Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disease that is characterized by a deficit in social interactions and communication, as well as repetitive and restrictive behaviors. Increasing lines of evidence suggest an important role for immune dysregulation and/or inflammation in the development of ASD. Recently, a relationship between inflammation, oxidative stress, and mitochondrial dysfunction has been reported in the brain tissue of individuals with ASD. Some recent studies have also reported oxidative stress and mitochondrial abnormalities in animal models of maternal immune activation (MIA). This review is focused on the hypothesis that MIA induces microglial activation, oxidative stress, and mitochondrial dysfunction, a deleterious trio in the brain that can lead to neuroinflammation and neurodevelopmental pathologies in offspring. Infection during pregnancy activates the mother’s immune system to release proinflammatory cytokines, such as IL-6, TNF-α, and others. Furthermore, these cytokines can directly cross the placenta and enter the fetal circulation, or activate resident immune cells, resulting in an increased production of proinflammatory cytokines, including IL-6. Proinflammatory cytokines that cross the blood–brain barrier (BBB) may initiate a neuroinflammation cascade, starting with the activation of the microglia. Inflammatory processes induce oxidative stress and mitochondrial dysfunction that, in turn, may exacerbate oxidative stress in a self-perpetuating vicious cycle that can lead to downstream abnormalities in brain development and behavior.
1. Introduction
Autism spectrum disorder (ASD) is defined as a neurodevelopmental illness, the diagnosis of which is based on two fundamental components: deficiencies in social communication, and restricted repetitive behaviors. Deficits in social communication and social interaction include the lack of social–emotional reciprocity (e.g., problems with normal conversations and difficulty in initiating and responding to social interactions), trouble with nonverbal communicative behaviors (e.g., eye contact, body language, and gestures), and deficits in establishing and sustaining relationships (e.g., problems with imaginative play and making friends, as well as a lack of interest in other children). Restricted repetitive patterns of behavior or interests can manifest as stereotypical movements or speech (e.g., lining up toys and repetitive phrases), inflexibility of behavior (e.g., extreme difficulty accepting change or transition and specific rituals associated with regular activities), fixated interests (e.g., strong attachment to objects, sometimes unusual objects, and restricted interests), and atypical responses to sensory stimuli (e.g., hyperreactivity or hyporeactivity, such as intense sensitivity to sound or touch or indifference to pain and temperature) []. The first symptoms start to manifest in early childhood, causing impairment in many areas of societal functioning. What is important, however, is that individuals on the spectrum can exhibit different types and severities of symptoms.
The Autism and Developmental Disabilities Monitoring (ADDM) Network, which analyzes the prevalence of ASD in the U.S. among children aged eight years old, reported that about 1 in 54 children had been identified with ASD in the U.S. in 2016. Importantly, a dramatic increase in the number of reported cases has been observed in the last 16 years, from 67 out of 10,000 (0.67%) in 2000, to 185 out of 10,000 (1.85%) in 2016 [,]. Such an increase may be the result of increased public awareness and changes in the diagnostic standards, but an actual increase in risk factors is also a possibility. ASD is diagnosed in all racial, ethnic, and socioeconomic groups, but it has a stark difference in the rate of diagnoses when it comes to sex: ASD is four times more common in males than in females []. However, some research indicates that this number might not be accurate. Current theories suggest the existence of a female phenotype of ASD, which has a different presentation from that of the male phenotype, making it harder for females to receive a proper diagnosis []. The most interesting trait in the female phenotype of ASD is “camouflaging”. The camouflage hypothesis states that females with ASD are much better than males with ASD at imitating behavior that is considered socially acceptable []. Even though females with ASD seem to be functioning well in society as a result of the coping mechanisms they develop, it has been reported that camouflaging comes with the cost of exhaustion, stress, and anxiety. Females and males with ASD deserve a proper diagnosis and support without regard to their sex.
Regardless of the many theories on the etiology of ASD, the exact cause remains elusive. Currently, ASD is believed to result from the interaction between genetic and environmental factors []. Twin studies have shown a high concordance among monozygotic (MZ) twins that is much lower in dizygotic (DZ) twins, demonstrating that ASD has a strong genetic link []. It appears that de novo and inherited genetic variants are causal in 10–30% of patients with ASD. Some gene mutations that increase the risk of ASD have been known for a while, such as mutations in the TSC1/TSC2 (tuberous sclerosis complex) or FMR1 (fragile X mental retardation 1) genes, and new ones are being discovered (e.g., CHD8, chromodomain helicase DNA-binding protein 8; DYRK1A, dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1A; and SCN2A, sodium voltage-gated channel alpha subunit 2) []. Till date, thousands of genes, copy number variants (CNV), and de novo mutations (DNMs) have been associated with ASD, but no risk loci that are common in all ASD cases have been found. As a result, in addition to genetic research, a great deal of research has been dedicated to potential environmental factors, including chemicals, such as valproic acid (VPA), a well-known drug used in the treatment of epilepsy and migraines, the use of which during pregnancy can severely impact a child’s neurodevelopment []. Additional factors, such as exposure to air pollution and paternal age at conception, may increase the likelihood of ASD in offspring [,]. Considering the growing trend in the Western world of having children later in life, this research is vital in our understanding of the risk factors for ASD. Maternal diabetes may also be a risk factor for ASD. It has been shown that the risk of ASD in offspring was elevated in mothers with type 1 or type 2 diabetes and gestational diabetes mellitus, as compared to healthy mothers []. Finally, the role of maternal immune activation (MIA) during pregnancy in ASD development has also shown potential as a risk factor []. In addition, epidemiological studies have shown that there is an association between maternal infection during pregnancy and central nervous system (CNS) diseases with neurodevelopmental origins in offspring. Exposure to different pathogens, such as the rubella virus, Cytomegalovirus, or Toxoplasma gondii, have also been linked to a higher risk of neurodevelopmental and neuropsychiatric disorders in children, including ASD, schizophrenia, bipolar disorder, major depressive disorder, epilepsy, and cerebral palsy [,,,]. A connection between maternal infection and a child’s ASD diagnosis was first proposed after the U.S. rubella epidemic of the 1960s. Children prenatally (especially in the first trimester) exposed to rubella have often been born with so-called congenital rubella syndrome (CRS), and as much as 8–13% of these children were later diagnosed with ASD [,]. Interestingly, the activation of the maternal immune system, rather than the infection itself, seems to be related to the increased risk of ASD in offspring []. A meta-analysis by Jiang et al. indicated that maternal infection during pregnancy, whether it was bacterial, viral, or otherwise, was associated with a 12% increase in the risk of ASD in offspring []. In the same meta-analysis, the risk increased significantly if the infections occurred in the first or second trimester, but not for infections occurring in the third trimester. Moreover, fever, a common symptom of inflammation, has been associated with an increased ASD risk in offspring, especially if the fever occurred in the second trimester, which suggests that the timing of the MIA is relevant []. An analysis of data obtained from the Danish Medical Birth Register revealed that the admission of pregnant people to the hospital due to viral infections in the first trimester, or bacterial infections in the second trimester, was associated with a later diagnosis of ASD in the offspring [,]. Epidemiological observations are further supported by research on MIA in animal models, including rodents and nonhuman primates. The most popular way of inducing prenatal inflammation is with the administration of either double-stranded RNA polyinosinic–polycytidylic acid (poly (I:C), to mimic viral infection), or lipopolysaccharide (LPS, to mimic bacterial infection) []. ASD symptoms can also be induced by prenatal VPA injection [] or IL-6 administration []. Our recent results, with rodent models, on the maternal infection risk factor induced by LPS have revealed that the offspring display features of autism, as well as immune-related disruptions in the brain and periphery [,].
In this review, we focus on the possible links between MIA, inflammation, and the changes in the brain of the offspring that develop into ASD symptoms. We will analyze both animal and human studies completed in the last few years to summarize the existing knowledge about the role of MIA in the pathogenesis of ASD and the prospective for potential therapies.
2. Proinflammatory Cytokines as the Link between Maternal Inflammation and Autism Development in Offspring
Pregnancy is considered an immunologically unique state, characterized by the delicate balance between the toleration of a semi-allogeneic fetus, and the protection of both mother and offspring against pathogens. The maternal immune system is highly engaged in the successful progression of pregnancy from implantation to delivery []. The most important immune cells found in the maternal–fetal interface are uterine natural killer (uNK) cells, macrophages, T cells, B cells, and dendritic cells (DC) [,]. For pregnancy to advance, cytokines need to be secreted in large amounts by both the mother and the developing fetus. After fertilization, implantation of the newly forming embryo takes place: the blastocyst attaches to the endometrial epithelial cells and then invades the endometrial stroma. This is followed by the decidualization of the endometrium and, later, the proliferation of the trophoblast, which is responsible for establishing the blood supply for the embryo []. The combination of these processes triggers an inflammatory response that is controlled by cytokines, including leukemia inhibitory factor (LIF), IL-6, and IL-1β []. LIF belongs to the IL-6 family and, during implantation, it is secreted by the endometrial cells and orchestrates trophoblast cell adhesion, decidualization and, later, placental development [,]. The upregulation of IL-6 and IL-1β during implantation is an evolutionarily conserved mechanism []. During implantation, IL-6 is produced by the endometrial epithelium and stromal cells, as well as by the embryo []. Later, this cytokine takes part in placental formation, early gestation and, finally, partition []. The highest expression of maternal IL-1β is observed in the first trimester of pregnancy []. Both IL-1β and interleukin-1 receptor antagonists (IL-1Ra) play important roles in the implantation and development of the placenta. In vitro studies demonstrated that trophoblasts treated with IL-1β release human chorionic gonadotropin (hCG), a key player in early pregnancy development []. IL-1β was also recognized as a mediator of contractions during labor [,]. In addition, both IL-6 and IL-1β are involved in the development of the CNS, modulating neuronal and glial cell growth and survival [,].
As was mentioned before, successful pregnancy would not be possible without the tolerance of the maternal immune system for the developing fetus. Such tolerance is achieved because of a specific property of the extravillous trophoblast (EVT) cells. EVT cells do not express the classical MHC class I antigens, which in humans are known as human leukocyte antigens (HLA), except HLA-C, and instead express nonclassical MHC class I antigens: HLA-E, HLA-F, and HLA-G []. EVT cells are in direct contact with maternal uNK cells and macrophages, promoting maternal–fetal immune tolerance via interactions between HLA ligands and NK receptors [].
The exact molecular pathways that lead from MIA to ASD, and other neurodevelopmental disorders, are not fully understood. However, a fair amount of research indicates that cytokines and chemokines may play an important role in the process, especially since both are crucial for a successful pregnancy []. It appears that viral or bacterial maternal infections can alter the aforementioned state of balance and trigger acute immune activation and the transient upregulation of proinflammatory cytokines that may affect the development of the fetus. Therefore, it is suggested that the MIA-induced alterations of the maternal and fetal cytokine profiles play a key role in ASD development []. In this review, we will focus on IL-6 and IL-1β, as they are essential in physiological pregnancy and involved in ASD development [], as well as on IL-17, which has been considered especially influential in MIA-induced autism [] but is also necessary for the maintenance of a successful pregnancy [].
2.1. Interleukin-6
IL-6 has been excessively studied in the context of MIA, as it appears to be a key mediator of the effects of MIA []. Although this cytokine is upregulated in utero during normal implantation [], an excess of IL-6 can alter fetal development [,]. An elevated level of IL-6 was observed in the frontal cortex and cerebrospinal fluid (CSF) [], as well as in the cerebellum [], of autistic children. Moreover, research on animal models has shown an increase of IL-6 levels in the fetal brain shortly after the induction of inflammation in pregnant mothers [,,].
IL-6 activation in the placenta during MIA has been associated with impaired behavior in the offspring []. In an MIA rodent model, the elevated levels of IL-6 in the placenta were of maternal origin []. IL-6 can access the prenatal environment during MIA by more than one path. First, IL-6 may indirectly influence fetal development through a JAK/STAT3 signaling mechanism []. When the maternal immune system is activated, causing the elevation of the IL-6 levels in maternal blood and, therefore, in the placenta, IL-6 activates resident immune cells in the decidua, resulting in IL-6 production in situ. Thereafter, IL-6 binds to its receptor complex at the cell surface, which contains either one membrane-associated subunit (IL-6R), or a soluble form of IL-6R (sIL-6R) and two gp130 subunits [,], and activates the JAK/STAT3 pathway. The activation of JAK leads to the recruitment and the phosphorylation of STAT3 and its subsequent dimerization. Later, this newly created complex translocates to the nucleus and regulates gene transcription, along with other genes responsible for the proinflammatory response [,]. Hsiao and Patterson demonstrated that maternal IL-6 activates JAK/STAT3 in a fetal part of the placenta, specifically in its spongiotrophoblast layer. As a result, they observed an increased expression of acute-phase genes, including SOCS3 []. Moreover, the authors demonstrated that IL-6 caused the downregulation of placental growth hormone (GH) production and signaling, which, in turn, led to the reduced expression of insulin-growth factor 1 (IGF-1) []. Both of these hormones are crucial for proper fetal development [], and IGF-1 has been associated with different neuropsychiatric diseases, including ASD []. Wu et al. demonstrated that the activation of IL-6 in the placenta is required in order for MIA-induced abnormalities in the fetal brain to occur. Using knockout mice lacking IL-6Ra specifically in the placental trophoblasts, they demonstrated that the lack of IL-6 signaling in the trophoblasts prevented MIA-induced inflammatory responses, both in the placenta and the fetal brain []. Second, it has been suggested that maternal IL-6 can cross the placenta, both in humans [] and rodents []. Given the immaturity of the blood–brain barrier (BBB) at this time [], we can hypothesize that maternal IL-6 may be able to act directly on the developing fetal brain by inducing the synthesis of fetal IL-6 and other proinflammatory cytokines, possibly through the aforementioned JAK/STAT3 pathway. Studies on transgenic mice with chronic elevated IL-6 expression in their CNSs showed signs of astrogliosis and altered neuronal function in their brains, especially in the hippocampus and cerebellum, that was accompanied by changes in their behavior []. Moreover, elevated IL-6 levels have been associated with the alteration of excitatory and inhibitory synaptic formations and functions in the brains of animal models []. With that in mind, it may be logical to conclude that excessive amounts of IL-6 during the delicate stages of brain development may impact the functioning of the CNS. However, it is important to emphasize that there is still uncertainty concerning the origin of the IL-6 that has such a critical influence on the fetal brain during MIA, and whether it originates in the mother, the placenta, the fetal brain, or the fetal periphery.
A single injection of IL-6 in pregnant mice caused behavioral changes, including impaired social interactions in the offspring [,]. Moreover, prenatal exposure to this cytokine resulted in impaired spatial learning []. Importantly, in the MIA model induced by poly (I:C) injection, the simultaneous administration of an IL-6-neutralizing antibody, along with the poly (I:C) injection, was enough to attenuate behavioral changes [,]. These results demonstrate the importance of IL-6 in MIA-induced neurodevelopmental disorders.
2.2. Interleukin-17
IL-17A (commonly called IL-17), is a proinflammatory cytokine expressed by TH17 cells (IL-17A+CD4+ T), the differentiation of which depends on a whole set of transcription factors, including STAT3 and the retinoic acid receptor-related orphan nuclear receptor, γt (RORγt) []. Using an MIA animal model, Choi et al. demonstrated that maternal CD4+ T cells expressing RORγt are essential for the ASD-like phenotype to occur in offspring []. Both of these receptors are regulated by IL-6 and, therefore, an increase in the level of IL-6 in the placenta and/or the fetal brain leads to the upregulation of IL-17A, which is known to promote the production of a variety of cytokines, including IL-6, IL-1β, and TNF-α, as well as a granulocyte-macrophage colony-stimulating factor (GM-CSF), and a granulocyte colony-stimulating factor (G-CSF) []. Elevated levels of IL-17A were observed in the serum of ASD children [] and in the neutrophils of ASD patients [], where it was accompanied by the upregulation of phospho-nuclear factor-kappa B (p-NFκB), IL-6, and NADPH oxidase 2 (NOX2)/ROS, which suggests that this cytokine plays a substantial role in the modulation of inflammation. High levels of IL-17A have been associated with several autoimmune diseases, and they were also linked to brain pathology in disorders such as epilepsy, ischemia, and multiple sclerosis (MS) [].
A few mechanisms have been proposed for the way by which IL-17A can find the fetal brain, although the exact process has not been confirmed. MIA-induced IL-6 expression in the placenta is followed by the differentiation of maternal TH17 cells and the increased secretion of IL-17A, which appears to be capable of crossing the placental barrier []. In addition, the transmigration of activated maternal TH17 cells across the placenta [] allows the secretion of IL-17A directly into the prenatal environment (Figure 1). IL-17A may also be induced in the fetal brain by maternal IL-6 that has crossed the placental barrier. After secretion, it could influence cells that express its receptor in the developing CNS.
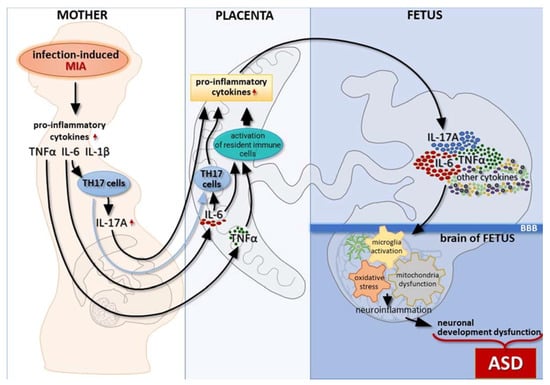
Figure 1.
Immunological changes in the placenta and fetal brain in response to systemic inflammation during pregnancy resulting in neuronal development dysfunction in offspring. Infection during pregnancy activates the mother’s immune system, which releases proinflammatory cytokines, such as IL-6, Il-1β, TNF-α, and others. The elevated level of IL-6 leads to the activation of maternal TH17 cells. As a consequence, IL-17A is released and, together with IL-6 and TNF-α, may reach the placenta, where they additionally activate resident immune cells, resulting in an increased production of proinflammatory cytokines, including IL-6. Moreover, activated maternal TH17 cells also transmigrate through the placenta and enhance cytokine production, which affects placenta function and causes damage. This allows the cytokines to pass through to the developing fetus and enhance the production of fetus-derived cytokines. Then, proinflammatory cytokines cross the BBB and initiate a neuroinflammation cascade. This leads to the abnormal development of neurons, as well as alterations in synaptic transmission, which may further lead to the development of ASD in subsequent stages of growth.
The exact molecular pathways that lead from IL-17A to MIA-triggered abnormalities are not yet known, though there are a few possibilities worth consideration. After the binding of IL-17A, interleukin 17 receptor A (IL-17RA) joins with the IL-17RC subunit to form a receptor complex []. Afterward, adaptor molecule Act1, a protein critical for the mediation of IL-17A signaling, is recruited []. Act1 (sometimes referred to as CIKS) is known to be an NF-κB activator [], allowing it (in a cascade of events) to rapidly translocate into the nucleus, which is followed by inflammatory gene transcription. It is also required for the IL-17-induced expression of the inhibitor of nuclear factor-kappa B zeta-(IκB-ζ) [], a member of the NF-κB transcription factor family. IL-17 induces IκB-ζ mRNA and protein expression, which allows for the IκB-ζ-dependent activation of various IL-17 target genes in cooperation with NF-κB []. NF-κB plays a role in controlling the growth of neural processes in the developing peripheral nervous system (PNS) and the CNS [] and, furthermore, increased expression and enriched NF-κB signaling has been found in some studies performed on the peripheral blood samples and postmortem brains of ASD patients, as well as in studies on animal models []. That said, it seems that NF-κB pathways are a promising research area within the context of ASD etiology. IL-17 also activates the mitogen-activated protein kinase (MAPK) pathways, which include the extracellular signal-regulated kinase (ERK), p38, and JUN N-terminal kinase (JNK) pathways [,]. The ERK/MAPK signaling is known to play a critical role in brain development, as well as in learning, memory, and cognition []. Therefore, it may be possible that MIA, through excessive IL-17A expression in the fetal brain, might impact ASD symptoms through the dysregulation of this pathway. Importantly, mutations in ERK-pathway genes have been observed in patients with ASD [], and the dysregulation of ERK signaling has been observed in animal models and the lymphocytes of ASD patients [].
Choi et al. proved that maternal IL-17 promoted behavioral abnormalities in MIA offspring when the pretreatment of MIA mothers with IL-17A-neutralizing antibodies prevented the behavioral abnormalities. Moreover, atypical cortical development, which has been observed in MIA offspring, was averted by the administration of anti-IL-17A to the mothers. Finally, the authors showed that IL-17A administration directly into the fetal brain promoted atypical cortical development and ASD-like behavioral abnormalities. These same symptoms did not occur when IL-17A was injected into the brains of knockout mouse fetuses lacking IL-17RA [].
2.3. Interleukin-1β
Interleukin-1β (IL-1β) is a proinflammatory cytokine expressed by various types of cells, including macrophages, DCs, monocytes, and microglia []. It is first produced in an inactive form as pro-IL-1β, which maturates when processed by caspase-1 [], and after secretion, it initiates local and systemic inflammation. Increased levels of this cytokine have been found in the blood of patients with ASD []. Our group also observed elevated levels of IL-1β in the blood of rats prenatally exposed to MIA [], whereas others detected excessive amounts of this cytokine in the fetal brain of MIA animals [,]. In addition, in monocyte cultures from ASD pediatric patients, an increased expression of IL-1β in response to the stimulation of TLR4 (LPS) was observed, in comparison to the controls []. That considered, it is possible that this cytokine takes part in the etiology of MIA-induced FASD.
It is not clear whether the IL-1β that disrupts fetal brain development after MIA is of maternal or fetal origin. IL-1β does not cross the placenta, but it is capable of crossing the BBB []. Its expression, however, is triggered not only by TLR4 signaling, but also by the presence of other cytokines, including IL-6 [], IL-17 [], and IL-1β itself []. It is possible that the secretion of fetal IL-1β is being triggered in response to inflammatory processes developing in the placenta and the aforementioned expression of cytokines. This requires further investigation. Nevertheless, IL-1β is known to affect the developing brain. Crampton et al. tested the influence of this cytokine on neural progenitor cells isolated from the developing rat ventral mesencephalon. The authors observed that IL-1β caused the inhibition of neural progenitor cell proliferation, accompanied by differentiation, where it promoted gliogenesis rather than neurogenesis. This effect was mediated by interleukin 1 receptor type I (IL-1R1) and MAPK pathway signaling []. IL-1R1 can also mediate signals via TNF, receptor-associated factor 6 (TRAF6), and the IKK complex, which results in the activation of NF-κB and is followed by inflammatory gene transcription []. Chudnovets et al. demonstrated changes in the placenta and fetal brain of mice treated with intraperitoneal injections of different doses of IL-1β from embryonic days (E) 14–17. The authors observed increased IL-1β in the placentas, as well as increased p-NF-κB and caspase-1 in the placentas and in the fetal brains of IL-1β-treated animals, but not consistently in the spleens, suggesting the induction of intrinsic IL-1β production. This was accompanied by the placental disruption–distortion of the labyrinth structure, decreased numbers of mononuclear trophoblast giant cells, and reduced proportions of endothelial cells, as compared to the control placentas. Moreover, reduced fetal viability and a reduction in the cortical neuronal morphology in fetal brains were observed []. That placental and fetal brain disturbance, resulting from the IL-1β influence, is confirmed by the fact that the coadministration of the IL-1 receptor antagonist (IL-1Ra), together with LPS, to pregnant females, was demonstrated to have protective effects, as compared to the LPS injection alone []. Taken together, proinflammatory cytokines increase in the placenta, and then in the fetal brain, in response to systemic inflammation during pregnancy, and may affect neuronal development dysfunction in offspring. Proinflammatory cytokines that cross the BBB might initiate a neuroinflammation cascade as a result of microglia activation (Figure 1).
3. Microglial Abnormalities in MIA-Induced Autism Spectrum Disorders
Microglia are the tissue-resident macrophage-like immune cells of the CNS that play a significant role in brain development, maturation, and maintenance, both in embryonic and adult organisms. Interestingly, microglia from the prenatal brain differ from those in the adult brain, not only morphologically, but also in the expression signatures. Moreover, prenatal microglia appear to proliferate more than adult microglia []. During embryonic development, the microglia differentiate from the immune progenitors in the fetal yolk sac and are present in the developing CNS around embryonic day 9.5 (E9.5) []. In the CNS, they take part in directing the migration of neural precursor cells and influence their differentiation into mature neurons []. During later development, microglia interact with the neurons and the astrocytes involved in the synaptic pruning process that permits the elimination of nonfunctional, or weak, synaptic connections []. In addition, it has been suggested that the microglia support the oligodendrocyte precursor cells involved in the myelination processes and influence neovascularization in the developing retina []. Later in life, microglia are responsible for maintaining homeostasis in the CNS. In physiological conditions, microglia appear to remain in a resting state, but this calmness is misleading, as microglial processes are constantly expanding and contracting, scanning their surroundings, and engaging in multiple interactions with other cells []. The situation dramatically changes upon infection or injury. Microglia are rapidly activated to secrete effector molecules, including proinflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, IL-23, and IL-18) and ROS, causing neuroinflammation []. On the other hand, microglia are capable of secreting growth factors and anti-inflammatory cytokines that promote tissue repair []. Microglia-induced inflammation and degeneration, as well as the general dysfunction of microglia, are important factors in many CNS disorders, including ASD.
Evidence suggests that microglia are activated in the brains of ASD individuals []. Research conducted on postmortem brain samples revealed an increased microglial activation in the cerebellum and the white matter of ASD patients, as compared to the controls [], as well as in the cortical regions [,], where it is accompanied by a higher microglial density [] and increased microglial–neuronal spatial clustering [], indicating the neuronal recruitment of the microglia. The activation of microglia in the brain can lead to molecular changes that need to be studied, and since it is not possible to conduct microglia research on living patients, animal models are a good alternative. Unfortunately, however, the findings obtained using MIA animal models are not conclusive. While some researchers report increased microglia density [,], changes in microglia morphology [,,], or changes in microglia process motility [], others report findings that are inconclusive [,,,]. The differences in their results might be caused by the diverse protocols for inducing MIA, the variation of time points and brain regions chosen for analysis, and divergent approaches to the definition of microglial activation []. Nevertheless, a few possible pathways leading from the MIA, through the microglia, and finally to neurological alterations, have been proposed. Schaafsma et al. demonstrated that LPS-induced MIA results in long-term changes in microglia responsiveness into adulthood, and that fetal microglia produced proinflammatory cytokines that contribute to neuro-inflammation, and that might ultimately affect brain development []. Ozaki et al. reported that MIA caused sustained alterations in the patterns of microglial process motility, and that such motility changes in the offspring were observed as early as gestational day 18 (E18) and were sustained well into adolescence []. These results emphasize that changes in microglial functions may affect neuronal function and plasticity, as well as susceptibility to developmental disorders in the offspring. A growing body of evidence indicates that proper microglial–neuronal interactions are fundamental for brain development and homeostasis, and that MIA may be one of the factors affecting these interactions. The crucial regulatory systems for microglial–neuronal crosstalk are the CX3CL1-CX3CR1 and the CD200-CD200R, with CX3CL1 (also known as fractalkine) localized on the neurons and the only ligand to the CX3CR1 receptor that can be found on microglial cells []. The interaction between the two is involved in the maturation and plasticity of synapses, as well as in the regulation of the functioning of the neuronal network and the immune processes in the CNS []. In vitro studies on microglial cultures activated with LPS have demonstrated that treatment with CX3CL1 reduced the production of inflammatory mediators, such as NO, IL-6, TNFα, and IL-1β [], and it maintained the microglia in a quiescent state []. CD200 is a surface glycoprotein expressed on multiple cell types, including neurons [], whereas CD200R is expressed by cells of myeloid lineage in the CNS, which are predominantly microglia []. The binding of CD200 by CD200R induces a signaling cascade that blocks the proinflammatory response of the myeloid cells. Any disturbance in the CD200–CD200R signaling results in microglial activation and can lead to neuroinflammation []. Recent studies have shown that MIA provoked by LPS or poly (I:C) leads to microglial–neuronal communication alterations in young male rat offspring through the modulation of CX3CL1–CX3CR1 and/or CD200–CD200R signaling []. Altered CX3CL1-CX3CR1 or CD200-CD200R pathways, provoked by MIA, may be involved in the disturbance of bidirectional communication between the neurons and the microglia, as well as in the etiology of neurodevelopmental disorders, including autism. In conclusion, the MIA-provoked alterations of the microglial processes in the brains of offspring may contribute to the underlying pathophysiological mechanisms linking maternal immune activation to subsequent risks for ASD.
4. Oxidative Stress in ASD Individuals and MIA Models
The process of inflammation is often accompanied by oxidative stress, which can be defined as an imbalance between antioxidants and reactive oxygen species (ROS) levels that can result in cellular and molecular damage []. ROS, including superoxide radicals (O2•−), hydrogen peroxide (H2O2), hydroxyl radicals (•OH), and singlet oxygen (1O2), are byproducts mainly of mitochondrial metabolism during oxidative phosphorylation []. On one hand, moderate concentrations of ROS are beneficial in physiological conditions, serving as signaling molecules in redox signaling [], and taking part in the host defense against pathogens in an oxidative burst []. On the other hand, ROS tend to act as a double-edged sword: in pathology, ROS increase oxidative stress attacks on the double bonds of lipids, proteins, and DNA bases, leading to cellular damage. The brain, with its immense demand for energy and high lipid content, is especially vulnerable to oxidative stress [].
A growing body of evidence links ASD to increased levels of oxidative stress and lower antioxidant capacity [,]. Markers of oxidative stress have also been observed in the postmortem brain samples of ASD patients, along with a decreased antioxidant capacity (GSH/GSSG ratio) and an increase in 3-nitrotyrosine (3-NT, marker of oxidative protein damage), 3-chlorotyrosine (3-CT, marker of inflammation), and 8-oxo-deoxyguanosine (8-oxo-dG, marker of oxidative DNA damage) []. In addition, a lower total antioxidative status, and a higher oxidative stress index, including decreased GSH concentration and increased catalase (CAT) activity, as well as higher levels of malondialdehyde (MDA) and protein carbonyl content, have been observed in the blood of ASD children [,,]. Different animal studies found a redox imbalance after prenatal exposure to MIA. After prenatal exposure to LPS, an increase in the GSSG level, and a decrease in the GSH level and the GSH/GSSG ratio, followed by an increase in the level of lipid peroxide, were observed in the brains of MIA rats [,]. In an LPS-triggered MIA model, increased oxidative stress and the expression of metalloproteinase in the amniotic fluid and fetal brain were indicated [].
Inflammation and oxidative stress are closely related. The two create what can be characterized as a self-perpetuating vicious cycle. Chronic inflammation is connected to oxidative stress and neurodegeneration, whereas oxidative stress is known to induce inflammation []. The presence of proinflammatory cytokines and damage-associated molecular patterns (DAMPs) induces phagocytic cells (such as macrophages in the PNS, and microglia in the CNS) to perform an oxidative burst. The deliberate production of ROS in those cells is stimulated by NOX2 activity []. As was mentioned earlier, ROS serve as signaling molecules and impact a variety of molecular pathways, including NF-κB and MAPKs [], which leads to the upregulation of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α [].
5. Mitochondrial Dysfunction in MIA-Evoked Autism Spectrum Disorders
High levels of oxidative stress can be connected with mitochondrial dysfunction. However, these organelles play a major role in ROS production in cells, mostly via the enzymes of the electron transport chain (ETC), especially complex I (NADH dehydrogenase) and complex III (cyt bc1), but also via several matrix proteins and complexes, including enzymes of the tricarboxylic acid (TCA) cycle (e.g., aconitase, pyruvate dehydrogenase, and α-ketoglutarate dehydrogenase), as well as the inner and outer mitochondrial membrane proteins, such as cytochrome P450 enzymes and monoamine oxidase (MAO) []. A high concentration of steady-state O2− (~5–10× higher than in the nucleus or the cytosol) makes mitochondria a target for ROS-induced damage [], which can lead to an exacerbation of ROS production, resulting in the aforementioned vicious cycle []. The mitochondria in the CNS are damaged not only by the overproduction of ROS locally, but also by the ROS produced by activated microglia elsewhere []. The reciprocal relationship between mitochondria and the immune response has been well documented, as mitochondria can impact the immune response, and vice versa []. Deficits in bioenergetics and mitochondrial dysfunction have been reported in ASD patients. About 4% of patients with ASD are diagnosed with classic mitochondrial disease (MD). However, the latest research examining the biomarkers of mitochondrial dysfunction suggest that abnormalities of mitochondrial function could affect a much higher percentage of children with ASD, perhaps up to 80% []. Observed mitochondrial disturbances include alterations in the levels of respiratory chain proteins, such as a reduced expression of the genes of complex I, complex III, complex IV, and complex V in a brain-region-specific manner in ASD brains [], as well as decreases in the protein levels of the subunits of these complexes [,]. Alterations of protein levels are reflected in the reduced activity of the ETC complexes, mostly complexes I and V, but also complex III in the frontal cortex of individuals with ASD []. Considering that complexes I and III are responsible for a major part of ROS production, and that complex V is directly involved in the conversion of ADP into ATP, dysfunction of the said complexes can both reduce energy production and increase ROS levels, both of which contribute to oxidative stress. These pathological changes are accompanied by alterations in the genes involved with mitochondrial transport, membrane potential, and dynamics (fusion and fission) [,].
Research performed on the lymphoblastoid cell lines (LCLs) derived from children with ASD confirmed mitochondrial dysfunction, characterized by increased respiration, and accompanied by higher sensitivity to oxidative stress. About one-third of ASD-LCLs showed elevated respiratory rates while, at the same time, being more sensitive to ROS exposure []. The same research group, comparing mitochondrial function in the LCLs of ASD patients, their unaffected siblings, and unrelated healthy controls demonstrated that ASD LCLs exhibited significantly higher respiratory rates, glycolysis, and glycolytic reserves, as compared to siblings, as well as greater sensitivity to ROS, when compared to both siblings and the controls, which suggests that the overactivity of mitochondria, and the sensitivity to ROS, can be linked directly with ASD []. Pecorelli et al. analyzed dermal fibroblasts collected from ASD patients and discovered a higher level of metabolic activity (e.g., oxygen-consuming ratio), accompanied by higher proton-leak values, in ASD fibroblasts, as compared to the control cells. The ASD cells exhibited increased basal and maximal glycolytic rates, and an increased expression of the subunits of all the mitochondrial complexes. Finally, the mitochondria in ASD fibroblasts had an atypical morphology, which was significantly different from the mitochondria in the control cells []. Abnormalities in respiratory function were also indicated in the peripheral blood mononuclear cells (PBMCs) that were derived from the blood samples of ASD children, and demonstrated that ASD patients diagnosed with developmental regression (DR) had a much higher maximal oxygen consumption rate (OCR), as compared to the controls [], which suggests a direct link between the presence of mitochondrial dysfunction and the severity of the observed ASD symptoms. Although the relationships between mitochondrial dysfunction, oxidative stress, and neuroinflammation have been well documented, the etiology of mitochondrial dysfunction in MIA-induced autism remains unclear. There also has not been much research regarding mitochondrial dysfunction in MIA animal models. Giulivi et al. showed that MIA activation by poly (I:C) at early gestation is associated with mitochondrial changes in the immune cells of adult offspring. They evaluated mitochondrial function in splenocytes derived from the spleens of 12-week-old mice prenatally exposed to poly (I:C), which showed decreased oxygen consumption and deficits in complex I in the MIA animals, as compared to the controls []. This suggests that prenatal immune changes following the maternal poly (I:C) administration are likely to imprint long-lasting changes in the bioenergetics of the adult offspring. Our investigations indicated that mitochondrial alterations in the MIA-affected brains of the offspring, which included changes in the ultrastructure of the synaptic mitochondria, were accompanied by the impairment of ETC gene expression and its functioning. We also observed decreased mitochondrial membrane potential, as well as increased generation of ROS [,].
Mitochondrial dysfunction and mitochondrial ROS (mtROS) production may trigger the activation of proinflammatory signaling pathways and, in particular, induce the activation of NF-κB, HIF, and AP-1, which contribute to the production of proinflammatory cytokines, including IL-1β and IL-6 []. However, mitochondria are susceptible to the influence of proinflammatory cytokines and chronic inflammation [] as well as elevated ROS production []. IL-1β and TNF-α disrupt the functioning of mitochondria in human chondrocytes, resulting in mitochondrial DNA (mtDNA) damage, decreased energy production, and enhanced ROS production []. Moreover, these cytokines have been connected with ETC inhibition, an increase in the permeability of the mitochondrial membrane, the suppression of pyruvate dehydrogenase activity, and the inhibition of oxidative phosphorylation []. In addition, mitochondrial dysfunction can result in the release of DAMPs that may be involved in the activation of proinflammatory processes via multiple mechanisms, and the propagation of mitochondria-dependent inflammation, a process called “mitoflammation”. However, many questions surrounding the molecular functions of mtROS and mitoflammation in MIA-dependent autism development remain.
6. Potential Treatment Strategies Targeting Inflammatory Pathways in ASD
Understanding the role of inflammation in the pathophysiology of ASD may provide new insights into the therapeutic strategies for this disorder. The maternal infection risk factor is currently being studied in animal models. The experiments involve the infection of pregnant mice or rats by infecting the mother, or by simply activating its immune system, in the absence of pathogens. The most popular studies have been those that involve the maternal injection of poly (I:C) to provoke an antiviral inflammatory response, or the maternal injection of LPS to provoke an antibacterial inflammatory response. Although these approaches activate different molecular mechanisms, the analyses of the offspring have thus far revealed considerable overlap in behavioral abnormalities. In addition, similar results have been obtained in both the mouse and rat models of MIA. The findings summarized in Table 1 demonstrate that immune-related therapies can prevent the development of abnormal behaviors in the offspring in the poly (I:C) and LPS models.

Table 1.
The summary of the studies on potential therapeutic strategies for the MIA treatment.
A growing body of literature has also examined the anti-inflammatory therapies for autism and related neurodevelopmental disorders. In Table 2, a review of the studies concerning medications, both with primary anti-inflammatory actions and those with additional anti-inflammatory properties besides their primary mechanisms of action, has been presented (in alphabetical order).
Table 2.
Summary of the studies on the role of anti-inflammatory medications in ASD treatment.
Amantadine is an antiviral medication that is widely used in the management of central nervous system disorders. Its anti-inflammatory effect involves the inhibition of the release of proinflammatory factors [,,]. A double-blind placebo-controlled study of amantadine hydrochloride in the treatment of children with ASD indicated a significant improvement in the absolute scores of hyperactivity and inappropriate speech, according to the clinician-rated ABC (Aberrant Behavior Checklist-Community version). Amantadine also improved the CGI score (Clinical Global Impressions), as compared to the placebo []. In another double-blind placebo-controlled trial on patients with severe behavioral issues, such as disruptive symptoms related to ASD, amantadine was added to risperidone for treatment. The data showed a significant improvement in hyperactivity and irritability measured by the ABC. In addition, the CGI-I scores showed significant improvement in the amantadine group, as compared to the placebo []. An NMDA receptor antagonist drug with anti-inflammatory and neuroprotective properties is memantine []. In a randomized controlled trial comparing treatment with risperidone with memantine, and risperidone with a placebo, in children with ASD, significant improvements were reported in irritability, stereotypic behaviors, and hyperactivity, as measured by the ABC []. Similarly, Joshi et al., in an open-label study, evaluated the efficacy and tolerability of memantine, which also resulted in a significant improvement in autism severity. The 12-week study on adult patients with ASD found that memantine treatment was associated with improvement in the CGI, SRS (Social Responsiveness Scale), and brief psychiatric rating scale scores, as well as in anxiety and nonverbal communication (which were measured using the Diagnostic Analysis of Nonverbal Accuracy Scale). Moreover, the applied treatment was not associated with any serious adverse effects [].
As a nonsteroidal anti-inflammatory selective inhibitor of cyclooxygenase-2 (COX-2), celecoxib has been widely used as an adjuvant therapy in several psychiatric disorders. In a randomized double-blind controlled trial on children with ASD, treatment with celecoxib as an adjuvant therapy presented significant improvements in irritability, social withdrawal/lethargy, and stereotypic behaviors []. Similar results were obtained in the studies using corticosteroids, a class of steroid hormones that exert anti-inflammatory properties []. The effects of corticosteroids, or adrenocorticotrophic hormones (ACTH), in regressive ASD, were investigated in two different case studies on a 6-year-old patient and an 18-month-old patient. The research showed that low-dose therapy with corticosteroids significantly improved spontaneous speech, responsiveness to verbal communications, social relatability, receptiveness, and expressive vocabulary [,]. The role of ORG 2766, an ACTH analog, in the improvement of ASD behaviors was analyzed by Buitelaar and colleagues in a series of analyses. In a double-blind crossover study, the authors showed that the four-week ORG 2766 administration significantly improved clinical symptoms (e.g., irritability, stereotypic behaviors, hyperactivity, and excessive speech) as measured by the parent-reported ABC []. The positive effects of ORG 2766 were also reported in a second crossover trial when administrated for eight weeks. The authors reported significant improvements in stereotypic behaviors and social interactions (e.g., in play behaviors). Furthermore, the adverse effects were minimal []. Corticosteroid therapy was also used in a case study on a 6-year-old boy with regressive ASD, over 28 months. Stefanatos et al. described a child whose language and behavior had regressed at 22 months of age, and in whom a pervasive developmental disorder was later diagnosed. The application of corticosteroid therapy resulted in significant improvements in language, social abilities, and stereotypic behaviors []. Corticosteroid therapy was also evaluated by Shenoy and colleagues in a case study of an 18-month-old child with regressive ASD who also developed autoimmune lymphoproliferative syndrome (ALPS). This research showed that, after a month of corticosteroid therapy, the patient started regaining his language and communication abilities. After 26 months of therapy, all the laboratory values returned to normal []. According to this data, corticosteroids may have potential as a treatment for ASD-like behavior; however, rigorously controlled trials are still needed [].
In the search for an anti-inflammatory therapy for ASD, the possible protective effect of lenalidomide, the immunomodulatory drug derivative of thalidomide and widely used in the treatment of many hematologic disorders, has been considered []. Lenalidomide can alter TNF-α with less toxicity than thalidomide. In the open-label pilot study, lenalidomide was administered to seven patients for 12 weeks. The patients showed significant improvements in ASD symptoms, which was measured by the Childhood Autism Rating Scale (CARS), as well as in expressive language, which was measured by the CGI. However, among the seven children enrolled, two developed a rash and had to withdraw from the study []. This study suggests that, if tolerated, lenalidomide may promote improvement in behavioral symptoms.
Recent data have demonstrated that flavonoids, the polyphenolic compounds widely found in fruits and vegetables, can inhibit regulatory enzymes or transcription factors important in controlling proinflammatory mediators []. Luteolin and quercetin are plant-derived flavonoids that show a broad range of effects, including antioxidant, anti-inflammatory, anticancer, and neuroprotective properties []. Luteolin is capable of inhibiting proinflammatory cytokine expression, NF-kB signaling, and TLR4 signaling, as well as weakening microglial activation []. Therefore, in a series of trials, the flavonoid properties were analyzed for ASD treatment. In an open-label pilot study, Tsilioni et al. analyzed the effects of luteolin and quercetin treatments on 50 children for 26 weeks. A total of 40 children completed the protocol, with significant improvements in adaptive functioning, which was measured using the Vineland Adaptive Behavior Scale (VABS), and in overall behavior, as indicated by the reduction (26.6–34.8%) according to the ABC subscale scores []. In an open-label case series of 37 children with ASD, Theoharides et al. evaluated the lutein formulation, NeuroProtek, a mixture of luteolin, quercetin, and rutin that is known for its antioxidant, anti-inflammatory, and neuroprotective properties, as a potential treatment for ASD. The results showed improvements in eye contact and attention to directions (in 50% of the patients), as well as in the retention of learned tasks and social interactions (in 30% and 50% of the patients, respectively), and speaking skills (in 10% of the patients) []. Additional blood samples from children who received NeuroProtek for four months showed a significant decrease in the mean serum IL-6 and TNF levels at the end of the treatment period. These effects were strongly associated with children whose behavior improved after NeuroProtek treatment.
Regarding the effectiveness of anti-inflammatory interventions in ASD, attention has been focused on a group of antibiotics in the tetracycline class, including minocycline, with its well-known anti-inflammatory properties. Pardo et al. investigated the effect of minocycline for six months. The open-label study with 10 patients showed protective changes in the profiles of brain-derived neurotrophic factor (BDNF) in CSF and blood, hepatic growth factor (HGF) in CSF, and CXCL8 (IL-8) in serum. However, minocycline did not have a significant effect on core autism symptoms. It was suggested that the group of children may have been too small to observe clinical improvement []. Ghaleiha and et al. analyzed minocycline as an adjunctive therapy to risperidone. In the 10-week randomized placebo-controlled study, minocycline showed a significant improvement in the irritability and hyperactivity/noncompliance scores measured by the ABC. Unfortunately, the treatment did not have any significant effect on lethargy/social withdrawal, stereotypic behavior, and inappropriate speech []. Thus far, there has been insufficient evidence to support the use of minocycline in the treatment of the core autism symptoms [].
The presented studies show that anti-inflammatory therapy for ASD has potential. However, the limited data suggest it may only be effective for a certain subset of individuals with ASD. Large-scale randomized controlled trials are needed to provide robust evidence for its efficacy as a therapeutic treatment.
7. Conclusions
The evidence reviewed in this paper strongly suggests a causal relationship between MIA in early gestation and ASD development in offspring (Figure 1). The injection of a single inflammatory cytokine (e.g., IL-6 or IL-17) is sufficient to induce several ASD-like behaviors in offspring, and MIA may induce alterations in multiple cytokines in the fetal brain within a matter of hours. Prenatal immune challenges and proinflammatory cytokines crossing the fetus’ BBB promote the microglial cells to their proinflammatory phenotype. Inflammatory processes in the brain induce oxidative stress and mitochondrial dysfunction that, in turn, may exacerbate oxidative stress in a self-perpetuating vicious cycle, leading to downstream abnormalities in brain development. Thus, MIA-induced immune dysregulation/inflammation, oxidative stress, and mitochondrial dysfunction in the brain of offspring, as well as the linkage between these abnormalities, could be the primary molecular pathways downstream from maternal infection. However, other molecular mechanisms should be taken into consideration. One possibility is that proinflammatory cytokines lead to long-lasting changes in the expression of other classes of immune molecules, including major histocompatibility complex I (MHCI), and molecules that are known to regulate synapse formation, synaptic plasticity, and synaptic pruning, as well as neural connectivity and function in the brains of offspring. It is also possible that immune signaling may converge upon the mammalian target of rapamycin (mTOR) signaling, which has been shown to be altered in the brains of MIA offspring, as well as in individuals with ASD. In summary, while the molecular mechanisms of MIA as an ASD primer requires further research, there is growing evidence that microglial activation, oxidative stress, and mitochondrial dysfunction in the brain of offspring could mediate the neuropathology and behaviors associated with ASD.
Author Contributions
Conceptualization, M.C. and A.A.; writing—original draft preparation, A.Z.; writing—review and editing M.C. and A.A.; supervision A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre (http://www.ncn.gov.pl), Grant 2016/23/D/NZ4/03572, and by POWER Och!Dok.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Edition, F. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association: Washington, DC, USA, 2013; Volume 21. [Google Scholar]
- Rice, C.E.; Baio, J.; Van Naarden Braun, K.; Doernberg, N.; Meaney, F.J.; Kirby, R.S. A public health collaboration for the surveillance of autism spectrum disorders. Paediatr. Perinat. Epidemiol. 2007, 21, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Young, H.; Oreve, M.J.; Speranza, M. Clinical characteristics and problems diagnosing autism spectrum disorder in girls. Arch. Pédiatr. 2018, 25, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Allely, C.S. Understanding and recognising the female phenotype of autism spectrum disorder and the “camouflage” hypothesis: A systematic PRISMA review. Adv. Autism 2019, 5, 14–37. [Google Scholar] [CrossRef]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry Allied Discip. 2016, 57, 585–595. [Google Scholar] [CrossRef]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef]
- Gentile, S. Risks of neurobehavioral teratogenicity associated with prenatal exposure to valproate monotherapy: A systematic review with regulatory repercussions. CNS Spectr. 2014, 19, 305–315. [Google Scholar] [CrossRef]
- Lam, J.; Sutton, P.; Kalkbrenner, A.; Windham, G.; Halladay, A.; Koustas, E.; Lawler, C.; Davidson, L.; Daniels, N.; Newschaffer, C.; et al. A Systematic Review and Meta-Analysis of Multiple Airborne Pollutants and Autism Spectrum Disorder. PLoS ONE 2016, 11, e0161851. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Advanced parental age and autism risk in children: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2017, 135, 29–41. [Google Scholar] [CrossRef]
- Xiang, A.H.; Wang, X.; Martinez, M.P.; Page, K.; Buchanan, T.A.; Feldman, R.K. Maternal Type 1 Diabetes and Risk of Autism in Offspring. JAMA 2018, 320, 89–91. [Google Scholar] [CrossRef]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Fujimoto, C.; Nakajima, E.; Isagai, T.; Matsuishi, T. Possible association between congenital cytomegalovirus infection and autistic disorder. J. Autism Dev. Disord. 2003, 33, 455–459. [Google Scholar] [CrossRef]
- Severance, E.G.; Xiao, J.; Jones-Brando, L.; Sabunciyan, S.; Li, Y.; Pletnikov, M.; Prandovszky, E.; Yolken, R. Toxoplasma gondii-A Gastrointestinal Pathogen Associated with Human Brain Diseases. Int. Rev. Neurobiol. 2016, 131, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Mawson, A.R.; Croft, A.M. Rubella Virus Infection, the Congenital Rubella Syndrome, and the Link to Autism. Int. J. Environ. Res. Public Health 2019, 16, 3543. [Google Scholar] [CrossRef]
- Boulanger-Bertolus, J.; Pancaro, C.; Mashour, G.A. Increasing Role of Maternal Immune Activation in Neurodevelopmental Disorders. Front. Behav. Neurosci. 2018, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Xu, L.L.; Shao, L.; Xia, R.M.; Yu, Z.H.; Ling, Z.X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hornig, M.; Bresnahan, M.A.; Che, X.; Schultz, A.F.; Ukaigwe, J.E.; Eddy, M.L.; Hirtz, D.; Gunnes, N.; Lie, K.K.; Magnus, P.; et al. Prenatal fever and autism risk. Mol. Psychiatry 2018, 23, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Atladottir, H.O.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef]
- Arsenault, D.; St-Amour, I.; Cisbani, G.; Rousseau, L.S.; Cicchetti, F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav. Immun. 2014, 38, 77–90. [Google Scholar] [CrossRef]
- Gąssowska-Dobrowolska, M.; Cieślik, M.; Czapski, G.A.; Jęśko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef] [PubMed]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Gąssowska-Dobrowolska, M.; Jęśko, H.; Czapski, G.A.; Wilkaniec, A.; Zawadzka, A.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Boguszewski, P.M.; et al. Maternal Immune Activation Induces Neuroinflammation and Cortical Synaptic Deficits in the Adolescent Rat Offspring. Int. J. Mol. Sci. 2020, 21, 4097. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, M.; Gassowska-Dobrowolska, M.; Zawadzka, A.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Czapski, G.A.; Adamczyk, A. The Synaptic Dysregulation in Adolescent Rats Exposed to Maternal Immune Activation. Front. Mol. Neurosci. 2020, 13, 555290. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Defining pregnancy phases with cytokine shift. J. Pregnancy Reprod. 2017, 1, 1–3. [Google Scholar] [CrossRef]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 2014, 72, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zheng, Q.; Jin, L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front. Immunol. 2019, 10, 2317. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Bernal, M.A.; Fazleabas, A.T. Physiologic Events of Embryo Implantation and Decidualization in Human and Non-Human Primates. Int. J. Mol. Sci. 2020, 21, 1973. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Wu, L.; Hu, W. The regulation of leukemia inhibitory factor. Cancer Cell Microenviron. 2015, 2. [Google Scholar] [CrossRef]
- Yockey, L.J.; Iwasaki, A. Interferons and Proinflammatory Cytokines in Pregnancy and Fetal Development. Immunity 2018, 49, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Prins, J.R.; Gomez-Lopez, N.; Robertson, S.A. Interleukin-6 in pregnancy and gestational disorders. J. Reprod. Immunol. 2012, 95, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Equils, O.; Kellogg, C.; McGregor, J.; Gravett, M.; Neal-Perry, G.; Gabay, C. The role of the IL-1 system in pregnancy and the use of IL-1 system markers to identify women at risk for pregnancy complications. Biol. Reprod. 2020, 103, 684–694. [Google Scholar] [CrossRef]
- Zhao, B.; Schwartz, J.P. Involvement of cytokines in normal CNS development and neurological diseases: Recent progress and perspectives. J. Neurosci. Res. 1998, 52, 7–16. [Google Scholar] [CrossRef]
- Parker-Athill, E.C.; Tan, J. Maternal immune activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neuro-Signals 2010, 18, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Hackmon, R.; Pinnaduwage, L.; Zhang, J.; Lye, S.J.; Geraghty, D.E.; Dunk, C.E. Definitive class I human leukocyte antigen expression in gestational placentation: HLA-F, HLA-E, HLA-C, and HLA-G in extravillous trophoblast invasion on placentation, pregnancy, and parturition. Am. J. Reprod. Immunol. 2017, 77. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhou, Q.; Li, L.; Wang, S. HLA-C promotes proliferation and cell cycle progression in trophoblast cells. J. Matern. Fetal Neonatal Med. 2021, 34, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Ravaccia, D.; Ghafourian, T. Critical Role of the Maternal Immune System in the Pathogenesis of Autism Spectrum Disorder. Biomedicines 2020, 8, 557. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef]
- Wong, H.; Hoeffer, C. Maternal IL-17A in autism. Exp. Neurol. 2018, 299, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, V.L.; Ellwanger, J.H.; Matte, M.C.C.; Savaris, R.F.; Vianna, P.; Chies, J.A.B. IL-17 blood levels increase in healthy pregnancy but not in spontaneous abortion. Mol. Biol. Rep. 2018, 45, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.M.; Cowan, M.; Moonah, S.N.; Petri, W.A., Jr. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol. Med. 2018, 24, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Chavan, A.R.; Protopapas, S.; Maziarz, J.; Romero, R.; Wagner, G.P. Embryo implantation evolved from an ancestral inflammatory attachment reaction. Proc. Natl. Acad. Sci. USA 2017, 114, E6566–E6575. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Jennische, E.; Hansson, H.A.; Holmäng, A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1345–R1356. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Wei, H.; Zou, H.; Sheikh, A.M.; Malik, M.; Dobkin, C.; Brown, W.T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Vuillermot, S.; Luan, W.; Meyer, U.; Eyles, D. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol. Autism 2017, 8, 9. [Google Scholar] [CrossRef]
- Simões, L.R.; Sangiogo, G.; Tashiro, M.H.; Generoso, J.S.; Faller, C.J.; Dominguini, D.; Mastella, G.A.; Scaini, G.; Giridharan, V.V.; Michels, M.; et al. Maternal immune activation induced by lipopolysaccharide triggers immune response in pregnant mother and fetus, and induces behavioral impairment in adult rats. J. Psychiatr. Res. 2018, 100, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Zhang, H.; Yu, J.; Yao, Z. Oral probiotic administration during pregnancy prevents autism-related behaviors in offspring induced by maternal immune activation via anti-inflammation in mice. Autism Res. 2019, 12, 576–588. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, X.; Zhong, Y. Inflammatory cytokines: Potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediat. Inflamm. 2015, 2015, 531518. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.B.; Mendes, A.P.D.; Marchetto, M.C. IGF-1 treatment causes unique transcriptional response in neurons from individuals with idiopathic autism. Mol. Autism 2020, 11, 55. [Google Scholar] [CrossRef]
- Wu, W.L.; Hsiao, E.Y.; Yan, Z.; Mazmanian, S.K.; Patterson, P.H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 2017, 62, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, J.; Samuelsson, A.M.; Jansson, T.; Holmäng, A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatric Res. 2006, 60, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Conroy, S.M.; Nguyen, V.; Quina, L.A.; Blakely-Gonzales, P.; Ur, C.; Netzeband, J.G.; Prieto, A.L.; Gruol, D.L. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J. Neuroimmunol. 2004, 155, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Alberts, I.; Li, X. Brain IL-6 and autism. Neuroscience 2013, 252, 320–325. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Al-Ayadhi, L.Y.; Mostafa, G.A. Elevated serum levels of interleukin-17A in children with autism. J. Neuroinflamm. 2012, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Attia, S.M.; Al-Ayadhi, L.Y.; Bakheet, S.A.; Al-Harbi, N.O. Oxidative and inflammatory mediators are upregulated in neutrophils of autistic children: Role of IL-17A receptor signaling. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 90, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wienecke, J.; Hebel, K.; Hegel, K.J.; Pierau, M.; Brune, T.; Reinhold, D.; Pethe, A.; Brunner-Weinzierl, M.C. Pro-inflammatory effector Th cells transmigrate through anti-inflammatory environments into the murine fetus. Placenta 2012, 33, 39–46. [Google Scholar] [CrossRef]
- Toy, D.; Kugler, D.; Wolfson, M.; Vanden Bos, T.; Gurgel, J.; Derry, J.; Tocker, J.; Peschon, J. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 2006, 177, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zepp, J.; Li, X. Function of Act1 in IL-17 family signaling and autoimmunity. Adv. Exp. Med. Biol. 2012, 946, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Commane, M.; Nie, H.; Hua, X.; Chatterjee-Kishore, M.; Wald, D.; Haag, M.; Stark, G.R. Act1, an NF-kappa B-activating protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10489–10493. [Google Scholar] [CrossRef] [PubMed]
- Ohgakiuchi, Y.; Saino, Y.; Muromoto, R.; Komori, Y.; Sato, A.; Hirashima, K.; Kitai, Y.; Kashiwakura, J.I.; Oritani, K.; Matsuda, T. Dimethyl fumarate dampens IL-17-ACT1-TBK1 axis-mediated phosphorylation of Regnase-1 and suppresses IL-17-induced IκB-ζ expression. Biochem. Biophys. Res. Commun. 2020, 521, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, H.; Hale, V.A.; Dolcet, X.; Davies, A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 2005, 132, 1713–1726. [Google Scholar] [CrossRef]
- Liao, X.; Li, Y. Nuclear Factor Kappa B in Autism Spectrum Disorder: A Systematic Review. Pharmacol. Res. 2020, 159, 104918. [Google Scholar] [CrossRef]
- Samuels, I.S.; Saitta, S.C.; Landreth, G.E. MAP’ing CNS development and cognition: An ERKsome process. Neuron 2009, 61, 160–167. [Google Scholar] [CrossRef]
- Kalkman, H.O. Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci. Biobehav. Rev. 2012, 36, 2206–2213. [Google Scholar] [CrossRef]
- Rosenzweig, J.M.; Lei, J.; Burd, I. Interleukin-1 receptor blockade in perinatal brain injury. Front. Pediatr. 2014, 2, 108. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. J. Psychiatr. Res. 2019, 115, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; Onore, C.E.; Van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef]
- Sadowska, G.B.; Chen, X.; Zhang, J.; Lim, Y.P.; Cummings, E.E.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Padbury, J.F.; Banks, W.A.; et al. Interleukin-1β transfer across the blood-brain barrier in the ovine fetus. J. Cereb. Blood Flow Metab. 2015, 35, 1388–1395. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Chudnovets, A.; Lei, J.; Na, Q.; Dong, J.; Narasimhan, H.; Klein, S.L.; Burd, I. Dose-dependent structural and immunological changes in the placenta and fetal brain in response to systemic inflammation during pregnancy. Am. J. Reprod. Immunol. 2020, 84, e13248. [Google Scholar] [CrossRef]
- Crampton, S.J.; Collins, L.M.; Toulouse, A.; Nolan, Y.M.; O’Keeffe, G.W. Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation—A role for maternal inflammation? J. Neurochem. 2012, 120, 964–973. [Google Scholar] [CrossRef]
- Scholz, C.C.; Cavadas, M.A.; Tambuwala, M.M.; Hams, E.; Rodríguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β-induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.; Tremblay, L.; Lepage, M.; Sébire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Aarum, J.; Sandberg, K.; Haeberlein, S.L.; Persson, M.A. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. USA 2003, 100, 15983–15988. [Google Scholar] [CrossRef]
- Cowan, M.; Petri, W.A., Jr. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef]
- Smolders, S.; Notter, T.; Smolders, S.M.T.; Rigo, J.M.; Brône, B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav. Immun. 2018, 73, 51–65. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef]
- Li, W.Y.; Chang, Y.C.; Lee, L.J.; Lee, L.J. Prenatal infection affects the neuronal architecture and cognitive function in adult mice. Dev. Neurosci. 2014, 36, 359–370. [Google Scholar] [CrossRef]
- Gumusoglu, S.B.; Fine, R.S.; Murray, S.J.; Bittle, J.L.; Stevens, H.E. The role of IL-6 in neurodevelopment after prenatal stress. Brain Behav. Immun. 2017, 65, 274–283. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflamm. 2017, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Corradini, I.; Focchi, E.; Rasile, M.; Morini, R.; Desiato, G.; Tomasoni, R.; Lizier, M.; Ghirardini, E.; Fesce, R.; Morone, D.; et al. Maternal Immune Activation Delays Excitatory-to-Inhibitory Gamma-Aminobutyric Acid Switch in Offspring. Biol. Psychiatry 2018, 83, 680–691. [Google Scholar] [CrossRef]
- Ozaki, K.; Kato, D.; Ikegami, A.; Hashimoto, A.; Sugio, S.; Guo, Z.; Shibushita, M.; Tatematsu, T.; Haruwaka, K.; Moorhouse, A.J.; et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci. Rep. 2020, 10, 21378. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [PubMed]
- Garay, P.A.; Hsiao, E.Y.; Patterson, P.H.; McAllister, A.K. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav. Immun. 2013, 31, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Giovanoli, S.; Notter, T.; Richetto, J.; Labouesse, M.A.; Vuillermot, S.; Riva, M.A.; Meyer, U. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflamm. 2015, 12, 221. [Google Scholar] [CrossRef]
- Mouihate, A. Prenatal Activation of Toll-Like Receptor-4 Dampens Adult Hippocampal Neurogenesis in An IL-6 Dependent Manner. Front. Cell. Neurosci. 2016, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.W.G.M.; Eggen, B.J.L. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef]
- Lauro, C.; Chece, G.; Monaco, L.; Antonangeli, F.; Peruzzi, G.; Rinaldo, S.; Paone, A.; Cutruzzolà, F.; Limatola, C. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front. Cell. Neurosci. 2019, 13, 414. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, C.; Liu, F. CD200-CD200R signaling and diseases: A potential therapeutic target? Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 297–309. [Google Scholar] [PubMed]
- Shrivastava, K.; Gonzalez, P.; Acarin, L. The immune inhibitory complex CD200/CD200R is developmentally regulated in the mouse brain. J. Comp. Neurol. 2012, 520, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Fonken, L.K.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Stress disinhibits microglia via down-regulation of CD200R: A mechanism of neuroinflammatory priming. Brain Behav. Immun. 2018, 69, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Chamera, K.; Kotarska, K.; Szuster-Głuszczak, M.; Trojan, E.; Skórkowska, A.; Pomierny, B.; Krzyżanowska, W.; Bryniarska, N.; Basta-Kaim, A. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: A link to schizophrenia-like behaviours. J. Neuroinflamm. 2020, 17, 247. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive oxygen species in the immune system. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, X.-J.; Liu, H.; Mao, X.; Gui, L.-N.; Wang, H.; Cheng, Y. Oxidative stress marker aberrations in children with autism spectrum disorder: A systematic review and meta-analysis of 87 studies (N = 9109). Transl. Psychiatry 2021, 11, 15. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Lanté, F.; Meunier, J.; Guiramand, J.; Maurice, T.; Cavalier, M.; de Jesus Ferreira, M.C.; Aimar, R.; Cohen-Solal, C.; Vignes, M.; Barbanel, G. Neurodevelopmental damage after prenatal infection: Role of oxidative stress in the fetal brain. Free. Radic. Biol. Med. 2007, 42, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Hooker, B.S.; Herbert, M.R. Bridging from Cells to Cognition in Autism Pathophysiology: Biological Pathways to Defective Brain Function and Plasticity. Am. J. Biochem. Biotechnol. 2008, 4, 167–176. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Matsuzaki, H.; Miyachi, T.; Tsujii, M.; Iwata, Y.; Suzuki, K.; Sugiyama, T.; Mori, N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013, 23, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; DiMauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Kaur, K.; Brown, W.T.; LaFauci, G.; Wegiel, J.; Chauhan, A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl. Psychiatry 2013, 3, e299. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Yamada, K.; Iwayama, Y.; Toyota, T.; Matsuzaki, H.; Miyachi, T.; Yamada, S.; Tsujii, M.; et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol. Autism 2012, 3, 12. [Google Scholar] [CrossRef]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Bennuri, S.C.; Wynne, R.; Melnyk, S.; James, S.J.; Frye, R.E. Mitochondrial and redox abnormalities in autism lymphoblastoid cells: A sibling control study. FASEB J. 2017, 31, 904–909. [Google Scholar] [CrossRef]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of mitochondrial bioenergetics, dynamics, and morphology support the theory of oxidative damage involvement in autism spectrum disorder. FASEB J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Singh, I.N.; Diggins, E.; Connors, S.L.; Karim, M.A.; Lee, D.; Zimmerman, A.W.; Frye, R.E. Developmental regression and mitochondrial function in children with autism. Ann. Clin. Transl. Neurol. 2020, 7, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Napoli, E.; Schwartzer, J.; Careaga, M.; Ashwood, P. Gestational exposure to a viral mimetic poly(i:C) results in long-lasting changes in mitochondrial function by leucocytes in the adult offspring. Mediat. Inflamm. 2013, 2013, 609602. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.; Grishko, V. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in human OA chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef]
- Pineda, E.; Shin, D.; You, S.J.; Auvin, S.; Sankar, R.; Mazarati, A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann. Neurol. 2013, 74, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Coiro, P.; Padmashri, R.; Suresh, A.; Spartz, E.; Pendyala, G.; Chou, S.; Jung, Y.; Meays, B.; Roy, S.; Gautam, N.; et al. Impaired synaptic development in a maternal immune activation mouse model of neurodevelopmental disorders. Brain Behav. Immun. 2015, 50, 249–258. [Google Scholar] [CrossRef]
- Ruskin, D.N.; Murphy, M.I.; Slade, S.L.; Masino, S.A. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS ONE 2017, 12, e0171643. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- King, B.H.; Wright, D.M.; Handen, B.L.; Sikich, L.; Zimmerman, A.W.; McMahon, W.; Cantwell, E.; Davanzo, P.A.; Dourish, C.T.; Dykens, E.M.; et al. Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 658–665. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Yadegari, N.; Hassanzadeh, E.; Farokhnia, M.; Yekehtaz, H.; Mirshafiee, O.; Akhondzadeh, S. Double-blind, placebo-controlled trial of risperidone plus amantadine in children with autism: A 10-week randomized study. Clin. Neuropharmacol. 2013, 36, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Asadabadi, M.; Mohammadi, M.R.; Ghanizadeh, A.; Modabbernia, A.; Ashrafi, M.; Hassanzadeh, E.; Forghani, S.; Akhondzadeh, S. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Psychopharmacology 2013, 225, 51–59. [Google Scholar] [CrossRef]
- Stefanatos, G.A.; Grover, W.; Geller, E. Case study: Corticosteroid treatment of language regression in pervasive developmental disorder. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 1107–1111. [Google Scholar] [CrossRef]
- Shenoy, S.; Arnold, S.; Chatila, T. Response to steroid therapy in autism secondary to autoimmune lymphoproliferative syndrome. J. Pediatr. 2000, 136, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, N.; Pacini, S.; Ruggiero, M. Clinical Experience of Integrative Autism treatment with a Novel type of Immunotherapy. J. Vaccines 2019, 3, 71–76. [Google Scholar] [CrossRef][Green Version]
- Chez, M.; Low, R.; Parise, C.; Donnel, T. Safety and observations in a pilot study of lenalidomide for treatment in autism. Autism Res. Treat. 2012, 2012, 291601. [Google Scholar] [CrossRef] [PubMed]
- Taliou, A.; Zintzaras, E.; Lykouras, L.; Francis, K. An open-label pilot study of a formulation containing the anti-inflammatory flavonoid luteolin and its effects on behavior in children with autism spectrum disorders. Clin. Ther. 2013, 35, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Taliou, A.; Francis, K.; Theoharides, T.C. Children with autism spectrum disorders, who improved with a luteolin-containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl. Psychiatry 2015, 5, e647. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Asadi, S.; Panagiotidou, S. A case series of a luteolin formulation (NeuroProtek®) in children with autism spectrum disorders. Int. J. Immunopathol. Pharmacol. 2012, 25, 317–323. [Google Scholar] [CrossRef]
- Ghaleiha, A.; Asadabadi, M.; Mohammadi, M.R.; Shahei, M.; Tabrizi, M.; Hajiaghaee, R.; Hassanzadeh, E.; Akhondzadeh, S. Memantine as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Int. J. Neuropsychopharmacol. 2013, 16, 783–789. [Google Scholar] [CrossRef]
- Joshi, G.; Wozniak, J.; Faraone, S.V.; Fried, R.; Chan, J.; Furtak, S.; Grimsley, E.; Conroy, K.; Kilcullen, J.R.; Woodworth, K.Y.; et al. A Prospective Open-Label Trial of Memantine Hydrochloride for the Treatment of Social Deficits in Intellectually Capable Adults with Autism Spectrum Disorder. J. Clin. Psychopharmacol. 2016, 36, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Buckley, A.; Thurm, A.; Lee, L.C.; Azhagiri, A.; Neville, D.M.; Swedo, S.E. A pilot open-label trial of minocycline in patients with autism and regressive features. J. Neurodev. Disord. 2013, 5, 9. [Google Scholar] [CrossRef]
- Ghaleiha, A.; Alikhani, R.; Kazemi, M.R.; Mohammadi, M.R.; Mohammadinejad, P.; Zeinoddini, A.; Hamedi, M.; Shahriari, M.; Keshavarzi, Z.; Akhondzadeh, S. Minocycline as Adjunctive Treatment to Risperidone in Children with Autistic Disorder: A Randomized, Double-Blind Placebo-Controlled Trial. J. Child Adolesc. Psychopharmacol. 2016, 26, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Buitelaar, J.K.; van Engeland, H.; van Ree, J.M.; de Wied, D. Behavioral effects of Org 2766, a synthetic analog of the adrenocorticotrophic hormone (4-9), in 14 outpatient autistic children. J. Autism Dev. Disord. 1990, 20, 467–478. [Google Scholar] [CrossRef]
- Buitelaar, J.K.; van Engeland, H.; de Kogel, K.H.; de Vries, H.; van Hooff, J.A.; van Ree, J.M. The use of adrenocorticotrophic hormone (4-9) analog ORG 2766 in autistic children: Effects on the organization of behavior. Biol. Psychiatry 1992, 31, 1119–1129. [Google Scholar] [CrossRef]
- Bradstreet, J.J.; Smith, S.; Granpeesheh, D.; El-Dahr, J.M.; Rossignol, D. Spironolactone might be a desirable immunologic and hormonal intervention in autism spectrum disorders. Med. Hypotheses 2007, 68, 979–987. [Google Scholar] [CrossRef]
- Hafizi, S.; Tabatabaei, D.; Lai, M.C. Review of Clinical Studies Targeting Inflammatory Pathways for Individuals with Autism. Front. Psychiatry 2019, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Ossola, B.; Schendzielorz, N.; Chen, S.H.; Bird, G.S.; Tuominen, R.K.; Männistö, P.T.; Hong, J.S. Amantadine protects dopamine neurons by a dual action: Reducing activation of microglia and inducing expression of GDNF in astroglia. Neuropharmacology 2011, 61, 574–582. [Google Scholar] [CrossRef]
- Kubera, M.; Maes, M.; Budziszewska, B.; Basta-Kaim, A.; Leśkiewicz, M.; Grygier, B.; Rogóz, Z.; Lasoń, W. Inhibitory effects of amantadine on the production of pro-inflammatory cytokines by stimulated in vitro human blood. Pharmacol. Rep. 2009, 61, 1105–1112. [Google Scholar] [CrossRef]
- Ma, H.M.; Zafonte, R.D. Amantadine and memantine: A comprehensive review for acquired brain injury. Brain Inj. 2020, 34, 299–315. [Google Scholar] [CrossRef]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Braidy, N.; Gortzi, O.; Sobarzo-Sanchez, E.; Daglia, M.; Skalicka-Woźniak, K.; Nabavi, S.M. Luteolin as an anti-inflammatory and neuroprotective agent: A brief review. Brain Res. Bull. 2015, 119, 1–11. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).