
Molecular Simulation and Statistical Learning Methods toward Predicting Drug–Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design

 and
and
Abstract
:1. Introduction
2. Theoretical Background
2.1. Solubility Parameters, δ
2.2. Flory–Huggins (FH) Interaction Parameter, χ
3. Molecular Modeling Approaches
3.1. QM Calculations for Elucidating Non-Bonding Interactions
3.2. Molecular Mechanics and Molecular Dynamics Methods
3.2.1. Overview of Molecular Dynamics
3.2.2. Docking Studies of API with Polymer Carrier
3.2.3. Theory of MD-Derived Solubility and FH Interaction Parameters
3.2.4. Applications of MD-Derived Solubility and FH Interaction Parameters
3.3. Mechanistic Insights from Molecular Modeling
4. Machine Learning Approaches
4.1. Overview of Machine Learning
4.2. ML Applications to General ASD Systems
4.3. ML Models of ASD Properties and Phenomena
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.M.; Dressman, J.B. The developability classification system: Application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 2010, 99, 4940–4954. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Walker, M.A. Novel tactics for designing water-soluble molecules in drug discovery. Expert Opin. Drug Discov. 2014, 9, 1421–1433. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Palucki, M.; Higgins, J.D.; Kwong, E.; Templeton, A.C. Strategies at the interface of drug discovery and development: Early optimization of the solid state phase and preclinical toxicology formulation for potential drug candidates. J. Med. Chem. 2010, 53, 5897–5905. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, W.W., 3rd; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in polymer design for enhancing oral drug solubility and delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharm. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Rasenack, N.; Muller, B.W. Micron-size drug particles: Common and novel micronization techniques. Pharm. Dev. Technol. 2004, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Aaltonen, J.; Tian, F.; Saville, D.J.; Rades, T. Influence of particle size and preparation methods on the physical and chemical stability of amorphous simvastatin. Eur. J. Pharm. Biopharm. 2009, 71, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems–An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, H.; Bala, R.; Arora, S. Lipid-Based drug delivery systems. J. Pharm. 2014, 2014, 801820. [Google Scholar] [CrossRef] [Green Version]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef] [Green Version]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Dai, W.G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharm. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Crystallization of amorphous solid dispersions of resveratrol during preparation and storage-Impact of different polymers. J. Pharm. Sci. 2013, 102, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Van Zee, N.J.; Hillmyer, M.A.; Lodge, T.P. Role of polymer excipients in the kinetic stabilization of drug-rich nanoparticles. ACS Appl. Bio Mater. 2020, 3, 7243–7254. [Google Scholar] [CrossRef]
- Ma, X.; Williams, R.O. Characterization of amorphous solid dispersions: An update. J. Drug Deliv. Sci. Technol. 2019, 50, 113–124. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent advances in understanding the micro-and nanoscale phenomena of amorphous solid dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef]
- Shi, Q.; Li, F.; Yeh, S.; Wang, Y.; Xin, J. Physical stability of amorphous pharmaceutical solids: Nucleation, crystal growth, phase separation and effects of the polymers. Int. J. Pharm. 2020, 590, 119925. [Google Scholar] [CrossRef]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Yang, F.; Su, Y.; Small, J.; Huang, C.; Martin, G.E.; Farrington, A.M.; DiNunzio, J.; Brown, C.D. Probing the molecular-level interactions in an active pharmaceutical ingredient (API)–Polymer dispersion and the resulting impact on drug product formulation. Pharm. Res. 2020, 37, 94. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Han, R.; Chen, W.; Zhang, W.; Li, Y.; Ji, Y.; Chen, L.; Pan, H.; Yang, X.; Pan, W.; et al. Analysis of the literature and patents on solid dispersions from 1980 to 2015. Molecules 2018, 23, 1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Johnston, B.F.; Mackay, S.P.; Schatzlein, A.G.; Gellert, P.; Sengupta, D.; Uchegbu, I.F. In silico modelling of drug-polymer interactions for pharmaceutical formulations. J. R. Soc. Interface 2010, 7, S423–S433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, S.; Kabedev, A.; Parrow, A.; Bergstrom, C.A.S.; Larsson, P. Molecular simulation as a computational pharmaceutics tool to predict drug solubility, solubilization processes and partitioning. Eur. J. Pharm. Biopharm. 2019, 137, 46–55. [Google Scholar] [CrossRef]
- Das, T.; Mehta, C.H.; Nayak, U.Y. Multiple approaches for achieving drug solubility: An in silico perspective. Drug Discov. Today 2020, 25, 1206–1212. [Google Scholar] [CrossRef]
- DeBoyace, K.; Wildfong, P.L.D. The application of modeling and prediction to the formation and stability of amorphous solid dispersions. J. Pharm. Sci. 2018, 107, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Hancock, B.C.; York, P.; Rowe, R.C. The use of solubility parameters in pharmaceutical dosage form design. Int. J. Pharm. 1997, 148, 1–21. [Google Scholar] [CrossRef]
- Mollet, H.; Grubenmann, A. Chapter 8: Solubility parameters, log, P., LSER, M numbers. In Formulation Technology: Emulsions, Suspensions, Solid Forms; Wiley-VCH Verlag GMBH: Weinheim, Germany, 2000; pp. 265–294. [Google Scholar]
- Li, C.; Strachan, A. Cohesive energy density and solubility parameter evolution during the curing of thermoset. Polymer 2018, 135, 162–170. [Google Scholar] [CrossRef]
- Ahmad, H.; Yaseen, M. Application of a chemical group contribution technique for calculating solubility parameters of polymers. Polym. Eng. Sci. 1979, 19, 858–863. [Google Scholar] [CrossRef]
- Fedors, R. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Krevelen-Van, D.W.; Hoftyzer, P.J. Their Correlation with Chemical Structure, Their Numerical Estimation and Prediction from Additive Group Contributions; Elsevier: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Telang, C.; Mujumdar, S.; Mathew, M. Improved physical stability of amorphous state through acid base interactions. J. Pharm. Sci. 2009, 98, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Maniruzzaman, M.; Morgan, D.J.; Mendham, A.P.; Pang, J.; Snowden, M.J.; Douroumis, D. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions. Int. J. Pharm. 2013, 443, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Pang, J.; Morgan, D.J.; Douroumis, D. Molecular modeling as a predictive tool for the development of solid dispersions. Mol. Pharm. 2015, 12, 1040–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, H.; Mo, H.; Zhang, M.; Song, Y.; Fang, K.; Taylor, L.S.; Li, T.; Byrn, S.R. Investigating the interaction pattern and structural elements of a drug-polymer complex at the molecular level. Mol. Pharm. 2015, 12, 2459–2468. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Barmpalexis, P.; Karagianni, A.; Katopodis, K.; Vardaka, E.; Kachrimanis, K. Molecular modelling and simulation of fusion-based amorphous drug dispersions in polymer/plasticizer blends. Eur. J. Pharm. Sci. 2019, 130, 260–268. [Google Scholar] [CrossRef]
- Shenogin, S.; Ozisik, R. Xenoview: Visualization for Atomistic Simulations. 2004. Available online: http://www.vemmer.org/xenoview/xenoview.html (accessed on 5 November 2020).
- Kapourani, A.; Chatzitheodoridou, M.; Kontogiannopoulos, K.N.; Barmpalexis, P. Experimental, thermodynamic, and molecular modeling evaluation of amorphous simvastatin-poly(vinylpyrrolidone) solid dispersions. Mol. Pharm. 2020, 17, 2703–2720. [Google Scholar] [CrossRef]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar]
- Mosquera-Giraldo, L.I.; Borca, C.H.; Meng, X.; Edgar, K.J.; Slipchenko, L.V.; Taylor, L.S. Mechanistic design of chemically diverse polymers with applications in oral drug delivery. Biomacromolecules 2016, 17, 3659–3671. [Google Scholar] [CrossRef]
- Biovia. Biovia Materials Studio: An Integrated, Multi-Scale Modeling Environment. 2020. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 5 November 2020).
- Erlebach, A.; Ott, T.; Otzen, C.; Schubert, S.; Czaplewska, J.; Schubert, U.S.; Sierka, M. Thermodynamic compatibility of actives encapsulated into PEG-PLA nanoparticles: In Silico predictions and experimental verification. J. Comput. Chem. 2016, 37, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Iesavand, H.; Rahmati, M.; Afzali, D.; Modiri, S. Investigation on absorption and release of mercaptopurine anticancer drug from modified polylactic acid as polymer carrier by molecular dynamic simulation. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 105, 110010. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.K.; Larson, R.G. Assessing the efficiency of polymeric excipients by atomistic molecular dynamics simulations. Mol. Pharm. 2014, 11, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Macháčková, M.; Tokarsky, J.; Capkova, P. A simple molecular modeling method for the characterization of polymeric drug carriers. Eur. J. Pharm. Sci. 2013, 48, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razmimanesh, F.; Amjad-Iranagh, S.; Modarress, H. Molecular dynamics simulation study of chitosan and gemcitabine as a drug delivery system. J. Mol. Model. 2015, 21, 165. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, Y.; Sohail, S.; Arshad, M.S.; Hussain, T.; Shah, S.N. Development of solid dispersions of artemisinin for transdermal delivery. Int. J. Pharm. 2013, 457, 197–205. [Google Scholar] [CrossRef]
- Yani, Y.; Kanaujia, P.; Chow, P.S.; Tan, R.B.H. Effect of API-Polymer miscibility and interaction on the stabilization of amorphous solid dispersion: A molecular simulation study. Ind. Eng. Chem. Res. 2017, 56, 12698–12707. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wires Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Chan, T.; Ouyang, D. Investigating the molecular dissolution process of binary solid dispersions by molecular dynamics simulations. Asian J. Pharm. Sci. 2018, 13, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Huang, T.; Liu, X.; Yin, X.; Li, H.; Lu, J.; Ji, Y.; Sun, H.; Ouyang, D. Insight into the dissolution molecular mechanism of ternary solid dispersions by combined experiments and molecular simulations. AAPS Pharmscitech 2019, 20, 274. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.X.; Anderson, B.D. Molecular dynamics simulation of amorphous indomethacin-poly(vinylpyrrolidone) glasses: Solubility and hydrogen bonding interactions. J. Pharm. Sci. 2013, 102, 876–891. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.X.; Anderson, B.D. Molecular dynamics simulation of amorphous hydroxypropylmethylcellulose and its mixtures with felodipine and water. J. Pharm. Sci. 2017, 106, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Brunsteiner, M.; Khinast, J.; Paudel, A. Relative contributions of solubility and mobility to the stability of amorphous solid dispersions of poorly soluble drugs: A molecular dynamics simulation study. Pharmaceutics 2018, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Eslami, M.; Nikkhah, S.J.; Hashemianzadeh, S.M.; Sajadi, S.A. The compatibility of Tacrine molecule with poly(n-butylcyanoacrylate) and Chitosan as efficient carriers for drug delivery: A molecular dynamics study. Eur. J. Pharm. Sci. 2016, 82, 79–85. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Olsen, K.W. Drug-polymer interactions at water-crystal interfaces and implications for crystallization inhibition: Molecular dynamics simulations of amphiphilic block copolymer interactions with tolazamide crystals. J. Pharm. Sci. 2015, 104, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2020.
- Fule, R.; Meer, T.; Sav, A.; Amin, P. Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation. J. Pharm. Investig. 2013, 43, 305–321. [Google Scholar] [CrossRef]
- Gangurde, A.B.; Kundaikar, H.S.; Javeer, S.D.; Jaiswar, D.R.; Degani, M.S.; Amin, P.D. Enhanced solubility and dissolution of curcumin by a hydrophilic polymer solid dispersion and its insilico molecular modeling studies. J. Drug Deliv. Sci. Technol. 2015, 29, 226–237. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2020; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020.
- Jadhav, P.; Gokarna, V.; Deshpande, V.; Vavia, P. bioavailability enhancement of olmesartan medoxomil using hot-melt extrusion: In-silico, in-vitro, and in-vivo evaluation. AAPS Pharmscitech 2020, 21, 254. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.H.; Szeleszczuk, L.; Pisklak, D.M. Periodic DFT calculations-review of applications in the pharmaceutical sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, W.; Curioni, A.; Mordasini, T. DFT-based molecular dynamics as a new tool for computational biology: First applications and perspective. IBM J. Res. Dev. 2001, 45, 397–407. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, A.; Szeleszczuk, L.; Pisklak, D.M. Can we predict the pressure induced phase transition of urea? Application of quantum molecular dynamics. Molecules 2020, 25, 1584. [Google Scholar] [CrossRef] [Green Version]
- Van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Meng, F.; Trivino, A.; Prasad, D.; Chauhan, H. Investigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersions. Eur. J. Pharm. Sci. 2015, 71, 12–24. [Google Scholar] [CrossRef]
- Wang, B.; Wang, D.; Zhao, S.; Huang, X.; Zhang, J.; Lv, Y.; Liu, X.; Lv, G.; Ma, X. Evaluate the ability of PVP to inhibit crystallization of amorphous solid dispersions by density functional theory and experimental verify. Eur. J. Pharm. Sci. 2017, 96, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Bookwala, M.; DeBoyace, K.; Buckner, I.S.; Wildfong, P.L.D. Predicting density of amorphous solid materials using molecular dynamics simulation. AAPS Pharmscitech 2020, 21, 96. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.X.; Anderson, B.D. Molecular dynamics simulation of amorphous indomethacin. Mol. Pharm. 2013, 10, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Kasimova, A.O.; Pavan, G.M.; Danani, A.; Mondon, K.; Cristiani, A.; Scapozza, L.; Gurny, R.; Moller, M. Validation of a novel molecular dynamics simulation approach for lipophilic drug incorporation into polymer micelles. J. Phys. Chem. B 2012, 116, 4338–4345. [Google Scholar] [CrossRef]
- Chen, S.; Li, J.; Wei, L.; Jin, Y.; Khosla, T.; Xiao, J.; Cheng, B.; Duan, H. A molecular modeling study for miscibility of polyimide/polythene mixing systems with/without compatibilizer. J. Polym. Eng. 2018, 38, 891–898. [Google Scholar] [CrossRef]
- Andrews, A.; Handler, R.A.; Blaisten-Barojas, E. Structure, energetics and thermodynamics of PLGA condensed phases from Molecular Dynamics. Polymer 2020, 206, 122903. [Google Scholar] [CrossRef]
- Muljajew, I.; Erlebach, A.; Weber, C.; Buchheim, J.R.; Sierka, M.; Schubert, U.S. A polyesteramide library from dicarboxylic acids and 2,2′-bis(2-oxazoline): Synthesis, characterization, nanoparticle formulation and molecular dynamics simulations. Polym. Chem. 2020, 11, 112–124. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. Molecular dynamics simulation of amorphous hydroxypropyl-methylcellulose acetate succinate (HPMCAS): Polymer model development, water distribution, and plasticization. Mol. Pharm. 2014, 11, 2400–2411. [Google Scholar] [CrossRef]
- Adhikari, U.; Goliaei, A.; Tsereteli, L.; Berkowitz, M.L. Properties of poloxamer molecules and poloxamer micelles dissolved in water and next to lipid bilayers: Results from computer simulations. J. Phys. Chem. B 2016, 120, 5823–5830. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Jonnalagadda, S. A molecular dynamics approach for predicting the glass transition temperature and plasticization effect in amorphous pharmaceuticals. Mol. Pharm. 2013, 10, 4136–4145. [Google Scholar] [CrossRef]
- Ma, S.M.; Zhao, L.; Wang, Y.L.; Zhu, Y.L.; Lu, Z.Y. The coarse-grained models of poly(ethylene oxide) and poly(propylene oxide) homopolymers and poloxamers in big multipole water (BMW) and MARTINI frameworks. Phys. Chem. Chem. Phys. 2020, 22, 15976–15985. [Google Scholar] [CrossRef] [PubMed]
- Rigby, D.; Sun, H.; Eichinger, B.E. Computer simulations of poly(ethylene oxide): Force field, pvt diagram and cyclization behaviour. Polym. Int. 1997, 44, 311–330. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applicationsoverview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Maple, J.R.; Dinur, U.; Hagler, A.T. Derivation of force fields for molecular mechanics and dynamics from ab initio energy surfaces. Proc. Natl. Acad. Sci. USA 1988, 85, 5350–5354. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Suryanarayanan, R. Local mobility in amorphous pharmaceuticals–Characterization and implications on stability. J. Pharm. Sci. 2009, 98, 2935–2953. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Influence of molecular mobility on the physical stability of amorphous pharmaceuticals in the supercooled and glassy States. Mol. Pharm. 2014, 11, 3048–3055. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Taskinen, M.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory-Huggins interaction parameter and phase diagram. Mol. Pharm. 2010, 7, 795–804. [Google Scholar] [CrossRef]
- Turpin, E.R.; Taresco, V.; Al-Hachami, W.A.; Booth, J.; Treacher, K.; Tomasi, S.; Alexander, C.; Burley, J.; Laughton, C.A.; Garnett, M.C. In silico screening for solid dispersions: The trouble with solubility parameters and chiFH. Mol. Pharm. 2018, 15, 4654–4667. [Google Scholar] [CrossRef]
- Paus, R.; Prudic, A.; Ji, Y. Influence of excipients on solubility and dissolution of pharmaceuticals. Int. J. Pharm. 2015, 485, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.; Duncan, Q. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Puncochova, K.; Ewing, A.V.; Gajdosova, M.; Sarvasova, N.; Kazarian, S.G.; Beranek, J.; Stepanek, F. Identifying the mechanisms of drug release from amorphous solid dispersions using MRI and ATR-FTIR spectroscopic imaging. Int. J. Pharm. 2015, 483, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Gao, W.; Taylor, L.S. Impact of Eudragit EPO and hydroxypropyl methylcellulose on drug release rate, supersaturation, precipitation outcome and redissolution rate of indomethacin amorphous solid dispersions. Int. J. Pharm. 2017, 531, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Bhugra, C.; Pikal, M.J. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J. Pharm. Sci. 2008, 97, 1329–1349. [Google Scholar] [CrossRef]
- Kalinin, A.A.; Higgins, G.A.; Reamaroon, N.; Soroushmehr, S.; Allyn-Feuer, A.; Dinov, I.D.; Najarian, K.; Athey, B.D. Deep learning in pharmacogenomics: From gene regulation to patient stratification. Pharmacogenomics 2018, 19, 629–650. [Google Scholar] [CrossRef]
- Kastrin, A.; Ferk, P.; Leskosek, B. Predicting potential drug-drug interactions on topological and semantic similarity features using statistical learning. PLoS ONE 2018, 13, e0196865. [Google Scholar] [CrossRef]
- Zuvela, P.; David, J.; Wong, M.W. Interpretation of ANN-based QSAR models for prediction of antioxidant activity of flavonoids. J. Comput. Chem. 2018, 39, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Alberi, K.; Nardelli, M.B.; Zakutayev, A.; Mitas, L.; Curtarolo, S.; Jain, A.; Fornari, M.; Marzari, N.; Takeuchi, I.; Green, M.L.; et al. The 2019 materials by design roadmap. J. Phys. D: Appl. Phys. 2019, 52, 013001. [Google Scholar] [CrossRef]
- Goldman, B.B.; Walters, W.P. Chapter 8. Machine learning in computational chemistry. In Annual Reports in Computational Chemistry; Spellmeyer, D.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 127–140. [Google Scholar]
- Mendyk, A.; Szlȩk, J.; Jachowicz, R. 3-ME_expert 2.0: A heuristic decision support system for microemulsions formulation development. In Formulation Tools for Pharmaceutical Development; Aguilar, J.E., Ed.; Woodhead Publishing: Cambridge, UK, 2013; pp. 39–71. [Google Scholar] [CrossRef]
- Rowe, R.C.; Roberts, R.J. Artificial intelligence in pharmaceutical product formulation: Knowledge-based and expert systems. Pharm. Sci. Technol. Today 1998, 1, 153–159. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Pan, W.-S. 4–Expert system for the development and formulation of push–pull osmotic pump tablets containing poorly water-soluble drugs. In Formulation Tools for Pharmaceutical Development; Aguilar, J.E., Ed.; Woodhead Publishing: Cambridge, UK, 2013; pp. 73–108. [Google Scholar] [CrossRef]
- Gubaev, K.; Podryabinkin, E.V.; Hart, G.L.W.; Shapeev, A.V. Accelerating high-throughput searches for new alloys with active learning of interatomic potentials. Comput. Mater. Sci. 2019, 156, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Hase, F.; Roch, L.M.; Kreisbeck, C.; Aspuru-Guzik, A. Phoenics: A Bayesian optimizer for chemistry. ACS Cent. Sci. 2018, 4, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Ulissi, Z.W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 2018, 1, 696–703. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Chakraborty, T. ANN in Pharmaceutical Product and Process Development. In Artificial Neural Network for Drug Design, Delivery and Disposition; Academic Press: Cambridge, MA, USA, 2016; pp. 277–293. [Google Scholar] [CrossRef]
- Han, R.; Yang, Y.; Li, X.; Ouyang, D. Predicting oral disintegrating tablet formulations by neural network techniques. Asian J. Pharm. Sci. 2018, 13, 336–342. [Google Scholar] [CrossRef]
- Han, R.; Xiong, H.; Ye, Z.; Yang, Y.; Huang, T.; Jing, Q.; Lu, J.; Pan, H.; Ren, F.; Ouyang, D. Predicting physical stability of solid dispersions by machine learning techniques. J. Control. Release 2019, 311–312, 16–25. [Google Scholar] [CrossRef]
- Katoch, S.; Chauhan, S.S.; Kumar, V. A review on genetic algorithm: Past, present, and future. Multimed. Tools Appl. 2020, 2020, 1–36. [Google Scholar] [CrossRef]
- Boateng, E.Y.; Abaye, D.A. A review of the logistic regression model with emphasis on medical research. J. Data Anal. Inf. Process. 2019, 7, 190–207. [Google Scholar] [CrossRef] [Green Version]
- Kotsiantis, S.B. Decision trees: A recent overview. Artif. Intell. Rev. 2013, 39, 261–283. [Google Scholar] [CrossRef]
- Fawagreh, K.; Gaber, M.M.; Elyan, E. Random forests: From early developments to recent advancements. Syst. Sci. Control. Eng. 2014, 2, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z. Introduction to machine learning: K-nearest neighbors. Ann. Transl. Med. 2016, 4, 218. [Google Scholar] [CrossRef] [Green Version]
- Rajiv, S.; Sourish, D.; Sujit, K.S. A Bayesian perspective of statistical machine learning for big data. Comput. Stat. 2020, 35, 893–930. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobot. 2013, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Barmpalexis, P.; Kachrimanis, K.; Georgarakis, E. Solid dispersions in the development of a nimodipine floating tablet formulation and optimization by artificial neural networks and genetic programming. Eur. J. Pharm. Biopharm. 2011, 77, 122–131. [Google Scholar] [CrossRef]
- Moore, M.D.; Wildfong, P.L. Informatics calibration of a molecular descriptors database to predict solid dispersion potential of small molecule organic solids. Int. J. Pharm. 2011, 418, 217–226. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Koutsidis, I.; Karavas, E.; Louka, D.; Papadimitriou, S.A.; Bikiaris, D.N. Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur. J. Pharm. Biopharm. 2013, 85, 1219–1231. [Google Scholar] [CrossRef]
- Medarević, D.P.; Kleinebudde, P.; Djuris, J.; Djuric, Z.; Ibric, S. Combined application of mixture experimental design and artificial neural networks in the solid dispersion development. Drug Dev. Ind. Pharm. 2016, 42, 389–402. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergstrom, C.A. Computational predictions of glass-forming ability and crystallization tendency of drug molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Nurzyńska, K.; Booth, J.; Roberts, C.J.; McCabe, J.; Dryden, I.; Fischer, P.M. Long-Term amorphous drug stability predictions using easily calculated, predicted, and measured parameters. Mol. Pharm. 2015, 12, 3389–3398. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Jeliński, T.; Cysewski, P. Application of multivariate adaptive regression splines (MARSplines) for predicting hansen solubility parameters based on 1D and 2D molecular descriptors computed from SMILES string. J. Chem. 2019, 2019, 9858371. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Yang, W. Molecular dynamics simulations with quantum mechanics/molecular mechanics and adaptive neural networks. J. Chem. Theory Comput. 2018, 14, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
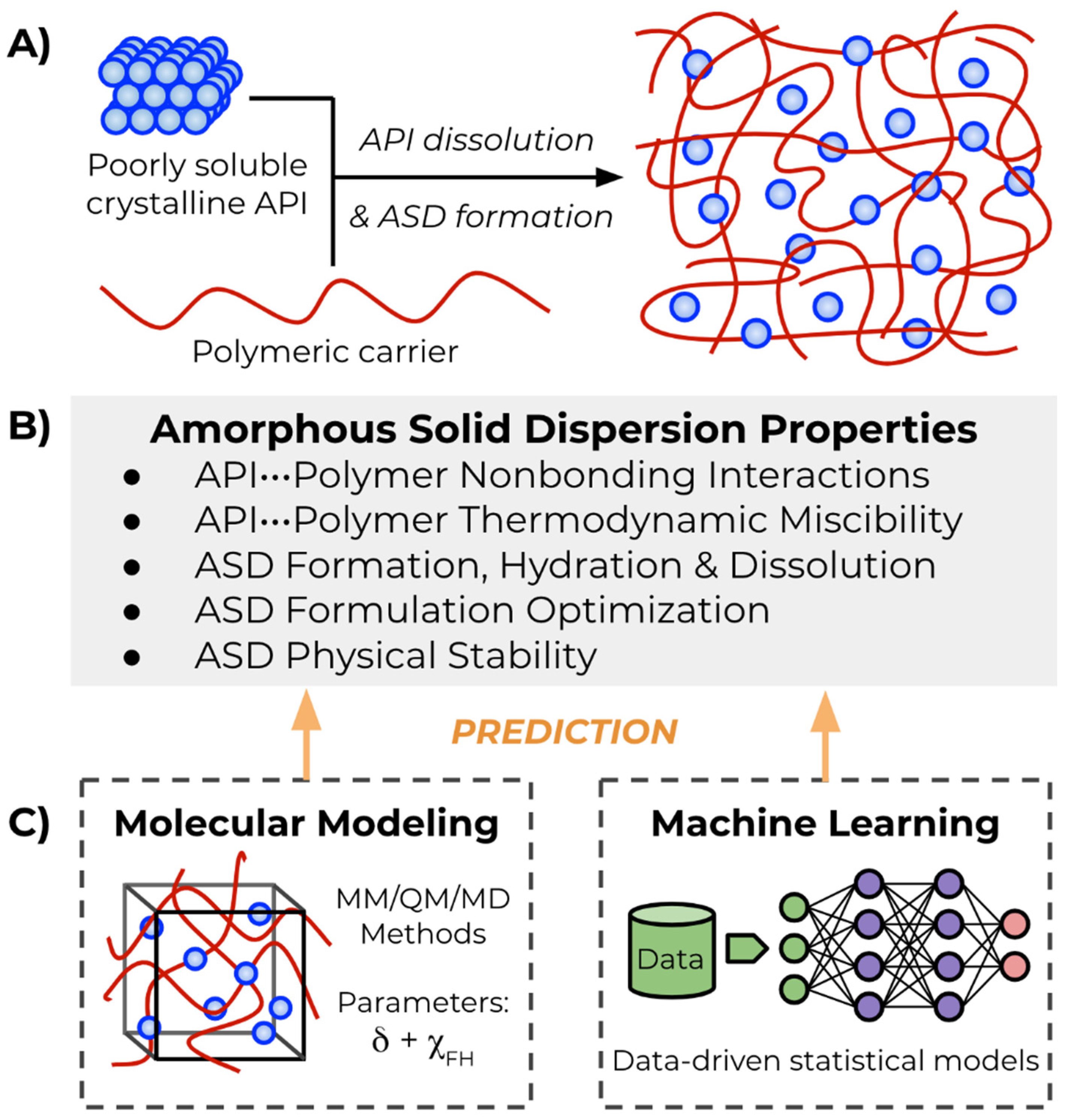

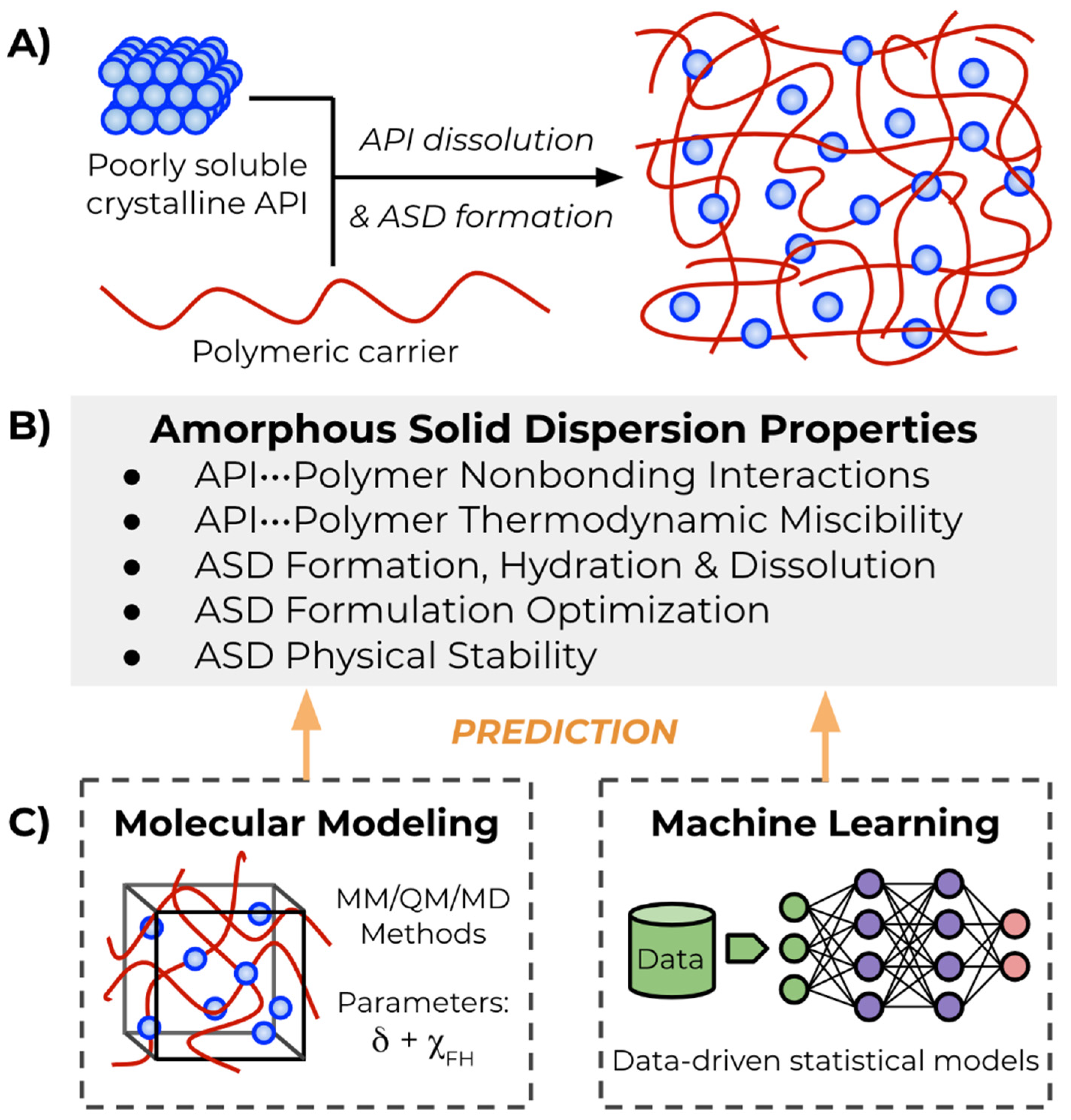{kind=link}
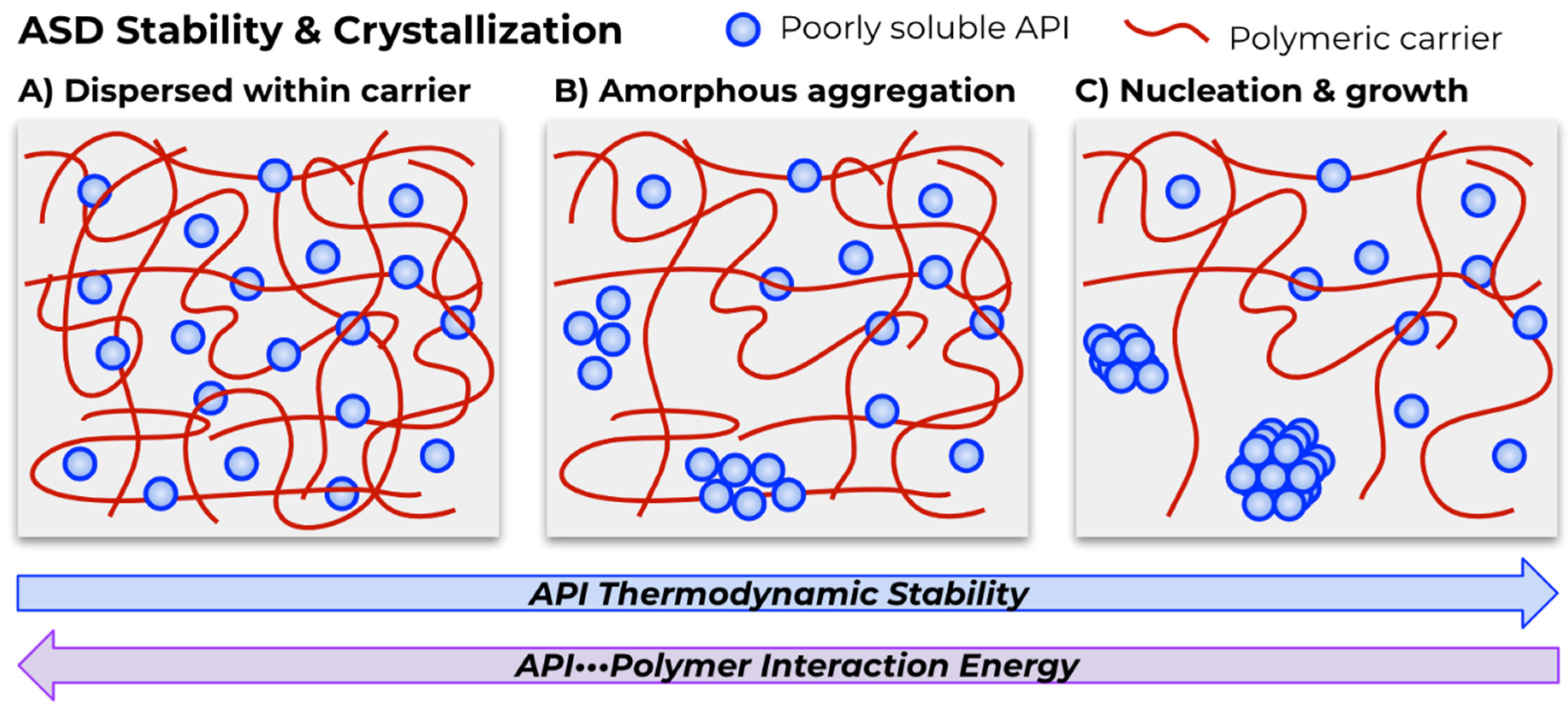
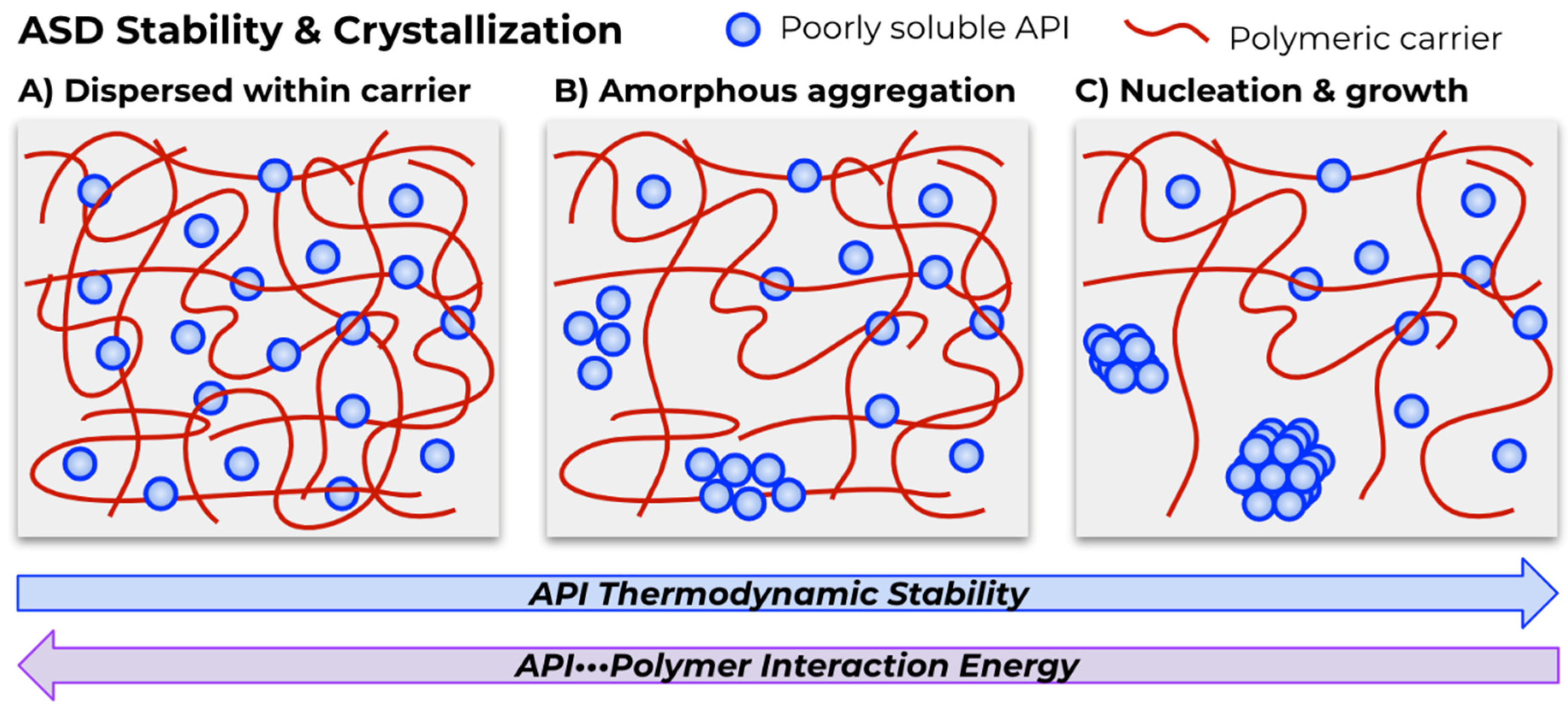{kind=link}
| Product | API | Carrier | Dosage Form |
|---|---|---|---|
| Afeditab | Nifedipine | Poloxamer/PVP | Tablet |
| Afinitor | Everolimus | HPMC | Tablet |
| Certican | Everolimus | HPMC | Tablet |
| Cesamet | Nabilone | PVP | Tablet |
| Crestor | Rosuvastatin | HPMC | Tablet |
| Cymbalta | Duloxetine | HPMC-AS | Capsule |
| Fenoglide | Fenofibrate | PEG | Tablet |
| Florfenicol | Florfenicol | Cellulose acetate phthalate | Powder |
| Gris-PEG | Griseofulvin | PEG | Tablet |
| Incivek | Teleprevir | HPMC-AS | Tablet |
| Incivo | Etravirine | HPMC | Tablet |
| Intelence | Etravirin | HPMC | Tablet |
| Isoptin | Verapamil | HPC/HPMC | Tablet |
| Kaletra | Lopinavir | PVP | Capsule |
| Kalydeco | Ivacaftor | HPMC-AS | Tablet |
| Nimotop | Nimodipine | PEG | Capsule |
| Nivadil | Nivaldipine | HPMC | Tablet |
| Novir | Ritonavir | PVP | Tablet |
| Onmel | Itraconazole | HPMC | Tablet |
| Prograf | Tacrolimus | HPMC | Capsule |
| Rezulin | Troglitazone | HPMC | Tablet |
| Shuilinjia | Silibinin | Lecithin | Capsule |
| Sporanox | Itraconazole | HPMC | Capsule |
| Stivarga | Regorafenib | HPMC | Tablet |
| Votubia | Everolimus | HPMC | Tablet |
| Zelboraf | Vemurafenib | Hypromellose acetate succinate | Tablet |
| Zortess | Everolimus | HPMC | Tablet |
| Software | Applicability | License | Reference(s) |
|---|---|---|---|
| Gaussian [45] | MM and QM computations | Commercial | [46,47,48] |
| AutoDock Vina [49] | MM conformational sampling docking | Apache License | [50] |
| XenoView [51] | MM and MD simulations | Non-commercial | [50,52] |
| HyperChem [53] | MM, QM, and MD simulations | Commercial | [54] |
| Materials Studio (BIOVIA) [55] | MM, QM, and MD | Commercial | [56,57,58,59,60,61,62,63] |
| Amber [64,65] | MM and MD simulations | Proprietary 1 | [66,67,68,69] |
| GROMACS [70,71] | MM and MD simulations | LGPL | [54,59,72] |
| LAMMPS [73] | MM and MD simulations | GPL | [74] |
| NAMD [75] | MM and MD simulations | Proprietary, free for noncommercial use | [50,76] |
| Maestro (Schrödinger) [77] | Molecular modeling | Commercial | [78,79] |
| Desmond (Schrödinger Materials Science Suite) [80] | MM and MD simulations | Commercial | [81] |
| APIs | Polymer Carriers | Miscibility Parameter(s) Investigated | Force Field | Brief Simulation Overview 1 | Experimental Miscibility Comparison | Reference |
|---|---|---|---|---|---|---|
| Indomethacin | PEO, glucose, sucrose | δ2 | COMPASS | 2 ns NVT 3/NPT 4 equilibration, 200–500 ps NVT production (298 K, 1 fs/step) | PEO (miscible), glucose (immiscible), sucrose (borderline) predictions in agreement with thermal analysis experiments. | [57] |
| Artemisinin | PVP/PEG | COMPASS | 500 ps NPT equilibration, 200 ps production (298 K, 1 fs/step) | Predicted PVP and PEG miscibility in agreement with observed drug dispersion from thermal analysis. | [62] | |
| Gemcitabine | Chitosan | COMPASS | 200 ps NPT equilibration, 800 ps production (298 K, 1 fs/step) | N/A | [61] | |
| Telaprevir | Cellulose derivatives | CHARMM | 50 ps NVE (0.5 fs/step), 5 ns NVT/NPT (310 K, 1 fs/step) equilibration, 40 ns NPT production (310 K, 1 fs/step) | N/A | [54] | |
| Clonazepam, ibuprofen, fenofibrate, alprazolam | PVP–VA 64, HPMC, and Eudragit EPO | COMPASS | 2 ns NPT equilibration, 500 ps NVT production (298 K, 1 fs/step) | Predicted fenofibrate/PVP-VA 64 weaker intermolecular interactions in agreement with observed recrystallization during stability experiments. | [63] | |
| Ibuprofen, carbamazepine | SOL/PEG | CHARMM | 2 ns NPT relaxation (393 K cooled to 298 K for ibuprofen, 474 K cooled to 298 K for carbamazepine, 10 K/step), 100 ps NPT equilibration/300 ps production at each temperature. | Both ibuprofen and carbamazepine predicted as miscible with SOL/PEG in agreement with observed single Tg values from DSC experiments. | [50] | |
| 6-Mercaptopurine | PLA, PEG-modified PLA | PCFF | 2 ns NPT dynamics (298 K, 1 fs/step) | N/A | [58] | |
| Olmesartan medoxomil | PVP–VA 64, SOL | OPLS | 5 ns NPT dynamics (300 K, 1 fs/step) | Predicted high miscibility with PVP–VA 64 carrier reflected in crystallography and thermal analysis experiments. | [81] | |
| Cyclosporin A | l/d–polylactide, chitosan, polyglycolic acid, PEG, cellulose | χ5 | PCFF | 1.5 ns NPT dynamics (298 K) | N/A | [60] |
| Indomethacin | PEG, PLA | COMPASS | 5 ps NVT equilibration at each temperature step (298 K heated to 500 K in three steps, then cooled back to 298 K in three steps). 30 ns equilibration at the last step. 1 ns NPT production (298 K, 1 fs/step) | Predicted significant miscibility (negative interaction parameters) for indomethacin with both PEG and PLA as carriers in agreement with encapsulation efficiency experiments. | [56] | |
| Felodipine | HPMC | , | Amber/GLYCAM | 10 ns equilibration (500–700 K), then cooled to 200 K (0.03 K/ps). 30–100 ns production (298 K, 1 fs/step). | Predicted miscibility from solubility and interaction parameters at all HPMC concentrations in agreement with observed single Tg values from DSC. | [69] |
| Indomethacin | PVP | , | Amber | 10 ns equilibration (600 K), then cooled to 200 K (0.03 K/ps). 100 ns production runs (298 K, 1 fs/step). | N/A | [68] |
| Tacrine | Chitosan, PBCA | , | PCFF | 100 ps equilibration (300 K), 5 ns NPT (298 K, 1 fs/step) | N/A | [74] |
| Simvastatin | PVP | , | PCFF | 5 ns NPT relaxation (600 K cooled to 200 K, 10 K/step, 1 fs/step). 400 ps NPT runs at each temperature. (1 fs/step) | Predicted miscibility from MD–based interaction parameter calculation in close agreement with measured value derived from melting point depression experiments. | [52] |
| Aspirin, caffeine, carbamazepine, finasteride, flufenamic acid, flutamide, mefenamic acid, salicylamide, theophylline | PVP–VA 64, poly (glycerol adipate) and derivatives | , | CHARMM | Iterate cell volume prior to arriving at target density. At each step of the cycle, minimization, then 200 ps NVT dynamics (700 K, 1 fs/step). Cell then underwent annealing from 750 K to 300 K (0.1 K/ps). Minimization then 10 ns NPT dynamics (300 K). | Predicted solubility and interaction parameters showed no correlation to measured miscibility limits. Opposite to experimental values, MD-derived FH interaction parameters for six of nine API–PGA polymers predicted complete miscibility. | [110] |
| Year | Target Features | Input Features | Algorithms | Dataset | Model Evaluations | Reference |
|---|---|---|---|---|---|---|
| 2011 | API percent release after 60 min, time for 90% dissolved, tablet floating strength and time | Ratios of API/polymers/effervescent agents | ANN, GA | 25 mixture proportions | Root mean squared error of prediction across each parameter ranges from 0.0184 to 0.0782 as evaluated on external validation set. | [138] |
| 2011 | Solid dispersion potential, P(Y) (miscibility) | Molecular and topological indices from atom connectivity and three-dimensional coordinates | LR | 12 compounds co-solidified with polymer carrier | Best univariate regression model: logit P(Y) = −1.927 + 0.208T(O···Cl) giving deviance of 6.513, likelihood ratio of 10.86, and leave-one-out cross-validation error of 0.3841. | [139] |
| 2013 | Percent API dissolved after 30 min | Polymer molecular weight, temperature of melt mixing, total mixing time, percent API in the ASD | ANN | 36 combinations of input features | R2 = 0.9896, p-value < 0.001, lack of fit p-value = 0.456, coefficient of variation = 3.53 | [140] |
| 2015 | API percent release after 10 and 20 min | Ratios within ternary system of API and two polymers | ANN | 25 mixture proportions | R2 = 0.978 (observed versus predicted) | [141] |
| 2019 | ASD physical stability after 3 and 6 months | Molecular weight, melting point, XlogP3, hydrogen bond donors and acceptors, rotatable bonds, polar surface area, heavy atoms, complexity, intrinsic solubility | ANN, DNN SVM, RF, DT, LGBM, kNN, NB | 50 compounds with 646 ASD physical stability data | Best model (RF) gave test set prediction accuracy of 82.5% (3 and 6 months). NB was the least accurate model (46.67%), with all other models ranging from 70.83% to 80.83% for test set accuracy (3 and 6 months) | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walden, D.M.; Bundey, Y.; Jagarapu, A.; Antontsev, V.; Chakravarty, K.; Varshney, J. Molecular Simulation and Statistical Learning Methods toward Predicting Drug–Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design. Molecules 2021, 26, 182. https://doi.org/10.3390/molecules26010182
Walden DM, Bundey Y, Jagarapu A, Antontsev V, Chakravarty K, Varshney J. Molecular Simulation and Statistical Learning Methods toward Predicting Drug–Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design. Molecules. 2021; 26(1):182. https://doi.org/10.3390/molecules26010182
Chicago/Turabian StyleWalden, Daniel M., Yogesh Bundey, Aditya Jagarapu, Victor Antontsev, Kaushik Chakravarty, and Jyotika Varshney. 2021. "Molecular Simulation and Statistical Learning Methods toward Predicting Drug–Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design" Molecules 26, no. 1: 182. https://doi.org/10.3390/molecules26010182