126.2, 123.4, 122.1, 121.7, 113.6, 86.2 (C-I), 53.0, 26.4. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C17H14INNaO4S+ ([M + Na]+), 477.9580, found 477.9594.
- (E)-2-(2-iodo-1-(4-methoxyphenyl)prop-1-en-1-yl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide 3d′
1-4 (0.2 mmol, 29.2 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3d′ (PE/EtOAc = 5:1, Rf = 0.32) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), colorless oil (14.6 mg, 16%). NMR spectroscopy: 1H NMR (500 MHz, Chloroform-d) δ 8.08 (d, J = 6.7 Hz, 1H), 7.96–7.79 (m, 3H), 7.48 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 3.80 (s, 3H), 2.71 (s, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 159.9, 157.2 (C=O), 138.1, 135.0, 134.4, 131.6, 131.3, 126.7, 125.6, 121.1, 113.5, 109.7 (C-I), 54.4, 30.5. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C17H14INNaO4S+ ([M + Na]+), 477.9580, found 477.9580.
- (E)-3-((2-iodo-1-(4-(trifluoromethyl)phenyl)pent-1-en-1-yl)oxy)benzo[d]isothiazole 3e
1-5 (0.2 mmol, 42.4 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3e (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow oil (70.8 mg, 68%). NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 7.5 Hz, 1H), 7.79 (t, J = 7.5 Hz, 1H), 7.76–7.72 (m, 3H), 7.64 (d, J = 8.0 Hz, 2H), 2.59 (t, J = 7.5 Hz, 2H), 1.73–1.63 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 145.4, 143.8, 138.8, 134.6, 133.7, 131.5 (q, J = 33.0 Hz), 130.9, 125.8, 125.3 (q, J = 4.5 Hz), 123.7 (q, J = 271.5 Hz), 123.2, 122.2, 100.1, 39.8, 22.3, 13.1. 19F NMR (565 MHz, CDCl3) δ −62.92. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C19H15F3INNaO3S+, ([M + Na]+), 543.9662, found, 543.9661.
- (E)-3-((2-iodo-1-(4-(trifluoromethoxy)phenyl)pent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3f
1-6 (0.2 mmol, 25.6 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3f (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow oil (80.9 mg, 75%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.87 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.2 Hz, 1H), 7.79 (t, J = 7.8 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 2.58 (t, J = 7.2 Hz, 2H), 1.71–1.65 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150MHz, CDCl3) δ 167.3, 149.9, 145.6, 143.8, 134.5, 133.8, 133.7, 132.2, 125.9, 123.3, 122.2, 120.4, 120.3 (q, J = 256.5 Hz) 99.8, 39.8, 22.3, 13.1. 19F NMR (565 MHz, CDCl3) δ −57.64. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C19H25F3INNaO4S+, ([M + Na]+), 559.9611, found, 559.9624.
- methyl (E)-4-(1-((1,1-dioxidobenzo[d]isothiazol-3-yl)oxy)-2-iodopent-1-en-1-yl)benzoate 3g
1-7 (0.2 mmol, 40.4 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3g (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow oil (75.6 mg, 74%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 8.05 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 7.2 Hz, 1H), 7.84 (d, J = 7.2 Hz, 1H), 7.79 (t, J = 7.2 Hz, 1H), 7.75 (t, J = 7.2 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 3.91 (s, 3H), 2.60 (t, J = 7.2 Hz, 2H), 1.73–1.64 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 166.3, 145.8, 143.7, 139.5, 134.5, 133.7, 131.0, 130.4, 129.5, 125.8, 123.2, 122.2, 99.7, 52.2, 39.8, 22.2, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C20H18INNaO5S+ ([M + Na]+), 533.9843, found, 533.9848.
- (E)-3-((1-(3-bromophenyl)-2-iodopent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3h
1-12 (0.2 mmol, 44.4 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3h (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow solid (73.2 mg, 69%), crystallization in CDCl3, mp. 111–113 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.8Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.79 (td, J = 7.8, 1.2 Hz, 1H), 7.74 (td, J = 7.2, 1.2 Hz, 1H), 7.70 (t, J = 1.8 Hz, 1H), 7.59–7.57 (m, 1H), 7.50–7.48 (m, 1H), 7.26–7.25 (m, 1H), 2.58–2.56 (m, 2H), 1.70–1.64 (m, 2H), 0.97 (q, J = 7.8 Hz, 3H).13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 145.4, 143.8, 137.2, 134.5, 133.6, 132.8, 132.7, 129.8, 129.5, 125.9, 123.3, 122.2, 122.0, 99.9, 39.8, 22.3, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C18H15BrINNaO3S+ ([M + Na]+), 553.8893, found, 553.8899.
- (E)-3-((1-(2-fluorophenyl)-2-iodopent-1-en-1-yl)oxy)benzo[d]isothiazole 3i
1-13 (0.2 mmol, 32.4 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3i (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (76.1 mg, 81%), crystallization in CDCl3, mp. 106–108 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.77 (td, J = 7.2, 0.6 Hz, 1H), 7.72 (td, J = 7.8, 0.6 Hz, 1H), 7.67 (td, J = 7.2, 1.8 Hz, 1H), 7.40–7.36 (m, 1H), 7.19 (td, J = 7.8, 0.6 Hz, 1H), 7.09 (t, J = 9.6 Hz, 1H), 2.60 (t, J = 7.2 Hz, 2H), 1.72–1.66 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.2, 159.9 (d, J = 250.0 Hz), 159.1, 143.8, 142.4, 134.4, 133.6 (d, J = 21.0 Hz), 132.0 (d, J = 8.1 Hz), 126.0, 124.0 (d, J = 3.5 Hz), 123.4, 123.3, 122.1, 115.8 (d, J = 21.0 Hz), 102.5, 39.5, 22.2, 12.9. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C18H15FINNaO3S+ ([M + Na]+), 493.9694, found, 493.9677.
- (E)-3-((1-(2,4-difluorophenyl)-2-iodopent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3j
1-14 (0.2 mmol, 36.0 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3j (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (71.6 mg, 73%), crystallization in CDCl3, mp. 109–111 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.79 (td, J = 7.2, 1.2 Hz, 1H), 7.74 (td, J = 7.2, 1.2 Hz, 1H), 7.67 (td, J = 8.4, 6.6 Hz, 1H), 6.93 (td, J = 8.0, 2.4 Hz, 1H), 6.84 (td, J = 8.4, 2.4 Hz, 1H), 2.60 (t, J = 7.2 Hz, 2H), 1.71–1.63 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.2, 164.0 (dd, J = 253.1, 11.9 Hz), 160.5 (dd, J = 253.4, 12.1 Hz), 143.7, 141.6, 135.0 (dd, J = 10.1, 3.2 Hz), 134.5, 133.7, 125.9, 123.4, 122.1, 119.7 (dd, J = 15.1, 3.6 Hz), 111.5 (dd, J = 21.3, 3.5 Hz), 104.3 (t, J = 25.1 Hz), 103.3, 39.4, 22.2, 12.9. 19F NMR (565 MHz, CDCl3) δ −105.54–−105.48 (m, 1F), −107.30–−105.25 (m, 1F). Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C18H14F2INNaO3S+ ([M + Na]+), 511.9599, found, 511.9610.
- (E)-3-((2-iodo-1-(naphthalen-2-yl)pent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3k
1-15 (0.2 mmol, 38.8 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3k (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (65.6 mg, 65%), crystallization in CDCl3, mp. 146–148 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 8.10 (s, 1H), 7.86–7.80 (m, 5H), 7.74–7.70 (m, 2H), 7.67 (dd, J = 8.4, 1.2 Hz, 1H), 7.51–7.47 (m, 2H), 2.62 (t, J = 7.2 Hz, 2H), 1.74–1.68 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.4, 146.9, 143.7, 134.3, 133.6, 133.5, 132.6, 132.5, 130.3, 128.5, 127.9, 127.7, 127.2, 127.1, 126.4, 126.0, 123.3, 122.1, 99.1, 39.9, 22.3, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C22H18INNaO3S+ ([M + Na]+), 525.9944, found, 525.9949.
- (E)-3-((2-iodo-1-(thiophen-2-yl)pent-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3l
1-16 (0.2 mmol, 30.0 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3l (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), browm solid (56.3 mg, 61%), crystallization in CDCl3, mp. 103–105 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.89 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.80 (td, J = 7.8, 1.2 Hz, 1H), 7.76 (td, J = 7.8, 1.2 Hz, 1H), 7.49 (dd, J = 4.2, 1.2 Hz, 1H), 7.38 (dd, J = 4.8, 1.2 Hz, 1H), 7.03 (dd, J = 4.8, 4.2 Hz, 1H), 2.61–2.59 (m, 2H), 1.71–1.64 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.4, 144.0, 141.5, 135.6, 134.5, 133.7, 131.1, 127.9, 126.8, 125.9, 123.3, 122.2, 99.9, 40.7, 22.5, 13.1. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C16H14INNaO3S2+ ([M + Na]+), 481.9352, found, 481.9355.
- (E)-3-((2-iodo-1-phenylprop-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3m
1-17 (0.2 mmol, 42.7 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3m (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (60.9 mg, 72%), crystallization in CDCl3, mp. 154–156 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.85 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.76 (td, J = 7.2, 1.2 Hz, 1H), 7.72 (td, J = 7.8, 1.2 Hz, 1H), 7.61–7.59 (m, 2H), 7.39–7.36 (m, 3H), 2.59 (s, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.2, 147.1, 143.8, 135.0, 134.4, 133.6, 130.1, 129.8, 128.2, 126.0, 123.4, 122.1, 89.0, 27.3. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C16H12INNaO3S+ ([M + Na]+), 447.9475, found, 447.9468.
- (E)-3-((6-chloro-2-iodo-1-(p-tolyl)hex-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3n
1-20 (0.2 mmol, 41.3 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3n (PE/EtOAc = 5:1, Rf = 0.3) wa purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow oil (77.3 mg, 75%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.85 (d, J = 7.2 Hz, 1H), 7.82 (d, J = 7.2 Hz, 1H), 7.77 (td, J = 7.2, 1.2 Hz, 1H), 7.72 (td, J = 7.2, 1.8 Hz, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 7.8 Hz, 2H), 3.55 (t, J = 6.0 Hz, 2H), 2.63–2.62 (m, 2H), 2.35 (s, 3H), 1.84–1.82 (m, 4H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 147.4, 143.8, 130.0, 134.4, 133.6, 132.2, 130.1, 128.9, 126.0, 123.3, 122.1, 97.2, 44.6, 37.3, 31.2, 26.2, 21.4. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C20H19ClINNaO3S+ ([M + Na]+), 537.9711, found, 537.9705.
- (E)-3-((3-chloro-2-iodo-1-phenylprop-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3o
1-21 (0.2 mmol, 30.0 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3o (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (84.4 mg, 92%), crystallization in CDCl3, mp. 146–148 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.88 (t, J = 7.8 Hz, 2H), 7.80–7.79 (m, 1H), 7.76 (dt, J = 7.2, 1.2 Hz, 1H), 7.64–7.62 (m, 2H), 7.41–7.39 (m, 3H), 4.53 (s, 2H).13C NMR {1H} (150 MHz, CDCl3) δ 167.2, 150.5, 143.8, 134.6, 133.8, 133.8, 130.6, 129.9, 128.4, 125.6, 123.5, 122.3, 90.5, 48.3. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C16H11ClINNaO3S2+ ([M + Na]+), 480.9085, found 481.9071.
- (E)-3-((2-cyclopropyl-2-iodo-1-(p-tolyl)vinyl)oxy)benzo[d]isothiazole 1,1-dioxide 3p
1-23 (0.2 mmol, 42.7 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3p (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (80.4 mg, 86%), crystallization in CDCl3, mp. 103–105 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 7.2 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.76 (t, J = 7.2 Hz, 1H), 7.72 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 2H), 7.18 (d, J = 7.8 Hz, 2H), 2.35 (s, 3H), 1.54–1.49 (m, 1H), 0.83 (d, J = 6.6 Hz, 4H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 147.8, 143.9, 139.8, 134.2, 133.5, 132.9, 130.1, 128.9, 126.4, 123.4, 122.1, 103.3, 21.4, 16.9, 10.0. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C19H16INNaO3S+ ([M + Na]+), 487.9788, found, 487.9790.
- (E)-3-((3-ethoxy-2-iodo-1-phenylprop-1-en-1-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3q
1-24 (0.2 mmol, 32.0 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3q (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (80.0 mg, 85%), crystallization in CDCl3, mp. 162–164 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.87 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.78 (td, J = 7.8, 1.2 Hz, 1H), 7.73 (td, J = 7.2, 1.2 Hz, 1H), 7.63–7.60 (m, 2H), 7.41–7.38 (m, 3H), 4.33 (s, 2H), 3.54 (q, J = 7.2 Hz, 2H), 1.20 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.4, 148.9, 143.9, 134.7, 134.5, 133.6, 130.1, 130.0, 128.3, 126.0, 123.3, 122.2, 95.2, 71.9, 66.0, 15.0. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C18H16INNaO4S+ ([M + Na]+), 491.9737, found 491.9750.
- (E)-3-((2-iodo-1,2-diphenylvinyl)oxy)benzo[d]isothiazole 1,1-dioxide 3r
1-25 (0.2 mmol, 35.6 mg) and 2a (0.24 mmol, 74.1 mg) were employed. 3r (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (51.3 mg, 53%), crystallization in CDCl3, mp. 172–174 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.76 (d, J = 7.2 Hz, 3H), 7.66 (t, J = 7.2 Hz, 1H), 7.57 (t, J = 7.2 Hz, 1H), 7.52 (d, J = 7.2 Hz, 1H), 7.48 (d, J = 7.2 Hz, 2H), 7.44–7.40 (m, 3H), 7.28–7.26 (m, 2H), 7.17 (t, J = 7.2 Hz, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.7, 147.5, 143.6, 139.6, 134.8, 134.1, 133.3, 130.2, 130.1, 128.8, 128.42, 128.39, 128.2, 125.9, 123.1, 121.9, 91.2. Mass spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C21H14INNaO3S+ ([M + Na]+), 509.9631, found, 509.9638.
- (E)-3-((4-iodohex-3-en-3-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3s
1-26 (0.24 mmol, 19.7 mg) and 2a (0.2 mmol, 61.8 mg) were employed. 3s (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), yellow oil (51.0 mg, 65%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 7.2 Hz, 1H), 7.82 (t, J = 7.2 Hz, 2H), 7.77 (t, J = 7.2 Hz, 1H), 2.79 (q, J = 7.2 Hz, 2H), 2.42 (q, J = 7.2 Hz, 2H), 1.12 (t, J = 7.2 Hz, 3H), 1.07 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.3, 149.7, 143.9, 134.4, 133.6, 126.1, 123.3, 122.1, 97.8, 31.2, 28.9, 13.9, 10.9. Spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C13H14INNaO3S+ ([M+ Na]+), 413.9631, found, 413.9638.
- (E)-3-((5-iodooct-4-en-4-yl)oxy)benzo[d]isothiazole 1,1-dioxide 3t
1-27 (0.24 mmol, 26.4 mg) and 2a (0.2 mmol, 61.8 mg) were employed. 3t (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (55.9 mg, 67%), crystallization in CDCl3, mp. 58–60 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 7.8 Hz, 1H), 7.83–7.80 (m, 2H), 7.76 (t, J = 7.2 Hz, 1H), 2.78 (t, J = 7.2 Hz, 2H), 2.39 (t, J = 7.2 Hz, 2H), 1.62–1.53 (m, 4H), 0.99 (t, J = 7.2 Hz, 3H), 0.88 (t, J = 7.8 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.1, 149.6, 143.9, 134.4, 133.6, 126.2, 123.2, 122.2, 97.1, 39.2, 36.9, 22.2, 20.1, 13.4, 12.9. Spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C15H18INNaO3S+ ([M+ Na]+), 441.9944, found, 441.9930.
- (E)-3-((1-(4-(tert-butyl)phenyl)-2-iodovinyl)oxy)benzo[d]isothiazole 1,1-dioxide 3u
1-29 (0.24 mmol, 37.9 mg) and 2a (0.2 mmol, 61.8 mg) were employed. 3u (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (56.9 mg, 61%), crystallization in CDCl3, mp. 151–153 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.78 (td, J = 7.2, 1.8 Hz, 1H), 7.74 (td, J = 7.8, 1.2 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 9.0 Hz, 2H), 6.77 (s, 1H), 1.32 (s, 9H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.9, 153.6, 151.7, 143.6, 134.4, 133.6, 129.4, 128.9, 126.5, 125.4, 123.4, 122.1, 68.7, 34.9, 31.1. Mass Spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C19H18INNaO3S+ ([M + Na]+), 489.9944, found, 489.9953.
- (E)-3-((1-([1,1′-biphenyl]-4-yl)-2-iodovinyl)oxy)benzo[d]isothiazole 1,1-dioxide 3v
1-30 (0.24 mmol, 42.7 mg) and 2a (0.2 mmol, 61.8 mg) were employed. 3v (PE/EtOAc = 5:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), white solid (66.1 mg, 68%), crystallization in CDCl3, mp. 161–162 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 7.5 Hz, 1H), 7.86 (d, J = 7.5 Hz, 1H), 7.78–7.74 (m, 4H), 7.64 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 7.9 Hz, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.37 (t, J = 7.2 Hz, 1H), 6.85 (s, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.9, 151.5, 143.6, 143.1, 140.1, 134.4, 133.6, 131.2, 129.6, 128.9, 127.9, 127.2, 127.1, 126.4, 123.4, 122.2, 69.5. Mass Spectrometry: HRMS (ESI-TOF) (m/z): Calcd for C21H14INNaO3S+ ([M + Na]+), 509.9631, found, 509.9640.
- 3-((1-iodo-4-phenylbut-3-yn-2-yl)oxy)benzo[d]isothiazole 1,1-dioxide 4a
1-31 (0.24 mmol, 30.7 mg) and 2a (0.2 mmol, 61.8 mg) were employed. 4a (PE/EtOAc = 5:1, Rf = 0.32) was purified by column chromatography on silica gel (PE/EtOAc = 10:1), colorless oil (36.4 mg, 42%). NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 7.6 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.80 (t, J = 7.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 7.51 (d, J = 6.6 Hz, 2H), 7.41–7.31 (m, 3H), 6.04 (t, J = 5.8 Hz, 1H), 3.73 (d, J = 5.9 Hz, 2H). 13C NMR {1H} (150 MHz, CDCl3) δ 167.91, 143.66, 134.42, 133.61, 132.17, 129.55, 128.44, 126.51, 123.72, 122.10, 120.98, 89.09, 82.67, 71.25, 3.99. HRMS (ESI-TOF) (m/z): C17H12INNaO3S+ Calcd for, ([M + Na]+), 459.9480 found 459.9474.
The reaction tube equipped with a magnetic stir bar was charged with 1-32 (0.2 mmol, 20.8 mg), 1-17 (0.2 mmol, 23.2 mg), 2a (0.2 mmol, 61.8 mg), and DCM (2 mL). The test tube was then sealed off with a screw cap, and the reaction mixture was stirred at room temperature for 6.0 or 24 h. After the reaction was completed, as indicated by TLC analysis, the mixture was extracted with DCM (2 × 10 mL). The combined organic phases were dried over anhydrous Na2SO4, and the solvent was evaporated under a vacuum. The residue was purified by column chromatography (petroleum ether/ethyl acetate 15:1 (v/v)) to give the corresponding product 4b (36.4 mg, 54%) and 4b′ (16.2 mg, 20%).
- 3-(2-iodo-1-phenylethoxy)benzo[d]isothiazole 1,1-dioxide 4b
4b (PE/EtOAc = 5:1, Rf = 0.28), colorless oil. NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 7.91–7.82 (m, 2H), 7.79–7.72 (m, 2H), 7.51–7.38 (m, 5H), 6.28–6.16 (m, 1H), 3.77 (dd, J = 10.8, 7.6 Hz, 1H), 3.66 (dd, J = 10.8, 5.7 Hz, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 168.1, 143.5, 136.1, 134.3, 133.5, 129.7, 128.9, 126.8, 126.7, 123.4, 122.0, 82.7, 5.5. HRMS (ESI-TOF) (m/z): C15H12INNaO3S+ Calcd for, ([M + Na]+), 435.9475 found 435.9485.
- 2-(2-iodo-1-phenylethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide 4b′
4b′ (PE/EtOAc = 5:1, Rf = 0.32), colorless oil. NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 6.8 Hz, 1H), 7.90–7.80 (m, 3H), 7.60 (d, J = 6.6 Hz, 2H), 7.47–7.31 (m, 3H), 5.42 (t, J = 8.2 Hz, 1H), 4.32 (dd, J = 10.5, 8.5 Hz, 1H), 4.03 (dd, J = 10.5, 7.8 Hz, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 158.6, 137.3, 135.6, 134.9, 134.4, 129.2, 128.8, 128.5, 127.0, 125.3, 120.9, 59.1, 2.3. HRMS (ESI-TOF) (m/z): C15H12INNaO3S+ Calcd for, ([M + Na]+), 435.9475 found 435.9468.
4.3. The General Procedure for the Synthesis of 5
In a nitrogen-filled glove box, a flame-dried screw cap reaction tube equipped with a magnetic stir bar was charged with NCBSI (0.4 mmol, 132.4 mg), CHCl3 (3 mL), and 1 (0.2 mmol). The test tube was then sealed off with a screw cap and removed from the glove box, and the reaction mixture was stirred at room temperature for 24 h. After the reaction was completed, as indicated by TLC analysis, the mixture was extracted using DCM (3 × 5.0 mL). The combined organic phases were dried over anhydrous Na2SO4, and the solvent was evaporated under a vacuum. The residue was purified via column chromatography (petroleum ether/ethyl acetate 30:1 (v/v)) to provide the corresponding product 5.
- (E)-2-chloro-1-phenylprop-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5a
1-17 (0.2 mmol, 23.2 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5a (PE/EtOAc = 10:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (76.7 mg, 86%), crystallization in CDCl3, mp. 87–88 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.96 (d, J = 7.2 Hz, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.48–7.44 (m, 3H), 7.25 (d, J = 7.8 Hz, 2H), 7.17–7.14 (m, 2H), 7.13 (t, J = 7.8 Hz, 1H), 7.06 (t, J = 7.8 Hz, 2H), 2.41 (s, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.8, 142.3, 135.9, 134.3, 132.5, 131.1, 129.5, 129.5, 129.0, 128.8, 128.7, 127.9, 127.7, 126.7, 21.8. HRMS (ESI-TOF) (m/z): Calcd for C21H18ClNNaO4S2+, ([M + Na]+), 470.0258, found 470.0265.
- (E)-2-chloro-1-phenylbut-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5b
1-18 (0.2 mmol, 26.0 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5b (PE/EtOAc = 10:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (74.0 mg, 80%), crystallization in CDCl3, mp. 98–99 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.96 (d, J = 7.2 Hz, 2H), 7.60 (d, J = 7.2 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.48–7.44 (m, 3H), 7.27–7.23 (m, 2H), 7.17–7.14 (m, 2H), 7.13 (t, J = 7.2 Hz, 1H), 7.06 (t, J = 7.2 Hz, 2H), 2.89–2.83 (m, 1H), 2.72–2.66 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 142.8, 141.4, 136.0, 135.4, 134.3, 132.5, 131.2, 129.7, 129.0, 128.8, 128.7, 127.9, 127.7, 126.7, 27.3, 11.9. HRMS (ESI-TOF) (m/z): Calcd for C22H20ClNNaO4S2+, ([M + Na]+), 484.0414, found 484.0426.
- (E)-2-chloro-1-phenylpent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5c
1-1 (0.2 mmol, 28.8 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5c (PE/EtOAc = 10:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (76.6 mg, 81%), crystallization in CDCl3, mp. 101–102 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.98–7.94 (m, 2H), 7.59 (dd, J = 8.4, 0.6 Hz, 2H), 7.55–7.51 (m, 1H), 7.48–7.44 (m, 3H), 7.26–7.23 (m, 2H), 7.17–7.14 (m, 2H), 7.14–7.11 (m, 1H), 7.05 (t, J = 7.2 Hz, 2H), 2.78–2.67 (m, 2H), 1.73–1.61 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.9, 142.1, 136.2, 134.3, 134.0, 132.4, 131.2, 129.7, 129.0, 128.8, 128.7, 127.9, 127.7, 126.7, 35.3, 20.5, 13.3. HRMS (ESI-TOF) (m/z): Calcd for C23H22ClNNaO4S2+, ([M + Na]+), 498.0571, found 498.0571.
- (E)-2-chloro-3,3-dimethyl-1-phenylbut-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5d
1-19 (0.2 mmol, 31.6 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5d (PE/EtOAc = 10:1, Rf = 0.33) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (81.5 mg, 83%), crystallization in CDCl3, mp. 101–102 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.91–7.88 (m, 2H), 7.73–7.69 (m, 2H), 7.53–7.50 (m, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.34 (t, J = 7.6 Hz, 2H), 7.26 (d, J = 7.2 Hz, 1H), 7.21 (d, J = 7.2 Hz, 2H), 7.14 (t, J = 7.2 Hz, 2H), 1.02 (s, 9H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.9, 141.9, 140.6, 137.9, 134.0, 132.7, 132.2, 131.2, 129.8, 128.8, 128.5, 127.8, 127.7, 126.8, 38.2, 30.5. HRMS (ESI-TOF) (m/z): Calcd for C24H24ClNNaO4S2+, ([M + Na]+), 512.0727, found 512.0723.
- (E)-2-chloro-2-cyclopropyl-1-phenylvinyl N-phenylsulfonylbenzenesulfonimidate 5e
1-22 (0.2 mmol, 28.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5e (PE/EtOAc = 10:1, Rf = 0.26) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), yellow solid (61.1 mg, 65%), crystallization in CDCl3, mp. 99–100 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.96 (dd, J = 7.8, 1.2 Hz, 2H), 7.65–7.62 (m, 2H), 7.55–7.53 (m, 1H), 7.49–7.45 (m, 3H), 7.26 (t, J = 7.8 Hz, 2H), 7.18–7.15 (m, 2H), 7.13–7.11 (m, 1H), 7.07 (t, J = 7.2 Hz, 2H), 2.64–2.60 (m, 1H), 0.97–0.93 (m, 1H), 0.92–0.86 (m, 2H), 0.82–0.78 (m, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.9, 142.1, 136.1, 135.2, 134.3, 132.4, 131.8, 129.6, 128.8, 128.7, 128.0, 127.7, 126.7, 13.2, 6.5, 6.2. HRMS (ESI-TOF) (m/z): Calcd for C23H20ClNNaO4S2+, ([M + Na]+), 496.0414, found 496.0413.
- (E)-2-chloro-2-cyclopropyl-1-(p-tolyl)vinyl N-(phenylsulfonyl)benzenesulfonimidate 5f
1-23 (0.2 mmol, 31.2 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5f (PE/EtOAc = 10:1, Rf = 0.28) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), yellow oil (56.7 mg, 58%). NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 7.5 Hz, 2H), 7.64 (d, J = 7.5 Hz, 2H), 7.54 (t, J = 7.5 Hz, 1H), 7.51–7.42 (m, 3H), 7.28 (t, J = 7.5 Hz, 2H), 7.05 (d, J = 8.0 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 2.60–2.55 (m, 1H), 2.24 (s, 3H), 0.97–0.81 (m, 3H), 0.80–0.74 (m, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 143.0, 142.3, 138.9, 136.3, 134.6, 134.1, 132.4, 129.6, 129.0, 128.8, 128.7, 128.4, 128.1, 126.8, 21.3, 13.2, 6.5, 6.1. C24H22ClNNaO4S2+ Calcd for, ([M + Na]+), 510.0571, found 510.0588.
- (E)-2-chloro-1-phenylvinyl N-phenylsulfonylbenzenesulfonimidate 5g
1-28 (0.2 mmol, 20.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5g (PE/EtOAc = 10:1, Rf = 0.25) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (69.8 mg, 81%), crystallization in CDCl3, mp. 66–67 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 8.01 (d, J = 7.8 Hz, 2H), 7.72 (d, J = 7.2 Hz, 2H), 7.58–7.53 (m, 2H), 7.50 (t, J = 7.8 Hz, 2H), 7.36–7.30 (m, 4H), 7.23 (t, J = 7.2 Hz, 1H), 7.16 (t, J = 7.8 Hz, 2H), 6.68 (s, 1H). 13C NMR {1H} (150 MHz, CDCl3) δ 147.2, 142.7, 135.2, 134.7, 132.6, 129.8, 129.8, 129.0, 128.8, 128.6, 128.0, 128.0, 126.8, 116.0. HRMS (ESI-TOF) (m/z): Calcd for C20H16ClNNaO4S2+, ([M + Na]+), 456.0101, found 456.0102.
- (E)-2-chloro-1-(p-tolyl)pent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5h
1-2 (0.2 mmol, 31.6 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5h (PE/EtOAc = 10:1, Rf = 0.3) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), colorless liquid (86.2 mg, 88%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.95 (d, J = 7.2 Hz, 2H), 7.60 (dd, J = 8.4, 1.2 Hz, 2H), 7.53 (t, J = 7.2 Hz, 1H), 7.49–7.44 (m, 3H), 7.26 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 7.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 2.72–2.63 (m, 2H), 2.23 (s, 3H), 1.70–1.59 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.9, 142.3, 139.1, 136.4, 134.0, 133.3, 132.4, 129.7, 128.8, 128.7, 128.3, 127.9, 126.7, 35.3, 21.3, 20.5, 13.3. HRMS (ESI-TOF) (m/z): Calcd for C24H24ClNNaO4S2+, ([M + Na]+), 512.0727, found 512.0727.
- (E)-1-([1,1′-biphenyl]-4-yl)-2-chloropent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5i
1-9 (0.2 mmol, 44.0 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5i (PE/EtOAc = 10:1, Rf = 0.32) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (96.7 mg, 88%), crystallization in CDCl3, mp. 99–100 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.2 Hz, 2H), 7.62 (d, J = 7.2 Hz, 2H), 7.53 (t, J = 7.2 Hz, 1H), 7.47–7.42 (m, 7H), 7.38–7.35 (m, 1H), 7.26–7.21 (m, 6H), 2.82–2.71 (m, 2H), 1.76–1.65 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.8, 142.0 141.7, 140.1, 136.3, 134.1, 134.0, 132.5, 130.2, 130.1, 128.9, 128.8, 128.7, 128.0, 127.8, 126.9, 126.7, 126.3, 35.4, 20.5, 13.3. HRMS (ESI-TOF) (m/z): Calcd for C29H26ClNNaO4S2+, ([M + Na]+), 574.0884, found 574.0893.
- (E)-2-chloro-1-(4-chlorophenyl)pent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5j
1-10 (0.2 mmol, 35.6 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5j (PE/EtOAc = 10:1, Rf = 0.32) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (72.7 mg, 71%), crystallization in CDCl3, mp. 104–105 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.94 (d, J = 7.2 Hz, 2H), 7.63 (d, J = 7.2 Hz, 2H), 7.57–7.53 (m, 2H), 7.47 (t, J = 7.8 Hz, 2H), 7.32 (t, J = 7.2 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 2.75–2.63 (m, 2H), 1.69–1.59 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.7, 141.0, 136.2, 135.0, 134.5, 134.4, 132.5, 131.1, 129.8, 129.0, 128.7, 127.9, 127.9, 126.7, 35.3, 20.5, 13.2. HRMS (ESI-TOF) (m/z): Calcd for C23H21Cl2NNaO4S2+, ([M + Na]+), 532.0181, found 532.0185.
- (E)-2-chloro-1-(4-bromophenyl)pent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5k
1-11 (0.2 mmol, 44.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5k (PE/EtOAc = 10:1, Rf = 0.32) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), yellow solid (79.4 mg, 72%), crystallization in CDCl3, mp. 101–102 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.94 (d, J = 7.8 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.47 (t, J = 7.8 Hz, 2H), 7.33 (t, J = 7.8 Hz, 2H), 7.21–7.17 (m, 2H), 7.04 (d, J = 8.4 Hz, 2H), 2.76–2.63 (m, 2H), 1.71–1.59 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.7, 141.1, 136.1, 134.5, 134.4, 132.5, 131.3, 130.9, 130.3, 129.0, 128.7, 127.9, 126.7, 123.4, 35.3, 20.5, 13.2. HRMS (ESI-TOF) (m/z): Calcd for C23H21BrClNNaO4S2+, ([M + Na]+), 575.9676, found 575.9680.
- (E)-tert-butyl 4-(2-chloro-1-((N-(phenylsulfonyl)phenylsulfonimidoyl)oxy)pent-1-en-1-yl)benzoate 5l
1-8 (0.2 mmol, 48.8 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5l (PE/EtOAc = 10:1, Rf = 0.30) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (53.2 mg, 46%), crystallization in CDCl3, mp. 103–104 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.93 (d, J = 7.2 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 7.8 Hz, 2H), 7.56–7.50 (m, 2H), 7.46 (t, J = 7.8 Hz, 2H), 7.32–7.29 (m, 2H), 7.25 (d, J = 8.4 Hz, 2H), 2.73–2.62 (m, 2H), 1.70–1.61 (m, 2H), 1.59 (s, 9H), 0.97 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 164.9, 142.7, 141.2, 136.1, 135.3, 135.0, 134.5, 132.5, 132.2, 129.5, 129.0, 128.7, 128.7, 127.9, 126.7, 81.3, 35.5, 28.1, 20.5, 13.3. HRMS (ESI-TOF) (m/z): Calcd for C28H30ClNNaO6S2+, ([M + Na]+), 598.1095, found 598.1094.
- (E)-2-chloro-1-(4-(trifluoromethyl)phenyl)pent-1-en-1-yl N-phenylsulfonylbenzene sulfonimidate 5m
1-5 (0.2 mmol, 42.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5m (PE/EtOAc = 10:1, Rf = 0.35) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (44.9 mg, 41%), crystallization in CDCl3, mp. 89–90 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.95 (d, J = 7.2 Hz, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.50–7.46 (m, 3H), 7.31–7.25 (m, 6H), 2.80–2.69 (m, 2H), 1.74–1.62 (m, 2H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 142.6, 140.6, 136.0, 135.5, 134.9, 134.6, 132.6, 130.7 (q, J = 32.5 Hz), 130.2, 129.0, 128.8, 127.8, 126.7, 124.6 (q, J = 3.5 Hz), 123.5 (q, J = 270.8 Hz), 35.4, 20.5, 13.3. 19F NMR (565 MHz, CDCl3) δ −63.10. HRMS (ESI-TOF) (m/z): Calcd for C24H21ClF3NNaO4S2+, ([M + Na]+), 566.0445, found 566.0453.
- (E)-2-chloro-1-(4-(trifluoromethoxy)phenyl)pent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5n
1-6 (0.2 mmol, 45.6 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5n (PE/EtOAc = 10:1, Rf = 0.35) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (50.2 mg, 45%), crystallization in CDCl3, mp. 86–87 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.2 Hz, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.49–7.46 (m, 3H), 7.28–7.25 (m, 2H), 7.22–7.17 (m, 2H), 6.88 (d, J = 7.8 Hz, 2H), 2.82–2.71 (m, 2H), 1.73–1.62 (m, 2H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (125 MHz, CDCl3) δ 149.1, 142.7, 140.7, 136.0, 135.0, 134.5, 132.6, 131.5, 130.0, 128.9, 128.7, 127.9, 126.7, 120.2 (q, J = 256.6 Hz), 120.1, 35.4, 20.5, 13.2. 19F NMR (565 MHz, CDCl3) δ −57.65. HRMS (ESI-TOF) (m/z): Calcd for C24H21ClF3NNaO5S2+, ([M + Na]+), 582.0394, found 582.0403.
- (E)-1-(3-bromophenyl)-2-chloropent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5o
1-12 (0.2 mmol, 44.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5o (PE/EtOAc = 10:1, Rf = 0.30) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), colorless liquid (60.1 mg, 54%). NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.2 Hz, 2H), 7.62 (dd, J = 8.4, 1.2 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.52–7.47 (m, 3H), 7.33–7.30 (m, 2H), 7.25–7.22 (m, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.17–7.16 (m, 1H), 6.97 (t, J = 7.8 Hz, 1H), 2.79–2.70 (m, 2H), 1.73–1.62 (m, 2H), 1.00 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 142.7, 140.6, 135.8, 135.2, 134.7, 133.2, 132.6, 132.4, 132.0, 129.2, 128.9, 128.8, 128.5, 127.8, 126.7, 121.7, 35.4, 20.5, 13.3. HRMS (ESI-TOF) (m/z): Calcd for C23H21BrClNNaO4S2+, ([M + Na]+), 575.9676, found 575.9684.
- (E)-2-chloro-1-(2-fluorophenyl)pent-1-en-1-yl N-phenylsulfonylbenzenesulfonimidate 5p
1-13 (0.2 mmol, 32.4 mg) and 2b (0.4 mmol, 132.4 mg) were employed. 5p (PE/EtOAc = 10:1, Rf = 0.35) was purified by column chromatography on silica gel (PE/EtOAc = 30:1), white solid (45.1 mg, 46%), crystallization in CDCl3, mp. 83–84 °C. NMR spectroscopy: 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.2 Hz, 2H), 7.64 (d, J = 7.8 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.47 (t, J = 7.2 Hz, 3H), 7.29–7.26 (m, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.17–7.13 (m, 1H), 6.93 (t, J = 7.2 Hz, 1H), 6.70 (t, J = 8.4 Hz, 1H), 2.78–2.69 (m, 2H), 1.72–1.61 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR {1H} (150 MHz, CDCl3) δ 159.4 (J = 251.1 Hz), 142.8, 136.9 (J = 24.5 Hz), 135.7, 134.3, 132.5, 132.1, 131.6 (J = 8.3 Hz), 130.9 (J = 5.1 Hz), 128.7 (J = 20.4 Hz), 127.8, 126.7, 123.5 (J = 3.5 Hz), 119.4 (J = 14.6 Hz), 115.4 (J = 21.2 Hz), 34.9, 20.5, 13.1. 19F NMR (565 MHz, CDCl3) δ −109.46 (bs). HRMS (ESI-TOF) (m/z): Calcd for C23H21ClFNNaO4S2+, ([M + Na]+), 516.0477, found 516.0494.
5. Conclusions
We achieved the robust trans-selective oxyiodination and oxychlorination of alkynes, employing N-iodosaccharin or N-chlorobenzenesulfonimide as the halogenation and oxygenation sources. This versatile approach tolerates many alkynes, including electron-rich and electron-deficient aryl-, bi-aryl, bi-alkyl, and terminal alkynes. The features of this transformation are as follows: easy operation, excellent functional group tolerance, broad substrate scope, and excellent trans-selectivity. Therefore, this method is an attractive alternative for synthesizing versatile halogenated enol esters and ethers. Employing highly electrophilic bifunctional reagents was key to achieving the general and practical halogenation of alkynes. To the best of our knowledge, this methodology represents the first oxyhalogenation of alkynes employing bifunctional N–X (halogen) reagents. Also, further applications in organic synthesis are ongoing in our lab.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28217420/s1, X-ray crystallographic data of 3i and 5j, 1H, 13C, 19F NMR spectra for all new compounds are included in the Supplementary Materials.
Author Contributions
J.S., Y.G., J.X. and G.Z. performed the experiments. J.S., G.Z. and Q.Z. conceived the concept, directed the project, and wrote the paper. All the authors participated in the analysis of the experimental data. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the Natural Science Foundation of Jilin Province (20230101047JC, YDZJ202201ZYTS338), NSFC (22001157, 21831002, 22193012, and 22201033), Jilin Educational Committee (JJKH20231295KJ, JJKH20231302KJ), and the Fundamental Research Funds for the Central Universities (2412022ZD012, 2412022QD016, 2412021QD007) for generous financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the supplementary materials.
Acknowledgments
The authors are grateful for the support from Northeast Normal University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takahashi, A.; Kirio, Y.; Sodeoka, M.; Sasai, H.; Shibasaki, M. Highly stereoselective synthesis of exocyclic tetrasubstituted enol ethers and olefins. A synthesis of nileprost. J. Am. Chem. Soc. 1989, 111, 643–647. [Google Scholar] [CrossRef]
- Lin, S.; Liu, H.; Svenningsen, E.B.; Wollesen, M.; Jacobsen, K.M.; Andersen, F.D.; Moyano-Villameriel, J.; Pedersen, C.N.; Nørby, P.; Tørring, T.; et al. Expanding the antibacterial selectivity of polyether ionophore antibiotics through diversity-focused semisynthesis. Nat. Chem. 2021, 13, 47–55. [Google Scholar] [CrossRef]
- Igarashi, Y.; Matsuoka, N.; In, Y.; Kataura, T.; Tashiro, E.; Saiki, I.; Sudoh, Y.; Duangmal, K.; Thamchaipenet, A. Nonthmicin, a Polyether Polyketide Bearing a Halogen-Modified Tetronate with Neuroprotective and Antiinvasive Activity from Actinomadura sp. Org. Lett. 2017, 19, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.B. Total Synthesis of Natural Products Containing Enamine or Enol Ether Derivatives. Acc. Chem. Res. 2021, 54, 1830–1842. [Google Scholar] [CrossRef]
- Fischer, P. Enol Ethers—Structure, Synthesis and Reactions. In Patai’s Chemistry of Functional Groups; Patai, S., Ed.; John Wiley & Sons: Chichester, UK, 1980; Volume 2, Chapter 17. [Google Scholar]
- Lempenauer, L.; Lemière, G.; Duñach, E. Cyclisation Reactions Involving Alkyl Enol Ethers. Adv. Synth. Catal. 2019, 361, 5284–5304. [Google Scholar] [CrossRef]
- Hansen, D.W., Jr.; Pappo, R.; Garland, R.B. A stereospecific total synthesis of the anthracyclinones (.+-.)-daunomycinone and (.+-.)-isodaunomycinone. J. Org. Chem. 1988, 53, 4244–4253. [Google Scholar] [CrossRef]
- Duan, H.; Sun, X.; Liao, W.; Petersen, J.L.; Shi, X. Proline as Lewis Base Catalyst: Diastereoselective Synthesis of Isoxazoline-N-oxide through [3 + 2] Cycloaddition. Org. Lett. 2008, 10, 4113–4116. [Google Scholar] [CrossRef]
- Tang, W.; Liu, D.; Zhang, X. Asymmetric Hydrogenation of Itaconic Acid and Enol Acetate Derivatives with the Rh-TangPhos Catalyst. Org. Lett. 2003, 5, 205–207. [Google Scholar] [CrossRef]
- Behenna, D.C.; Stoltz, B.M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef]
- Trost, B.M.; Xu, J. Palladium-Catalyzed Asymmetric Allylic α-Alkylation of Acyclic Ketones. J. Am. Chem. Soc. 2005, 127, 17180–17181. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodriguez, A.; Campos, P.J. Synthesis of 2-Functionalized 1,1-Diiodo-1 Alkenes. Generation and Reactions of 1-Iodo-1-Lithio-1 Alkenes and 1,1-Dilithio-1-Alkenes. J. Am. Chem. Soc. 1988, 110, 5567–5568. [Google Scholar] [CrossRef]
- Reppe, W. Vinylation. 1. Vinyl Ethers and Vinyl Esters. Liebigs Ann. Chem. 1956, 601, 84–111. [Google Scholar]
- Agarwal, V.; Miles, Z.D.; Winter, J.M.; Eustáquio, A.S.; El Gamal, A.A.; Moore, B.S. Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev. 2017, 117, 5619–5674. [Google Scholar] [CrossRef] [PubMed]
- Varenikov, A.; Shapiro, E.; Gandelman, M. Decarboxylative Halogenation of Organic Compounds. Chem. Rev. 2021, 121, 412–484. [Google Scholar] [CrossRef] [PubMed]
- China, H.; Kumar, R.; Kikushima, K.; Dohi, T. Halogen-Induced Controllable Cyclizations as Diverse Heterocycle Synthetic Strategy. Molecules 2020, 25, 6007. [Google Scholar] [CrossRef]
- Dohi, T. Recent Topics in Organohalogen Reagents and Compounds. Curr. Org. Chem. 2020, 24, 2029–2030. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Jothish, S.; Dohi, T.; Singh, F.V. Iodine(V)-Based Oxidants in Oxidation Reactions. Molecules 2023, 28, 5250. [Google Scholar] [CrossRef]
- Song, S.; Li, X.; Wei, J.; Wang, W.; Zhang, Y.; Ai, L.; Zhu, Y.; Shi, X.; Zhang, X.; Jiao, N. DMSO-catalysed late-stage chlorination of (hetero)arenes. Nat. Catal. 2020, 3, 107–115. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Dai, R.; Yan, Z.; Wei, J.; Dou, X.; Qiu, X.; Zhang, H.; Wang, C.; Liu, Y.; et al. Catalytic Electrophilic Halogenation of Arenes with Electron-Withdrawing Substituents. J. Am. Chem. Soc. 2022, 144, 13415–13425. [Google Scholar] [CrossRef]
- Wang, W.; Wang, H.; Dai, R.; Wang, Y.; Li, Z.; Yang, X.; Lu, B.; Jiao, N.; Song, S. Organocatalytic Deoxyhalogenation of Alcohols with Inorganic Halides. ACS Catal. 2023, 13, 9033–9040. [Google Scholar] [CrossRef]
- Kutsumura, N.; Niwa, K.; Saito, T. Novel One-Pot Method for Chemoselective Bromination and Sequential Sonogashira Coupling. Org. Lett. 2010, 12, 3316. [Google Scholar] [CrossRef] [PubMed]
- Reiser, O. Palladium-Catalyzed Coupling Reactions for the Stereoselective Synthesis of Tri- and Tetrasubstituted Alkenes. Angew. Chem. Int. Ed. 2006, 45, 2838. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Shimizu, M.; Hiyama, T. Straightforward Synthesis of CF3-Substituted Triarylethenes by Stereoselective Threefold Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2007, 46, 8659. [Google Scholar] [CrossRef]
- Kitamura, T.; Furuki, R.; Taniguchi, H.; Stang, P.J. Electrophilic additions of iodosylbenzene activated by trifluoromethanesulfonic acid, [PhIO-TfOH], to alkynes. Tetrahedron 1992, 48, 7149–7156. [Google Scholar] [CrossRef]
- Muraki, T.; Togo, H.; Yokoyama, M. Reactivity and Synthetic Utility of 1-(Arenesulfonyloxy)benziodoxolones. J. Org. Chem. 1999, 64, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, J.; Jiang, H.; Zhu, S.; Li, Y.; Qi, C. Silver-Catalyzed Difunctionalization of Terminal Alkynes: Highly Regio- and Stereoselective Synthesis of (Z)-β-Haloenol Acetates. Org. Lett. 2010, 12, 3262–3265. [Google Scholar] [CrossRef]
- Okamoto, N.; Miwa, Y.; Minami, H.; Takeda, K.; Yanada, R. Regio- and Stereoselective Multisubstituted Enol Ester Synthesis. J. Org. Chem. 2011, 76, 9133–9138. [Google Scholar] [CrossRef]
- Priebbenow, D.L.; Gable, R.W.; Baell, J. Regio- and Stereoselective Iodoacyloxylations of Alkynes. J. Org. Chem. 2015, 80, 4412–4418. [Google Scholar] [CrossRef]
- Ding, W.; Chai, J.; Wang, C.; Wu, J.; Yoshikai, N. Stereoselective Access to Highly Substituted Vinyl Ethers via trans-Difunctionalization of Alkynes with Alcohols and Iodine(III) Electrophile. J. Am. Chem. Soc. 2020, 142, 8619–8624. [Google Scholar] [CrossRef]
- Kikuchi, J.; Maesaki, K.; Sasaki, S.; Wang, W.; Ito, S.; Yoshikai, N. Stereoselective Synthesis of β-Alkoxy-β-amido Vinylbenziodoxoles via Iodo(III)etherification of Ynamides. Org. Lett. 2022, 24, 6914–6918. [Google Scholar] [CrossRef]
- Liu, M.; Sun, J.; Zhang, T.; Ding, Y.; Han, Y.-Q.; Martín-Montero, R.; Lan, Y.; Shi, B.-F.; Engle, K.M. Regio- and Stereoselective 1,2-Oxyhalogenation of Non-Conjugated Alkynes via Directed Nucleopalladation: Catalytic Access to Tetrasubstituted Alkenes**. Angew. Chem. Int. Ed. 2022, 61, e202209099. [Google Scholar] [CrossRef] [PubMed]
- Chemler, S.R.; Bovino, M.T. Catalytic Aminohalogenation of Alkenes and Alkynes. ACS Catal. 2013, 3, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Wille, U.; Krüger, O.; Kirsch, A.; Lüning, U. Radical Addition of N-Bromophthalimide to Linear and Cyclic Alkynes. Eur. J. Org. Chem. 1999, 1999, 3185–3189. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Gao, P.; Shen, Y.-W.; Liang, Y.-M. Copper-Catalyzed Chloroamination of Alkynes: Highly Regio- and Stereoselective Synthesis of (E)-β-Chloro-Enesulfonamides. Adv. Syn. Catal. 2011, 353, 3157–3160. [Google Scholar] [CrossRef]
- Li, M.; Yuan, H.; Zhao, B.; Liang, F.; Zhang, J. Alkyne aminohalogenation enabled by DBU-activated N-haloimides: Direct synthesis of halogenated enamines. Chem. Commun. 2014, 50, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- Dolenc, D. N-Iodosaccharin—A New Reagent for Iodination of Alkenes and Activated Aromatics. Synlett 2000, 2000, 544–546. [Google Scholar]
- Aloui, M.; Fairbanks, A.J. N-Iodosaccharin: A Potent New Activator of Thiophenylglycosides. Synlett 2001, 2001, 0797–0799. [Google Scholar] [CrossRef]
- Dolenc, D. Iodination of Enol Acetates and 1,3-Diones Using N-Iodosaccharin. Synth. Commun. 2003, 33, 2917–2924. [Google Scholar] [CrossRef]
- Bailey, L.; Handy, S.T. Aromatic iodination using N-iodosaccharin in room temperature ionic liquids. Tetrahedron Lett. 2011, 52, 2413–2414. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, X.; Li, J.; Chen, W.; Wang, W.; Lin, L.; Feng, X. Asymmetric Iodoamination of Chalcones and 4-Aryl-4-oxobutenoates Catalyzed by a Complex Based on Scandium(III) and a N,N′-Dioxide Ligand. Chem. Eur. J. 2011, 17, 14916–14921. [Google Scholar] [CrossRef]
- Dutta, H.S.; Khan, B.; Khan, A.A.; Raziullah; Ahmad, A.; Kant, R.; Koley, D. Metal-Free, Oxidant-Free, Site-Selective C–H Halogenations to Aminoquinolines at Room Temperature using N-Halosaccharins. ChemistrySelect 2017, 2, 6488–6492. [Google Scholar] [CrossRef]
- Davis, F.A.; Towson, J.C.; Vashi, D.B.; ThimmaReddy, R.; McCauley, J.P., Jr.; Harakal, M.E.; Gosciniak, D.J. Chemistry of oxaziridines. 13. Synthesis, reactions, and properties of 3-substituted 1,2-benzisothiazole 1,1-dioxide oxides. J. Org. Chem. 1990, 55, 1254–1261. [Google Scholar] [CrossRef]
- Fukuzumi, T.; Bode, J.W. A Reagent for the Convenient, Solid-Phase Synthesis of N-Terminal Peptide Hydroxylamines for Chemoselective Ligations. J. Am. Chem. Soc. 2009, 131, 3864–3865. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Zhao, J.; Li, Z.; Zhang, Q.; Sun, J.; Sun, H.; Zhang, Q. Highly Regio- and Stereoselective Intermolecular Seleno- and Thioamination of Alkynes. Chem. Eur. J. 2016, 22, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, R.; Sun, J.; Zheng, G.; Zhang, Q. NHC and visible light-mediated photoredox co-catalyzed 1,4-sulfonylacylation of 1,3-enynes for tetrasubstituted allenyl ketones. Chem. Sci. 2022, 13, 3169–3175. [Google Scholar] [CrossRef]
- Wang, L.; Ma, R.; Xia, J.; Liu, X.; Sun, J.; Zheng, G.; Zhang, Q. DBU-Mediated Isomerization/6-pi Electro-Cyclization/Oxidation Cascade of Sulfonyl-Substituted Allenyl Ketones for the Construction of Hetero-1,3,5-Trisubstituted Benzene. Chem. Eur. J. 2023, 29, e202203309. [Google Scholar] [CrossRef]
- Wang, L.; Sun, J.; Xia, J.; Li, M.; Zhang, L.; Ma, R.; Zheng, G.; Zhang, Q. Visible light-mediated NHCs and photoredox co-catalyzed radical 1,2-dicarbonylation of alkenes for 1,4-diketones. Sci. China Chem. 2022, 65, 1938–1944. [Google Scholar] [CrossRef]
- Wu, Y.; Li, M.; Sun, J.; Zheng, G.; Zhang, Q. Synthesis of Axially Chiral Aldehydes by N-Heterocyclic-Carbene-Catalyzed Desymmetrization Followed by Kinetic Resolution. Angew. Chem. Int. Ed. 2022, 61, e202117340. [Google Scholar] [CrossRef]
- Wang, L.; Sun, J.; Xia, J.; Ma, R.; Zheng, G.; Zhang, Q. Visible light-mediated NHC and photoredox co-catalyzed 1,2-sulfonylacylation of allenes via acyl and allyl radical cross-coupling. Org. Chem. Front. 2023, 10, 1047–1055. [Google Scholar] [CrossRef]
- Liang, T.; Wu, Y.; Sun, J.; Li, M.; Zhao, H.; Zhang, J.; Zheng, G.; Zhang, Q. Visible Light-Mediated Cobalt and Photoredox Dual-Catalyzed Asymmetric Reductive Coupling for Axially Chiral Secondary Alcohols. Chin. J. Chem. 2023, 41, 3253–3260. [Google Scholar] [CrossRef]
- Sun, J.; Wang, L.; Zheng, G.; Zhang, Q. Recent advances in three-component radical acylative difunctionalization of unsaturated carbon–carbon bonds. Org. Chem. Front. 2023, 10, 4488–4515. [Google Scholar] [CrossRef]
- Palav, A.; Misal, B.; Ganwir, P.; Badani, P.; Chaturbhuj, G. Rapid, chemoselective and mild oxidation protocol for alcohols and ethers with recyclable N-chloro-N-(phenylsulfonyl)benzenesulfonamide. Tetrahedron Lett. 2021, 73, 153094. [Google Scholar] [CrossRef]
- Palav, A.; Misal, B.; Chaturbhuj, G. NCBSI/KI: A Reagent System for Iodination of Aromatics through In Situ Generation of I-Cl. J. Org. Chem. 2021, 86, 12467–12474. [Google Scholar] [CrossRef] [PubMed]
- Misal, B.; Palav, A.; Ganwir, P.; Chaturbhuj, G. Activator free, expeditious and eco-friendly chlorination of activated arenes by N-chloro-N-(phenylsulfonyl)benzene sulfonamide (NCBSI). Tetrahedron Lett. 2021, 63, 152689. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).